
Abstract
Wild wheat relatives have been explored in plant breeding to increase the genetic diversity of bread wheat, one of the most important food crops. Aegilops umbellulata is a diploid U genome-containing grass species that serves as a genetic reservoir for wheat improvement. In this study, we report the construction of a chromosome-scale reference assembly of Ae. umbellulata accession TA1851 based on corrected PacBio HiFi reads and chromosome conformation capture. The total assembly size was 4.25 Gb with a contig N50 of 17.7 Mb. In total, 36,268 gene models were predicted. We benchmarked the performance of hifiasm and LJA, two of the most widely used assemblers using standard and corrected HiFi reads, revealing a positive effect of corrected input reads. Comparative genome analysis confirmed substantial chromosome rearrangements in Ae. umbellulata compared to bread wheat. In summary, the Ae. umbellulata assembly provides a resource for comparative genomics in Triticeae and for the discovery of agriculturally important genes.
Similar content being viewed by others

Background & Summary
The genus Aegilops contains several grass species, commonly referred to as goatgrass. The genus comprises at least 23 diploid and polyploid species and six different genomes (C, D, M, N, S, and U)1,2,3,4. Aegilops species belong to the same tribe as the major cereal crops bread wheat (Triticum aestivum, 2n = 6x = 42; AABBDD genome), durum wheat (Triticum durum, 2n = 4x = 28; AABB genome) and barley (Hordeum vulgare, 2n = 2x = 14). The genus has thus been explored to increase genetic diversity of wheat via wide hybridization and chromosome recombination5,6.
Aegilops umbellulata (2n = 2x = 14, UU genome) is the only diploid Aegilops species containing the U genome (Fig. 1a). Compared to the bread wheat A, B and D genomes, the U genome contains several large chromosome rearrangements. In particular, chromosomes 4U, 6U,and 7U show multiple reciprocal translocations, inversions and intra-chromosomal translocations7,8. The U genome is a source of disease resistance genes that have been transferred into wheat, including Lr9, Lr76, Yr70 and PmY399,10,11. Recently, the leaf rust resistance gene Lr9 has been cloned and found to encode an unusual kinase fusion protein. Ae. umbellulata accession TA1851 was identified as the probable donor of Lr912. In this previous analysis, a contig-level assembly of TA1851 was generated to evaluate the Lr9 translocation in bread wheat. The TA1851 contig-level assembly was based on ~157 Gb (~35-fold coverage) of HiFi reads13.

Construction of an Aegilops umbellulata chromosome-scale assembly. (a) An Ae. umbellulata plant (left) is shown next to a bread wheat plant (right) (b) Circos plot of Ae. umbellulata genome with (1) collinearity blocks against Ae. tauschii, (2) gene density and (3) repeat density along each pseudomolecule.
In this current study, we first polished the TA1851 HiFi reads using the DeepConsensus14 pipeline in order to increase read accuracy and to improve the primary contig-level assembly. We then assembled an Ae. umbellulata chromosome-scale reference genome by integrating chromatin conformation capture (Omni-C) data. CpG methylation along the chromosomes was inferred from the PacBio CCS data. The high-quality Ae. umbellulata assembly obtained in this study provides a reference for the U genome of the Triticeae tribe. It will serve as the basis to study chromosome rearrangements across different Triticeae species and can be explored to detect U genome introgressions in durum and bread wheat.
Methods
Plant material, DNA extraction and sequencing
The DNA extraction and generation of PacBio HiFi reads was described previously12. In brief, high molecular weight (HMW) DNA was extracted from young seedlings of Ae. umbellulata accession TA1851 using a modified Qiagen Genomic DNA extraction protocol (https://doi.org/10.17504/protocols.io.bafmibk6)15. DNA was sheared to the appropriate size range (15–20 kb) using Megaruptor 3 (Diagenode) for the construction of PacBio HiFi sequencing libraries. Library preparation was done with the Express Template Prep Kit 2.0 (100-938-900 + Enzyme Clean up 2.0 (101-932-600)), and size was selected with a PippinHT System (Sage Science, HTP0001). Sequencing was performed on PacBio Sequel II systems. The Omni-C library was prepared and sequenced at Cantata Bio using the Dovetail® Omni-C® Kit for plant tissues according to the manufacturer’s protocol. One library was sequenced on an Illumina MiSeq platform to generate ~776 million 2 × 150 bp read pairs for Ae. umbellulata accession TA1851.
Contig-level assembly benchmarking
We first compared contig-level assemblies generated by hifiasm16 and the La Jolla Assembler (LJA)17 using standard HiFi reads and corrected HiFi reads generated with DeepConsensus14. The raw subreads from five SMRT cells were processed using the ccs software (https://github.com/PacificBiosciences/ccs) or DeepConsensus (Table 1). The correction with DeepConsensus produced fewer HiFi data (~157 Gb and ~150 Gb for ccs and DeepConsensus, respectively), but resulted in an increase of the mean read QV (29.9 and 33.1 for ccs and DeepConsensus, respectively) (Table 1).
Contig-level assemblies generated with the different assemblers and data sets were assessed using the basic summary statistics (Table 2). All four assemblies had similar total assembly sizes. For hifiasm, we observed marked increases of contig N50 (11.1 Mb to 14 Mb; + 26%) and contig N90 (3.2 Mb to 3.8 Mb; + 20%) when using corrected HiFi reads (Table 2). Overall, LJA outperformed hifiasm in terms of contiguity. In comparison to hifiasm, DeepConsensus did not result in a considerable increase of contig N50 with LJA, while the contig N90 increased by 16% (4.5 Mb to 5.2 Mb). The highest contiguity was observed with LJA and DeepConsensus, showing a 59% and 63% increase in contig N50 and contig N90, respectively, compared to the hifiasm assembly with standard HiFi reads (Table 2). In terms of computational resources, all the contig-level assemblies were performed on a single AMD node using 120 cores. We observed that the memory usage was higher with LJA with an increase of 61% and 20% with the standard and corrected HiFi reads, respectively. The computing time was also considerably higher with LJA (Table 2). Based on the overall performance, the LJA-DeepConsensus contig-level assembly was used to construct a chromosome-scale Ae. umbellulata assembly.
Chromosome-scale assembly
Construction of the pseudomolecules was performed by integrating Omni-C read data using Juicer (v2; https://github.com/aidenlab/juicer)18 and the 3D-DNA pipeline (https://github.com/aidenlab/3d-dna)19. First, to generate the contact maps, Omni-C Illumina short reads were preprocessed with juicer.sh (parameters: -s none–assembly). The output file “merged_nodups.txt” and the primary assembly were then used to produce an assembly with 3D-DNA19 (using run-asm-pipeline.sh with -r 0 parameter). We used Juicebox (v2.14.00)20 to visualize the Hi-C contact matrix along the assembly, and to manually curate the assembly. The orientation and the chromosome number of each pseudomolecule were determined based on an existing assembly of Ae. tauschii21, a close relative of Ae. umbellulata, using a dotplot comparison produced with chromeister (https://github.com/estebanpw/chromeister)22. There has been some inconsistency in naming the highly rearranged chromosomes 4U and 6U. We decided to follow the most common nomenclature used in the recent publication of Said, et al.8. Contigs not anchored in the pseudomolecules were concatenated into an “unanchored chromosome”. The final Hi-C contact maps and assemblies were saved using run-asm-pipeline-post-review.sh from the 3D-DNA pipeline. The genome assembly resulted in seven pseudomolecules and one unanchored chromosome (Fig. 1b; Table 3).
Repeat annotation and gene model prediction
Transposable element annotation was performed using EDTA23 (v2.0.0; parameters: --sensitive 1 --anno 1 --evaluate 1) using the current version of the TREP database (v19)24 as a curated input library. Overall, 82.30% of the assembly was classified as repetitive sequences (Table 4).
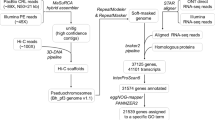
Gene model prediction was performed by combining a lifting approach using liftoff (v1.6.3)25 and a genome-guided approach using transcriptomics data with HISAT2 (v2.2.1)26, StringTie (2.1.7)27 and Transdecoder (v5.7.0)28. Post-processing of gff3 files and filtering were performed using AGAT (https://github.com/NBISweden/AGAT)29 and gffread (v0.11.7)30. For the gene lifting, gene models of hexaploid wheat line Chinese Spring31, Ae tauschii21, and Triticum monoccocum accession TA29932 were independently transferred using liftoff (parameters: -a 0.9 -s 0.9 -copies -exclude_partial -polish). For the genome-guided approach, we used publicly available RNA-Seq data of 12 representative Ae. umbellulata accessions33 and the RNA-Seq data of two bulks representing Ae. umbellulata leaf tissues34. All the RNA-Seq data were mapped individually against the reference sequence using HISAT2 (parameters: --dta --very-sensitive) and the transcripts were assembled using StringTie (parameters: -m 200 -f 0.3) and merged into a single gtf file. The Transdecoder.LongOrfs script was used to identify open reading frames (ORF) of at least 100 amino acids from the merged gtf file. The predicted protein sequences were compared to the UniProt (2021_03) and Pfam35 databases using BLASTP36 (parameters: -max_target_seqs 1 -outfmt 6 -evalue 1e−5) and hmmer337 (v3.3.2 - parameters: hmmsearch -E 1e-10). The Transdecoder.Predict script was used with the BLASTP and hmmer results to select the best translation per transcript. Finally, the annotation gff3 file was computed using the perl script “cdna_alignment_orf_to_genome_orf.pl” provided in the Transdecoder package.
All the output gff files from the lifting and genome-guided approaches were merged into a single file using the perl script “agat_sp_merge_annotations.pl”. The merged file was then post-processed using gffread tools (parameters:–keep-genes -N -J) to retain transcripts with start and stop codons, and to discard transcripts with 1) premature stop codons and/or 2) having introns with non-canonical splice sites. In total, 36,268 gene models were predicted for which the putative functional annotations were assigned using a protein comparison against the UniProt database (2021_03) using DIAMOND38 (parameter: -f 6 -k 1 -e 1e-6). PFAM domain signatures and GO were assigned using InterproScan version 5.55–88.039.
The synteny analysis against Ae. tauschii was computed using MCScanX40 with defaults parameters, which allowed us to identify the main translocation events within the Ae. umbellulata genome (Fig. 1b).
PacBio DNA methylation profile
Methylation in CpG context was inferred with ccsmeth (v0.3.2)41, a deep-learning method to detect DNA 5mCpGs by using kinetics features from PacBio CCS reads. The methylation prediction for CCS reads were called using the model “model_ccsmeth_5mCpG_call_mods_attbigru2s_b21.v1.ckpt”. Then, the reads with the MM + ML tags were aligned to the pseudomolecules using BWA (v0.7.17)42 and the subsequent BAM file was filtered for hard/soft clips and quality (MAPQ ≥ 60) using SAMtools (v1.8)43. The methylation frequency was calculated at genome level with the modbam files and the aggregate mode of ccsmeth with the model “model_ccsmeth_5mCpG_aggregate_attbigru_b11.v2.ckpt”.
Genome visualization
The genome of Ae. umbellulata accession TA1851 was uploaded into the Persephone® multi-genome browser (https://web.persephonesoft.com/?data=genomes/TA1851). The data tracks available are the DNA sequence, gene model prediction, and the CpG methylation. A BLAST36 search and synteny analysis with the hexaploid wheat line Chinese Spring (v.2.1)44 are also available (Fig. 2).

Genome visualization with Persephone. (a) Persephone genome browser visualization. The upper panel represents the position along chromosome 3U. The middle panel shows an example of three gene models with their predicted isoforms. In the lower panel, the CpG methylation profile is represented in blue and red for the unmethylated and methylated bases, respectively. (b) Synteny matrix between the seven Ae. umbellulata chromosomes (x-axis) and the 21 chromosomes of the bread wheat line Chinese spring v2.1 (y-axis) (c) Synteny comparison of the highly rearranged Ae. umbellulata chromosome 6U (in central position) in comparison to bread wheat chromosomes 1D, 2D, 4D, 6D and 7D. The links between chromosomes represented orthologous gene relationships.
Data Records
The corrected HiFi reads and the raw Omni-C reads were deposited in the Sequence Read Archive at NCBI under accession number ERP14784445. The final chromosome assembly was deposited at NCBI under the accession number GCA_032464435.146.
The Ae. umbellulata assembly, gene model prediction, repeat annotations, methylation profile and Hi-C contact map are available on DRYAD Digital Repository47 (https://doi.org/10.5061/dryad.05qfttf82).
Technical Validation
Assessment of genome assembly and annotation
The Hi-C contact map was manually curated and assessed with Juicebox and showed a dense pattern along the diagonal revealing no potential mis-assemblies (Fig. 3). The anti-diagonals are typical for Triticeae genomes and correspond the Rabl configuration of Triticeae chromosomes48,49. Chromosome 6U does not show the anti-diagonal, which is most likely due to the extreme acrocentric nature of this chromosome50,51 (Fig. 3).

Contact map after the integration of the Omni-C data and manual correction. Green and blue boxes represent contigs and pseudomolecules, respectively.
The BUSCO52 (v5.4.5 – poales_odb10) score of 98% (0.4% fragmented and 1.6% missing BUSCOs) at the genome level indicates a high completeness of the TA1851 assembly. The quality of the Ae. umbellulata assembly was assessed with Merqury53 based on the PacBio HiFi reads using 19-mers. The QV (consensus quality value) and k-mer completeness scores were 59.3 and 98.1%, respectively. We further determined the LTR Assembly Index (LAI) and obtained a value of 16.42, which corresponds to a reference quality genome54. Telomeric repeats (TTTAGGG)n55,56 were found at the extremities of all the pseudomolecules, except the short arms of chromosomes 1U and 5U,which corresponds to the location of the rDNA loci in Ae. umbellulata57.
Completeness of the gene model prediction was evaluated using BUSCO and produced a score of 98.1% (0.3% fragmented and 1.6% missing BUSCOs). The number of predicted gene models (36,268) is in the range of a diploid Triticeae species (34,000–43,000 high-confidence gene models per haploid genome)58.
Code availability
All software and pipelines were executed according to the manual and protocol of published tools. No custom code was generated for these analyses.
References
Molnar-Lang, M., Ceoloni, C. & Dolezel, J. Alien introgression in wheat. (Springer, 2015).
Van Slageren, M. Wild wheats: a monograph of Aegilops L. and Amblyopyrum (Jaub. & Spach) Eig (Poaceae). (Agricultural University Wageningen, 1994).
Kimber, G. Genome symbols and plasma types in the wheat group. in Proc. 7th Intl. Wheat Genet. Symp. 1209–1211 (1988).
Kishii, M. An Update of Recent Use of Aegilops Species in Wheat Breeding. Front Plant Sci 10, 585 (2019).
Kilian, B. et al. Aegilops, wild crop relatives, genomic and breeding resources. Cereal (Ed Kole, C), 1–76 (2011).
Schneider, A., Molnar, I. & Molnar-Lang, M. Utilisation of Aegilops (goatgrass) species to widen the genetic diversity of cultivated wheat. Euphytica 163, 1–19 (2008).
Molnár, I. et al. Dissecting the U, M, S and C genomes of wild relatives of bread wheat (Aegilops spp.) into chromosomes and exploring their synteny with wheat. The Plant Journal 88, 452–467 (2016).
Said, M. et al. Development of DNA Markers From Physically Mapped Loci in Aegilops comosa and Aegilops umbellulata Using Single-Gene FISH and Chromosome Sequences. Front Plant Sci 12, 689031 (2021).
Sears, E. R. Brookhaven Symposia in Biology 9, 1–21 (1956).
Bansal, M. et al. Aegilops umbellulata introgression carrying leaf rust and stripe rust resistance genes Lr76 and Yr70 located to 9.47-Mb region on 5DS telomeric end through a combination of chromosome sorting and sequencing. Theor Appl Genet 133, 903–915 (2020).
Zhu, Z. D. et al. Microsatellite marker identification of a Triticum aestivum - Aegilops umbellulata substitution line with powdery mildew resistance. Euphytica 150, 149–153 (2006).
Wang, Y. et al. An unusual tandem kinase fusion protein confers leaf rust resistance in wheat. Nature Genetics (2023).
Wenger, A. M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature Biotechnology 37, 1155–1162 (2019).
Baid, G. et al. DeepConsensus improves the accuracy of sequences with a gap-aware sequence transformer. Nature Biotechnology 41, 232–238 (2023).
Driguez, P. et al. LeafGo: Leaf to Genome, a quick workflow to produce high-quality de novo plant genomes using long-read sequencing technology. Genome Biol 22, 256 (2021).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Bankevich, A., Bzikadze, A. V., Kolmogorov, M., Antipov, D. & Pevzner, P. A. Multiplex de Bruijn graphs enable genome assembly from long, high-fidelity reads. Nature Biotechnology 40, 1075–1081 (2022).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101 (2016).
Luo, M. C. et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551, 498–502 (2017).
Perez-Wohlfeil, E., Diaz-Del-Pino, S. & Trelles, O. Ultra-fast genome comparison for large-scale genomic experiments. Sci Rep 9, 10274 (2019).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol 20, 275 (2019).
Wicker, T., Matthews, D. E. & Keller, B. TREP: a database for Triticeae repetitive elements. Trends Plant Sci 7, 561–562 (2002).
Shumate, A. & Salzberg, S. L. Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643 (2021).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33, 290–295 (2015).
Haas, B. & Papanicolaou, A. TransDecoder (find coding regions within transcripts). http://transdecoder.github.io.
Dainat, J. AGAT: Another Gff Analysis Toolkit to handle annotations in any GTF/GFF format. Version v0 4, 10.5281 (2020).
Pertea, G. & Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Res 9, 304 (2020).
International Wheat Genome Sequencing, C. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361 (2018).
Ahmed, H. I. et al. Einkorn genomics sheds light on history of the oldest domesticated wheat. Nature (2023).
Okada, M. et al. RNA-seq analysis reveals considerable genetic diversity and provides genetic markers saturating all chromosomes in the diploid wild wheat relative Aegilops umbellulata. BMC plant biology 18, 1–13 (2018).
Edae, E. A. & Rouse, M. N. Bulked segregant analysis RNA-seq (BSR-Seq) validated a stem resistance locus in Aegilops umbellulata, a wild relative of wheat. PLoS One 14, e0215492 (2019).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic acids research 49, D412–D419 (2021).
Camacho, C. et al. BLAST+: architecture and applications. BMC bioinformatics 10, 1–9 (2009).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic acids research 39, W29–W37 (2011).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60 (2015).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res 33, W116–120 (2005).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49 (2012).
Ni, P. et al. DNA 5-methylcytosine detection and methylation phasing using PacBio circular consensus sequencing. Nature Communications 14, 4054 (2023).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Zhu, T. et al. Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J 107, 303–314 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:ERP147844 (2023).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_032464435.1 (2023).
Abrouk, M. et al. Data from:Chromosome-scale assembly of the wild wheat relative Aegilops umbellulata. Dryad Digital Repository. https://doi.org/10.5061/dryad.05qfttf82 (2023).
Tiang, C. L., He, Y. & Pawlowski, W. P. Chromosome organization and dynamics during interphase, mitosis, and meiosis in plants. Plant Physiol 158, 26–34 (2012).
Mascher, M. et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433 (2017).
Friebe, B., Jiang, J., Tuleen, N. & Gill, B. Standard karyotype of Triticum umbellulatum and the characterization of derived chromosome addition and translocation lines in common wheat. Theoretical and Applied Genetics 90, 150–156 (1995).
Zhang, H., Jia, J., Gale, M. & Devos, K. Relationships between the chromosomes of Aegilops umbellulata and wheat. Theoretical and Applied Genetics 96, 69–75 (1998).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome biology 21, 1–27 (2020).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res 46, e126 (2018).
Richards, E. J. & Ausubel, F. M. Isolation of a higher eukaryotic telomere from Arabidopsis thaliana. Cell 53, 127–136 (1988).
Peska, V. & Garcia, S. Origin, Diversity, and Evolution of Telomere Sequences in Plants. Front Plant Sci 11, 117 (2020).
Castilho, A. & Heslop-Harrison, J. S. Physical mapping of 5S and 18S–25S rDNA and repetitive DNA sequences in Aegilops umbellulata. Genome 38, 91–96 (1995).
Poretti, M., Praz, C. R., Sotiropoulos, A. G. & Wicker, T. A survey of lineage‐specific genes in Triticeae reveals de novo gene evolution from genomic raw material. Plant Direct 7, e484 (2023).
Acknowledgements
We thank the KAUST Bioscience Core Laboratory for sequencing support, Lingli Zou (KAUST) for greenhouse support, and the KAUST supercomputing facilities (https://www.hpc.kaust.edu.sa) for providing computing resources. This publication is based upon work supported by the King Abdullah University of Science and Technology.
Author information
Authors and Affiliations
Contributions
M.A. and S.G.K. designed the study. Y.W. performed the DNA extraction. M.A. and E.C-G. analyzed the data. M.T. and M.K. managed the visualization platform. M.A. and S.G.K. wrote the initial manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abrouk, M., Wang, Y., Cavalet-Giorsa, E. et al. Chromosome-scale assembly of the wild wheat relative Aegilops umbellulata. Sci Data 10, 739 (2023). https://doi.org/10.1038/s41597-023-02658-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02658-2
This article is cited by
-
Discrepancy of flowering time between genetically close sublineages of Aegilops umbellulata Zhuk.
Scientific Reports (2024)
