
Abstract
Oryza coarctata (2n = 4X = 48, KKLL) is an allotetraploid, undomesticated relative of rice and the only species in the genus Oryza with tolerance to high salinity and submergence. Therefore, it contains important stress and tolerance genes/factors for rice. The initial draft genome published was limited by data and technical restrictions, leading to an incomplete and highly fragmented assembly. This study reports a new, highly contiguous chromosome-level genome assembly and annotation of O. coarctata. PacBio high-quality HiFi reads generated 460 contigs with a total length of 573.4 Mb and an N50 of 23.1 Mb, which were assembled into scaffolds with Hi-C data, anchoring 96.99% of the assembly onto 24 chromosomes. The genome assembly comprises 45,571 genes, and repetitive content contributes 25.5% of the genome. This study provides the novel identification of the KK and LL genome types of the genus Oryza, leading to valuable insights into rice genome evolution. The chromosome-level genome assembly of O. coarctata is a valuable resource for rice research and molecular breeding.
Similar content being viewed by others

Background & Summary
Oryza coarctata is the only halophyte species in the genus Oryza1, exhibiting distinct natural traits, including high tolerance to submersion and salinity2 (Fig. 1), as well as unique leaf anatomical features, such as the presence of Kranz anatomy (Fig. 2). These features are a result of high selection pressure, allowing its propagation in a wide range of ecological conditions, from submerged saline to non-saline terrestrial soils3. Oryza coarctata is primarily found in coastal areas across Bangladesh, India, Sri Lanka, and Malaysia1,2,4,5.

Photographs of rice plants. (a) Oryza sativa. (b) Oryza coarctata.

Transverse section of young leaf of O. coarctata under light microscope.
The history of research on Oryza coarctata is complex. Until 1999, it was excluded from the rice genus due to some morphological differences and was classified as Porteresia coarctata. However, a study on the evolutionary relationship between various species and genome types of the rice genus demonstrated that it belongs to the genus Oryza, and it was named Oryza coarctata. It was also determined to be allotetraploid6. A subsequent study identified the genome types, KK and LL, from its allotetraploid genome7.
Numerous studies have demonstrated that Oryza coarctata offers a wealth of genetic resources for rice breeding research, including salt resistance, drought resistance, and improved photosynthetic efficiency2,3,8,9. Therefore, sequencing a high-quality chromosome-level genome of Oryza coarctata is essential for genomics research and can provide new insights into the evolutionary studies of rice. In our study, we sequenced a high-quality chromosome-level genome of Oryza coarctata using PacBio HiFi reads (~59.99 X) and Hi-C data. We also identified the genome types, KK and LL, from its allotetraploid genome, which can provide new insights into the evolution of genus Oryza.
Methods
Staining method of O. coarctata leaf
Fresh leaf samples from three-leaf-stage plants were selected and snipped into 1cm-by-1cm squares. Immediately, these samples were placed in Carnoy’s fixative (a mixture of ethanol and acetic acid in a 3:1 ratio). After being fixed at room temperature for 48 hours, the samples were shifted to 75% ethanol for permanent preservation. If proceeding with subsequent experiments, slices were manually prepared using a double blade, perpendicular to the leaf veins. The prepared filamentous sections were stained using 0.1% methylene blue for 3 minutes. Once stained, excess dye was rinsed off, and the samples were set on microscope slides for observation under a light microscope.
Genome sequencing
We began with fresh Oryza coarctata seedlings, sourced from the Koyra Riverbank in Khulna district, Bangladesh (22.77 N latitude and 89.48 E longitude), and utilized them for superior DNA extraction. Our extraction protocol involved initial fragmentation of DNA samples via a g-TUBE, subsequent repair of damaged DNA, end repair, and ligation with dumbbell-shaped adapters. After an exonuclease digestion, we screened the DNA fragments using BluePippin, forming the PacBio sequencing library. For the Hi-C library, we employed formaldehyde for crosslinking cells, thereby maintaining both intra- and intermolecular interactions, and preserving the cell’s 3D structure. Following crosslinking, we employed the restriction enzyme HindIII for DNA digestion and incorporated biotin-labeled nucleotides during the end repair stage. After ligation of the repaired ends, we circularized the DNA, which enabled the identification of interactive DNA positions in further sequencing and analyses. We then decrosslinked and purified the DNA, fragmenting it into 300–700 bp lengths. Interaction-representing biotin-labeled DNA fragments were captured with streptavidin magnetic beads, thereby facilitating library construction. We sequenced the PacBio library on the PacBio Sequel II system (CCS mode), generating ~34.4 Gb clean data (~59.99 × ), and all the CCS reads exhibited an N50 of ~15.2 kb. The Hi-C library, sequenced on the Illmina NovaSeq 6000 (PE150), produced ~73.76 Gb clean data (Table 1).
RNA sequencing
RNA was extracted from the root, stem, and leaf tissues of Oryza coarctata plants. Following extraction, these RNA samples were combined in equal measures, from which an RNA-seq library was prepared. The transcriptomes were sequenced on the Illmina NovaSeq. 6000 platform, operated by the Biomarker Technology Company, Beijing, China. The sequencing process produced 13.23 Gb of short-read RNA-seq data (Table 1), which was used for predicting whole-genome protein-coding genes.
Genome assembly
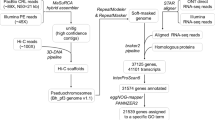
We used hifiasm software10 to assemble the high-quality HiFi reads, which yielded a total of 460 contigs with a total length of 573.4 Mb and an N50 of 23.1 Mb (Table 2). Using Hi-C data, more than 96% of the contigs have been anchor to 24 chromosomes (Fig. 3). Subsequently, we joined contigs into scaffolds using Hi-C clean data. The 46.55% of unique mapped read pairs were valid interaction pairs and were used for correction of scaffolds and clustered, ordered and orientated scaffolds onto chromosomes by LACHESIS11. Before the assembly of chromosomes, we first executed a preassembly phase to correct errors in scaffolds, requiring the division of scaffolds into average segments of 50 kb. The Hi-C data were then mapped to these segments using the BWA (version 0.7.10-r789)12 software. We preserved uniquely mapped data for assembly operations using LACHESIS software. We manually checked any pair of segments that exhibited inconsistent connection with the raw scaffold data. These corrected scaffolds were subsequently assembled using LACHESIS. After this process, we manually adjusted any placement and orientation errors that exhibited distinct chromatin interaction patterns. In the end, we anchored 24 scaffolds, amounting to 96.99% total length, to the chromosomes (Fig. 4, Table 3).

Genome features of the O. coarctata. (a) Chromosome ideograms of O. coarctata genome. (b) TE density. (c) Gene density. (d) GC content. (e) Syntenic blocks of genome sequence.

Hi-C contact map of the chromosome-level assembly of O. coarctata. The intensity of interactions was calculated using a bin size of 300,000 bp.
Repeat annotation
Transposon element (TE) and tandem repeat were masked and annotated by the following workflows. TE were identified by a combination of homology-based and de novo approaches. We first customized a de novo repeat library of the genome using RepeatModeler13, which can automatically execute two de novo repeat finding programs, including RECON (version 1.08)14 and RepeatScout15.Then full-length long terminal repeat retrotransposons (fl-LTR-RTs) were identified using both LTRharvest16 and LTR_finder17. The high-quality intact fl-LTR-RTs and non-redundant LTR library were then produced by LTR_retriever18. Non-redundant species-specific TE library was constructed by combining the de novo TE sequences library above with the known Repbase (version 19.06)19, REXdb (V3.0)20 and Dfam (v3.2)21 database. Final TE sequences in the Oryza coarctata genome were identified and classified by homology search against the library using RepeatMasker v4.1022. Tandem repeats were annotated by Tandem Repeats Finder23 and MIcroSAtellite identification tool (MISA v2.1)24 (Tables 4, 5).
Gene prediction annotation of the genome
Gene prediction is typically performed using three methods: homology-based prediction, de novo prediction, and transcriptome-based prediction. De novo prediction was performed using Augustus v2.425 and SNAP (2006-07-28)26. Homology-based prediction was performed using GeMoMa v1.727 based on homologous species. Transcriptome-based prediction was conducted using both reference-based and de novo transcriptome assembly. Reference-based transcriptome assembly was performed using Hisat v2.0.428 and Stringtie v1.2.329, and GeneMarkS-T v5.130 was used for gene prediction. De novo transcriptome assembly was performed using Trinity v2.1131, and gene prediction was conducted using PASA v2.0.232. Finally, EVM v1.1.133 was used to integrate the results from the three methods, and PASA v2.0.2 was used for annotation, resulting in 45,571 predicted genes (Fig. 5).

The genes that are integrated originated from the distribution maps of three prediction methods.
In order to evaluate the accuracy of gene prediction, we compared the length distributions of protein-coding genes, coding sequences (CDS), exons, and introns of our study species with those from four additional reference species (A. thaliana34, O. brachyantha35, O. punctata36, and O. sativa37). Notably, we did not observe any significant differences in the length distribution of gene features among these species (Fig. 6, Table 6).

Comparisons of gene features among O. coarctata and the four other species (A. thaliana, O. brachyantha, O. punctata and O. sativa). Gene features include gene length, CDS length, exon length and intron length.
Noncoding RNAs annotation
Non-coding RNA (ncRNA) refers to RNA that does not encode proteins, including various types of functional RNAs such as microRNA, rRNA, and tRNA. Different strategies were used to predict different ncRNAs based on their structural characteristics. The tRNA was identified using tRNAscan-SE v1.3.138. The rRNA prediction was mainly based on the Rfam(v 12.0)39 database and predicted using barrnap(v 0.9)40. miRNA was identified using the miRBase41 database, while snoRNA and snRNA were predicted based on the Rfam(v 12.0) database and using Infenal 1.142. A total of 2,804 tRNAs, 9,075 rRNAs, and 157 miRNAs were predicted (Table 7).
Pseudogene prediction
Pseudogenes are sequences similar to functional genes, but they have lost their original function due to mutations such as insertions or deletions. We used GenBlastA v1.0.443 to compare the genome after masking the loci of real genes, in order to identify homologous gene sequences (putative genes). We then used GeneWise v2.4.144 to detect premature stop codons and frameshift mutations in these sequences, and ultimately predicted 28 pseudogenes (Table 8).
Functional annotation of the genome
To annotate the predicted gene sequences, we performed searches against the NR (202009, ftp://ftp.ncbi.nlm.nih.gov/blast/db), EggNOG45, GO46, SWISS-PROT47, and Pfam48 databases. Overall, 96.59% of the genes were annotated in these databases (Table 9).
Discovery of genomic variations among K and L
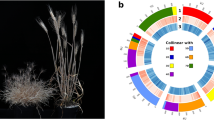
We utilized the syntenic blocks between Oryza coarctata (KKLL) and its related species Oryza puctata (BB) (Fig. 7) to uncover a pairing relationship among the 24 chromosomes. Then, using Subphaser49 based on the principle of K-mer frequency difference between genomes of different species, we successfully separated the heterozygous chromosomes KK (~271 Mb) and LL (~261 Mb) from the Oryza coarctata genome (Fig. 8). A whole-genome synteny analysis was conducted between Subgenome K and Subgenome L using MUMmer, which, as shown in Fig. 9a, revealed a high-level of overall concordance between the K type and L type genomes. To further investigate genomic variations and local differences between the two assemblies, we employed SyRI v1.5 software50. This analysis led to the identification of several Mb-sized structural variations such as inversions, translocations, and duplications (Fig. 9b)

Syntenic blocks between O. coarctata and O. punctata, represented through a linear collinear graph (a) and a dot plot (b).

Phased subgenomes of allotetraploid Oryza coarctata. (a) Chromosomal characteristics (window size: 1 Mb). Rings from outer to inner:(1) Subgenome assignments by a k-Means algorithm. (2) Significant enrichment of subgenome-specific k-mers (blank for non-enriched windows). (3) Normalized proportion of subgenome-specific k-mers. (4–6) Density distribution (count) of each subgenome-specific k-mer set. (7) Density distribution (count) of subgenome-specific LTR-RTs and other LTR-RTs (the most outer, in grey color). (8) Homoeologous blocks of each homoeologous chromosome set. (b) Heatmap and clustering of differential k-mers. (c) Insertion time of subgenome-specific LTR-RTs. (d) Principal component analysis (PCA) of differential k-mers. Points indicate chromosomes.

Whole-genome comparison of the Subgenome1 with Subgenome2 assembly. (a) Dot plot for the syntenic blocks. (b) Chromosome-level local sequence differences.
Data Records
The sequencing data, genome assembly and annotation data reported in this paper have been deposited in the Genome Warehouse in National Genomics Data Center (NGDC), Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation51 under the BioProject accession number PRJCA016514 that is publicly accessible at https://ngdc.cncb.ac.cn/gwh. All the clean genome sequencing data including PacBio long-read data52, Illumina short-read DNA-seq53,54. and Hi-C data55, as well as Illumina short-read RNA sequencing data56 were deposited in the Genome Sequence Archive (GSA)57 of NGDC under the accession number CRA011195. The genome assembly and annotation data have been deposited in the Genome Assembly Sequences and Annotations (GWH) of NGDC under accession number GWHCBHR00000000. The assembled genome has also been deposited in the NCBI assembly with the accession number JAULJY00000000058. The annotation results of repeated sequences, gene structure and functional prediction were deposited in the Figshare database59.
Technical Validation
Assessment of the genome assembly
To evaluate the quality of the assembly, we assessed it from three different perspectives: second-generation data alignment rate, CEGMA evaluation, and BUSCO evaluation. The second-generation data alignment rate was over 99%, indicating the high accuracy of our assembly. Furthermore, the CEGMA evaluation showed that over 98% of the genes and more than 95% of the highly conserved genes were present in the assembly. The BUSCO evaluation also demonstrated the completeness of the assembly, with a score of 97.83% (Tables 10–12).
Moreover, we evaluated the result of Hi-C based pseudo-chromosomes construction. LACHESIS software was utilized to divide and sequence the genome sequences into groups, while also orienting them. Manual mapping and inspection were then performed to obtain the chromosome level genome version. Our manual checks entailed re-examining the raw Hi-C data, confirming the inconsistency, and determining the correct alignment or orientation based on the highest number of supporting read pairs. Furthermore, the adjustment of placement and orientation errors exhibiting obvious discrete chromatin interaction patterns was performed when the chromatin interaction patterns indicated an arrangement inconsistent with the majority of the data. These adjustments were made based on the same principle of choosing the alignment or orientation that was supported by the highest number of read pairs. After the Hi-C assembly and manual heat map adjustments, it was determined that the 24 chromosomes contained a total of 556,116,023 bp genome sequence, accounting for 96.99% of the sequences located on the chromosomes. Among those sequences located on the chromosomes, the sequence and direction could be determined in 554,379,116 bp, accounting for 99.69% of the total sequence located on the chromosomes.
Assessment of the genome annotation
The number of genes supported by each prediction method was counted, and the majority of the genes were predicted using transcriptome-based and homology-based methods, indicating the high quality of the predictions. The embryophyta database of BUSCO contains 1,614 conserved core genes. We used BUSCO v5.0 software to evaluate the completeness of gene prediction, and 96.22% of BUSCO genes were found in our predicted genes, indicating high completeness (Table 13). The accuracy and completeness of gene prediction were evaluated from the overall level by mapping RNA-seq clean data to the assembled genome using Hisat2 software and calculating and summarizing the coverage of annotated exons, introns, and intergenic regions. In this genome, 87.64% of the transcriptome data mapped to the annotated exons, demonstrating the high accuracy of our prediction model (Fig. 10).

Transcriptome Mapping Statistics.
Code availability
The versions, settings and options of software tools used in this work are described below:
(1) Hifiasm: v0.12, default parameters;
(2) CEGMA: v2.5, default parameters;
(3) BUSCO: v5, default parameters;
(4) HiC-Pro: v2.10.0, default parameters;
(5) BWA: 0.7.10-r789, default parameters;
(6) LACHESIS, parameters: CLUSTER_MIN_RE_SITES = 78 CLUSTER_MAX_LINK_DENSITY = 2 ORDER_MIN_N_RES_IN_TRUNK = 15 ORDER_MIN_N_RES_IN_SHREDS = 15;
(7) Circlize: 0.4.10, default parameters;
(8) Diamond v0.9.29.130, default parameters;
(9) MCScanX, default parameters;
(10) JCVI: v0.9.13, default parameters;
(11) VGSC: v2.0, default parameters;
(12) RepeatModeler2: v2.0.1, default parameters;
(13) RECON: v1.0.8, default parameters;
(14) RepeatScout: v1.0.6, default parameters;
(15) LTR_retriever: v2.8, default parameters;
(16) LTRharvest: v1.5.9, default parameters;
(17) LTR_FINDER: v1.1, default parameters;
(18) RepeatMasker: v4.1.0, default parameters;
(19) MISA: v2.1, default parameters;
(20) TRF: v409, parameters:1 1 2 80 5 200 2000 –d -h;
(21) Augustus: v2.4, default parameters;
(22) SNAP: v2006-07-28, default parameters;
(23) GeMoMa: v1.7, default parameters;
(24) Hisat: v2.0.4, default parameters;
(25) Stringtie: v1.2.3, default parameters;
(26) GeneMarkS-T: v5.1, default parameters;
(27) Trinity: v2.11, default parameters;
(28) PASA: v2.0.2, default parameters;
(29) EVM: v1.1.1, default parameters;
(30) EggNOG-mapper: v2, default parameters;
(31) tRNAscan-SE: v1.3.1, default parameters;
(32) Barrnap v0.9, default parameters;
(33) Infenal v1.1, default parameters;
(34) GenBlastA: v1.0.4, default parameters;
(35) GeneWise: v2.4.1, default parameters;
(36) InterProScan: v5.34-73.0, default parameters;
(37) Subphaser: v1.2, parameters: -q 90;
(38) SyRi: v1.5, default parameters;
(39) Plotsr: v1.0.0, default parameters;
No customized code was developed by the authors.
References
Chowrasia, S. et al. Oryza coarctata roxb. The wild Oryza genomes, 87–104 (2018).
Bansal, J., Gupta, K., Rajkumar, M. S., Garg, R. & Jain, M. Draft genome and transcriptome analyses of halophyte rice Oryza coarctata provide resources for salinity and submergence stress response factors. Physiol Plant 173, 1309–1322 (2021).
Chowrasia, S., Nishad, J., Pandey, R. & Mondal, T. K. Oryza coarctata is a triploid plant with initial events of C4 photosynthesis evolution. Plant Sci 308, 110878 (2021).
Bal, A. & Dutt, S. Mechanism of salt tolerance in wild rice (Oryza coarctata Roxb). Plant and soil 92, 399–404 (1986).
Sengupta, S. & Majumder, A. L. Porteresia coarctata (Roxb.) Tateoka, a wild rice: a potential model for studying salt‐stress biology in rice. Plant, cell & environment 33, 526–542 (2010).
Lu, B. R. & Ge, S. Oryza coarctata: the name that best reflects the relationships of Porteresia coarctata (Poaceae: Oryzeae). Nordic Journal of Botany 23, 555–558 (2003).
Lu, F. et al. Comparative sequence analysis of MONOCULM1-orthologous regions in 14 Oryza genomes. Proceedings of the National Academy of Sciences 106, 2071–2076 (2009).
Mondal, T. K., Rawal, H. C., Gaikwad, K., Sharma, T. R. & Singh, N. K. First de novo draft genome sequence of Oryza coarctata, the only halophytic species in the genus Oryza. F1000Res 6, 1750 (2017).
Mondal, T. K. et al. Draft genome sequence of first monocot-halophytic species Oryza coarctata reveals stress-specific genes. Sci Rep 8, 13698 (2018).
Lieberman-Aiden, E. et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 326, 289–293 (2009).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology 31, 1119−+ (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America 117, 9451–9457 (2020).
Bao, Z. R. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Research 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, I351–I358 (2005).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. Bmc Bioinformatics 9 (2008).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Ou, S. J. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiology 176, 1410–1422 (2018).
Bao, W.D., Kojima, K.K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6 (2015).
Neumann, P., Novak, P., Hostakova, N. & Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mobile DNA 10 (2019).
Wheeler, T. J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Research 41, D70–D82 (2013).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4 10 1–4 10 14 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–80 (1999).
Beier, S., Thiel, T., Munch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–44 (2008).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res 44, e89 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–60 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–5 (2015).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43, e78 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644–U130 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31, 5654–5666 (2003).
Haas, B.J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology 9 (2008).
de, A.G.I.g.t.o.g.g. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. nature 408, 796–815 (2000).
Chen, J. et al. Whole-genome sequencing of Oryza brachyantha reveals mechanisms underlying Oryza genome evolution. Nature Communications 4, 1595 (2013).
Stein, J. C. et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nature genetics 50, 285–296 (2018).
Goff, S. A. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100 (2002).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–64 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Research 33, D121–D124 (2005).
Loman, T. A novel method for predicting ribosomal RNA genes in prokaryotic genomes. (2017).
Griffiths-Jones, S., Grocock, R. J., van Dongen, S., Bateman, A. & Enright, A. J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34, D140–4 (2006).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
She, R., Chu, J. S. C., Wang, K., Pei, J. & Chen, N. S. genBlastA: Enabling BLAST to identify homologous gene sequences. Genome Research 19, 143–149 (2009).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Research 14, 988–995 (2004).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research 47, D309–D314 (2019).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research 44, D457–D462 (2016).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research 31, 365–370 (2003).
Finn, R. D. et al. Pfam: clans, web tools and services. Nucleic Acids Research 34, D247–D251 (2006).
Jia, K. H. et al. SubPhaser: a robust allopolyploid subgenome phasing method based on subgenome-specific k-mers. New Phytol 235, 801–809 (2022).
Goel, M., Sun, H., Jiao, W.-B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome biology 20, 1–13 (2019).
Chen, M. et al. Genome Warehouse: a public repository housing genome-scale data. Genomics, proteomics & bioinformatics 19, 584–589 (2021).
NGDC Genome Sequence Archive (GSA) https://ngdc.cncb.ac.cn/gsa/browse/CRA011195/CRR792593 (2023).
NGDC Genome Sequence Archive (GSA) https://ngdc.cncb.ac.cn/gsa/browse/CRA011195/CRR792595 (2023).
NGDC Genome Sequence Archive (GSA) https://ngdc.cncb.ac.cn/gsa/browse/CRA011195/CRR792596 (2023).
NGDC Genome Sequence Archive (GSA) https://ngdc.cncb.ac.cn/gsa/browse/CRA011195/CRR792594 (2023).
NGDC Genome Sequence Archive (GSA) https://ngdc.cncb.ac.cn/gsa/browse/CRA011195/CRR792597 (2023).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics Proteomics Bioinformatics (2021).
GenBank https://identifiers.org/ncbi/insdc:JAULJY000000000.1 (2023).
Zhao, H. A high-quality chromosome-level wild rice genome of Oryza coarctata. figshare. https://doi.org/10.6084/m9.figshare.23938590.v1 (2023).
Acknowledgements
This research was funded by National Key Research and Development Program of China (2022YFF1001700). We thank Dr. Hongbing Liu for his comments and suggestions of improvement to the manuscript.
Author information
Authors and Affiliations
Contributions
M.H.A.R., M.K., M.S.K., Z.I.S. and M.S.R. contribute to the collection, preservation, and characterisation of Oryza coarctata germplasm. H.Z., W.W. and Y.Y. performed data analysis and drafted the manuscript. Z.W., J.S. and K.Y. performed RNA preparation, sequencing and data analysis. The idea was developed by Z.Z. and M.S.R., who also designed the experiment and wrote and edited the manuscript. All authors provided feedback on the draft manuscript and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, H., Wang, W., Yang, Y. et al. A high-quality chromosome-level wild rice genome of Oryza coarctata. Sci Data 10, 701 (2023). https://doi.org/10.1038/s41597-023-02594-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02594-1
