
Abstract
Cold seeps harbor abundant and diverse microbes with tremendous potential for biological applications and that have a significant influence on biogeochemical cycles. Although recent metagenomic studies have expanded our understanding of the community and function of seep microorganisms, knowledge of the diversity and genetic repertoire of global seep microbes is lacking. Here, we collected a compilation of 165 metagenomic datasets from 16 cold seep sites across the globe to construct a comprehensive gene and genome catalog. The non-redundant gene catalog comprised 147 million genes, and 36% of them could not be assigned to a function with the currently available databases. A total of 3,164 species-level representative metagenome-assembled genomes (MAGs) were obtained, most of which (94%) belonged to novel species. Of them, 81 ANME species were identified that cover all subclades except ANME-2d, and 23 syntrophic SRB species spanned the Seep-SRB1a, Seep-SRB1g, and Seep-SRB2 clades. The non-redundant gene and MAG catalog is a valuable resource that will aid in deepening our understanding of the functions of cold seep microbiomes.
Similar content being viewed by others


Background & Summary
Cold seeps occur on continental margins worldwide. At these sites, methane-rich fluids migrate from the deep subsurface to the sediment-water interface1. Methane is a climate-active greenhouse gas that is approximately 30 times more potent than carbon dioxide2. In seep sediments, methane can be consumed through the process of anaerobic oxidation of methane (AOM). This process removes approximately 90% of the methane produced globally in marine sediments, acting as an efficient methane filter3,4. As a consequence, these seeps are critical in regulating the amount of methane released into the overlying waters and atmosphere, and they play a vital role in mitigating global warming. AOM is performed by anaerobic methanotrophic archaea (ANME). Normally, ANME rely on a syntrophic partner to couple CH4 oxidation to the reduction of terminal electron acceptors, such as sulfate, iron, nitrate, and manganese5,6. AOM coupled to sulfate reduction is the primary biological process in seep sediments since sulfate is the dominant anion present at the marine sediment-water interface. High rates of AOM fueled by near-saturated methane concentrations would rapidly consume sediment pools of any individual electron acceptor, creating unique geobiological engines that contribute significantly to local and global biogeochemical cycles1.
Cold seeps are deep-sea oases that support immense biodiversity and where specialization and adaptation create extraordinary lifestyles1. However, the majority of microorganisms found in seeps have not yet been characterized7. Culture-independent metagenomic techniques are the key to unraveling the genetic diversity and metabolic potential of uncharacterized microbes and have been applied to identify thousands of microorganisms and their metabolic versatility. Recently, the microbial community and function of cold seep sediments have been increasingly studied with metagenomes obtained from different sea areas7,8,9,10. However, there are no large-scale gene and genome catalogs available for the microbiome of global cold seeps. A comprehensive gene and genome catalog of cold seeps could serve as a reference for mining novel genetic resources in the deep sea, including various natural products with diverse bioactivities (e.g., antibiotic dixiamycins and immune-enhancing exopolysaccharides)11,12.
ANME and their syntrophic sulfate-reducing bacteria (SRB) partners play a crucial role in the regulation of both the carbon and sulfur cycles of seeps. Through their mutualistic interactions, they perform AOM, leading to a reduction in methane release and the generation of inorganic carbon and sulfide. These processes are of significant importance for both local and global biogeochemical cycles, underscoring the essential role of these microorganisms in deep-sea ecosystems. Although previous findings have revealed various lineages of ANME and SRB in seep sediments13,14,15, there is currently no a comprehensive genome catalog of these lineages in cold seep sediments globally. Extensive, high-quality reference genomes of the global seep microbiome could improve the resolution and accuracy of taxonomic and functional analyses and provide the opportunity for large-scale comparative genomics16,17,18,19, especially for elucidating the physiological basis of ANME-SRB interactions.
Here, we collected metagenomic sequence data from 165 sediment samples at 16 cold seeps across the Pacific, Atlantic, and Arctic Oceans (Fig. 1), encompassing gas hydrates (n = 4), methane seeps (n = 14), oil and gas seeps (n = 4), mud volcanoes (n = 2) and asphalt volcanoes (n = 1). The sediment samples span different depths and redox conditions, from the oxic sediment-water interface to anoxic layers down to 68.55 m below the sea floor (mbsf) (Supplementary Table 1). The non-redundant gene catalog was constructed from these metagenomes, comprising a total of 147,289,169 protein clusters (Fig. 2). The mapping ratios of the non-redundant gene catalog to clean reads of the 165 metagenomes averaged 62%. This is the most comprehensive gene catalog generated from the cold seep sediment microbiome to date, corresponding to half the size of the global microbial gene catalog (GMGC v1; 303 million)16, the size of the global topsoil microbiome gene catalog (~160 million)20, three times the size of the ocean microbial reference gene catalog (OM-RGC v2; ~47 million)21, and six times the size of the Tibetan Glacier gene catalog (TG2G; ~25 million)17.

Overview of the studied areas and bioinformatics workflow. (a) Geographic distribution of the 16 global cold seep sites where metagenomic sequencing data were collected. The map was drawn using the maptools and ggplot2 packages in R v4.0.3. (b) Numbers and proportions of cold seep samples classified according to their types and depths. (c) Overview of the computational pipeline used to generate the non-redundant gene and MAG catalogs.

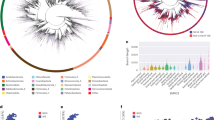
Functional and taxonomic characterization of the non-redundant gene catalog. (a) An overview of annotations for the non-redundant gene catalog. Non-annotation indicates that these genes were not annotated in at least one of the following databases: eggNOG, Pfam, KEGG, EC, GO and CAZy. (b) Number of genes with functional annotations across the six functional databases. Vertical bars represent the number of genes unique (color) to each functional database or shared (black) between different functional databases. Horizontal bars in the lower panel indicate the total number of genes with functional annotations in each database. (c) Functional annotations at the COG category level. S: Function unknown. (d) Breakdown of taxonomic classifications for the non-redundant gene catalog.
A total of 3,164 species-level MAGs were recovered in this study. The total mapping ratios of all these MAGs to clean reads of the 165 metagenomes averaged 27%. These MAGs covered various prokaryotic lineages spanning 113 phyla (97 bacterial and 16 archaeal). The phyla with the largest diversity of recovered species included Chloroflexota (n = 371), Proteobacteria (n = 335), Desulfobacterota (n = 306), Planctomycetota (n = 190), Patescibacteria (n = 152) and Bacteroidota (n = 151) and the archaeal phyla Halobacteriota (n = 129), Thermoplasmatota (n = 108), Thermoproteota (n = 98), Asgardarchaeota (n = 95) and Nanoarchaeota (n = 47) (Fig. 3b). Overall, ~94% of the recovered species are not represented in current databases (Fig. 3c), suggesting that cold seep sediments harbor a rich diversity of previously undescribed microbes. The non-redundant MAG catalog considerably expands the phylogenetic diversity and is an unparalleled genome resource of the cold seep microbiome. The compendium of ANME (Fig. 4) and syntrophic SRB MAGs (Fig. 5) expands the currently known diversity of these groups in cold seeps and will aid in expanding our understanding of the physiological basis of their interactions and their evolutionary histories.

Quality and novelty of non-redundant MAGs. (a) Genome statistics for the representative species of non-redundant MAGs. (b) Taxonomic classification (domain and phylum levels) of the species-level representative MAGs. (c) Taxonomic novelty of the representative species.

Phylogenetic tree of ANME genomes and related archaea. The phylogenetic tree was constructed from 41 previously published ANME genomes and 135 MAGs belonging to Halobacteriota from this study. The tree was constructed by the maximum likelihood method using a concatenated alignment of 53 conserved archaeal single-copy marker genes.

Phylogenetic tree of syntrophic SRB genomes. The tree was constructed by using 60 reference SRB genomes collected from previous studies and 327 MAGs assigned to Desulfobacterota from this study. The tree was constructed by the maximum likelihood method using a concatenated alignment of 120 conserved bacterial single-copy marker genes.
Methods
Collection of metagenomes
Metagenomic datasets comprised 165 sediment samples (0 to 68.55 mbsf) collected from 16 globally distributed cold seep sites (Fig. 1a; Supplementary Table 1). These sites are as follows: Eastern North Pacific (ENP), Santa Monica Mounds (SMM), Western Gulf of Mexico (WGM), Eastern Gulf of Mexico (EGM), Northwestern Gulf of Mexico (NGM), Scotian Basin (SB), Haakon Mosby mud volcano (HM), Mediterranean Sea (MS), Laptev Sea (LS), Jiaolong cold seep (JL), Shenhu area (SH), Haiyang4 (HY4), Qiongdongnan Basin (QDN), Xisha Trough (XST), Haima seep (HM1, HM3, HM5, HM_SQ, S11, SY5, and SY6) and site F cold seep (RS, SF, FR, and SF_SQ). Paired-end sequencing data from ENP, SMM, WGM, NGM, HM, MS, LS and part of site F (RS and FR) were downloaded from the National Center for Biotechnology Information-Sequence Read Archive (NCBI-SRA) and European Bioinformatics Institute-European Nucleotide Archive (EBI-ENA) according to the accession numbers published in each study8,9,10,22,23,24,25,26. The remaining 106 metagenomic datasets used in this study were obtained from our previous publications7,14,27,28,29,30,31,32,33,34. Detailed sequencing information is available in Supplementary Table 1. These metagenomic samples were collected from a range of cold seeps, including oil and gas seeps, methane seeps, gas hydrates, asphalt volcanoes, and mud volcanoes. The samples were taken at various depths and under different redox conditions, from the oxic sediment-water interface to anoxic layers as deep as 68.55 meters below the sea floor.
Contig assembly, gene prediction and gene catalog construction
Metagenomic sequence data were quality controlled using the Read_QC module (parameters: --skip-bmtagger) within the metaWRAP (v1.3.2) pipeline35 and fastp (v0.23.2; default parameters)36. After quality control, 9.5 Tb of clean reads remained for subsequent analyses. Clean reads from each cold seep sediment metagenome were assembled using MEGAHIT (v1.1.3 and v1.2.9, default parameters)37. In addition, co-assemblies were performed by combining metagenomes from all depths of each cold seep sediment using MEGAHIT (v1.1.3; parameters: --k-min 27 --kmin-1pass --presets meta-large)37. The assembly parameters are summarized in Supplementary Table 1. Contigs (length > 500 bp, n = 225,026,054) from individual assemblies and co-assemblies were used to predict protein-coding sequences (CDSs) with Prodigal (v2.6.3; parameter: -meta)38, which generated 373,051,862 protein sequences. These sequences were then clustered at 95% amino acid identity using CD-HIT (v4.8.1; parameters: -c 0.95 -aS 0.9 -g 1 -d 0)39. The cutoff of 95% amino acid identity was adopted to be consistent with the fact that members of the same microbial species generally share more than 95% average amino acid identity40. It should also be noted that the mixed-assembly approach used here, which combines data from single assemblies and co-assemblies, may enrich artificially long proteins to a certain extent41. This resulted in a non-redundant gene catalog comprising 147,289,169 representative clusters. The mapping-based mode of Salmon (v1.10.2)42 with a “meta-flag” was used to calculate the mapping rate of the non-redundant gene catalog in each metagenome.
Functional annotation and taxonomic classification of the non-redundant gene catalog
The representative amino acid sequences from each cluster were functionally annotated using eggNOG-mapper (v2.1.9; default parameters)43,44. The functional annotations, including those for eggNOG 5.0, Pfam 33.1, KEGG, EC, GO, and CAZy, were derived from the eggNOG-mapper results. We found that 64% of the non-redundant genes had a hit in at least one of the following databases: eggNOG (n = 88,929,242; ~60%), Pfam (n = 85,404,569; ~58%), KEGG (n = 48,756,524; ~33%), EC (n = 27,619,712; ~19%), GO (n = 5,966,227; ~4%) and CAZy (n = 1,514,988; ~1%) (Fig. 2a,b). After analyzing the annotated genes based on the eggNOG database (Fig. 2c), the predominant category was “Function unknown” (n = 17,018,774). This category includes proteins that have not yet been characterized or for which there is insufficient information to assign a specific function. A total of ~40% of genes (n = 58,359,927; Fig. 2b) could not be assigned to an eggNOG orthologous group, similar to the percentage observed in the OM-RGC v2 (~39%)21 and higher than that in the GMGC v1 (~27%)16. According to the eggNOG database annotation, half of the genes (~51%), including 58,359,927 unannotated genes and 17,018,774 genes labeled as “Function unknown”, were functionally unidentified, suggesting that cold seeps harbor numerous unknown functional genes.
MMseqs2 taxonomy (v13.45111; parameter: --tax-lineage 1)45 was used to assign taxonomic labels to each representative amino acid sequence, using the GTDB R207 as a reference database46. The MMseqs2 taxonomy uses an approximate 2bLCA (lowest common ancestor, LCA) approach (--lca-mode: 2bLCA). A notable percentage of the non-redundant sequences (n = 44,441,531; ~30%) could not be classified as belonging to any prokaryotes in the GTDB, suggesting that these sequences may be attributed to novel prokaryotes (Fig. 2d). Approximately 9% (n = 13,154,825) of the non-redundant sequences could be identified only as either bacteria or archaea and could not be further classified at the phylum level (Fig. 2d). The results of taxonomic classification further confirm that this gene catalog contains many untapped genetic resources.
Metagenomic binning and non-redundant MAG catalog construction
Assembled contigs were filtered by length (>1000 bp) for subsequent binning. BWA software (v0.7.17; BWA-MEM algorithm)47 was used to align short reads back to filtered contigs, with the alignment being sorted by SAMtools (v1.9)48. The contig depth profiles were produced using jgi_summarize_bam_contig_depths for running metabat2, maxbin2, SemiBin, Rosella and VAMB, while for running concoct, concoct_coverage_table.py was used. The binning process was performed using the metaWRAP binning module (v1.3.2; parameters: -metabat2, -maxbin2, -concoct, -universal)35, SemiBin with single_easy_bin mode (v1.4.0; default parameters)49, and Rosella (v0.4.1; default parameters; https://github.com/rhysnewell/rosella). The number of metagenomic samples collected from S11 (n = 13) and RS (n = 19) was larger than that obtained from other sites, making it computationally challenging to bin the co-assemblies of the samples from these sites. Thus, individual assemblies from the S11 and RS sites were concatenated and binned separately using the VAMB tool in “bin-split” mode (v3.0.2; parameters: --minfasta 200000 -o C)50. Afterward, the bins obtained with each binning tool were integrated and refined using the Bin_refinement module of the metaWRAP pipeline (v1.3.2; parameters: -c 50 -x 10)35. The completeness and contamination of refined bins were evaluated with CheckM (v1.2.1)51. Then, the resulting 8,654 MAGs were checked by GUNC (v1.0.5; default parameters)52 to remove genomes potentially containing chimerism based on “pass.GUNC”. All MAGs were dereplicated at the species level using dRep (v3.4.0; parameters: -comp 50 -con 10)53 with an average nucleotide identity (ANI) cutoff value of 95%. Representative genomes were selected based on the dRep scores derived from genome completeness, contamination and N50. A total of 3,164 MAGs with the highest dRep score from each species cluster were selected as the species representatives. MAGpurify software (v2.1.1; default parameters)54 was used to identify and remove putative contaminant contigs from each MAG based on the clade-markers, tetra-freq, gc-content, and clean-bin modules. Importantly, the resulting representative genomes should be considered population genomes within species55.
MAGs were taxonomically classified using the GTDB-Tk toolkit (v2.1.1)56,57 with default parameters against the R207 database. According to the taxonomic classification, four species clusters, with medium- or high-quality representatives (CSMAG_1499, CSMAG_2247, CSMAG_2329, and CSMAG_3128), were not assigned to any existing phylum. They did not cluster together and were included in different clades, exhibiting low relative evolutionary divergence values ranging from 0.32 to 0.43. These results suggest that these species belong to undescribed phyla. Additionally, 44 classes, 184 orders, 412 families, 1,043 genera and 2,984 species lacked classification assignments based on the GTDB R207 (Fig. 3c), representing potential novel lineages.
The coverage of each MAG was calculated using CoverM in genome mode (v0.6.1; https://github.com/wwood/CoverM; parameters: -min-read-percent-identity 0.95 -min-read-aligned-percent 0.75 -trim-min 0.10 -trim-max 0.90 -m relative_abundance) by mapping clean reads from the 165 metagenomes to all MAGs.
Genomes for ANME and their syntrophic SRB
To explore the diversity of ANME lineages in global cold seep sediments, a phylogenetic tree was constructed that included 41 previously published ANME genomes58,59,60,61,62,63,64,65,66 and 135 MAGs belonging to Halobacteriota from this study. These published ANME genomes cover all of the currently described subclades: ANME-1, ANME-2a, ANME-2b, ANME-2c, ANME-2d, and ANME-3. To identify their syntrophic SRB, we constructed a phylogenetic tree of concatenated marker genes from 60 reference SRB genomes6,23,67,68 (including syntrophic SRB, namely, HotSeep-1, Seep-SRB2, Seep-SRB1a and Seep-SRB1g, and non-syntrophic SRB) and 327 MAGs assigned to Desulfobacterota from this study. The concatenated multiple sequence alignment of genomes based on 53 archaeal and 120 bacterial single-copy marker genes was produced via the identify and align workflow of GTDB-Tk (v2.1.0)56. The maximum likelihood tree was constructed using IQ-TREE (v2.2.0.3; parameters: -m MFP -B 1000)69. All produced trees were visualized using iTOL (v6)70.
A total of 81 ANME genomes were identified, namely, ANME-1 (n = 38), ANME-2a (n = 16), ANME-2b (n = 1), ANME-2c (n = 24), and ANME-3 (n = 2) (Fig. 4). In comparison, Chen et al.15 assessed the phylogenetic diversity of ANME MAGs from global methane seeps, which resulted in 47 species clustered into three subclades, including ANME-1a/b (n = 21), ANME-2a/b (n = 11), and ANME-2c (n = 15). The higher diversity of ANME captured here reflects the incorporation of all seep environments, not only those characterized by methane seepage. We also identified 23 syntrophic SRB MAGs (Fig. 5) spanning three clades (Seep-SRB2, n = 8; Seep-SRB1a, n = 14, and Seep-SRB1g, n = 1).
Data Records
Details for the non-redundant gene catalog, the functional annotation and taxonomic classification for gene clusters, non-redundant MAGs, and phylogenetic trees are available in the Figshare repository71. All non-redundant MAGs are deposited in the NCBI database under BioProject PRJNA950938 (ref. 72) with the accession numbers detailed in Supplementary Table 2.
Technical Validation
To maximize the number of genes and ensure the quality of the genes, we selected assembled contigs with a length greater than 500 bp to predict CDSs, as suggested in previous studies17,73,74. Then, we selected assembled contigs by length (>1000 bp) for metagenomic binning. The quality of MAGs was strictly controlled according to the following standards: (1) completeness >50% and contamination <10%; (2) genome sequences without potential chimerism (details in Supplementary Table 2); and (3) genome sequences without potential misassigned contigs.
Usage Notes
The dataset compiled and analyzed in this study is the largest of its kind from cold seep sediment environments. Researchers could use the gene catalog of seeps to compare genes of interest to those in other habitats, such as glaciers, polar regions and hydrothermal vents, to study the habitat specificity of genes. The compendium of ANME could be used to investigate the distributional pattern of ANME archaeal communities in global cold seeps and ecological niche partitioning. Furthermore, the evolutionary and physiological basis of ANME-SRB interactions could also be explored.
Code availability
The present study did not use custom scripts to generate the dataset. The parameters and versions of all the bioinformatics tools used for the analysis are described in the Methods section. The code used to run each of the tools is available in the Figshare repository71.
References
Joye, S. B. The geology and biogeochemistry of hydrocarbon seeps. Annu. Rev. Earth Pl. Sci. 48, 205–231 (2020).
Al-Shayeb, B. et al. Borgs are giant genetic elements with potential to expand metabolic capacity. Nature 610, 731–736 (2022).
Benito Merino, D., Zehnle, H., Teske, A. & Wegener, G. Deep-branching ANME-1c archaea grow at the upper temperature limit of anaerobic oxidation of methane. Front. Microbiol. 13, 988871 (2022).
Regnier, P. et al. Quantitative analysis of anaerobic oxidation of methane (AOM) in marine sediments: A modeling perspective. Earth-Sci. Rev. 106, 105–130 (2011).
Leu Andy, O. et al. Lateral gene transfer drives metabolic flexibility in the anaerobic methane-oxidizing archaeal family Methanoperedenaceae. mBio 11, e01325–20 (2020).
Murali, R. et al. Physiological adaptation of sulfate reducing bacteria in syntrophic partnership with anaerobic methanotrophic archaea. bioRxiv, 2022.11.23.517749 (2022).
Dong, X. et al. Metabolic potential of uncultured bacteria and archaea associated with petroleum seepage in deep-sea sediments. Nat. Commun. 10, 1816 (2019).
Zhao, R., Summers, Z. M., Christman, G. D., Yoshimura, K. M. & Biddle, J. F. Metagenomic views of microbial dynamics influenced by hydrocarbon seepage in sediments of the Gulf of Mexico. Sci. Rep 10, 5772 (2020).
Li, L. et al. Bacteria and archaea synergistically convert glycine betaine to biogenic methane in the Formosa cold seep of the South China Sea. mSystems 6, e0070321 (2021).
Savvichev, A. S. et al. Biogeochemical activity of methane-related microbial communities in bottom sediments of cold seeps of the Laptev Sea. Microorganisms 11, 250 (2023).
Cong, M. et al. Deep-Sea Natural Products from Extreme Environments: Cold Seeps and Hydrothermal Vents. Marine Drugs 20, 404 (2022).
Jin, E., Li, H., Liu, Z., Xiao, F. & Li, W. Antibiotic Dixiamycins from a Cold-Seep-Derived Streptomyces olivaceus. Journal of Natural Products 84, 2606–2611 (2021).
Vigneron, A. et al. Contrasting pathways for anaerobic methane oxidation in Gulf of Mexico cold seep sediments. mSystems 4, e00091–18 (2019).
Li, W.-L., Wu, Y.-Z., Zhou, G.-W., Huang, H. & Wang, Y. Metabolic diversification of anaerobic methanotrophic archaea in a deep-sea cold seep. Mar. Life Sci. Tech. 2, 431–441 (2020).
Chen, J. et al. Genomic insights into niche partitioning across sediment depth among anaerobic methane-oxidizing archaea in global methane seeps. mSystems 8, e01179–22 (2023).
Coelho, L. P. et al. Towards the biogeography of prokaryotic genes. Nature 601, 252–256 (2022).
Liu, Y. et al. A genome and gene catalog of glacier microbiomes. Nat. Biotechnol. 40, 1341–1348 (2022).
Zeng, S. et al. A compendium of 32,277 metagenome-assembled genomes and over 80 million genes from the early-life human gut microbiome. Nat. Commun. 13, 5139 (2022).
Nayfach, S. et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 39, 499–509 (2021).
Bahram, M. et al. Structure and function of the global topsoil microbiome. Nature 560, 233–237 (2018).
Salazar, G. et al. Gene Expression Changes and Community Turnover Differentially Shape the Global Ocean Metatranscriptome. Cell 179, 1068–1083 (2019).
Glass, J. B. et al. Microbial metabolism and adaptations in Atribacteria-dominated methane hydrate sediments. Environ. Microbiol. 23, 4646–4660 (2021).
Yu, H. et al. Sulfate differentially stimulates but is not respired by diverse anaerobic methanotrophic archaea. ISME J. 16, 168–177 (2022).
Laso-Perez, R. et al. Anaerobic degradation of non-methane alkanes by “Candidatus Methanoliparia” in hydrocarbon seeps of the Gulf of Mexico. mBio 10, e01814–19 (2019).
Ruff, S. E. et al. In situ development of a methanotrophic microbiome in deep-sea sediments. ISME J. 13, 197–213 (2019).
Zhang, H. et al. Metagenome sequencing and 768 microbial genomes from cold seep in South China Sea. Sci. Data 9, 480 (2022).
Dong, X. et al. Thermogenic hydrocarbon biodegradation by diverse depth-stratified microbial populations at a Scotian Basin cold seep. Nat. Commun. 11, 5825 (2020).
Dong, X. et al. Phylogenetically and catabolically diverse diazotrophs reside in deep-sea cold seep sediments. Nat. Commun. 13, 4885 (2022).
Jiang, Q., Jing, H., Jiang, Q. & Zhang, Y. Insights into carbon-fixation pathways through metagonomics in the sediments of deep-sea cold seeps. Mar. Pollut. Bull. 176, 113458 (2022).
Li, J. et al. Deep sea cold seep is an atmospheric Hg sink and MeHg source. Research Square https://doi.org/10.21203/rs.3.rs-2323106/v1 (2022).
Lu, R. et al. Asgard archaea in the haima cold seep: Spatial distribution and genomic insights. Deep-Sea Res. Pt. I 170, 103489 (2021).
Li, W. L. et al. Microbial ecology of sulfur cycling near the sulfate-methane transition of deep-sea cold seep sediments. Environ. Microbiol. 23, 6844–6858 (2021).
Zhang, C. et al. The majority of microorganisms in gas hydrate-bearing subseafloor sediments ferment macromolecules. Microbiome 11, 37–37 (2023).
Xiao, X. et al. Metal-driven anaerobic oxidation of methane as an important methane sink in methanic cold seep sediments. Microbiol. Spectr. 11, e05337–22 (2023).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Konstantinidis, K. T. & Tiedje, J. M. Towards a Genome-Based Taxonomy for Prokaryotes. J. Bacteriol. 187, 6258–6264 (2005).
Delgado, L. F. & Andersson, A. F. Evaluating metagenomic assembly approaches for biome-specific gene catalogues. Microbiome 10, 72 (2022).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Mirdita, M., Steinegger, M., Breitwieser, F., Soding, J. & Levy Karin, E. Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics 37, 3029–3031 (2021).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2022).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–9 (2009).
Pan, S., Zhu, C., Zhao, X.-M. & Coelho, L. P. A deep siamese neural network improves metagenome-assembled genomes in microbiome datasets across different environments. Nat. Commun. 13, 2326 (2022).
Nissen, J. N. et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 39, 555–560 (2021).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–55 (2015).
Orakov, A. et al. GUNC: detection of chimerism and contamination in prokaryotic genomes. Genome Biol. 22, 178 (2021).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Nayfach, S., Shi, Z. J., Seshadri, R., Pollard, K. S. & Kyrpides, N. C. New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510 (2019).
Saheb Kashaf, S., Almeida, A., Segre, J. A. & Finn, R. D. Recovering prokaryotic genomes from host-associated, short-read shotgun metagenomic sequencing data. Nat. Protoc. 16, 2520–2541 (2021).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2019).
Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086 (2020).
Chadwick, G. L. et al. Comparative genomics reveals electron transfer and syntrophic mechanisms differentiating methanotrophic and methanogenic archaea. PLoS Biol. 20, e3001508 (2022).
Meyerdierks, A. et al. Metagenome and mRNA expression analyses of anaerobic methanotrophic archaea of the ANME-1 group. Environ. Microbiol. 12, 422–439 (2010).
Krukenberg, V. et al. Gene expression and ultrastructure of meso- and thermophilic methanotrophic consortia. Environ. Microbiol. 20, 1651–1666 (2018).
Wang, F. P. et al. Methanotrophic archaea possessing diverging methane-oxidizing and electron-transporting pathways. ISME J. 8, 1069–1078 (2014).
Haroon, M. F. et al. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500, 567–570 (2013).
Yu, H. et al. Comparative genomics and proteomic analysis of assimilatory sulfate reduction pathways in anaerobic methanotrophic archaea. Front. Microbiol. 9, 2917 (2018).
Wilkins, D., Leung, M. H. & Lee, P. K. Microbiota fingerprints lose individually identifying features over time. Microbiome 5, 1 (2017).
Parks, D. H. et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542 (2017).
Arshad, A. et al. A metagenomics-based metabolic model of nitrate-dependent anaerobic oxidation of methane by Methanoperedens-like archaea. Front. Microbiol. 6, 1423 (2015).
Krukenberg, V. et al. Candidatus Desulfofervidus auxilii, a hydrogenotrophic sulfate-reducing bacterium involved in the thermophilic anaerobic oxidation of methane. Environ. Microbiol. 18, 3073–3191 (2016).
Skennerton, C. T. et al. Methane-fueled syntrophy through extracellular electron transfer: uncovering the genomic traits conserved within diverse bacterial partners of anaerobic methanotrophic archaea. mBio 8, e00530–17 (2017).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A Fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Han, Y. A comprehensive gene and genomic catalog from global cold seeps. figshare https://doi.org/10.6084/m9.figshare.22568107 (2023).
NCBI Bioproject https://identifiers.org/ncbi/bioproject:PRJNA950938 (2023).
Xie, F. et al. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome 9, 137 (2021).
Sánchez, P. et al. Marine picoplankton metagenomes from eleven vertical profiles obtained by the Malaspina Expedition in the tropical and subtropical oceans. bioRxiv, 2023.02.06.526790 (2023).
Acknowledgements
We thank all authors from the originating and submitting laboratories who contributed to the generation of the metagenomic sequence data and all persons who developed the software used in this study. The work was supported by the Scientific Research Foundation of Third Institute of Oceanography, MNR (No. 2022025 and No 2023022), State Key Laboratory of Marine Geology, Tongji University (No. MGK202303), and China Postdoctoral Science Foundation (2022M723709 and 2023M734096).
Author information
Authors and Affiliations
Contributions
X.D. designed this study. Y.H. and X.D. performed the analyses. C.Z. contributed to assembly of part of samples. X.D., Y.H. and Z.S. interpreted the data. Z.Z., Y.P., J.L., Q.J. and Q.L. contributed to the data collection. Y.H. and X.D. wrote the paper, with input from all other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, Y., Zhang, C., Zhao, Z. et al. A comprehensive genomic catalog from global cold seeps. Sci Data 10, 596 (2023). https://doi.org/10.1038/s41597-023-02521-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02521-4
This article is cited by
-
Going to extremes: progress in exploring new environments for novel antibiotics
npj Antimicrobials and Resistance (2024)
