
Abstract
Pampus argenteus (Euphrasen, 1788) is one of the major fishery species in coastal China. Pampus argenteus has a highly specialized morphology, and its declining fishery resources have encouraged massive research efforts on its aquacultural biology. In this study, we reported the first high-quality chromosome-level genome of P. argenteus obtained by integrating Illumina, PacBio HiFi, and Hi-C sequencing techniques. The final size of the genome was 518.06 Mb, with contig and scaffold N50 values of 20.47 and 22.86 Mb, respectively. The sequences were anchored and oriented onto 24 pseudochromosomes based on Hi-C data corresponding to the 24-chromatid karyotype of P. argenteus. A colinear relationship was observed between the P. argenteus genome and that of a closely related species (Scomber japonicus). A total of 24,696 protein-coding genes were identified from the genome, 98.9% of which were complete BUSCOs. This report represents the first case of high-quality chromosome-level genome assembly for P. argenteus and can provide valuable information for future evolutionary, conservation, and aquacultural research.
Similar content being viewed by others

Background & Summary
Pampus argenteus (Euphrasen, 1788; Fishbase ID: 491), also known as the silver pomfret, is a commercially important fish in the Northwest Pacific area that is widely distributed throughout the South China Sea to coastal Japan, Korea, and Russia1,2. It belongs to the family Stromateidae of the suborder Stromateoidei3, which was identified in Scombriformes according to a recent phylogenetic study4. This species is one of the major fishery species in coastal China, with harvests exceeding three million tons in 20165. Overfishing and environmental changes have resulted in a noticeable decline in P. argenteus fishery resources in recent years6,7. The aquaculture of P. argenteus has made substantial progress, which in some ways compensates for the decline in fishery resources8,9. However, the industry is still facing many restrictions and issues owing to the high sensitivity of P. argenteus to injury and pathogenic infection during aquaculture and transportation10. Due to the medusivorous habit of P. argenteus11, its aquaculture greatly relies on fish bait composed of jellyfish and minced fish meat. Using fish bait leads to higher costs in water quality control and risking outbreaks of harmful pathogens, which have become one of the major bottlenecks in P. argenteus aquaculture, necessitating substitution with better formulated feeds12. However, the digestive and immune systems of P. argenteus are considered specialized for the digestion of jellyfish and tolerance of medusocongestin13,14. The inclusion of jellyfish in an artificial diet can significantly improve the growth performance and survival rate of P. argenteus larvae and juveniles15. The impact of changing fish bait to formulated feed on P. argenteus at different growth stages still requires further clarification. Clarifying the genetic basis of the physiological process of P. argenteus, particularly those related to the immune response16, intestinal enzyme activities14, stress responses17, etc., is becoming increasingly important for the future prospects of the aquaculture industry. However, the genome of P. argenteus, which represents the foundation of physiological responses18, has not yet been completely sequenced.
In addition to its fishery importance, P. argenteus is considered one of the most advanced species within Stromateoidei19. The dorsal and anal fin spines of P. argenteus are reduced into small blades, with a pelvic fin absent from its abdominal region. Stromateoidei is distinct from other Actinopterygii by having a unique pharyngeal sac immediately behind the last gill arch, which functions to fragmentize food19. The saccular structure of P. argenteus, which primarily feeds on small crustaceans and medusae, is smaller, more elongated, and densely covered within elongated tooth-like papillae; additionally, this species ably adapts to shredding rubbery tissue of jellyfish19. The pharyngeal sac is believed to be a key innovation for stromateiods, while the specialized shape of pharyngeal sac in the genus Pampus might bring further advantages and lead to its broad success in the Indo-Pacific region19. Clarifying the genetic basis for the formation of the pharyngeal sac is crucial to understanding the evolution of the genus Pampus.
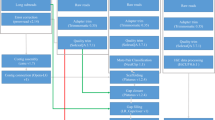
In this study, a high-quality chromosome-level genome assembly of P. argenteus was generated by integrating multiple sequencing technologies, including Illumina sequencing, PacBio circular consensus sequencing (CCS), and Hi-C techniques (Fig. 1). The final assembly size for the P. argenteus genome was 518.06 Mb, with 97.30% of the contigs anchored to 24 chromosomes (Table 1 & Fig. 2). The contig and scaffold N50 lengths for the genome were 20.47 and 22.86 Mb, respectively. The genome consisted of 13.45% repeated sequences and 17.18% nod-coding genes. A total of 24,696 protein-coding genes were predicted, 93.38% of which were functionally annotated. Compared to the Pampus genome reported by AlMomin et al.20, the genome of P. argenteus generated herein was assembled into fewer and longer contigs and scaffolds (Table 1). More genes and repetitive regions were identified from this genome, with an average protein-coding gene length 7.5-fold greater than that of the previous version20. These results suggested that the genome developed in this study has a much higher integrity and quality. The chromosome-level genome assembly of P. argenteus will provide valuable information for establishing effective molecular markers for future conservation and aquaculture goals. The genome also represents the first case of high-quality chromosome-level genome assembly for stromateoids; this information could be an important reference for whole-genome sequencing of its close relatives, and, foreseeably, could become one of the foundations for exploring the genomic evolution and phylogeny of the Stromateoidei.

Workflow overview for the P. argenteus chromosome-level genome assembly.

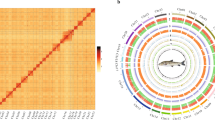
(a) A photo of P. argenteus; (b) Circos plot indicating gene density, repetitive sequences, GC content, and colinear relationship among chromosomes of the P. argenteus genome assembly.
Methods
Sample collection
In 2021, a female P. argenteus specimen was caught from the wild for whole-genome sequencing using a fishing boat in Shengsi, Zhejiang Province, China. The specimen was identified based on the morphological descriptions of P. argenteus in Liu et al.3, who designated the P. argenteus neotype. Eye, muscle, ovary, heart and liver samples for DNA and RNA sequencing were isolated from the specimens immediately after they were caught. The samples were washed three times with phosphate-buffered saline (PBS), frozen in liquid nitrogen for three hours, and subsequently stored at −80 °C until DNA extraction. All the experiments were conducted under the approval and regulations of the Institutional Animal Care and Use Committee of the Institute of Oceanology, Chinese Academy of Science.
Library construction, sequencing and data preparation
The Illumina, PacBio HiFi, and Hi-C data were obtained and used for generating a chromosome-level genome assembly of P. argenteus. For Illumina sequencing, total genomic DNA was isolated from muscle samples using the cetyltrimethylammonium bromide (CTAB) method21. The quality of the extracted DNA was assessed using a Qubit 2.0 (Thermo Fisher Scientific, USA) and a NanoDrop® Series (Thermo Fisher Scientific, USA). For Illumina sequencing, a short-fragment library with an insert size of 300–500 base pairs (bp) was prepared using the NEBNext®ΜLtra™ DNA Library Prep Kit (New England Biolabs, USA) following the manual instructions. The library was purified with AMPure XP Beads (Beckman Coulter, USA) and then subjected to sequencing on an Illumina NovaSeq 6000 platform (Illumina, USA) to generate 150-bp paired-end (PE150) reads. After filtering in Fastp (v0.20.0)22, a total of 75.52 Gb of clean Illumina PE150 data were obtained, with Q20 and Q30 being 97.2% and 92.5%, respectively (Table 2). For PacBio CCS, total genomic DNA total genomic was extracted from muscle samples using the sodium dodecyl sulfate (SDS) method23. The high-molecular-weight gDNA was sheared to 8–10 kb using g-TUBEs (Covaris, USA). The HiFi library was then prepared using the SMRTbell prep kit 3.0 and sequenced in CCS mode on the PacBio Sequel II system (Pacific Biosciences, USA) following the manufacturer’s protocols. After the removal of low-quality reads and adaptors from the raw data, 63.80 Gb of clean HiFi data was retained, with Q20 and Q30 values of 99.9% and 54.78%, respectively (Table 2). Hi-C library preparation was performed with muscle tissue using a Frasergen Hi-C Kit (Frasergen, China) following the protocol instructions, which included crosslinking, lysis, fragmentation, repairing, biotin labeling, ligation, extraction, purification, and library construction. All the purification steps were performed using AMPure XP beads (Beckman Coulter, USA), while the biotin-labeled DNA was enriched with Pierce™ Streptavidin Magnetic Beads (Thermo Fisher Scientific, USA). The library was assessed with an Agilent 2100 Bioanalyzer (Agilent, USA) to determine a sufficient concentration and an insert size of 300–800 bp. The Hi-C library was subjected to sequencing on an Illumina HiSeq X Ten platform (Illumina, USA) to generate PE150 reads. After filtering in Fastp22, a total of 138.39 Gb of clean Hi-C data were obtained, for which the Q20 and Q30 were 96.57% and 90.54%, respectively (Table 2).
To assist in gene prediction, muscle, eye, ovary, heart, and liver tissues were pooled to obtain the transcriptome of P. argenteus. Total RNA was extracted from the pooled sample using a TRIzol reagent kit (Invitrogen, USA) following the manufacturer’s instructions. The quality and concentration of the extracted RNA were assessed using a NanoDrop® Series (Thermo Scientific, USA) and an Agilent 2100 Bioanalyzer. For RNA-seq data, three cDNA libraries (i.e., Pa-op1, Pa-op2, and Pa-op3; Table 3) were prepared via total RNA extraction using the NEBNext® Ultra™ RNA Library Prep Kit (New England Biolabs, USA) and subsequently subjected to sequencing on an Illumina NovaSeq 6000 platform (Illumina, USA). After filtering via Fastp22, a total of 20.91 Gb of clean RNA-seq data were obtained from the five tissue samples (Table 3). For isoform data, a single cDNA library was reverse transcribed from the total RNA using the Clontech SMARTer PCR cDNA Synthesis Kit (Takara Bio, USA) following the manufacturer’s instructions. The PCR products were purified using AMPure XP Beads (Beckman Coulter, USA) and used for SMRTbell library construction via the SMRTbell Prep Kit 3.0. The library was sequenced and processed with the PacBio Sequel II system (Pacific Biosciences, USA). After filtering, a total of 34.96 Gb of isoform data were obtained (Table 3). The reference genome and protein-coding gene data of closely related species of P. argenteus [i.e., Dunckerocampus dactyliophorus (Bleeker, 1853)24, Hippocampus zosterae Jordan & Gilbert, 188225, Scomber japonicus Houttuyn, 178226, Thunnus albacares (Bonnaterre, 1788)27, and T. maccoyii (Castelnau, 1872)28] were downloaded from GenBank and subsequently used for gene prediction and comparisons.
Genome survey
A survey of the P. argenteus genome was performed using the k-mer method. K-mer analysis was conducted using jellyfish (v2.2.6)29 with 75.52 Gb of Illumina data and the best K value of 17. After the removal of abnormal k-mers, 60,502,700,002 k-mers were yielded with a k-mer peak at a depth of 126.64 (Table 4 & Fig. 3). The genome size, heterozygosity rate, repetition rate, and GC content estimated from GenomeScope (v1.0.0)30 were 463.10 Mb, 1.55%, 29.89% and 39.45%, respectively (Table 4).

17-mer frequency distribution in the P. argenteus genome, the numbers of k-mers of each sequencing depth are indicated.
Chromosome-level genome assembly
The genome of P. argenteus was first assembled into 416 contigs with HiFi long-read data using the default parameters in Hifiasm (v0.16.1)31. The Illumina PE150 data were used to correct the contig assemblies in Pilon (v1.23)32. Finally, 74 non-redundant contigs with a total length of 518.04 Mb were obtained in Redundans (v14a)33. The contig N50 and maximum length were 20.47 and 25.41 Mb, respectively (Table 1). The clean Hi-C data were aligned to the genome assembly using BWA (v0.7.12)34. Reading depth and coverage were calculated in Picard (v1.129)35 and BEDtools (v2.25.0)36. To obtain the chromosome-level genome, clean Hi-C data were assembled with 74 contigs and adjusted using Juicer (v1.6)37, 3D-DNA (v180114)38 and JuiceBox39. Finally, the assembled sequences were anchored and oriented to 24 pseudochromosomes, ranging in size from 9.80–27.76 Mb (Table 1), which is congruent with the 24-chromatid karyotype reported by Liu et al.40. The total length of the chromosome-level assembly was 518.06 Mb, with a scaffold N50 of 22.86 Mb (Table 1 & Fig. 1). Therefore, the clean Illumina, PacBio HiFi, and Hi-C data had 145.77-, 123.15-, and 267.13-fold coverage of the P. argenteus genome, respectively (Table 2). A collinearity dot plot generated using MCScanX41 and SynVisio42 indicated clear genomic collinearity between P. argenteus and the scombriform species S. japonicus26 but scattered collinearity with the syngnathiform H. zosterae27, supporting the closer affinity of P. argenteus to the scombriform species (Fig. 4).

Dot plots showing the collinearities of P. argenteus with the syngnathiform (H. zosterae) and scombriform (S. japonicus) species.
Repeated sequence annotations
Tandem repeats were predicted using Tandem Repeats Finder (v4.10.0) (TRF)43. Transposable elements (TEs) were identified by a combination of signature-based, de novo, and homology-based approaches. De novo prediction was performed using RepLoc (v2021-3-12)44, and TEs shorter than 30 bp were removed from the results. Long terminal repeat retrotransposons (LTR-RTs) were identified using both LTR_FINDER (v1.0.2)45 and LTRharvest (v1.6.2)46. Miniature inverted-repeat transposable element (MITE) sequences were found by MiteFinder (v1.0.006)47. Helitron sequences were scanned by a HelitronScanner (v1.0)48. TIRvish (v1.6.2)49 was used to find terminal inverted repeat (TIR) sequences. All the predicted repetitive sequences were combined with the known repetitive sequence data in the Repbase database50 to generate the non-redundant repeat sequence library for P. argenteus using AB-BLAST (v3.0)51, MCL (v14–137)52, MMseqs. 2 (v13.45111)53 and CD-HIT54. The final repeat sequences in the P. argenteus genome were identified and classified by homology searching against the library using RepeatMasker (v4.1.2-p1)55 and RepeatProteinMask (v4.1.2-p1)56. In brief, 13.57% of the P. argenteus genome was annotated as repetitive elements, with 9.39% 9.39% TEs (48.64 Mb) and 8.51% tandem repeats (44.10 Mb) (Table 5).
Non-coding RNA annotation
For non-coding RNA annotation, tRNA and rRNA were predicted by tRNAScan-SE (v2.0.9)57 and barrnap (v0.9)58, respectively, while snRNA and miRNA were identified by aligning to the Rfam database59 with Infernal cmscan (v1.1.4)60. A total of 10,650 tRNAs, 634 rRNAs, 1,514 snRNAs, and 675 miRNAs were identified, comprising 17.18% of the P. argenteus genome (Table 6).
Protein-coding gene prediction and annotation
The protein-coding genes were predicted based on four different strategies, namely, RNA-seq-based, isoform-based, homology-based, and de novo predictions. The clean RNA-seq data were assembled into the P. argenteus genome using two different methods: (i) assembly with the reference genome using HISAT (v2.1.1)61, StringTie (v2.2.0)62, and Scallop2 (v1.1.1)63; and (ii) de novo assembly using RNA-Bloom (v2.0.1)64, Evigene65, minimap2 (v2.26)66 and miniprot (v0.12)67. For isoform-based prediction, SMRT-link (PacBio, USA) was used to generate isoforms and ESTs, and the transcriptome and protein sequences were generated with the Evigene65 platform; these sequences were subsequently mapped onto the P. argenteus genome using minimap266 and miniprot67. Protein sequences from D. dactyliophorus24, H. zosterae25, S. japonicus26, T. albacares27, T. maccoyii28 and the Swissprot protein database68 were obtained from their genomes and aligned to the P. argenteus genome for homology-based gene prediction in miniprot66. The predicted gene models of RNA-seq, isoform, and homology-based strategies were used as training datasets in AUGUSTUS (v3.4.0)69 and SNAP (v2.0)70 for de novo prediction. Finally, all the predicted gene models were integrated into a single, non-redundant, and complete gene set using EvidenceModeler (v1.1.1)71. The untranslated region (UTR) and alternative splicing of these genes were annotated in Mikado (v2.3.2)72. The gene statistics, including gene length, coding sequence (CDS), intron length, and exon length, were similar between the reference24,25,26,27,28 and P. argenteus genomes (Fig. 5). The predicted protein-coding genes were annotated by searching the GenBank Non-Redundant (Nr) (ftp://ftp.ncbi.nih.gov/pub/nrdb/), SwissProt68, eukaryotic orthologous groups (KOG)73 and Kyoto Encyclopedia of Genes and Genomes (KEGG)74 protein databases using Diamond (v0.7.9)75, with an E-value threshold of 1e−5. EggNOG-mapper (v2.0)76 was used for gene ontology (GO) annotation in combination with the eggNOG database (v5.0)77. A total of 24,696 genes were predicted in the P. argenteus genome (Table 7). Among these, 23,062 genes (93.38%) were annotated by at least one database, while 12,974 genes (52.53%) were supported by all five databases (Table 8 and Fig. 6).

Comparisons of gene, CDS, exon, and intron lengths of P. argenteus and the five closely related species (D. dactyliophorus, H. zosterae, S. japonicus, T. albacares and T. maccoyii).

Venn diagram indicating number of genes annotated by different gene databases.
Data Records
The Illumina (SRR27308594), PacBio HiFi (SRR27308592–SRR27308592), Hi-C (SRR27308591), RNA-seq (SRR27308587–SRR27308589) and isoform (SRR27308590) data used for the genome assembly of P. argenteus were deposited in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under sequence read project SRP47932578. The chromosome-level assembly of the P. argenteus genome was deposited in the NCBI genome database under accession number GCA_03632111579. The chromosome assembly of P. argenteus, genomic annotation results, and software settings can be found in the figshare database80.
Technical Validation
Evaluation of the genome assembly and annotation
The quality of this chromosome-level genome assembly was assessed using the following three criteria: (i) the mapping rate of Illumina PE150 reads, (ii) the Core Eukaryotic Genes Mapping Approach (CEGMA)81, and (iii) the Benchmarking Universal Single-Copy Orthologs (BUSCO) assessment82. In brief, 99.30% of the Illumina PE150 reads could be aligned to the P. argenteus genome using BWA (v0.7.12)34, for a coverage rate of 99.95%, which indicates high mapping efficiency and sufficient coverage. A total of 230 (92.74%) of the 248 highly conserved core genes for eukaryotes provided in CEGMA could be completely aligned with their homologous genes in the P. argenteus genome. In BUSCO (v4.1.2)82, 98.90% of the complete BUSCOs were detected in the P. argenteus genome, whereas fragmented and missing BUSCOs only comprised 1.08% of the total orthologs. This evidence indicated the high integrity and quality of the obtained chromosome-level genome assembly of P. argenteus.
References
Liu, J., Li, C. & Li, X. Studies on Chinese pomfret fishes of the genus Pampus (Pisces: Stromateidae). Stud. Mar. Sin. 44, 240–252 (2002).
Wei, J. et al. Species diversity and distribution of genus Pampus (Pelagiaria: Stromateidae) based on global mitochondrial data. Front. Mar. Sci. 9, 1050386, https://doi.org/10.3389/fmars.2022.1050386 (2022).
Liu, j, Li, C. S. & Ning, P. Identity of silver pomfret Pampus argenteus (Euphrasen, 1788) based on specimens from its type locality, with a neotype designation (Teleostei, Stromateidae). Acta Zootaxonomica Sin. 38, 171–177 (2013).
Hughes, L. C. et al. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. PNAS 115, 6249–6254, https://doi.org/10.1073/pnas.1719358115 (2018).
Wei, J. et al. Validity of Pampus liuorum Liu & Li, 2013, Revealed by the DNA Barcoding of Pampus Fishes (Perciformes, Stromateidae). Diversity 13, 618, https://doi.org/10.3390/d13120618 (2021).
Yang, W. T., Li, J. & Yue, G. H. Multiplex genotyping of novel microsatellites from silver pomfret (Pampus argenteus) and cross-amplification in other pomfret species. Mol. Ecol. Notes 6, 1073–1075, https://doi.org/10.1111/j.1471-8286.2006.01438.x (2006).
Zhao, F. et al. Genetic diversity of silver pomfret (Pampus argenteus) in the Southern Yellow and East China Seas. Biochem. Syst. Ecol. 39, 145–150, https://doi.org/10.1016/j.bse.2011.02.002 (2011).
Shi, Z. H., Zhao, F., Fu, R., Huang, X. & Wang, J. Study on artificial larva rearing techniques of silver pomfret (Pampus argenteus). Mar. Fish. 31, 53–57, https://doi.org/10.3969/j.issn.1004-2490.2009.01.008 (2009).
Hu, J. B. et al. Advances in the artificially reproductive and breeding studies of silver pomfret, Pampus argenteus. J. Biol. 33, 87–117, https://doi.org/10.3969/j.issn.2095-1736.2016.04.087 (2016).
Yu, N. et al. Reduced stress responses by MS-222 in juvenile silver pomfret (Pampus argenteus). J. World Aquac. Soc. 51, 1192–1207, https://doi.org/10.1111/jwas.12725 (2020).
Liu, C. et al. Medusa consumption and prey selection of silver pomfret Pampus argenteus juveniles. Chin. J. Oceanol. Limn. 32, 71–80, https://doi.org/10.1007/s00343-014-3034-5 (2014).
Wang, L., Ren, X. & Wang, Y. Feeding rates of juvenile silver pomfret in the East China Sea on different soft pellet diet. Fish. Sci. Tech. Info. 47, 1–5, https://doi.org/10.16446/j.cnki.1001-1994.2020.01.001 (2020).
Wang, Q. et al. Alternations in the liver metabolome, skin and serum antioxidant function of silver pomfret (Pampus Argenteus) is induced by jellyfish feeding. 3 Biotech 11, 192, https://doi.org/10.1007/s13205-021-02702-1 (2021).
Wang, Y. et al. Dietary jellyfish affect digestive enzyme activities and gut microbiota of Pampus argenteus. Comp. Biochem. Physiol. - D: Genom. Proteom. 40, 100923, https://doi.org/10.1016/j.cbd.2021.100923 (2021).
Liu, C. et al. Potential of utilizing jellyfish as food in culturing Pampus argenteus juveniles. Hydrobiologia 754, 189–200, https://doi.org/10.1007/s10750-014-1869-6 (2015).
Zhang, Y. et al. Immune response of silver pomfret (Pampus argenteus) to Photobacterium damselae subsp. Damselae: Virulence factors might induce immune escape by damaging phagosome. Aquaculture 578, 740014, https://doi.org/10.1016/j.aquaculture.2023.740014 (2024).
Sun, P., Tang, B. & Yin, F. Gene expression during different periods of the handling-stress response in Pampus argenteus. J. Oceanol. Limnol. 36, 1349–1359, https://doi.org/10.1007/s00343-018-7012-1 (2018).
Hilsdorf, A. W. S. et al. 49–74 (Academic Press, 2020).
Haedrich, R. L. The stromateoid fishes: systematics and a classification. Bull. Mus. Comp. Zool. 135, 31–139 (1967).
AlMomin, S. et al. Draft genome sequence of the silver pomfret fish, Pampus argenteus. Genome 59, 51–58, https://doi.org/10.1139/gen-2015-0056 (2015).
Richards, E., Reichardt, M. & Rogers, S. Preparation of Genomic DNA from Plant Tissue. Curr. Protoc. Mol. Biol. 27, 2–3, https://doi.org/10.1002/0471142727.mb0203s27 (1994).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Zhou, J., Bruns, M. A. & Tiedje, J. M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62, 316–322, https://doi.org/10.57760/sciencedb.04022 (1996).
NCBI GenBank assembly, https://identifiers.org/ncbi/insdc.gca:GCA_901007775.1 (2020).
NCBI GenBank assembly, https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_025434085.1/ (2022).
NCBI GenBank assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_027409825.1/ (2023).
NCBI GenBank assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_914725855.1/ (2021).
NCBI GenBank assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_910596095.1/ (2021).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Walker, B. J. et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLOS ONE 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Pryszcz, L. P. & Gabaldón, T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44, e113, https://doi.org/10.1093/nar/gkw294 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Broad Institute. Picard Toolkit. https://broadinstitute.github.io/picard/ (2019).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, https://doi.org/10.1093/bioinformatics/btq033 (2010).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Robinson, J. T. et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 6, 256–258, https://doi.org/10.1016/j.cels.2018.01.001 (2018).
Liu, K. et al. Chromosome Samples Preparation and Karyotype Analysis of Pomfret (Pampus argenteus). Progr. Fish. Sci. 38, 64–69, https://doi.org/10.11758/yykxjz.20161107001 (2017).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49, https://doi.org/10.1093/nar/gkr1293 (2012).
Bandi, V. et al. in Plant Bioinformatics: Methods and Protocols (ed. Edwards, D.). 285–308 (Springer US, 2022).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Feng, C., Dai, M., Liu, Y. & Chen, M. Sequence repetitiveness quantification and de novo repeat detection by weighted k-mer coverage. Brief. Bioinform. 22, bbaa086, https://doi.org/10.1093/bib/bbaa086 (2021).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268, https://doi.org/10.1093/nar/gkm286 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Hu, J., Zheng, Y. & Shang, X. MiteFinderII: a novel tool to identify miniature inverted-repeat transposable elements hidden in eukaryotic genomes. BMC Med. Genom. 11, 101, https://doi.org/10.1186/s12920-018-0418-y (2018).
Xiong, W., He, L., Lai, J., Dooner, H. K. & Du, C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc. Natl. Acad. Sci. USA 111, 10263–10268, https://doi.org/10.1073/pnas.1410068111 (2014).
Gremme, G., Steinbiss, S. & Kurtz, S. GenomeTools: A Comprehensive Software Library for Efficient Processing of Structured Genome Annotations. IEEE/ACM Trans. Comput. Biol. Bioinform. 10, 645–656, https://doi.org/10.1109/TCBB.2013.68 (2013).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Gish W. AB-BLAST, version 3.0 http://blast.advbiocomp.com/ (2009).
van Dongen, S. & Abreu-Goodger, C. in Bacterial Molecular Networks: Methods and Protocols (eds. Van Helden, J., Toussaint, A. & Thieffry, D.) 281–295 (Springer New York, 2012).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152, https://doi.org/10.1093/bioinformatics/bts565 (2012).
Steinegger, M. & Söding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028, https://doi.org/10.1038/nbt.3988 (2017).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinformatics 25, 4.10.11–4.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Tempel, S. in Mobile Genetic Elements: Protocols and Genomic Applications (ed Bigot, Y.) 29–51 (Humana Press, 2012).
Chan, P. P. & Lowe, T. M. in Gene Prediction: Methods and Protocols (ed. Kollmar, M.) 1–14 (Springer New York, 2019).
Seemann, T. Barrnap 0.9: Rapid ribosomal RNA prediction. Available at: https://github.com/tseemann/barrnap.
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200, https://doi.org/10.1093/nar/gkaa1047 (2021).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Zhang, Q., Shi, Q. & Shao, M. Accurate assembly of multi-end RNA-seq data with Scallop2. Nat. Comput. Sci. 2, 148–152, https://doi.org/10.1038/s43588-022-00216-1 (2022).
Nip, K. M. et al. RNA-Bloom enables reference-free and reference-guided sequence assembly for single-cell transcriptomes. Genome Res. 30, 1191–1200, https://doi.org/10.1101/gr.260174.119 (2020).
Gilbert, D. G. Genes of the pig, Sus scrofa, reconstructed with EvidentialGene. PeerJ 7, e6374, https://doi.org/10.7717/peerj.6374 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014, https://doi.org/10.1093/bioinformatics/btad014 (2023).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Venturini, L., Caim, S., Kaithakottil, G. G., Mapleson, D. L. & Swarbreck, D. Leveraging multiple transcriptome assembly methods for improved gene structure annotation. GigaScience 7, giy093, https://doi.org/10.1093/gigascience/giy093 (2018).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinform. 4, 41, https://doi.org/10.1186/1471-2105-4-41 (2003).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462, https://doi.org/10.1093/nar/gkv1070 (2016).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314, https://doi.org/10.1093/nar/gky1085 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP479325 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_036321115 (2024).
Wei, J. The first high-quality chromosome-level genome assembly of the silver pomfret (Pampus argenteus). figshare https://doi.org/10.6084/m9.figshare.24155052 (2023).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067, https://doi.org/10.1093/bioinformatics/btm071 (2007).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Nos. 31872195 and 32270472) and Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB42000000).
Author information
Authors and Affiliations
Contributions
Y.X. and J.L. conceived the research project. Y.X. and J.L. collected the samples. J.W. and Y.X. performed the analyses. J.W., Y.X., J.L., K.X., A.H. and K.L. wrote and revised the manuscript. Correspondence and requests for materials should be addressed to J.L. and K.X.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, J., Xiao, Y., Liu, J. et al. Chromosome-level genome assembly of the silver pomfret Pampus argenteus. Sci Data 11, 234 (2024). https://doi.org/10.1038/s41597-024-03070-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03070-0
