
Abstract
The green microalga Haematococcus pluvialis can synthesize high amounts of astaxanthin, which is a valuable antioxidant that has been utilized in human health, cosmetics, and aquaculture. To illustrate detailed molecular clues to astaxanthin yield, we performed PacBio HIFI along with Hi-C sequencing to construct an improved chromosome-level haplotypic genome assembly with 32 chromosomes and a genome size of 316.0 Mb. Its scaffold N50 (942.6 kb) and contig N50 (304.8 kb) have been upgraded remarkably from our previous genome draft, and a total of 32,416 protein-coding genes were predicted. We also established a high-evidence phylogenetic tree from seven representative algae species, with the main aim to calculate their divergence times and identify expanded/contracted gene families. We also characterized genome-wide localizations on chromosomes of some important genes such as five BKTs (encoding beta-carotene ketolases) that are putatively involved in astaxanthin production. In summary, we reported the first chromosome-scale map of H. pluvialis, which provides a valuable genetic resource for in-depth biomedical investigations on this momentous green alga and commercial astaxanthin bioproduction.
Similar content being viewed by others

Background & Summary
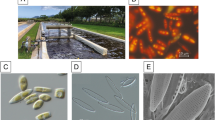
The freshwater unicellular green microalga Haematococcus pluvialis is well known as the best natural bioresource for production of the carotenoid astaxanthin. It has attracted a lot of attention frequently due to its high capacity to synthesize astaxanthin, which is of high value with strong pharmaceutical activity for commercial industries1. The intriguing life cycle of H. pluvialis includes four stages of distinguishable cellular morphologies, i.e., macrozooid, palmella, immature aplanospore and aplanospore (from left to right in Fig. 1a). It maintains the green motile stage at favorable environmental conditions. However, when it experiences unfavorable environmental or stress conditions, the cells of H. pluvialis change into red immobile cells (also named as cysts). Meanwhile, these cells also expand cell size, lose flagella, produce astaxanthin, and build thick cell walls2. On the other hand, during their vegetative growth, H. pluvialis cells are spherical, ellipsoidal, and pear-shaped with flagella and chloroplasts (see more details in the images of Fig. 1a).

H. pluvialis and its genome. (a) Images of the H. pluvialis life cycle, inclduing macrozooid, palmella, immature aplanospore and aplanospore (from left to right), respectively. (b) The heatmap view of Hi-C result. (c) The circos view of H. pluvialis genome. (d) Phylogeny and gene-family analysis of seven representative microalgae species.
Previous studies with transcriptomics, metabolomics and/or proteomics data have identified several important genes related to astaxanthin biosynthesis under stress conditions (such as high irradiation, nitrogen deprivation, and nutrient starvation)3,4,5. In our previous report6, a draft genome assembly was generated with assistance of only Illumina short-read sequencing. However, its contigs and scaffolds are fragmental, resulting in somewhat redundancy. That assembly is 669.0 Mb in length, with relatively low values of scaffold N50 (288.6 kb) and contig N50 (8.4 kb)6. Due to fragmental assembly and limited genome resources, details of molecular clues to astaxanthin biosynthesis in H. pluvialis remain elusive. Here, we performed long-read PacBio HIFI and high-resolution chromosome conformation capture (Hi-C) sequencing, with the main aim to obtain a high-quality and chromosome-level genome assembly of H. pluvialis.
Whole-genome sequencing, assembly, and annotation of this economically important microalga were fulfilled with a great improvement. In addition, carotene biosynthetic genes cooperate with β-carotene ketolase (CRTO) and hydroxylase (CRTR-B) to synthesize astaxanthin under high irradiation and salinity stress, which are the most common stressors during cultivation of H. pluvialis3,4,5. We therefore conducted additional transcriptome analysis on stressed cells to reveal differential astaxanthin production from certain critical genes, such as those encoding beta-carotene ketolases (BKTs)6. In the coming future, these valuable genomic resources will facilitate breeding of novel H. pluvialis strains to obtain higher astaxanthin yield.
Methods
Sample materials, whole genome sequencing and genome assembly
The H. pluvialis strain 192.80 was purchased from the SAG Culture Collection of Algae (Göttingen University, Göttingen, Germany). Microalga cells were cultivated in ESP Ag medium7. Genomic DNAs were extracted from cultivated cells using Qiagen GenomicTip100 (Qiagen, Germantown, MD, USA). A routine whole-genome shotgun sequencing strategy was applied. In brief, long SMRTbell libraries were constructed for the HiFi sequencing based on PacBio’s standard protocol (Pacific Biosciences, Menlo Park, CA, USA). These libraries were sequenced through a PacBio Sequel II System (Pacific Biosciences). About 1.6 million of consensus reads (26.1 Gb) with a mean length of 16.6 kb were generated. For the Hi-C sequencing, genomic DNAs were fixed with formaldehyde, sheared by a restriction enzyme (MboI) to build a Hi-C library, and then sequenced on a HiSeq-Xten sequencing platform (Illumina Inc., San Diego, CA, USA). A total of 98.0 Gb of 150-bp paired-end Hi-C data were generated.
The sequenced HIFI reads were initially assembled to be contigs by hifiasm (version 0.14-r312)8. This primary genome assembly contains 315.1 Mb of contigs, with a N50 length of 304.8 kb (Table 1). Subsequently, we integrated Hi-C data to construct a high-quality de novo assembly at the chromosome level. In brief, quality control was performed to filter the Hi-C raw reads, and then valid Hi-C connected reads were generated by Juicer version 1.59. The 3D de novo assembly (3D-DNA, version 180922) pipeline10 was employed to link the contig sequences into chromosome-level sequences. Haplotypic 32 chromosomes (chr; Fig. 1b) with a total length of 285.4 Mb (Table 2) were built by using these Hi-C sequences; they account for about 90.3% of the whole genome assembly (Fig. 1c), individually ranging from 1.2 Mb (Chr32) to 21.5 Mb (Chr8) in length (Table 2). The completeness of this final genome assembly was evaluated by Benchmarking Universal Single-Copy Orthologs (BUSCO)11 v5.2.2, revealing that 93.4% BUSCOs are complete. We also compared this assembly (~300 Mb) with our previous draft assembly6 (~600 Mb), and found a lot of 1:2 blocks (Fig. 2a) by using the i-ADHoRe v3.0 software12. For example, two scaffolds (7 and 81) in the draft genome correspond to the Chr14 in part (see Fig. 2b).

Synteny blocks between our current chromosome-level assembly and the previous 600-Mb draft6 of the H. pluvialis genome. (a) An overview of the total synteny blocks. Only partial blocks are visible due to the fragmental contigs in the previous draft assembly. (b) An example of the solid 1:2 blocks in Chr14.
Genome annotation
Repeat sequences of the assembled H. pluvialis genome were identified by employing several programs including Tandem Repeats Finder13, LTR_FINDER14, RepeatProteinMask and RepeatMasker15. Tandem Repeats Finder13 was employed to search for tandem repeats using optimized parameters (Match = 2, Mismatch = 7, Delta = 7, PM = 80, PI = 10, Minscore = 50, and MaxPerid = 2,000). A de novo repeat library was built by the LTR_FINDER (version 1.0.6; parameter: -w 2). RepeatMasker was then utilized to map the H. pluvialis assembly onto the Repbase TE library (version 3.2.9)16 so as to search for known repeat sequences as well as map it onto the de novo repeat libraries to identify novel types of repeat sequences.
We then performed annotation of the H. pluvialis genome assembly with two routine methods, including homology-based and transcriptome-based annotation. Eight representative species, including Chlamydomonas reinhardtii, Paramecium tetraurelia, Saccharomyces cerevisiae, Symbiodinium kawagutii, Symbiodinium minutum, Chlamydomonas eustigma, Chromochloris zofingiensis and Micromonas pusilla, were downloaded from NCBI and were selected for the homology annotation. Their protein sequences were mapped onto the H. pluvialis genome assembly utilizing TblastN17 with an E-value ≤ 1e−5. Genewise 2.2.018 was subsequently employed to predict gene structures based on these TblastN results. Total RNA was extracted from those control cells (sample ID: LLMT4, 5, and 6; see more details in the followed section on Total RNA Isolation) for subsequent transcriptome sequencing on an Illumina HiSeq4000 platform. These data were mapped onto the assembled genome using HISAT v2.0.419. We then utilized Cufflinks (version 2.2.1)20 to identify those preliminary genes. Finally, we applied Maker21 to integrate predicted genes from both annotation procedures.
The final gene set is composed of 32,416 protein-coding genes, with an average of 6.3 kb in length. Their deduced protein sequences were then mapped against public TrEMBL, Swiss-Prot22, InterProScan23,24, KEGG25 and NR databases using BlastP with an E-value ≤ 1e−5. Finally, approximately 97.2% of the predicted genes have at least one functional assignment from these public databases.
Transcriptome analysis
The H. pluvialis strain 192.80 was purchased and cultured by using Bold Basal Medium in 250-mL Erlenmeyer flasks at 22 °C under continuous fluorescent lamps (20 μmol·m−2·s−1) to the logarithmic phase (about 1 × 105 cells·mL−1)6. Algal cells were sub-cultured into 300 mL BBM medium and treated with salicylic acid (SA, 25 mg·L−1) and high light (HL, 350 μmol·m−2·s−1). The treatment of salicylic acid and high light was named as SAHL for short. Algal cells after treatments with at 0 h (Control), 1 h (SAHL 1), 6 h (SAHL 6), 12 h (SAHL 12), 24 h (SAHL 24), and 48 h (SAHL 48) were collected and used for RNA-seq sampling, respectively. These transcriptome reads were dowanloaded from previous study26 with the accession number PRJNA675306. Paired-end raw reads were then processed by removal of adapters and low-quality sequences using SOAPnuke (version 1.5.6)27 with default parameters. These clean data were then mapped onto the assembled genome using HISAT19.
Transcript quantification of fragments per kilobase per million (FPKM) in each sample was calculated by using Cufflink28. Differentially expressed genes (DEGs) between the treatment and control groups were identified using “edgeR” (version 3.15) in the R package29 with log2 (ratio) ≥ 1 and adjusted P-value ≤ 0.05 as the threshold. Finally, a pathway enrichment analysis was conducted on these up- and down-regulated DEGs according to the KEGG database25.
Evolutionary placement of H. pluvialis
A whole-genome phylogenetic analysis on H. pluvialis and other six related microalgae was performed to determine the evolutionary position of H. pluvialis. These examined species, including Chlorella sorokiniana, Chlamydomonas reinhardtii, Cladocopium goreaui, Breviolum minutum, Fugacium kawagutii and Symbiodinium microadriacticum, were downloaded from NCBI. The whole-genome gene sets from H. pluvialis and others were aligned by BLASTp (version 2.2.6) to check homology for generation of a sequence similarity matrix, which was then utilized to identify gene families by using OrthoMCL30 and Markov Chain Clustering (MCL) with default parameters. We identified single-copy orthologues among the seven species, and then these orthologues were aligned with MUSCLE version 3.731. All alignments were combined to obtain a super alignment sequence.
We first applied the maximum likelihood (ML) method to generate a phylogenetic tree (Fig. 1d), which was implemented in PhyML version 3.032. To confirm this topology, we also utilized Bayesian inference (BI) to establish the same phylogenetic tree by using MrBayes version 3.2.233. Meanwhile, we calculated the divergence time by MCMCtree in the PAML package34, with calibration references from the TIMETREE35. Expanded and contracted gene families were predicted by employing CAFE v4.2.136. In total, 41 single-copy gene families including 287 genes were identified from the seven representative microalgal species. These genes from each species were concatenated together and finally constituted a super-length nucleotide dataset to yield 165,027 aligned sites. Our phylogenetic analysis and divergence results discovered that H. pluvialis is close to C. reinhardtii, and their divergence time is at 520.4 million years ago (Mya). We found 4,592 expanded gene families and 26,300 contracted gene families in the H. pluvialis genome (Fig. 1d).
Localization of some astaxanthin biosynthesis related genes in the H. pluvialis genome
Protein sequences of BKT (accession no. CAA60478.1, Beta-carotene ketolase), PSY (CHLRE_02g095092v5, Phytoene synthase), PDS (CHLRE_12g509650v5, Phytoene desaturase), ZDS1 (CHLRE_07g314150v5, Zeta-carotene desaturase 1), LCYB (CHLRE_08g358538v5, Lycopene beta-cyclase), and CHYB (CHLRE_04g215050v5, Beta-carotenoid hydroxylase) were downloaded from the NCBI. We utilized tBLASTn (version 2.2.6) to search the coding regions of these putative astaxanthin biosynthesis related genes6, and their encoding sequences in the H. pluvialis genome were further predicted by Genewise2.2.018.
H. pluvialis is popular for its strong capacity to produce large amounts of astaxanthin that is a strong antioxidant for human health, cosmetics and aquaculture1.
Because about 1 kg of dry H. pluvialis cells can produce over 40 g astaxanthin37, this species is a great material for production of astaxanthin. Previous study has shown that CRTO and CRTR-b are two key enzymes for the astaxanthin biosynthesis pathway38. In this study, three BKT genes were identified to be up-regulated in H. pluvialis cells with diverse stress treatments39. However, in our current genome searching, we identified five BKT genes that were distributed in Chr10, Chr26 and scaffold206 (Fig. 3a). More excitingly, a tandem duplication of three BKT genes (BKT1, BKT2 and BKT3) were observed in the Chr26. These BKT genes have similar protein sequences (Figs. 3b), 3D structures (Fig. 3c) and gene structures (Fig. 3d), while with potentially different astaxanthin-producing capacity. For example, we qualified BKT3 with the highest transcription level (data not shown) under the SAHLtreatment (see the transcriptome sequencing section and the previous report26). We therefore propose that some of these duplicated BKT genes (BKT1, 2 and 3) are potetntially the major contributors to the rapid synthesis and accumulation of large amounts of astaxanthin. Conversely, the relative C. reinhardtii, without evidence of astaxanthin production, doesn’t have any functional BKT gene in its genome.

Summary of the five bkt genes in Haematococcus pluvialis. (a) The chromosome location of BKT1-5. (b) Alignment of the five BKT protein sequences. (c) 3D structures of the five BKT proteins. (d) Distribution of exons and introns.
On the other hand, we compared several other genes from the astaxanthin biosynthesis pathway, and observed that some in H. pluvialis are remarkably expanded than C. reinhardtii. For example, the H. pluvialis genome contains 8 CHYB, 3 PSY, 3 ZDS1, 2 LCYB and 1 PDS genes, corresponding to only one of each in the C. reinhardtii genome; these high copy numbers may provide additional support for the high astaxanthin production in H. pluvialis.
In summary, we reported the first chromosome-level whole-genome assembly for the attractive astaxanthin-producing green microalga H. pluvialis. This genome is a valuable material for deep understanding the molecular clue of algal astaxanthin yield. We found five BKT genes in H. pluvialis genome. These expanded genes may play a key role in the high astaxanthin production. Our genome and transcriptome data sets will facilitate molecular breeding or biosynthesis of novel strains with significantly improved astaxanthin contents.
Data Records
The PacBio long reads, Hi-C sequencing reads, and the final genome assembly were deposited at NCBI with the accession number PRJNA96447940. The annotation data and protein sequences were deposited at Figshare with doi number https://doi.org/10.6084/m9.figshare.2304708841. The raw reads were deposited at NCBI Sequence Read Archive with accession number SRR25425436 and SRR2542543642.
Technical Validation
The extracted DNA quality was examined by using the agarose gel electrophoresis with over 1.8 of the DNA spectrophotometer ratios (260/280) and around 20 kb main band. The Nanodrop ND-1000 spectrophotometer (RIN > 8.0; LabTech, Corinth, MS, USA) was utilized to check the purified RNA quality. The completeness of this H. pluvialis genome was validated by BUSCO v5.2.2. The final BUSCO result showed 93.4% completeness.
Code availability
The execution of all software and pipelines in this study strictly followed the manuals and protocols of the published bioinformatic tools. The versions of the software employed have been specified in the Methods section. If no parameter is provided, the default is used. No custom code was employed.
References
Ambati, R. R., Phang, S. M., Ravi, S. & Aswathanarayana, R. G. Astaxanthin: sources, extraction, stability, biological activities and its commercial applications–a review. Mar. Drugs 12, 128–152 (2014).
Shah, M. M., Liang, Y., Cheng, J. J. & Daroch, M. Astaxanthin-Producing Green Microalga Haematococcus pluvialis: From Single Cell to High Value Commercial Products. Front. Plant Sci. 7, 531 (2016).
Kim, D.-K. et al. Transcriptomic Analysis of Haematococcus lacustris during Astaxanthin Accumulation under High Irradiance and Nutrient Starvation. Biotechnol. Bioproc. E. 16, 698–705 (2011).
Su, Y. et al. Metabolomic and network analysis of astaxanthin-producing Haematococcus pluvialis under various stress conditions. Bioresour. Technol. 170, 522–529 (2014).
Gao, Z. et al. Transcriptome Analysis in Haematococcus pluvialis: Astaxanthin Induction by Salicylic Acid (SA) and Jasmonic Acid (JA). PLoS One 10, e0140609 (2015).
Luo, Q. et al. Genome and Transcriptome Sequencing of the Astaxanthin-Producing Green Microalga, Haematococcus pluvialis. Genome. Biol. Evol. 11, 166–173 (2019).
Grünewald, K., Hagen, C. & Braune, W. Secondary carotenoid accumulation in flagellates of the green alga Haematococcus lacustris. Eur. J. Phycol. 32, 387–392 (1997).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Proost, S. et al. i-ADHoRe 3.0–fast and sensitive detection of genomic homology in extremely large data sets. Nucleic. Acids Res. 40, e11 (2012).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic. Acids Res. 27, 573–580 (1999).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC bioinformatics 9, 18 (2008).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics Chapter 4, Unit 4.10 (2004).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Trapnell, C. et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53 (2013).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Bairoch, A. et al. The universal protein resource (UniProt). Nucleic Acids Res. 33, D154–D159 (2005).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, 353–361 (2017).
Hu, Q. et al. Transcriptome-based analysis of the effects of salicylic acid and high light on lipid and astaxanthin accumulation in Haematococcus pluvialis. Biotechnol. Biofuels 14, 82 (2021).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6 (2018).
Pollier, J., Rombauts, S. & Goossens, A. Analysis of RNA-Seq data with TopHat and Cufflinks for genome-wide expression analysis of jasmonate-treated plants and plant cultures. Methods Mol. Biol. 1011, 305–315 (2013).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 (2010).
Chen, F., Mackey, A. J., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34, 363–368 (2006).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Yang, Z. & Rannala, B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Molecular biology and evolution 23, 212–226 (2006).
Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 34, 1812–1819 (2017).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Lorenz, R. T. & Cysewski, G. R. Commercial potential for Haematococcus microalgae as a natural source of astaxanthin. Trends Biotechnol. 18, 160–167 (2000).
Grunewald, K., Hirschberg, J. & Hagen, C. Ketocarotenoid biosynthesis outside of plastids in the unicellular green alga Haematococcus pluvialis. J. Biol. Chem. 276, 6023–6029 (2001).
Huang, J. C., Chen, F. & Sandmann, G. Stress-related differential expression of multiple beta-carotene ketolase genes in the unicellular green alga Haematococcus pluvialis. J. Biotechnol. 122, 176–185 (2006).
Bian, C. GenBank https://identifiers.org/ncbi/insdc:JASKMD000000000 (2023).
Bian, C. Haematococcus pluvialis genome annotation and protein sequences. figshare https://doi.org/10.6084/m9.figshare.23047088 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP451499 (2023).
Acknowledgements
This work was supported by Chinese National Key R&D Project for Synthetic Biology (2018YFA0902500), National Natural Science Foundation of China (32273118), the Science and Technology Program of Guangdong Province of China (2022B1111070005, 2020B121202014), Shenzhen Special Fund for Sustainable Development (KCXFZ20211020164013021) and Shenzhen University 2035 Initiative (2022B010) to Dr. Zhangli Hu. This work was also supported by Key laboratory of Tropical and Subtropical Fishery Resources Application and Cultivation, Ministry of Agriculture and Rural Affairs, Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences (20220202).
Author information
Authors and Affiliations
Contributions
Zhangli Hu and Qiong Shi designed the research. Guiying Zhang and Ming Tao collected samples. Chao Bian, Chenglong Liu, Danqiong Huang, Chaogang Wang, Sulin Lou, Hui Li, Zhangli Hu and Qiong Shi performed experiments or date analyses; Chao Bian, Chenglong Liu, Zhangli Hu and Qiong Shi wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bian, C., Liu, C., Zhang, G. et al. A chromosome-level genome assembly for the astaxanthin-producing microalga Haematococcus pluvialis. Sci Data 10, 511 (2023). https://doi.org/10.1038/s41597-023-02427-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02427-1
