
Abstract
Five new physalins, including a novel 1,10-seco one, physalin V (1), a tricarboxylic acid cycle one, physalin VIII (5), a rare 11,15-cyclo one, physalin IX (6), and two new ones, physalins VI (2) and VII (4) were isolated from stems and leaves of Physalis angulata together with eleven known analogues (3 and 7–16). Their structures were established by MS, IR, UV, and NMR spectroscopic analysis, together with the X-ray diffraction analysis of neophysalin, physalin P (12), and the structure of physalin D1 (3) has been revised here. These isolated compounds were evaluated for their antiproliferative activities against human cancer cells (C4-2B, 22Rv1, 786-O, A-498, ACHN, and A375-S2) and inhibitory effects on nitric oxide production. Compounds 9 and 10 showed antiproliferative activities against all tested human cancer cells with IC50 values of 0.24–3.17 μM. Compounds 1, 3, 4, 9, 10, 13, 14, and 16 exhibited inhibitory activities against NO production. The IC50 values of compounds 9, 10, 13, and 16 were between 0.32 and 4.03 μM, while compounds 1, 3, 4, and 14 had IC50 values of 12.83–34.19 μM. Herein, plausible biosynthetic pathways for rare structures 1 and 6 and structure−activity relationships on the inhibition of NO production for all isolated compounds are discussed.
Similar content being viewed by others

Introduction
The withanolides are a group of natural C28 steroids with a γ- or δ-lactone based on an ergostane skeleton, which are derived from a parent 23-hydroxy-26-oic or 22-hydroxy-26-oic acid. They can be further divided into 22 subtypes based on the difference of the structural skeleton, such as normal withanolides, physalins, withaphysalins, neophysalins, jaborols, and so on1, 2. Physalins, commonly termed 16,24-cyclo-13,14-seco steroids, are classified as a group of withanolides with the most advanced oxidation level, from which they are formally derived from oxidative bond cleavage between C-13 and C-14 to produce a nine-membered ring, formation of a new six-membered carbocycle between C-16 and C-24, oxidation of the C-18 methyl group to a COOH group, which leads to 18,20-lactonization, and formation of an oxo bridge between C-14 and C-17, resulting in an oxygen heterocyclic system across rings C and D3,4,5.
The genus Physalis has attracted attention from scientists due to the occurrence of the first 16,24-cyclo-13,14-seco withanolide physalin A from Physalis alkekengi var. franchetii in 19696. Over the past 47 years, about 60 physalins have been isolated from this genus. The genus Physalis (Solanaceae) comprising approximately 120 species, is widely distributed in subtropical and tropical regions all over the world. Physalis angulata L., known as ku-zhi in China7, is a folk medicine that has been used to treat a variety of illnesses in many countries, such as dermatitis, trachitis, impaludism, rheumatism, and hepatitis. It is also used as a diuretic, antipyretic, antileukemic, anticancer, and immuno-modulatory agents8. Phytochemical investigations of P. angulata have led to the isolation of many physalins and normal withanolides, such as physalins A, B, D, E, F, G, I, and H, and physagulins A, B, C, and F, and some of them displayed remarkable anti-inflammatory9,10,11,12, antitumor13,14,15,16, antinociceptive17, and immunomodulatory18 activities. As part of our ongoing research on isolating bioactive physalins from the genus Physalis to provide potential anticancer and anti-inflammatory medicines11, 12, 15, 16, the EtOH extracts of the dried stems and leaves of P. angulata were isolated to afford a novel 1,10-seco physalin, physalin V (1), a tricarboxylic acid cycle one, physalin VIII (5), a rare 11,15-cyclo one, physalin IX (6), and two new ones, physalins VI (2) and VII (4), together with eleven known analogues (Fig. 1), and the structure of the known physalin D1 (3) was revised. In this paper, we describe the isolation and structural elucidation of these compounds together with their antiproliferative and anti-inflammatory evaluations in vitro. Furthermore, biosynthetic pathways for the rare physalins 1 and 6 are proposed, and structure–activity relationships for all isolated compounds are preliminarily discussed.

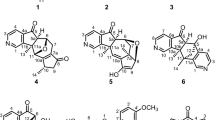
Chemical constituents of P. angulata.
Results and Discussion
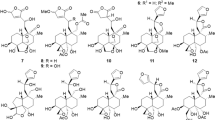
The EtOH extracts of stems and leaves of P. angulata were separated by silica gel, Sephadex LH-20, ODS open column chromatography, preparative TLC, and preparative HPLC to yield five new physalins, including an uncommon 1,10-seco one, physalin V (1), physalins VI (2), VII (4), and VIII (5), and a novel 11,15-cyclo one, physalin IX (6), together with eleven known analogues, 25β-hydroxyphysalin D (7)19, physalins D1 (3), D (8)20, B (9)21, F (10)22, G (11)23, P (12)24, H (13)25, I (14)20, and R (15)26, and isophysalin B (16)25 (Fig. 1).
Structure elucidation
Physalin V (1) was isolated as an amorphous powder, and HRESIMS spectrum (Supplementary Fig. 8) showed that the quasi-molecular peak at m/z 549.1750 [M + Na]+ (calcd for C28H30O10Na, 549.1737), requiring the molecular formula C28H30O10. The IR spectrum (Supplementary Fig. 9) of 1 showed the presence of hydroxy (3400 cm−1), carbonyl (1728 cm−1), and olefinic (1646 cm−1) functionalities. The 1H NMR spectrum (Supplementary Fig. 1) of 1 displayed signals for three methyl groups at δ H 1.83 (3 H, s, Me-19), 1.82 (3 H, s, Me-21), and 1.17 (3 H, s, Me-28), and two olefinic protons at δ H 6.49 (1 H, dd, J = 11.2, 2.0 Hz, H-4) and 5.49 (1 H, td, J = 9.3, 2.0 Hz, H-3). The 13C NMR data (Supplementary Fig. 2) showed 28 carbon resonances, including one carbonyl carbon (δ C 209.0), three hydroxycarbonyl carbons (δ C 171.80, 171.79, and 167.2), four olefinic carbons (δ C 141.0, 128.8, 125.7, and 118.3), one ketal carbon (δ C 105.2), six oxygenated carbons (δ C 80.5, 80.3, 78.8, 76.4, 71.2, and 61.2), and three methyl carbons (δ C 24.3, 21.3, and 17.1). The 1H and 13C NMR data of 1 were closely resembled those of physalin G (11) isolated from P. angulata 23, with the exception that signals of ring A [δ H 6.49 (1 H, dd, J = 11.2, 2.0 Hz, H-4), 5.49 (1 H, ddd, J = 11.2, 9.3, 2.0 Hz, H-3), and 5.25 (1 H, br s, H-6); δ C 171.8 (C-1), 141.0 (C-10), 128.8 (C-4), 125.7 (C-5), 118.3 (C-3), 71.2 (C-6), and 35.6 (C-2)] were present in 1, indicating that 1 was a 1,10-seco physalin27, 28. This conclusion was confirmed by the HMBC correlations from H-2a to C-1/C-3/C-4, H-3 to C-1/C-2/C-5, H-4 to C-2/C-5/C-6/C-10, and H-6 to C-1/C-5/C-8/C-10 (Supplementary Fig. 3–5), suggesting the presence of the seven-membered β,γ-unsaturated lactonic ring (Fig. 2). The relative configuration of 1 was established through the NOESY correlation of H-6 with H-2a (Fig. 3, Supplementary Fig. 6 and 7) in combination with the coupling constant (J 2a,3 = 4.7 Hz), requiring a-orientations of H-6 and H-2a27, 28. According to X-ray diffraction data (Cu Ka) of physalin P (12) (Fig. 4), an acid induced benzilic acid-type rearranged product of physalins29, in combination with the biogenetic grounds and previous literatures30,31,32,33,34, 1 was assigned as (6R,8R,9S,13S,14R,16S,17R,20S,22R,24S,25S)-1,10-seco physalin G.

Selected HMBC correlations of compounds 1–6.

Selected NOESY correlations of compounds 1–6.

ORTEP drawing of compound 12.
Physalin VI (2) was obtained as an amorphous powder with the molecular formula C28H32O10 based on HRESIMS m/z 527.1918 [M − H]− (calcd for C28H31O10, 527.1917; Supplementary Fig. 17) and 13C NMR data (Supplementary Fig. 11). The IR spectrum (Supplementary Fig. 18) displayed absorption bands corresponding to hydroxy (3397 cm−1), carbonyl (1716 cm−1), and olefinic (1646 cm−1) functionalities. The 1H NMR data (Supplementary Fig. 10) of 2 observed three olefinic protons at δ H 6.07 (1 H, br d, J = 11.2 Hz, H-4), 5.70 (1 H, br d, J = 4.8 Hz, H-6), and 5.67 (1 H, m, H-3), four methyl groups at δ H 1.75 (3 H, s, Me-21), 1.43 (3 H, s, Me-28), 1.36 (3 H, s, Me-27), and 1.17 (3 H, s, Me-19), and the reduced two geminal oxymethylene protons in conjunction with the 13C NMR data [δ C 209.6 (C-1), 140.4 (C-5), 128.0 (C-4), 126.6 (C-6), 122.6 (C-3), 101.0 (C-14), 22.4 (C-28), 20.7 (C-21), 19.4 (C-27), and 17.8 (C-19)], indicating the existence of a 1-oxo-3,5-diene unit and the nonexistence of a C(14)-O-C(27) cyclization moiety. The above NMR spectroscopic data of 2 were related to those of physalin M isolated from P. alkekengi L. var. franchetii 35, except for the presence of a methyl group [δ H 1.36 (3 H, s, Me-27); δ C 19.4 (C-27)] and an oxygenated carbon [δ C 72.6 (C-25)], with the assumption for the presence of a hydroxy group at C-25. The key HMBC correlations from Me-27 to C-24/C-25/C-26 and Me-28 to C-16/C-23/C-24/C-25 (Fig. 2 and Supplementary Fig. 12–14) confirmed the hypothesis. The orientation of OH-25 was deduced to be α by NOESY correlations of Me-27 with H-23a and Me-28 with H-23a (Fig. 3 and Supplementary Fig. 15 and 16). Therefore, 2 was determined as (8R,9S,10R,13S,14R,16S,17R,20S,22R,24S,25R)-25-hydroxyphysalin M.
Physalin D1 (3) had the molecular formula C28H32O11 identified by HRESIMS m/z 543.1870 [M − H]− (calcd for C28H31O11, 543.1866; Supplementary Fig. 26). Compound 3 exhibited the 1H and 13C NMR data (Supplementary Fig. 19–23) completely identical to those of physalin D1 isolated from P. alkekengi L. var. franchetii 36, indicating that they were the same compound. However, the NOESY spectrum of 3 displayed a key correlation between OH-5 and Me-19 (Fig. 3 and Supplementary Fig. 24 and 25), indicating a β-orientation of OH-5. This conclusion was further confirmed by the 13C NMR chemical shift value of the methyl group at C-10 [δ C 8.5 (C-19)], since this value in 5,6-dihydroxy or 4,5,6-trihydroxy withanolides could indicate the relationships between rings A and B to be cis- (around δ C 10) or trans-fusion (around δ C 15)37. Hence, its structure was revised as (5S,6S,8R,9S,10R,13S,14R,16S,17R,20S,22R,24S,25S)-5,6-dihydroxyphysalin D.
The molecular formula of physalin VII (4) was determined as C28H30O10 based on HRESIMS m/z 525.1769 [M − H]− (calcd for C28H29O10, 525.1761; Supplementary Fig. 35) and 13C NMR data (Supplementary Fig. 29). Comparison of the NMR data of 4 and isophysalin B (16)25 indicated that the proton at C-25 was replaced by a hydroxy group, since the chemical shift value of C-25 was deshielded from δ C 50.9 in 16 to δ C 73.6 in 4, which was further supported by the HMBC correlations from OH-25 to C-25, H-27a/H-27b to C-14/C-24/C-25/C-26, and Me-28 to C-16/C-23/C-24/C-25 (Fig. 2 and Supplementary Fig. 30–32). Furthermore, the NOESY correlations of OH-25 with H-23a/Me-28 (Fig. 3 and Supplementary Fig. 33 and 34) suggested that OH-25 has the same orientation as Me-28 with β. Thus, 4 was established as (8R,9S,10R,13S,14R,16S,17R,20S,22R,24S,25R)-25-hydroxyisophysalin B.
HRESIMS analysis of physalin VIII (5) established the molecular formula C33H38O15 [m/z 673.2127 [M−H]− (calcd for C33H37O15, 673.2132)] (Supplementary Fig. 45), indicating extra five carbons except for the C28 skeleton of the physalins. The detailed analysis of 1H and 13C NMR data (Supplementary Fig. 37 and 38) for 5 and physalin D (8)20 revealed that 5 possessed a similar structure to 8, and the only difference was the presence of five carbon resonances (δ C 170.9, 170.1, 67.0, 51.3, and 38.1) in 5. The HMBC correlations from OMe-4′ to C-4′, H-2′ to C-3′/C-4′, and OH-2′ to C-1′ (Fig. 2 and Supplementary Fig. 39–43) indicated 5 had a methyl malate moiety (Supplementary Fig. 44). Its linkage was deduced at C-5 based on the downfield shift of C-5 from δ C 76.5 in 8 to δ C 90.3 in 5. Moreover, its configuration was established as L-configuration by a polarimetric analyses for the hydrolyzed product of 5 with optical value of −2.0. Thus, 5 was characterized as (2' S,8R,9S,10R,13S,14R,16S,17R,20S,22R,24S,25S)-5-L-methyl malatephysalin D.
Physalin IX (6) was isolated as an amorphous powder, and the molecular formula was established as C28H32O11 according to HRESIMS m/z 567.1842 [M + Na]+ (calcd for C28H32O11Na, 567.1837; Supplementary Fig. 54) and 13C NMR data (Supplementary Fig. 48). The 13C NMR spectrum observed characteristic resonances at δ C 112.6 (C-14), 85.7 (C-15), 82.0 (C-17), 75.8 (C-13), and 47.1 (C-11), indicating that 6 was an unusual 11,15-cyclo physalin in which C-15 was an oxy-carbon rather than a ketonic carbon26. The long-range correlations from H-11 to C-9/C-10/C-13/C-14/C-15 and OH-15 to C-11/C-15/C-16 in the HMBC spectrum (Supplementary Fig. 49–51) confirmed the above deduction. A comparison of 1H and 13C NMR data for 6 and physalin R (15)26 indicated the absence of two carbons [δ C 135.5 (C-5) and 123.4 (C-6) in 15] and the presence of two oxygenated carbons [δ C 77.2 (C-5) and 74.1 (C-6)] in 6. The HMBC correlations from OH-5 to C-5, OH-6 to C-6, and H-6 to C-5/C-8/C-10 (Fig. 2) suggested that two hydroxy groups were linked at C-5 and C-6, respectively. The NOESY correlations of H-4a with Me-19, H-4b with OH-5, OH-6 with Me-19, OH-15 with H-11/H-16, Me-28 with H-16 (Fig. 3 and Supplementary Fig. 52 and 53) revealed an α-orientation of OH-5, and the β-orientations of OH-6, H-11, and OH-15. Accordingly, 6 was identified as (5R,6R,8R,9S,10R,11S,13S,14R,15S,16R,17R,20S,22R,24S,25S)-11-hydro-5,6,15-trihydroxyphysalin R.
Plausible biogenetic pathway
The discovery of compounds 1 and 6 in the genus Physalis is rather uncommon from the viewpoint of chemotaxonomy. Compounds 1 and 6 could have been produced from physalin B, one of the major constituents of P. angulata (Fig. 5). Epoxidation of physalin B followed by Michael addition reaction from C-3 could give intermediate i. Then nucleophile attack on intermediate i by C-1, followed by ring cleavage between C-1 and C-10, lactonization, and dehydration could yield 1 27. The cyclization of physalin B between C-11 and C-15 could produce intermediate ii as well as physalin R except for the orientations of H-11 and OH-15, which could be further epoxidized and hydrated to give 6.

A plausible biogenetic pathway for compounds 1 and 6.
Antiproliferative assay
The isolated compounds were examined for their antiproliferative activities against human prostate cancer cells (C4-2B and 22Rv1), human renal cancer cells (786-O, A-498, and ACHN), human melanoma cancer cells (A375-S2), and their inhibitory effects on NO production induced by LPS in macrophages. In the antiproliferative experiment (Table 3), 5-fluorouracil was used as a positive control drug. Compounds 9 and 10 displayed significant inhibitory effects against all tested cancer cells with IC50 values of 0.24 and 3.17 μM, respectively.
Inhibitory effects of all the compounds on NO production induced by LPS in macrophages
Nitric oxide (NO) is famous as a cellular signaling molecule, and considered as an important regulator in many physiological mechanisms38,39,40. Pharmacological studies have indicated that inflammation is related to overproduction of NO41. The inhibitory effects of all isolated compounds on NO production induced by LPS in macrophages were assayed (Table 4). As shown in Table 4, compounds 9, 10, 13, and 16 exhibited significant inhibitory activities against NO production with IC50 values of 0.32–4.03 μM, while compounds 1, 3, 4, and 14 showed moderate inhibitory activities with IC50 values of 12.83–34.19 μM. A comparison of the inhibitory efficiency of physalins revealed that the Δ5,6 double bond and the proton at C-25 were of pivotal importance. Compound 16 displayed significant inhibitory effect, while 4 showed moderate inhibitory activity, indicating that H-25 could increase inhibitory activity. An analogous case was observed for compounds 8 and 9, the former showed weak inhibitory effect since the 5-ene unit was hydroxylated. However, compound 3 displayed moderate inhibitory effect, indicating that the configurations of OH-5 and OH-6 could influence inhibitory activity. All isolated compounds were evaluated for their cytotoxic effects against RAW 264.7 macrophages, but did not exhibit any at their effective concentration.
Conclusion
In summary, a 1,10-seco physalin, physalin V (1), physalin VIII (5), a novel 11,15-cyclo physalin, physalin IX (6) and two other new ones (2 and 4) were isolated from the stems and leaves of P. angulata together with eleven known ones. The absolute configuration of physalin P (12) was established by single crystal X-ray crystallography, and it is the first report about the absolute configuration of the neophysalins. To our knowledge, the 1,10-seco normal withanolides were previously isolated from P. minmina, P. peruviana and Flos Daturae, while it is the first report about the presence of 1,10-seco physalin in nature and the genus Physalis. The inhibitory effects on nitric oxide production and antiproliferative activities against human cancer cells of the isolated compounds were evaluated. Compounds 9 and 10 showed significant antiproliferative activities against all tested human cancer cells. Compounds 9, 10, 13, and 16 showed significant inhibitory activities against NO production. These results indicated that they are promising candidates that could be further researched on and developed as antitumor and anti-inflammatory agents.
Methods
General experimental procedures
Optical rotations were recorded on a Perkin-Elmer 241 polarimeter. UV spectra were measured on a Shimadzu UV 2201 spectrophotometer. IR spectra were recorded on a Bruker IFS 55 spectrometer. Bruker AV-400 and AV-600 spectrometers were used in the NMR experiments. Chemical shift values were expressed in δ (ppm) using the peak signals of the solvent DMSO-d 6 (δ H 2.50 and δ C 39.51) as references, and coupling constants (J in Hz) were given in parentheses. HRESIMS data were acquired on an Agilent 6210 TOF mass spectrometer. Silica gel GF254 prepared for TLC was purchased from Qingdao Marine Chemical Factory (Qingdao, China). Silica gel (200–300 mesh, Qingdao Marine Chemical Factory, Qingdao, China), Sephadex LH-20 (Pharmacia, USA), and octadecyl silica gel (Merck Chemical Company Ltd., German) were used for column chromatography (CC). RP-HPLC separations were conducted using an LC-6AD liquid chromatograph and a SPD-20A UV detector (Shimadzu, Kyoto, Japan) with a RP-C18 column (250 × 20 mm, 120 Å, 5 μm, YMC Co. Ltd.).
Plant material
The stems and leaves of P. angulata were collected from Nanning, Guangxi Province, China, in July 2013, and identified by Jia-Fu Wei, Guangxi Institute for Food and Drug Control. A voucher specimen (PA-20130826) has been deposited in the herbarium of the Department of Natural Products Chemistry, Shenyang Pharmaceutical University.
Extraction and isolation
The dried stems and leaves of P. angulata (9.5 kg) were extracted with 75% EtOH (2 × 2 h × 110 L) and concentrated in vacuo. The resulting extracts (1.3 kg) were suspended in H2O (5 L), and partitioned successively with petroleum ether (3 × 5 L), EtOAc (3 × 5 L), and n-BuOH (3 × 5 L). The EtOAc extracts (116 g) were subjected to silica gel CC (10 × 80 cm) eluted with CH2Cl2–MeOH (100:1, 80:1, 60:1, 40:1, 20:1, 10:1, 8:1, 5:1, 3:1, 1:1, and 0:1, v/v) to afford compound 8 (500 mg) and six fractions (E1−E6). E3 (35 g) was subjected to silica gel CC (6 × 80 cm) eluted with petroleum ether–acetone (10:1 to 0:1) to produce seven subfractions (E31−E37). E33 (4.0 g) was separated by ODS CC (3 × 50 cm) using a gradient of increasing MeOH in H2O (1:9 to 1:0) to yield three subfractions (E331−E333). E331 (2 g) was chromatographed over silica gel CC (2 × 50 cm, CHCl3–MeOH, 80:1 to 1:1) and preparative TLC (CH2Cl2–acetone, 4:1), yielding compound 6 (12 mg). E332 (1.5 g) was separated by silica gel CC (2 × 50 cm, petroleum ether–acetone, 50:1 to 1:1), preparative TLC (CH2Cl2–acetone, 4:1), and preparative HPLC (60% MeOH–H2O, 6 mL min−1) to afford compounds 2 (10 mg, t R = 24 min) and 14 (21 mg, t R = 26 min). Compound 9 (140 mg) was recrystallized from E34 (4.2 g) using MeOH. E36 (3.8 g) was subjected to silica gel CC (5 × 70 cm), eluted with petroleum ether–acetone (80:1 to 1:1), to afford five subfractions (E361−E365). E363 (1.5 g) was subjected to silica gel CC (3.5 × 70 cm) using CH2Cl2–acetone (80:1 to 4:1) to produce compound 10 (200 mg). E364 (210 mg) was separated by silica gel CC (2 × 50 cm, CH2Cl2–acetone, 80:1 to 4:1) and preparative TLC (CH2Cl2–acetone, 4:1) to yield compound 12 (28 mg). E37 (3.2 g) was purified by preparative TLC (CH2Cl2–EtOAc, 1:1) to give compounds 1 (19 mg) and 11 (25 mg). E4 (15 g) was subjected to silica gel CC (5 × 70 cm), eluted with CHCl3–acetone (80:1 to 1:1), to afford five subfractions (E41−E45). E43 (1.0 g) was chromatographed over ODS CC (3 × 50 cm, MeOH–H2O, 1:9 to 1:0) and preparative HPLC (60% MeOH–H2O) to give compound 7 (10 mg, t R = 24 min). E45 (4 g) was separated by an ODS column (3 × 50 cm, MeOH–H2O, 1:9 to 1:0) and preparative TLC (CH2Cl2–acetone, 2:1), yielding compound 3 (50 mg) and an impure subfraction, which was further purified by preparative HPLC (65% MeOH–H2O) to afford compounds 5 (8 mg, t R = 20 min) and 15 (10 mg, t R = 23 min). E6 (7 g) was subjected to silica gel CC (5 × 70 cm) eluted with CHCl3–acetone (50:1 to 1:1) to afford four subfractions (E61−E64). E61 (900 mg) was separated by Sephadex LH-20 CC (3 × 80 cm, MeOH) and preparative HPLC (60% MeOH–H2O) to produce compound 13 (5 mg, t R = 16 min). E64 (1.5 g) was subjected to ODS CC (3 × 50 cm, MeOH–H2O, 1:9 to 1:0) and preparative TLC (CH2Cl2–acetone, 10:1), yielding compounds 4 (8 mg) and 16 (28 mg).
Spectroscopic data of 1–6
Physalin V (1): amorphous powder; \({[\alpha ]}_{{\rm{D}}}^{25}\) −12.0 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 234 (4.0) nm; IR (KBr) ν max 3400, 2921, 2850, 1782, 1765, 1728, 1646, 1385, 1143 cm−1; 1H (600 MHz, DMSO-d 6) and 13C NMR (150 MHz, DMSO-d 6) data, see Table 1; HRESIMS m/z 549.1750 [M + Na]+ (calcd for C28H30O10Na, 549.1737).
Physalin VI (2): amorphous powder; \({[\alpha ]}_{{\rm{D}}}^{25}\) −64.0 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 216 (3.8) nm; IR (KBr) ν max 3397, 2921, 2850, 1716, 1646, 1467, 1384, 1111 cm−1; 1H (400 MHz, DMSO-d 6) and 13C NMR (100 MHz, DMSO-d 6) data, see Table 1; HRESIMS m/z 527.1918 [M−H]− (calcd for C28H31O10, 527.1917).
Physalin D1 (3): amorphous powder; \({[\alpha ]}_{{\rm{D}}}^{25}\) –43.6 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 220 (3.9) nm; IR (KBr) ν max 3396, 2921, 2850, 1765, 1734, 1648, 1468, 1384, 1134 cm−1; 1H (400 MHz, DMSO-d 6) and 13C NMR (100 MHz, DMSO-d 6) data, see Table 1; HRESIMS m/z 543.1870 [M − H]− (calcd for C28H31O11, 543.1866).
Physalin VII (4): amorphous powder; \({[\alpha ]}_{{\rm{D}}}^{25}\) (c 0.055, MeOH) −138.2; UV (MeOH) λ max (log ε) 218 (4.0) nm; IR (KBr) ν max 3431, 2920, 2850, 1767, 1740, 1697, 1645, 1465, 1384, 1138 cm−1; 1H (600 MHz, DMSO-d 6) and 13C NMR (150 MHz, DMSO-d 6) data, see Table 2; HRESIMS m/z 525.1769 [M − H]− (calcd for C28H29O10, 525.1761).
Physalin VIII (5): amorphous powder; \({[\alpha ]}_{{\rm{D}}}^{25}\) −76.4 (c 0.055, MeOH); UV (MeOH) λ max (log ε) 216 (3.5) nm; IR (KBr) ν max 3442, 2921, 2850, 1791, 1753, 1729, 1687, 1647, 1441, 1383, 1260, 1167 cm−1; 1H (600 MHz, DMSO-d 6) and 13C NMR (100 MHz, DMSO-d 6) data, see Table 2; HRESIMS m/z 673.2127 [M − H]− (calcd for C33H37O15, 673.2132).
Physalin IX (6): amorphous powder; \({[\alpha ]}_{{\rm{D}}}^{25}\) –53.0 (c 0.055, MeOH); UV (MeOH) λ max (log ε) 218 (3.7) nm; 1H (600 MHz, DMSO-d 6) and 13C NMR (150 MHz, DMSO-d 6) data, see Table 2; HRESIMS m/z 567.1837 [M + Na]+ (calcd for C28H32O11Na, 567.1842).
X-ray crystal structure determination of compound 12
The data were collected on an Xcalibur, Eos, Gemini diffractometer using monochromatized Cu Kα radiation. The structure was solved by direct methods using SHELXL. Crystallographic data have been hosted in the Cambridge Crystallographic Data Centre (CCDC number 1465139). Copies of the data can be obtained, free of charge, from the CCDC website (www.ccdc.cam.ac.uk). Crystal Data: C29H35.30334O11.65167, M = 570.30, orthorhombic, size 0.16 × 0.09 × 0.05 mm3, a = 7.65075(19) Å, b = 17.3380(6) Å, c = 19.8734(5) Å, α = β = γ = 90°, V = 2636.18(13) Å3, T = 103.2, space group P212121 (no. 19), Z = 4, μ(Cu Kα) = 0.937, completeness θ max = 100.0%, F(000) = 1210, 2θ range for data collection from 6.766 to 143.782°, 9459 reflections measured, 5071 unique (R int = 0.0293) which were used in all calculations. The final wR(F 2) was 0.0974 (all data). The Flack parameter was 0.04(11). The largest difference peak and hole were 0.293 and −0.200 e Å−3.
Optical rotation analysis for hydrolyzed product of compound 5
Compound 5 (3 mg) and hydrochloric acid (2 M, 4 mL) were added into the flask (10 mL) with cover, and stirred at 90 °C for 3 h. The reaction mixture was extracted thrice with CHCl3, then the water layer was freeze-dried in vacuo to afford the residue. The hydrolyzed product (0.3 mg), purified over a Sephedax LH-20 column from the residue using CHCl3-MeOH (1:1), was analyzed by a Perkin-Elmer 241 polarimeter (Perkin-Elmer, Waltham, MA, USA) and an API3200 mass spectrometer (AB SCIEX, Framingham, MA, USA).
Antiproliferative assay
Compounds were evaluated by the MTT method for antiproliferative activities against human prostate cancer cells (C4-2B and 22Rvl), human renal carcinoma cells (786-O, A-498, and ACHN), and human melanoma cells (A375-S2)42. All these cells were incubated in RPMI-1640 or EMEM medium with 10% fetal bovine serum at a humidified atmosphere (5% CO2, 37 °C). Cells (1 × 104 cells/well) were added into the 96-well plates for 12 h before drug addition. The test compounds with various concentrations were added into the 96-well plates, then incubated for 48 h. 5-Fluorouracil was used as the positive control, and every assay was repeated three times. Cell viability was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay.
NO production bioassay
All compounds were assayed for the inhibition of NO production according to the Griess method43, 44. 1 × 106 Cells/well of RAW 264.7 cells were added into the 96-well plates, and incubated at 37 °C for 24 h by the stimulation of LPS (1 μg/mL) with or without test compounds. After the addition of Griess reagent [0.1% N-(1-naphthyl)-ethylenediamine (50 μL); 1% sulfanilamide in 5% H3PO4 (50 μL)], absorbance (540 nm) was recorded by using a microplate reader. The standard curve was used to calculate the NO concentrations and inhibitory rates.
References
Misico, R. I. et al. Withanolides and related steroids. Prog. Chem. Org. Nat. Prod 94, 127–229 (2011).
Chen, L. X., He, H. & Qiu, F. Natural withanolides: an overview. Nat. Prod. Rep. 28, 705–740 (2011).
Choudhary, M. I., Yousaf, S., Ahmed, S., Samreen & Yasmeen, K. & Atta-ur-Rahman. Antileishmanial physalins from. Physalis minima. Chem. Biodivers. 2, 1164–1173 (2005).
Veleiro, A. S., Oberti, J. C. & Burton, G. Chemistry and bioactivity of withanolides from south American Solanaceae. Studies in natural products chemistry, bioactive natural products 32, 1019–1052 (2005).
Anjaneyulu, A. S. R., Rao, D. S. & Lequesne, P. W. Withanolides, biologically active natural steroidal lactones: a review. Studies in natural products chemistry, bioactive natural products 20, 135–261 (1998).
Matsuura, T., Kawai, M., Nakashima, R. & Butsugan, Y. Bitter principles of Physalis alkekengi L. var. franchetii; structure of physalin A. Tetrahedron Lett. 14, 1083–1086 (1969).
Institute of Botany of the Chinese Academy of Sciences, and Kunming Institute of Botany of the Chinese Academy of Sciences, Chinese Flora (Zhongguo Zhiwu Zhi) 67, 50–59 (1978).
Damu, A. G. et al. Isolation, structures, and structure-cytotoxic activity relationships of withanolides and physalins from. Physalis angulata. J. Nat. Prod. 70, 1146–1152 (2007).
Sun, C. P., Kutateladze, A. G., Zhao, F., Chen, L. X. & Qiu, F. A novel withanolide with an unprecedented carbon skeleton from Physalis angulata. Org. Biomol. Chem. 15, 1110–1114 (2017).
Sun, C. P. et al. Antiproliferative and anti-inflammatory withanolides from Physalis angulata. J. Nat. Prod. 79, 1586−1597 (2016).
Qiu, L. et al. Steroids and flavonoids from Physalis alkekengi var. franchetii and their inhibitory effects on nitric oxide production. J. Nat. Prod. 71, 642–646 (2008).
He, H. et al. Nitric oxide induces apoptosis and autophagy; autophagy down-regulates NO synthesis in physalin A-treated A375-S2 human melanoma cells. Food Chem. Toxicol. 71, 128–135 (2014).
Xia, G. Y., et al. Withanolides from the stems and leaves of Physalis pubescens and their cytotoxic activity. Steroids 115, 136–146 (2016).
Yang, Y. K. et al. Six new physalins from Physalis alkekengi var. franchetii and their cytotoxicity and antibacterial activity. Fitoterapia 112, 144–152 (2016).
He, H. et al. Physalin A induces apoptosis via p53-Noxa-mediated ROS generation, and autophagy plays a protective role against apoptosis through p38-NF-kappa B survival pathway in A375-S2 cells. J. Ethnopharmacol. 148, 544–555 (2013).
He, H. et al. Physalin A induces apoptotic cell death and protective autophagy in HT1080 human fibrosarcoma cells. J. Nat. Prod. 76, 880–888 (2013).
Lima, M. D. S. et al. Antinociceptive properties of physalins from Physalis angulata. J. Nat. Prod. 77, 2397–2403 (2014).
Sun, L. J., Liu, J. W., Liu, P., Yu, Y. J., Ma, L. & Hu, L. H. Immunosuppression effect of withangulatin A from Physalis angulata via heme oxygenase 1-dependent pathways. Process Biochem. 46, 482–488 (2011).
Kawai, M. et al. Cytotoxic activity of physalins and related compounds against HeLa cells. Pharmazie 57, 348–350 (2002).
Row, L. R., Reddy, K. S., Sarma, N. S., Matsuura, T. & Nakashima, R. New physalins from Physalis angulata and Physalis lancifolia. Structure and reactions of physalins D, I, G and K. Phytochemistry 19, 1175–1181 (1980).
Matsuura, T. & Kawai, M. Physalis alkekengi L. var. franchetii; structure of physalin B. Tetrahedron Lett. 22, 1765–1766 (1969).
Row, L. R., Sarma, N. S., Reddy, K. S., Matsuura, T. & Nakashima, R. The structure of physalins F and J from Physalis angulata and P. Lancifolia. Phytochemistry 17, 1647–1650 (1978).
Chen, R., Liang, J. Y., Yang, Y. & Liu, R. Chemical constituents of Physalis alkekengi and structural revision of physalin G. Chin. J. Nat. Med. 5, 186–189 (2007).
Kawai, M. et al. The structure of physalin P, a neophysalin from Physalis alkekengi. Bull. Chem. Soc. Jpn. 66, 1299–1300 (1993).
Choudhary, M. I., Samreen, S. Y., Shah, S. A. A. & Ahmed, S. & Atta-ur-Rahman, Biotransformation of physalin H and leishmanicidal activity of its transformed products. Chem. Pharm. Bull. 54, 927–930 (2006).
Makino, B., Kawai, M., Kito, K., Yamamura, H. & Butsugan, Y. New physalins possessing an additional carbon-carbon bond from Physalis alkekengi var. franchetii. Tetrahedron 51, 12529–12538 (1995).
Fang, S. T., Liu, J. K. & Li, B. A novel 1,10-seco withanolide from Physalis peruviana. J. Asian Nat. Prod. Res. 12, 618–622 (2010).
Kazuo, I., Makoto, I. & Kinzo, W. Stoloniolide I and II, new marine lactonic steroids with an unprecedented 1,10-seco ergostane skeleton, isolated from the okinawan soft coral, Clavularia viridis. Chem. Lett. 24, 1109–1110 (1995).
Kawai, M., Ogura, T., Butsugan, Y., Taga, T. & Hayashi, M. Benzilic acid rearrangement-type reaction of physalins to neophysalins. Structural revision of one of the dehydration products of physalin A. Tetrahedron 47, 2103–2110 (1991).
Matsuura, T., Kawai, M., Nakashima, R. & Butsugan, Y. Structures of physalin A and physalin B, 13,14-seco-16,24-cyclo-steroids from Physalis alkekengi var. franchetii. J. Chem. Soc. 5, 664–670 (1970).
Kawai, M., Taga, T., Miwa, Y. & Butsugan, Y. 1H NMR spectral analysis of physalin A, a 13,14-seco-16,24-cyclo steroid, based on crystal structure of (25R)-2,3,25,27-tetrahydrophysalin A dimethanol solvate. J. Crystallogr. Spectrosc. Res. 22, 131–137 (1992).
Kawai, M. et al. Crystal structure of 5α,6α-epoxy and 2,3-dihydro derivatives of physalin B, a 13,14-seco-16,24-cyclo steroid, and their 1H NMR spectral analysis. Bull. Chem. Soc. Jpn. 67, 222–226 (1994).
Kimpende, P. M. et al. Isolation, pharmacological activity and structure determination of physalin B and 5β,6β-epoxyphysalin B isolated from Congolese Physalis angulata L. Acta Cryst. C 69, 1557–1562 (2013).
Men, R. Z. et al. Unprecedent aminophysalin from Physalis angulata. Steroids 88, 60–65 (2014).
Kawai, M. et al. Structure of physalin M isolated from Physalis alkekengi var. franchetii. Bull. Chem. Soc. Jpn. 61, 2696–2998 (1988).
Li, X. et al. Physalins and withanolides from the fruits of Physalis alkekengi L. var. franchetii (Mast.) Makino and the inhibitory activities against human tumor cells. Phytochem. Lett. 10, 95–100 (2014).
Shingu, K., Yahara, S., Okabe, H. & Nohara, T. Three new withanolides, physagulins E, F and G from Physalis angulata L. Chem. Pharm. Bull. 40, 2448–2451 (1992).
Turtay, M. G., Karabas, M., Parlakpinar, H., Colak, C. & Sagir, M. The analgesic effect of apelin-13 and its mechanism of action within the nitric oxide and serotonin pathways. Hippokratia 19, 319–323 (2015).
Bahnson, E. S., Havelka, G. E., Koo, N. C., Jiang, Q. & Kibbe, M. R. Periadventitial adipose tissue modulates the effect of PROLI/NO on neointimal hyperplasia. J. Surg. Res. 205, 440–445 (2016).
Ren, H., Bull, J. L. & Meyerhoff, M. E. Transport of nitric oxide (NO) in various biomedical grade polyurethanes: measurements and modeling impact on NO release properties of medical devices. ACS Biomater. Sci. Eng. 2, 1483–1492 (2016).
Mulligan, M. S., Hevel, J. M., Marletta, M. A. & Ward, P. A. Tissue injury caused by deposition of immune complexes is L-arginine dependent. Proc. Natl. Acad. Sci. USA. 88, 6338–6342 (1991).
Alley, M. C. et al. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 48, 589–601 (1988).
Li, J., Zhao, F., Li, M. Z., Chen, L. X. & Qiu, F. Diarylheptanoids from the Rhizomes of Curcuma kwangsiensis. J. Nat. Prod. 73, 1667–1671 (2010).
Dirsch, V. M., Stuppner, H. & Vollmar, A. M. The Griess assay: suitable for a bio-guided fractionation of anti-inflammatory plant extracts? Planta Med. 64, 423–426 (1998).
Acknowledgements
This work was financially supported by grants from the National Natural Science Foundation of China (NSFC) (Grant No. 81430095, 21472138, and 31270399), Fund of the Educational Department of Liaoning Province (Grant No. L2011177), Liaoning Baiqianwan Talents Program (Grant No. 2013921043), and 2015 Career Development Program for Young and Middle-aged Teachers of Shenyang Pharmaceutical University (ZQN2015015).
Author information
Authors and Affiliations
Contributions
F.Q. and L.X.C. initiated the project. F.Q. and L.X.C. designed and coordinated the project. C.P.S. and C.Y.Q. performed the extraction, isolation, and structural identification of compounds. F.Z. performed the anti-inflammatory assay. K.N. and L.X.C. performed the antiproliferative assay. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, CP., Qiu, CY., Zhao, F. et al. Physalins V-IX, 16,24-cyclo-13,14-seco withanolides from Physalis angulata and their antiproliferative and anti-inflammatory activities. Sci Rep 7, 4057 (2017). https://doi.org/10.1038/s41598-017-03849-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-03849-9
This article is cited by
-
A new 1,2-diketone physalin isolated from Physalis minima and TRAIL-resistance overcoming activity of physalins
Journal of Natural Medicines (2023)
-
Reproductive biology and hybridization of Physalis L. species
Brazilian Journal of Botany (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
