
Abstract
Triplophysa yarkandensis, a species of freshwater fish endemic to Xinjiang, China, is currently classified as endangered. The objective of this study was to obtain the chromosome-level genome of T. yarkandensis using PacBio and Hi-C techniques. The PacBio sequencing technology resulted in an assembly of 520.64 Mb, with a contig N50 size of 1.30 Mb. Hi-C data was utilized for chromosome mapping, ultimately yielding 25 chromosome sequences. The success rate of chromosome mapping was 93%, with a scaffold N50 of 19.14 Mb, and a BUSCO evaluation integrity of 94.1%. The genome of T. yarkandensis encompasses 25,505 predicted protein-coding genes, with a total of 30,673 proteins predicted. The BUSCO evaluation integrity for predicted protein-coding genes was found to be 91.5%. Additionally, the genome contained a genomic repeat sequence accounting for 27.29% of its total length. Future research employing comparative genomics holds considerable importance in elucidating the molecular mechanisms behind saline-alkali adaptation and ensuring the conservation of biological resources.
Similar content being viewed by others


Background & Summary
Food security is a fundamental challenge in the context of human survival and development. Aquatic foods, known as blue foods, are abundant in essential micronutrients and fatty acids while imposing lower environmental burdens. These foods offer protein and valuable nutrients to billions of people, particularly in developing countries1,2,3,4. In comparison to capture fisheries, aquaculture continues to dominate global blue food production and holds promise for meeting food demand and addressing malnutrition2,5. Inland aquaculture, excluding mariculture, significantly contributes to global food security, particularly in the global south6,7,8. However, freshwater resources and arable land pose primary constraints to the growth of inland aquaculture industries. Arid regions, covering approximately 6.1 billion hectares or 41% of the Earth’s land area, constitute a substantial part of the planet’s landmass9. Expanding inland aquaculture in arid areas represents a crucial pathway for industry development. Advancements in cultivation techniques have facilitated the robust development of aquaculture in arid regions, particularly in Africa. This not only mitigates food crises to some extent but also drives overall national and societal progress10.
The Tarim River, China’s longest inland river, serves as the main river of the southern Xinjiang Autonomous Region11. The Tarim River Basin is characterized by arid conditions, including limited precipitation, high evaporation rates, sparse vegetation, minimal runoff, severe water salinization, and a simple native fish fauna12. To utilize saline-alkaline water resources and diversify animal protein sources, several euryhaline non-native fish species, such as Ctenopharyngodon della, Cyprinus carpio, and Carassius auratus, have been introduced for saline-alkaline fisheries. Unfortunately, this practice poses significant ecological risks13,14,15. Furthermore, various water storage and diversion projects have been implemented along the upstream to downstream axis of the Tarim River Basin to alleviate water scarcity16,17. The combination of hydraulic engineering projects and the introduction of non-native fish species for aquaculture can facilitate fish invasions and lead to a drastic decline in indigenous fish populations and faunal homogeneity18,19,20.
Breeding euryhaline native fish species may offer an effective solution for mitigating the ecological impacts resulting from biological invasions and conserving native fish populations through artificial propagation and release15,21. T. yarkandensis, exclusively distributed in the Tarim River Basin, is an euryhaline indigenous fish species with aquaculture potential22,23,24. This species is currently under serious threat and has been included in the list of key protected wild animals for the Xinjiang Uygur Autonomous Region (https://www.xinjiang.gov.cn/xinjiang/zfgbml/202301/2dff780e69894c2cbe56a7b7866e58ca.shtml). Elucidating the complete genome of T. yarkandensis not only provides insights into breeding techniques but also offers valuable suggestions for its protection. Therefore, this study combines PacBio long-read sequencing and high-throughput chromosome conformation capture (Hi-C) technology to generate a high-quality, chromosome-level reference genome of T. yarkandensis. This achievement will assist in developing effective protection strategies for this species and serve as a basis for exploring adaptive evolution in arid regions.
Methods
Sample collection and sequencing
In August 2021, a female T. yarkandensis (Fig. 1) was captured at the sampling location of Yarkant River (76°30′56′′ E, 37°59′5′′ N). This species was identified according to their morphological features as described in Fauna Sinica (Osteichthyes: Cyprinifores) and The fishes of the Qinghai-Xizang Plateau. Following anesthesia with MS-222 and disinfection, white muscle tissue was preserved in liquid nitrogen for genomic DNA sequencing. The QIAGEN Genomic-tip 100/G kit was employed for genomic DNA extraction from T. yarkandensis, and high-quality DNA was utilized for subsequent library preparation and high-throughput sequencing.

The morphological image of female Triplophysa yarkandensis collected in the Yarkant River.
To construct a 20 kb long-read sequencing library (SMRT bell library), 10 µg of DNA was utilized. Once the library passed the quality assessment, PacBio Sequel was used for sequencing, following the desired data volume requirements25. Sequencing was conducted using the Sequel Binding Kit 2.0, Sequel Sequencing Kit 2.1, and Sequel SMRT Cell 1 M v2, and the resulting data was processed using SMRT LINK 5.0 software. For Hi-C sequencing, the process began with Hi-C biotin labeling and genomic DNA extraction26. The captured DNA was subjected to end repair, poly A tailing, adapter ligation, evaluation of PCR amplification cycles, and purification. After qualifying the library inspection, the library was pooled based on the effective concentration and the target offline data volume for HiSeq sequencing.
In order to assist in genomic annotation, total RNA was extracted from six tissues involving kidney, liver, gonad, muscle, brain and gill. The cDNA library was constructed using mixed RNA samples, and the Illumina HiSeq X-Ten platform was employed for sequencing.
Genome assembly
The SMRTbell libraries were subjected to sequencing on a PacBio Sequel II system. The consensus reads, also known as HiFi reads, were generated using the ccs software (https://github.com/pacificbiosciences/unanimity) with the parameter ‘-minPasses 3’. To enhance the quality and validate the assemblies, we generated 24.6 Gb of PacBio HiFi reads for this specific sample (Table 1). These HiFi reads, which are long (approximately 15 kb) and highly accurate ( > 99%), were assembled using Hifiasm27 (https://github.com/chhylp123/hifiasm). To rectify any errors in the primary assembly, Illumina-derived short reads were employed, and remaining errors were corrected using pilon28,29 (v1.23). As a result, the T. yarkandensis genome assembly reached a total length of approximately 520.6 Mb, consisting of 1707 contigs, with a ContigN50 value of 1.3 Mb (Table 2).
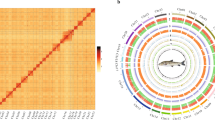
For the anchored contigs, a total of 130.5 Gb of clean read pairs was generated from the Hi-C library (Table 1). These reads were mapped to the polished T. yarkandensis genome using BWA (bwa-0.7.17) with default parameters. Paired reads in which the mate was mapped to a different contig were utilized for Hi-C-associated scaffolding. Various types of invalid reads, including self-ligation, non-ligation, Start NearRsite, PCR amplification, random break, Large Small Fragments, and Extreme Fragments, were filtered out using Hicup software. Subsequently, we successfully clustered 1354 contigs into 25 groups using the agglomerative hierarchical clustering method in 3d-DNA (Fig. 2). 3d-DNA was further employed to order and orient the clustered contigs30,31,32,33. Finally, we obtained the first high-quality assembly at the chromosomal level, with chromosomal lengths ranging from 13.6 Mb to 31.1 Mb, the T. yarkandensis genome was obtained with 245 scafolds and a total length of 521,366,357 bp, encompassing 93% of the total sequence (Table 2).

Triplophysa yarkandensis genome contig contact matrix using Hi-C data. LG1–25 represented for the 25 pseudo-chromosomes. The depth of red color shows the contact density.
Repetitive sequence annotation
In our study, we employed a combination of two methods, namely homology-based and de novo prediction, to identify repeat contents in the T. yarkandensis genome. For the homology-based analysis, we used RepeatMasker (open-4.0.9) with the Repbase TE library to identify known TEs within the genome. In addition, we employed RepeatModeler (http://www.repeatmasker.org) for de novo prediction. RepeatModeler integrates two core de novo repeat-finding programs, RECON (v1.08) and RepeatScout (v1.0.5), to comprehensively discover, refine, and classify consensus models of potential interspersed repeats in the T. yarkandensis genome34,35. Moreover, we conducted a de novo search specifically for long terminal repeat (LTR) retrotransposons using LTR FINDER (v1.0.7), LTR harvest (v1.5.11), and LTR_retriever (v2.7) against the T. yarkandensis genome sequences36,37. Tandem repeats were identified using the Tandem Repeat Finder38 (TRF) package, and Simple Sequence Repeats (SSRs) were detected using MISA (v1.0). Finally, we merged the library files generated from both methods and utilized RepeatModeler to determine the repeat contents. Based on these analyses, we identified a total of 149.99 Mb of repeats in the T. yarkandensis genome (Table 3).
Non-coding RNA annotation
To identify specific gene categories in the T. yarkandensis genome, we utilized various algorithms and databases. The tRNAscan-SE (v1.3.1) algorithm with default parameters was employed to detect tRNA genes. tRNA molecules act as adaptors in biological processes, bridging the three-letter genetic code in messenger RNA (mRNA) with the twenty-letter amino acid code in proteins. For the identification of rRNA genes, we used RNAmmer (v1.2) with the parameters “-S euk -m lsu,ssu,tsu”. rRNAs are integral components of the ribosome and play a crucial role in protein synthesis. snoRNAs, a class of small RNA molecules, guide chemical modifications of other RNAs, including ribosomal RNAs, transfer RNAs, and small nuclear RNAs. MiRNAs and snRNAs were identified by CMSAN (v1.1.2) software against the Rfam (v14.0) database with default parameters (Table 4)39,40,41.
Protein-coding gene prediction and annotation
To predict protein-coding genes in the T. yarkandensis genome, we employed three methods: ab initio gene prediction, homology-based gene prediction, and RNA-Seq-guided gene prediction. Prior to gene prediction, the assembled genome underwent hard and soft masking using RepeatMasker. Ab initio gene prediction was performed using Augustus (v. 3.3.3)42. The gene predictors’ models were trained using a set of high-quality proteins derived from the RNA-Seq dataset. For homology-based gene prediction, we utilized MAKER (v. 2.31.10)43. Protein and transcript sequences were aligned to the genome assembly, and coding genes were predicted using maker with default parameters. RNA-Seq-guided gene prediction involved aligning clean RNA-Seq reads to the genome using hisat2 (v2.0.0). Gene structures were generated using Trinity (v2.3.2), Transdecoder (v2.01), and MAKER. To integrate the predictions from the three methods and generate gene models, we employed EVidenceModeler (EVM, v1.1.1)44. The resulting output comprised consistent and non-overlapping sequence assemblies, which described the gene structures. Overall, a total of 25,505 protein-coding genes with an average length of 158,469 bp were predicted in the assembled T. yarkandensis genome. The predicted protein-coding gene BUSCO integrity using the Actinopterygii odb9 database was 91.5%. The number of predicted proteins was 30,673.
For inferring gene functions, we conducted alignments to various protein databases, including the National Center for Biotechnology Information (NCBI) Non-Redundant (NR), TrEMBL, KOG, and SwissProt, using BLASTP (NCBI BLAST v2.6.0+). Additionally, we utilized the Kyoto Encyclopedia of Genes and Genomes (KEGG) database with an E-value threshold of 1E-5. Protein domains were annotated using PfamScan (v1.6) based on the PFAM and InterPro protein databases. Gene Ontology (GO) IDs for each gene were obtained from Blast2GO. In total, approximately 23,288 (about 91%) of the predicted protein-coding genes in T. yarkandensis could be functionally annotated with known genes, conserved domains, and Gene Ontology terms (Table 5).
Data Records
All raw data of the whole genome have been deposited into the National Center for Biotechnology Information (NCBI) SRA database under BioProject accession number PRJNA995909 The genomic PacBio sequencing data were deposited in the SRA at NCBI SRR2535771245 and the Hi-C sequencing data were deposited in the SRA at NCBI SRR2534350746. The RNA sequencing data were deposited in the SRA at SRR2637750347. The assembled genome was deposited in the NCBI Genome with the accession number GCA_033220385.148. Genome annotations, along with predicted coding sequences and protein sequences, can be accessed through the Figshare49.
Technical Validation
Evaluation of the quality of genomic DNA and RNA
Before constructing the DNA library, we assessed the purity (OD260/280 and OD260/230) and concentration of the genomic DNA using Nanodrop (LabTech, USA). To precisely measure the concentration of genomic DNA, we employed Qubit (Thermo Fisher Scientific, USA). By comparing the Qubit concentration with the Nanodrop concentration, we determined the sample purity. Additionally, the integrity of the DNA was assessed through agarose gel electrophoresis (1%). RNA purity and integrity were determined using NanoPhotometer spectrophotometer (Implen, USA) and Agilent 2100 bioanalyzer (Agilent Technologies, USA).
Genome assembly integrity assessment
The assembled genome was subjected to BUSCO (v3.1) analysis using OrthoDB to assess its completeness50. Overall, the assembled genome identified 94.1% completeness of the BUSCOs (Actinopterygii odb9) (Fig. 3).

BUSCO assessment results of Triplophysa yarkandensis genome. C represents complete BUSCOs, S represents complete and single-copy BUSCOs, D represents complete and duplicated BUSCOs, F represents fragmented BUSCOs, M represents missing BUSCOs, n represents total BUSCO groups searched.
Code availability
No custom scripts or codes were used in the management and verification of the data sets in this study. All software and pipelines used for data processing were executed according to the manuals and protocols of the bioinformatics software cited above. The specific parameters were described if the default parameters were not applied for data analysis.
References
Gephart, J. A. et al. Environmental performance of blue foods. Nature 597, 360–365 (2021).
Golden, C. D. et al. Aquatic foods to nourish nations. Nature 598, 315–320 (2021).
Garlock, T. et al. Aquaculture: The missing contributor in the food security agenda. Glob. Food Sec. 32, 100620 (2022).
Tigchelaar, M. et al. The vital roles of blue foods in the global food system. Glob. Food Sec. 33, 100637 (2022).
FAO. The State of World Fisheries and Aquaculture 2022. Towards Blue Transformation (Food and Agriculture Organization of the United Nations, 2022).
Zhang, W. et al. Aquaculture will continue to depend more on land than sea. Nature 603, E2–E4 (2022).
Wang, Q. et al. Freshwater aquaculture in PR China: Trends and prospects. Rev. Aquac. 7, 283–302 (2015).
Subasinghe, R., Soto, D. & Jia, J. Global aquaculture and its role in sustainable development. Rev. Aquac. 1, 2–9 (2009).
Millennium Ecosystem Assessment. Ecosystems And Human Well-being: Wetlands and Water (World Resources Institute, 2005).
Mohamed, S., Magdy, E. M., Mona, S. & Mansour, E. M. Aquaculture in Egypt: Insights on the Current Trends and Future Perspectives for Sustainable Development. Rev. Fish. Sci. Aquac. 26, 99–110 (2018).
Tian, J. et al. A cooperative game model with bankruptcy theory for water allocation: a case study in China Tarim River Basin. Environ. Sci. Pollut. Res. 29, 2353–2364 (2022).
Qi, Y., Zhao, W., Li, Y. & Zhao, Y. Environmental and geological changes in the Tarim Basin promoted the phylogeographic formation of Phrynocephalus forsythii (Squamata: Agamidae). Gene 768, 145264 (2021).
Wang, X. N., Gu, Y. G. & Wang, Z. H. Fingerprint characteristics and health risks of trace metals in market fish species from a large aquaculture producer in a typical arid province in Northwestern China. Environ. Technol. Innov. 19, 100987 (2020).
Xu, W. L., Wang, H. Q. & Li, Y. H. [国内外盐碱水域分布及水产养殖应用] (Distribution and Aquaculture Application of Saline Alkali Water at Home and Abroad). [中国水产] (China Fish. 7, 50–53) (2021).
Mo, B. L., Ai, S. F. & Li, Y. H. [我国东北和西北地区不同类型盐碱水体养殖鱼类研究现状] (Research Status of Fish Culture in Different Types of Saline Alkali Water Bodies in Northeast and Northwest China). [中国水产] (China Fish. 6, 72–74) (2022).
Sun, Q. [塔里木河干流防洪与洪水资源利用探析] (Discussion on flood control and flood resources utilization in the main stream of Tarim river). [水利技术监督] (Tech. Superv. Water Resour. 2, 166–169) (2022).
Yan, L. L. [探讨塔里木河水利工程项目规划建设实施策略] (Disscusion on strategy of planning and construction of Tarim River water conservancy project). [水利科学与寒区工程] (Hydro Sci. Cold Zone Eng. 5, 126–128) (2022).
Sun, S. et al. Genetic Diversity and Population Structure of Hemiculter leucisculus (Basilesky, 1855) in Xinjiang Tarim River. Genes 13, 1790 (2022).
Li, G. G., Feng, C. G., Tang, Y. T., Zhang, R. Y. & Zhao, K. [新疆内陆河土著鱼类资源调查] (Survey of native fish resources in inland river system in Xinjiang). [甘肃农业大学学报] (Gansu Agric. Univ. 52, 22–27) (2017).
Chen, G. Z., Qiu, Y. P. & Li, L. P. [塔里木盆地鱼类入侵及区演演变趋势] (Fish invasions and changes in the fish fauna of the Tarim Basin). [生态学报] (Acta Ecol. Sin. 37, 700–714) (2017).
Chen, X. Z., Lai, Q. F., Mo, Z. L., Gao, H. Y. & Han, C. X. [盐碱水绿뉲养殖技术模式] (Saline alkali water green breeding technology model). [中国水产] (China Fish. 9, 61–63) (2020).
Chen, S. A. et al. [塔里木河叶尔羌高原鳅盐碱耐受性研究] (Studies on the Tolerance of Triplophysa (Hedinichthys) yarkandensis (Day) to Salinity and Alkalinity). [四川动物] (Sichuan Zool. 35, 523–527) (2016).
Zhou, X. Y. et al. Genetic Diversity and Population Differentiation of Kashgarian Loach (Triplophysa yarkandensis) in Xinjiang Tarim River Basin. Biology 10, 734 (2021).
Chen, S. A., Hou, J. L., Yao, N., Xie, C. X. & Li, D. Comparative transcriptome analysis of Triplophysa yarkandensis in response to salinity and alkalinity stress. Comp. Biochem. Physiol. Part D Genomics Proteomics 33, 100629 (2020).
Peng, Y. et al. Chromosome-level genome assembly of the Arctic fox (Vulpes lagopus) using PacBio sequencing and Hi-C technology. Mol. Ecol. Resour. 21, 2093–2108 (2021).
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Flusberg, B. A. et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 1–11 (2015).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 25, 955–964 (1997).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 18 (2008).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–8 (2007).
Chan, P. P. & Lowe, T. M. in Gene prediction: methods and protocols (ed. Kollmar, M.) Ch. 1 (Humana Press, 2019).
Nawrocki, E. P. & Eddy, S. C. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 491 (2011).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25357712 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25343507 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26377503 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_033220385.1 (2023).
She, J., Chen, S., Liu X. & Huo B. Genome annotations of Triplophysa yarkandensis. Figshare https://doi.org/10.6084/m9.figshare.c.6729378.v1 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This study is funded by the Finance Special Fund of the Ministry of Agriculture and Rural Affairs “Fisheries Resources and Environment Survey in the Key Water Areas of Northwest China” (No. 2130111), National Natural Science Foundation of China (No. 31360635) and Corps Science and Technology Bureau Key Areas of Science and Technology Public Relations Plan (No. 2022DB019). The funders didn’t have any role in study design, data collection and analysis, decision to publish, or preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
B.H. and S.C. conceived of the project. J.S. and X.L. collected the samples and extracted the genomic DNA. S.C., J.S. and X.L. performed the data analysis and wrote the manuscript. J.S. contributed to the data analyses. B.H. revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
She, J., Chen, S., Liu, X. et al. Chromosome-level assembly of Triplophysa yarkandensis genome based on the single molecule real-time sequencing. Sci Data 11, 39 (2024). https://doi.org/10.1038/s41597-023-02900-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02900-x
