
Abstract
This study presents eight new high-quality de novo transcriptomes from six co-occurring species of calanoid copepods, the first published for Neocalanus plumchrus, N. cristatus, Eucalanus bungii and Metridia pacifica and additional ones for N. flemingeri and Calanus marshallae. They are ecologically-important members of sub-arctic North Pacific marine zooplankton communities. ‘Omics data for this diverse and numerous taxonomic group are sparse and difficult to obtain. Total RNA from single individuals was used to construct gene libraries that were sequenced on an Illumina Next-Seq platform. Quality filtered reads were assembled with Trinity software and validated using multiple criteria. The study’s primary purpose is to provide a resource for gene expression studies. The integrated database can be used for quantitative inter- and intra-species comparisons of gene expression patterns across biological processes. An example of an additional use is provided for discovering novel and evolutionarily-significant proteins within the Calanoida. A workflow was designed to find and characterize unannotated transcripts with homologies across de novo assemblies that have also been shown to be eco-responsive.
Similar content being viewed by others

Background & Summary
Transcriptomics, the application of high-throughput sequencing of millions of short nucleotide sequences generated from mRNA, has transformed studies of the physiological ecology of marine organisms1,2,3,4,5. Gene expression profiles of one or more species have resulted in new insights into physiological acclimatization, metabolic trade-offs and resilience, causes of high-mortality events and niche partitioning between similar species. However, progress in the application of environmental transcriptomics has been hampered by a scarcity of genomic resources, especially for co-occurring zooplankton species. Furthermore, existing references for gene expression studies are of variable quality and many genes, including eco-responsive genes remain unannotated5. Here, we present eight de novo transcriptomes for six calanoid copepod species that co-occur across the Sub-arctic North Pacific. The goals were to (1) generate high-quality assemblies, (2) assess their value as references for cross-species comparisons; and (3) to develop a workflow to search for and identify novel predicted proteins that are unique to copepods.
Calanoida is an order within the Copepoda, the second largest sub-class of the Crustacea. Calanoids are small (≤15 mm) free-living, mostly planktonic organisms that inhabit marine and freshwaters worldwide6,7. Abundant and ecologically important, they are a key link between phytoplankton and the upper trophic levels, such as larval fishes and other planktivores including invertebrates, sea birds and marine mammals7. Relative to their importance they are understudied: their natural habitats are difficult to access and few species are amenable to laboratory cultivation. Nevertheless, calanoids are known for many unique adaptations (high transparency, fluorescent proteins, bioluminescence, myelin, unique sensory structures) that have set them apart from other better studied arthropods8,9,10,11,12. These cellular properties depend on the evolution of novel proteins. Thus, it is not surprising that de novo assemblies of copepods always include a significant number of predicted proteins that are unannotated with unknown function13,14,15. A significant number of these are differentially-expressed eco-responsive genes3,16, yet no studies have used available copepod transcriptomes to search for taxon-specific proteins with unknown function.
In contrast to terrestrial arthropods, genomes of only a few marine copepods have been fully sequenced, assembled and annotated17,18,19,20. However, de novo assemblies of expressed transcripts of non-model organisms provide an alternative solution to generate reference transcriptomes for gene expression studies and the discovery of novel proteins. The six species in this study are important members of sub-arctic pelagic communities, inhabiting both oceanic regions and coastal sounds and fjords in the North Pacific and in the Bering Sea. The species account for nearly 80% of the annual calanoid copepod production in the region21. Climatic events like heat waves and global warming have raised concerns about the stability of the zooplankton community. How the community might change requires a better understanding of the ecophysiology of all six species across a range of environmental conditions.
The six species in this study present a taxonomic range that increases opportunities for finding new proteins, both shared or group-specific. They include three species in the genus Neocalanus (N. flemingeri, N. plumchrus, N. cristatus), one additional member of the family Calanidae (Calanus marshallae), one member of the non-calanid myelinate calanoids (“Myelinata”; the eucalanid Eucalanus bungii) and a more basal and amyelinate species, Metridia pacifica (Table 1). Taxonomically, the species belong to three calanoid superfamilies, with different adaptations (Table 1)7,22,23,24. The four species in the family Calanidae belong to a group of lipid-rich copepods that are a key food source for upper trophic levels. Their complex life cycle includes a seasonal developmental arrest and dormancy (“diapause”) at depths of 200 m or deeper25. The amyelinate M. pacifica is known for its significant diel vertical migration and bioluminescence26. The sixth species, E. bungii, in addition to being myelinate, is characterized by its transparency and very long first antennae. Behaviourally E. bungii hovers in place generating feeding currents to bring food particles to its mouth27.
Methods
Work-flow
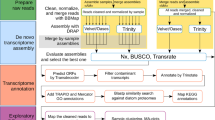
A diagram of the workflow used in this study is presented in Fig. 1. De novo transcriptomes were assembled from short-read sequences generated from RNA extracted from field-collected individuals. Each assembly was assessed for quality using multiple indicators, and assembled transcripts were annotated based on sequence similarity. Cross-species comparisons focused on identifying annotated and unannotated orthologous genes among the six species. In downstream analysis, selected unannotated genes were examined for their eco-responsiveness, their presence as orthologs across taxonomic groups and functional motifs in their predicted proteins.

Flow chart of the processing of individual copepods for RNA-seq and data analyses. After processing short sequence reads (150 bp, paired-end) for quality and Trinity assembly, de novo transcriptomes were assessed for quality (left), annotated (center) and compared across species (right). The flow on the right branch focuses on the analysis of full-length predicted proteins and includes the identification of orthologous genes and the search for novel proteins.
Sample collection
Zooplankton were collected from depth (2015, 2017, 2018, and 2019) during the cruises of the Seward Line Long-term Observation Program (LTOP) (http://www.sfos.uaf.edu/sewardline/) at two stations in Prince William Sound: “PWS2” (Lat: 60°32.1′ N, Long: 147°48.2′ W, depth 798 m) and “PWS3” (Lat: 60°40.0′ N, Long: 147°40.0′ W, depth 742 m). Table 1 lists the collection date, station and depth stratum for each individual. Zooplankton collections were made using either a QuadNet with two 150 µm and two 53 µm mesh nets (April collection), or a multiple opening and closing plankton net (0.25 m2 cross-sectional area; 150 μm mesh nets; Multinet-Midi, Hydro-Bios; September collections). The QuadNet net was towed vertically from 100 to 0 m, while the Multinet was towed either vertically or obliquely sampling depth strata between 700 m and the surface. The live zooplankton collections were immediately diluted into ambient seawater and maintained at collection temperatures (5 – 7 °C). Copepods were removed from the diluted samples using a small ladle, and sorted under a dissection microscope to select individuals from the target species (Table 1). Briefly, live and undamaged individuals were identified and staged using morphological criteria, with species identification being confirmed through the COI sequence in the assembled transcriptomes. Identified copepods were isolated using a wide-bore pipette into a dish with filtered seawater before transferring them into microcentrifuge tubes (1.6 ml) with 0.5 ml of RNAlater Stabilization Reagent (QIAGEN). Preserved copepods were frozen first in −20 °C during the cruises, and then transferred to −80 °C until further processing.
Total RNA extraction, library construction, RNA sequencing and quality control
For each target species, total RNA was extracted from individuals using QIAGEN RNeasy Plus Mini Kit (catalog # 74134) in combination with a Qiashredder column (catalog # 79654). Selection for sequencing was based on high RNA yields and purity of extraction (RIN >8). One individual was selected from each species, except for two where two individuals were sequenced from the same species. The final list included pre-adults (CV) for Neocalanus flemingeri (n = 2), N. cristatus (n = 1), C. marshallae (n = 2), E. bungii (n = 1), an adult male (developmental stage CVI) for N. plumchrus (n = 1) and an adult female for M. pacifica (n = 1). Total RNA was shipped on dry ice to the Georgia Genomics Bioinformatics Core (https://dna.uga.edu) for RNA-Seq. There, double-stranded cDNA libraries (KAPA Stranded mRNA-Seq Kit, with KAPA mRNA Capture Beads (cat KK8421) from each individual were multiplexed and sequenced using an Illumina Next-Seq. 500 instrument (High-Output Flow Cell, 150 bp, paired end). Quality of each RNA-Seq library was reviewed with the FastQC software28. From each RNA-Seq library, low quality reads were removed using FASTQ Toolkit (v. 2.2.5 within BaseSpace). Illumina adaptors, reads < 50 bp long, reads with an average Phred score < 30 and the first 12 bp from each read, were removed from each library. The same workflow was applied to all eight datasets.
De novo assembly, mapping, core-gene statistics
Individual de novo transcriptomes were generated from each dataset at the National Center for Genome Analysis Support’s (NCGAS; Indiana University, Bloomington, IN, USA) Mason Linux cluster using Trinity software (v. 2.4.0, except N. plumchrus, v. 2.0.6)29. Initial evaluation involved self-mapping of reads against the respective de novo assembly using Bowtie2 software (v. 2.3.5.1)30. Completeness of each de novo assembly was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO) software31 by searching each assembly for the presence of eukaryote “core” genes using the Arthropoda database as reference (BUSCO version 5.3.2, dataset: arthropoda_odb10 (2020-09-10, 90 genomes, 1,013 BUSCOs).
Functional annotation
Assemblies were functionally annotated against the NCBI Swiss-Prot protein and UniProt databases. Initial annotations were obtained by using the BLASTx algorithm on a local BLAST webserver with a Beowulf cluster using the Swiss-Prot protein database (downloaded February 2021) as reference and a threshold E-value of 10−5. Transcripts with BLAST annotations were then searched against the Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway databases using UniProt32.
Species confirmation and contamination testing
The annotation results were also used to confirm species identity and to provide an initial assessment of possible contaminants. Sequences annotated as “COX1” (cytochrome oxidase subunit 1 or “COI”) were retrieved from each file, followed by a confirmatory BLASTn into the NCBI nucleotide collection (nr/nt) database. BLAST results for each top hit were checked for a species match, transcript coverage, E-value and percent identity (>97% identity required). To search further for fragments of foreign COI sequences, an artificial “species filter” that quantifies such contaminants33, was constructed from the full-length COI sequences of each of the species presented here, as well as likely contaminants (See Supplementary Information SD1, Part II). Raw reads from each transcriptome were mapped against the artificial one, and the counts attracted to the COI of each species were used as a measure of contamination level. In addition, each assembly was searched manually for ribosomal RNA sequences. The BLASTn algorithm was run using species-specific 18 S rRNA sequences as queries to: (1) obtain the native sequence(s) most closely matching the rRNA reference; and (2) to search for rRNA sequences indicating possible foreign contamination. MAFFT (v. 7.511) alignments were used to identify sequences with identities in the low 90% range or below (“% ID to native” column in Supplementary Information SD2) as candidates for foreign rRNA. Such candidates were then checked for taxon identification and magnitude of contamination using a reciprocal BLAST (BLASTn) and relative abundance in the count file generated by Bowtie2.
Protein prediction
The assembled transcriptomes were further validated by tallying the numbers of predicted proteins they contained and the homologies among them, both within and between species. For each assembly, open reading frames (ORFs) were identified using TransDecoder (v. 5.5.0; setting: report only the single best ORF)29. The resulting translated transcriptomes were filtered for predicted full-length (“complete”) proteins.
Homolog clustering
Prevalence of homologs, especially among closely-related species, was used as a further validation of transcriptome quality. Groups of predicted proteins with homologies among selected combinations of the new transcriptomes were generated using OrthoVenn2, a web-based tool designed to find sets of homologous proteins, termed “clusters”34. When homology clusters needed a common reference, we included a vetted transcriptome previously-published for a stage CV N. flemingeri (NCBI accession series GHLB01000000, BioProject PRJNA496596)3. To estimate the reproducibility of separate assemblies of the same species, we generated two intraspecies cluster sets, one for N. flemingeri (n = 3) and one for C. marshallae (n = 2). To estimate the extent of homologies between transcriptomes of more distantly-related species, we generated heterospecific cluster sets for each of the assemblies paired with the N. flemingeri reference transcriptome. Finally, to survey the consistency of homologies across different phylogenetic breadths in these transcriptomes, we generated cluster sets from multi-species selections of transcriptomes representing four taxonomic categories: within genus (n = 3, Neocalanus), within family (n = 4, Calanidae: Neocalanus species plus C. marshallae), within the Myelinata (n = 5, Calanidae + E. bungii), and within order (n = 6, Calanoida: Myelinata + M. pacifica).
Bioprospecting for novel proteins
To illustrate the potential for these transcriptomes to be useful in discovering novel proteins responsive to changes in environmental conditions, we focused on multi-species clusters of homologs for which OrthoVenn2 failed to find annotations in SwissProt. Starting with the clusters in the 6-species Calanoida set just described, which we will refer to as the “primary set,” we selected three secondary subsets of clusters having the three taxonomic coverages just described: Neocalanus, Calanidae, and Myelinata. For each of these subsets, we retrieved from OrthoVenn2 a “clusters shared by all” list - a list of homologous clusters containing representation from all of the selected species (see Supplementary Information SF1 for examples). While these subsets contained all clusters in the primary set with homologs (orthologs) shared among the targeted secondary species, some of the clusters also included homologs from outside of the targeted taxa. To restrict the list of candidate clusters to a taxonomically homogeneous set that lacked such “foreign” homologs, we searched the lists and removed any such outsiders; i.e. from the Myelinata subset we removed clusters that contained homologs in the amyelinate M. pacifica transcriptome; from the Calanidae subset, M. pacifica and E. bungii-containing clusters; and from the Neocalanus subset, M. pacifica, E. bungii and C. marshallae clusters.
The resulting lists in each of the three taxonomic categories, which are enriched for potential novel and taxonomically-unique proteins, were then further refined by searching for environmentally-regulated proteins. Each list was compared with lists of translated non-annotated N. flemingeri genes that had been identified in previous studies as differentially expressed in response to environmental conditions3. To pinpoint where these candidate novel proteins might have evolved, we put each through a tBLASTn taxonomic scan of transcriptomes in the NCBI TSA databases, searching for similar sequences in transcriptomes in taxonomic categories with increasing phylogenetic distance from N. flemingeri. Two measures were employed to refine the assessment of novelty: first, the E-value of the best match (top hit) for each search was used as a measure of similarity for that target taxonomic category. Novel proteins were expected to have a high similarity among those taxa possessing them and a much lower similarity for taxa lacking them. Thus, a sharp rise in the similarity (i.e. decrease in E-value) with taxonomic proximity to N. flemingeri was taken as a likely phylogenetic point of emergence for the novel protein. Second, this predicted emergence-point was referenced to a cladogram constructed from the top hits, translated into proteins. The translated proteins were aligned using MAFFT software35,36 and then manually trimmed to remove non-conserved regions. The cladogram was constructed from these sequences with the software package for phylogenetic analysis using Bayesian Markov chain Monte Carlo methods, MrBayes 3.237,38,39, with two independent runs of four chains each and 10,000,000 generations (the initial 25% discarded as burn-in) using the WAG substitution model of protein evolution40 and a gamma distribution of rates. Maximum likelihood bootstrap values were obtained using RAxML 8 with 1,000 bootstrap replicates using the WAG substitution model and a gamma distribution of rates41. The consensus tree from MrBayes was visualized in FigTree v. 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and the bootstrap values were reported. The cladograms were compared with the known phylogenetic relationships among the component species. The preferred outgroup(s) for each cladogram was taken from the crustacean category (excluding Copepoda) or from the Arthropoda (excluding Crustacea). If neither category produced a satisfactory outgroup, one from a greater taxonomic distance was used. Selected sequences were examined for function-related motifs as a step toward determining possible roles for the non-annotated proteins. Motif searches were performed via three web-based search portals: UCL’s PSIPRED and MEMSAT (http://bioinf.cs.ucl.ac.uk/psipred/); Kyoto University Bioinformatics Center’s MotifSearch (https://www.genome.jp/tools/motif/), and Swiss Institute of Bioinformatics’ MotifScan (https://myhits.sib.swiss/cgi-bin/motif_scan).
Data Records
The raw full-length data (Table 1) were deposited in the NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP289633 (2020)42. The respective TSA accession numbers are N. plumchrus (GJRU00000000), N. flemingeri (GJRT01000000, GJSD01000000), N. cristatus (GJRH00000000), E. bungii (GJRG00000000), M. pacifica (GJAO00000000), C. marshallae (GJRL00000000, GJRF00000000)43,44,45,46,47,48,49,50. Additional metadata and functional annotations of each transcriptome assembly are available at BCO-DMO (Project: 720280)51.
Technical Validation
Quality control, de novo assembly, mapping and core gene statistic: Sequencing yields, assembly statistics and transcriptome completeness for the eight datasets are summarized in Table 2. The number of cleaned reads retained for the assembly exceeded 90% and ranged from 45 to 73 million (Table 2). Seven transcriptomes had N50 values that exceeded 1,000 bp. The longest transcript in all seven assemblies was at least 13,000 bp with transcripts >20,000 bp in C. marshallae, N. cristatus and N. plumchrus. The number of Trinity transcripts ranged from 66,838 to 119,923 (Table 2). While seven assemblies were very similar, the statistics for E. bungii suggests that the reads from this species did not assemble as well as the others: there were fewer transcripts and transcript lengths were shorter (N25, N50, N75, maximum length, see Table 2). Self-mapping rate for the assemblies averaged 89% and ranged from 84% to 92% (Table 2). Based on the BUSCO analysis, the number of “complete” orthologous BUSCO “core” genes ranged from 70% to 94%, with only one species (E. bungii) failing to attain at least 85% of “complete” core genes. The transcriptomes had an additional 3% to 14% “fragmented” “core” transcripts (Table 2).
Functional annotation
The functional annotation of the transcripts using the Swiss-Prot database identified over 30,000 significant BLAST hits for seven of the eight assemblies (Table 2). The assembly with the lowest number of assembled transcripts (E. bungii) returned fewer than 18,000 hits. The percentage of transcripts with significant Swiss-Prot hits was similar across all eight assemblies, ranging between 44% (M. pacifica, Mp2017) and 49% (N. flemingeri, Nf2019). Most of the transcripts (>95%) with BLAST hits were also annotated with gene ontology (GO) terms with a smaller number annotated with KEGG enzyme commission numbers (EC, Table 2). The distribution of GO terms across major biological processes was similar in the eight transcriptomes (Fig. 2), as expected based on other copepod de novo assemblies13,52. While the assembly for E. bungii was less complete, the transcripts were of high quality, and we retained this transcriptome in the downstream analyses.

Distribution of Gene Ontology terms (GO). Stacked bar plot showing for the eight transcriptomes relative % of transcripts annotated to GO terms within the biological process category. For each transcriptome, percentages were calculated as the number of transcripts annotated to a given GO term divided by the total number of GO annotated transcripts in the transcriptome.
Species confirmation
COI sequences from the transcriptomes (Supplementary Information SD1) received confirmatory top species hits with >97% identity for five species even when the assembled transcript was short (Table 3). The COI sequence of the sixth species, C. marshallae is very similar to that of C. glacialis (>97%), raising questions about whether they are separate species or members of a species complex53,54. While very similar, the sequences and other posted C. marshallae complete and partial sequences (e.g. accession numbers GJQX01213347, KX675691 and KP241590), showed consistent differences with respect to C. glacialis sequences, especially in two locations (nucleotides 489 and 627) within the full-length 1,556 bp mtCOI sequence (Supplementary Information SD1). Thus, while very similar there appeared to be consistent genetic differences between the two species. Our analysis is consistent with a recent phylogenetic analysis using 191 genes55.
Testing for contamination
Heterospecific contamination can occur in de novo assemblies from field collections and must be routinely checked for33. A cursory assessment of the 18 S ribosomal RNA sequences suggested some contamination from foreign sources (Supplementary Information SD2). The levels of contamination were extremely low (<1%), however, the level was 5.5% for reads mapping to a C. glacialis sequence present in the M. pacifica transcriptome. This led us to perform an additional analysis to check for cross-contamination between species by mapping the RNA-Seq data to a “species-filter” (see Methods). Bowtie mapping resulted in ca. a million reads from the E. bungii transcriptome mapping to its COI sequence, while none mapped to a human COI sequence or any other species in the species filter. Similarly, with almost half a million M. pacifica reads mapping to its COI sequence, only a total of 9 mapped to the suspected C. marshallae/C. glacialis COI sequences (see Supplementary Information SD1 Part II for filter composition and numerical details). We conclude that there was either no or negligible contamination from these sources with some counts even arising from cross-mapping.
Protein prediction
TransDecoder found open reading frames (ORFs) of 100 amino acids or longer in more than 78% of the transcripts in all transcriptomes, with 22% to 38% predicted to be full length (Table 2). The full-length proteins were analyzed further using the OrthoVenn2 clustering algorithms as described in the Methods and summarized in Table 4.
Intraspecific homologs
Within-species homologies among transcriptomes were expected to be high and an indicator of similarity across assemblies. As shown in Table 4, the percentage of the OrthoVenn2 clusters shared between the two C. marshallae transcriptomes approached 70%. A similar percentage was observed between at least two of the three N. flemingeri transcriptomes (Table 4; Supplementary Information SD3M). Interestingly, when partial-protein sequences were included in the OrthoVenn2 uploads, the shared fraction remained similar. Merging multiple transcriptomes with software such as CAP3 can be implemented by a user to produce a more complete reference at the expense of obscuring the hierarchical information provided by Trinity. Mergers also add genetic diversity owing to the use of more than one individual.
Interspecific comparisons
As described in the Methods section, the previously-vetted N. flemingeri transcriptome3 was translated and used as a common reference since it had been used in gene expression studies, allowing identification of genes responsive to changes in environmental conditions. The numbers of complete-protein homolog clusters that were shared between this reference and each of the new transcriptomes ranged from 40% to 70% (Supplementary Information SD3), with the highest percentage shared between the N. flemingeri reference and its and conspecifics and two congeners, and the lowest percentages with the two more distantly-related taxa (E. bungii and M. pacifica). In the survey of the overall taxonomic extent of homologies among the primary cluster sets from the four multi-species sets of transcriptomes (Table 4), the fractions of clusters shared by all of the members (“overlap clusters”) ranged from 7.2% for the set including all 6 species of the order Calanoida, to 35% for those comprising the three species of the Neocalanus genus. The fraction was greater the tighter the taxonomic coverage, as would be expected. Thus, while there were the expected differences among the assembled complete proteins correlating with phylogenetic distance, the large number of overlapping clusters indicates that the individual assemblies produced mutually-consistent transcriptomes.
Usage Notes
Reference transcriptomes for ecophysiology
The de novo transcriptomes were generated from individuals belonging to six key zooplankton species of the sub-arctic waters of the Gulf of Alaska: Neocalanus flemingeri, N. plumchrus, N. cristatus, Calanus marshallae, Eucalanus bungii and Metridia pacifica. The assemblies and their functional annotation provide references for future RNA-Seq studies focused either on single or multiple species, as well as in community studies based on metatranscriptomics17. The predicted sequences can be used to design species-specific primers for PCR-based studies focused on biomarkers56. Homologous genes identified across transcriptomes from these six co-occurring species can be used to generate a set of references for interspecies comparisons. While there are more studies that are generating RNA-Seq data for bulk samples of planktonic organisms, interspecies comparisons, especially for zooplankton, continue to be challenging given the absence of robust community-specific references. By focusing on key species from one region, these de novo transcriptomes open opportunities to examine species-specific responses and niche separation under natural and/or experimental conditions.
Resource for protein discovery
De novo transcriptomes are composed of predicted expressed transcripts assembled from short sequences. These transcripts are then translated into predicted proteins, which may or may not be real. In spite of this limitation, transcriptomes have been rich data sources for protein discovery57,58,59,60,61. Here, we illustrate how non-annotated transcripts with predicted homologous proteins in multiple transcriptomes might be used in the search for protein discovery (Table 5). In the single-species cluster sets for C. marshallae and N. flemingeri, around a third failed annotation through lack of significant BLAST hits in SwissProt. In the multi-species sets, the 6-calanoid primary set as well as the three secondary sets derived from it (see Supplementary Information ST1 for cluster compositions), the percentages of non-annotated clusters ranged from 15% to 20%. After removing clusters with membership outside of the selected secondary taxonomic category, as described in Methods, only a few hundred prime candidates remained, inviting further investigation (Table 5, “non-annotated taxonomically homogeneous” row).
Environmentally-responsive genes among the non-annotated predicted proteins
We compared the candidate genes to lists of environmentally-responsive transcripts of pre-adult N. flemingeri (DEGs) reported from a spatial transect in the Gulf of Alaska and an interannual comparison (2015–2017) within Prince William Sound3,5. Among the DEGs were 101 and 83 non-annotated transcripts/clusters along the spatial gradient and the interannual comparison, respectively (Table 5). Thirty transcripts were differentially-expressed under both conditions (Supplementary Information ST2), highlighting the genes’ likely importance in the ecophysiology of this species. The availability of homologs in the other transcriptomes not only means that these genes are “real”, but also raises the question whether they may be eco-responsive in the co-occurring species under similar conditions.
Comparisons with a rapidly-evolving gene
We took a further step toward testing ways of validating the novelty of these unannotated clusters of homologous environmentally-responsive predicted proteins by searching databases for similar proteins at different phylogenetic distances from the cluster. In order to provide some context, we first characterized the similarity profile of an annotated transcript from the glutathione S-transferases (GST) family as a reference. The GSTs belong to a gene family known to be rapidly evolving62. Using the BLAST-scan algorithm described in Methods, we took the omega GST from N. flemingeri as a query to identify the most similar homologous transcripts in calanoid and other copepods, crustaceans and arthropods. The bar graph in Fig. 3a shows that the similarity [ = -log10(E-value)] declines gradually as a function of phylogenetic distance from the N. flemingeri query. The cladogram in Fig. 3b, based on the same hits, shows nested clades with moderately-well-supported branching (bootstrap values >70) that includes homologs from all the calanoids as well as one from the harpacticoid, Tigriopus. Amino-acid substitution rates (branch lengths) between clades nested within this large clade are modest (~0.2–0.3). The cladogram largely parallels the evolutionary relationships among the Copepoda, especially the calanoid families, as would be expected for a broadly-distributed protein that changed in concert with the species’ evolution6,22,23.

Taxonomic-similarity profile (a) and corresponding MrBayes cladogram (b) for a rapidly-evolving protein (glutathione S-transferase: GST, omega variant). (a) Bar graph of similarity [defined as -log10(E-value)] of the N. flemingeri homolog of the translated GST omega reference sequence (accession # GHLB01049544) in a taxonomic BLAST-scan into NCBI’s TSA database with top-hit transcripts at different phylogenetic distances. (b) Cladogram generated by MrBayes for the top-hit sequences shown in (a). Numbers at nodes are bootstrap values (>50) computed by RAxML. Scale bar: 0.3 estimated substitutions per site. Despite the rapid evolution of this protein, differences between adjacent taxonomic categories in the bar graph are modest (<25 log units of E-value) and the stem-lengths in the cladogram are <0.2 within the Crustacea and 0.4 between the insect homolog and the Crustacea. Taxon codes for bars: Nf: Neocalanus flemingeri; Np: N. plumchrus; Nc: N. cristatus; Ca: family Calanidae (excluding Neocalanus) Calanus marshallae, C. hyperboreus; Eu: superfamily Eucalanoidea Rhincalanus gigas; Cn: superfamily Centropagoidea (Diaptomoidea) Hemidiaptomus amblyodon; Au: superfamily Augaptiloidea (Arieteloidea), Metridia pacifica; Ha: order Harpacticoida, Tigriopus japonicus; Cy: order Cyclopoida, Paracyclopina nana; Cr: class Crustacea (excluding Copepoda), Scylla olivacea; Ar: phylum Arthropoda (excluding Crustacea) Nionia palmeri. Sequences of predicted proteins, accession numbers and E-values are given in the Supplementary Information (SD4).
Environmentally-responsive predicted proteins of N. flemingeri that lack annotations can be compared with the GST pattern. As examples, taxonomic BLAST-scans into NCBI (TSA) of three such sequences are presented in Fig. 4. In contrast to the omega GST pattern, the predicted proteins were highly similar within a narrow taxonomic range, with a sharp decline in similarity [-log10(E-value)] outside of the group (Fig. 4 left panels, set 1). The high-similarity group for Fig. 4a1 included only hits from the order Calanoida. Significantly, the BLAST got no hits among the Harpacticoida or Cyclopoida, giving further emphasis to the uniqueness of that protein group. The high-similarity groups in Figure 4b1, c1 were even more restricted, being confined solely to calanid proteins. Most of the nodes leading to the N. flemingeri query sequence and its cluster of homologs received bootstrap support similar to that of the GST (>80). The support was particularly strong (100) for the clades that included the high-similarity (calanid) taxa in Figure 4b2, c2. For Fig. 4a2, while the branch pattern in the cladogram produced by MrBayes grouped the calanoids appropriately, bootstrap support was not strong. In all three cladogram examples, substitution rates (branch lengths) between the high-similarity taxa and those outside the group were notably longer than for the GST (>0.5). Thus both the sharp rises in the BLAST-scan similarity with taxonomic proximity to N. flemingeri and the long branch lengths for the corresponding points in the cladograms are consistent with a phylogenetic point of emergence of a novel protein within the Copepoda.

Taxonomic-similarity profiles and corresponding MrBayes cladograms of three predicted proteins without functional annotation. Left panels: Bar graphs of similarities in taxonomic BLAST-scans of the N. flemingeri query sequence with top-hit transcripts in available TSA transcriptomes [-log10(E-value)]. Abbreviations (x-axis) are the same as in Fig. 3. A sharp increase in similarity with increasing proximity to the query indicated at the top is hypothesized to be the taxon of evolutionary emergence of each non-annotated cluster (broken line). Right panels: Cladogram branch lengths are proportional to the evolutionary distance from the clade’s branch point (scale bars: 0.3 estimated substitutions per site). Taxonomic categories corresponding to the abrupt change in similarity on the left panel colored in red. Red branches indicate taxa proposed to possess a novel version of the set of homologs as identified by the corresponding similarity bar-plot profile on the left. Where possible, the outgroup used to root the tree was either the crustacean homolog (excluding Copepoda) or the arthropod homolog (excluding Crustacea). In (b), no adequate homolog was identified in either out-group category, so a more distant outgroup was used (a sponge). In Panels a and b, hits were missing for certain taxonomic categories: Harpacticoida and Cyclopoida for (a) and Augaptiloidea and Arthropoda for (b). In some cases, the top-hit sequence was for a partial protein that was too short to be included in a reliable cladogram, in which case the next most similar hit was used. Accession numbers for the N. flemingeri query sequence are given above each bar graph. Species and sequence accession numbers: (a) Nf: Neocalanus flemingeri; Np: Neocalanus plumchrus; Nc: Neocalanus cristatus, Ca: Calanus marshallae; Eu: Rhincalanus gigas; Cn: Labidocera madurae; Au: Metridia pacifica; Cr: Eogammarus possjeticus. (b) Nf: Neocalanus flemingeri; Np: Neocalanus plumchrus; Nc: Neocalanus cristatus, Ca: Calanus hyperboreus; Eu: Rhincalanus gigas; Cn: Temora stylifera; Ha: Platychelipus littoralis; Cy: Apocyclops royi; Sponge: Aplysina aerophoba. (c) Nf: Neocalanus flemingeri; Np: Neocalanus plumchrus; Nc: Neocalanus cristatus, Ca: Calanus hyperboreus; Eu: Rhincalanus gigas; Cn: Eurytemora affinis; Au: Metridia pacifica; Ha: Tigriopus californicus; Cy: Eucyclops serrulatus; Cr: Eurypanopeus depressus; Ar: Anoplogonius nigricollis. Sequences of predicted proteins and E-values are given in the Supplementary Information (SD4). Additional examples are provided in Supplementary Information SF2.
Functional motifs of candidate novel proteins
The presence of functional motifs — sequences of amino acids that have been shown in other studies to endow proteins with known properties — can provide some insights into function. All three putative novel proteins from Fig. 4 contained functional motifs as shown in Fig. 5. The protein of Fig. 5a appears to be a member the pacifastin family, as it contains the typical repeated 6-cysteine-residue motifs (Fig. 5a, violet highlight). The pacifastins are arthropod serine peptidase inhibitors that have been shown to have a role in the immune response63. This copepod sequence features four inhibitor motifs. Interestingly, the malacostracan (amphipod) homolog possesses many more (17; Supplementary Information SD4). The predicted protein of Fig. 5b includes a channel-pore-lining motif (green highlight), suggesting a role as a trans-membrane ion channel. Finally, in the protein of Fig. 5c, the N-terminal portion contains an ankyrin repeat motif (red highlights), which is highly conserved throughout the Arthropoda. A comparison across homologous sequences showed that the predicted proteins of the five myelinate species possess an extensive C-terminal segment that is missing from the amyelinates and more distantly-related taxa. This is shown diagrammatically in Fig. 5d which separates the conserved portion of the protein (red) from the C-terminal extension (blue) (see SD4 for all sequences).

Motifs contained in the sequences for three N. flemingeri proteins of Fig. 4 as identified by motif-search programs (see Methods). (a) Pacifastin-inhibiting motifs: residues 47–70; C80 & 97–124; C128 & 145–168, C200 & 211–241 (violet highlight); Pore-forming motif: residues 258–273 (green highlight). (b) Pore-lining motif: residues 15–30 (green). (c) Ankyrin repeat motif: residues 1–191 (red highlight). (d) Ankyrin motifs in the sequences of the different taxonomic categories of Fig. 4c. Lengths of the different regions in protein homologs containing ankyrin motifs (red portion of bar) in the taxonomic groups and sequences from Fig. 4c. Non-ankyrin regions in blue (residue scale in decades at bottom in light blue). Note the long C-terminal segments in the Myelinata (upper 5 bars), which are absent from all other taxonomic categories. Percentages indicate percent identity between the sequence and the translated N. flemingeri query.
Code availability
Parameters to software tools involved are described below:
FastQC: version 0.11.8
FASTQ Toolkit (BaseSpace): version 2.2.5, parameters: trim of first 12 bp, Quality cutoff (Phred score <30), adapter removal
Trinity: version 2.0.6 (N. plumchrus) and version 2.4.0 (all others), parameters: –seqType fq–CPU 32–max_memory 200 G –min_contig_length 300 –normalize_max_read_cov50
Bowtie: version 2.1.0, default parameters
BUSCO: version 5.3.2, dataset: arthropoda_odb10 (Creation date: 2020-09-10, number of genomes: 90, number of BUSCOs: 1013).
Sequence Manipulation Suite (SMS): (https://sites.ualberta.ca/~stothard/javascript/rev_comp.html)
TransDecoder: version 3.0.0, default parameters (open reading frame > 100 amino acid)
Transeq: Search and Sequence Analysis Tools Services from EMBL-EBI in 2022 https://europepmc.org/
MAFFT: version 7.511 (https://mafft.cbrc.jp/alignment/software/)
BLASTp: (local Beowulf Linux computer cluster) against NCBI Swiss-Prot database: version February 2015, parameters: E-value of 10−6 cutoff
UniProt: (http://www.uniprot.org/help/uniprotkb), February 2021, parameters: Gene Ontology and the KEGG (Kyoto Encyclopedia of Genes and Genomes; https://www.kegg.jp) databases
OrthoVenn2: version 2018. Inflation value 1.5; E-value 1e-15 except as indicated. https://orthovenn2.bioinfotoolkits.net/home
MrBayes: version 3.2, parameters: lset rates = gamma ngammacat = 4; prset aamodelpr = fixed(wag); mcmc ngen = 10000000 relburnin = yes burninfrac = 0.25 printfreq = 1000 samplefreq = 1000 nchains = 4 savebrlens = yes
RAxML: version 8.2.12, parameters: 1,000 bootstrap replicates (-# 1000), WAG substitution model and gamma distribution of rates (-m PROTGAMMAIWAGF)
FigTree: version 1.4.4 (url: http://tree.bio.ed.ac.uk/software/figtree/)
References
Alexander, H., Jenkins, B. D., Rynearson, T. A. & Dyhrman, S. T. Metatranscriptome analyses indicate resource partitioning between diatoms in the field. Proc. Natl. Acad. Sci. USA 112, E2182–E2190 (2015).
Rubin, E. T., Cheng, S., Montalbano, A. L., Menden-Deuer, S. & Rynearson, T. A. Transcriptomic response to feeding and starvation in a herbivorous dinoflagellate. Front. Mar. Sci. 6, 246 (2019).
Roncalli, V., Cieslak, M. C., Germano, M., Hopcroft, R. R. & Lenz, P. H. Regional heterogeneity impacts gene expression in the sub-arctic zooplankter Neocalanus flemingeri in the northern Gulf of Alaska. Commun. Biol. 2, 1–13 (2019).
Mojib, N., Thimma, M., Kumaran, M., Sougrat, R. & Irigoien, X. Comparative metatranscriptomics reveals decline of a neustonic planktonic population. Limnol. Oceanogr. 62, 299–310 (2017).
Roncalli, V. et al. Physiological acclimatization in high-latitude zooplankton. Mol. Ecol. 31, 1753–1765, https://doi.org/10.1111/mec.16354 (2022).
Huys, R. & Boxshall, G. A. Copepod evolution. (The Ray Society, Unwin Brothers, 1991).
Mauchline, J. The Biology of Calanoid Copepods. Vol. 33 (Academic Press, 1998).
Hunt, M. E., Scherrer, M. P., Ferrari, F. D. & Matz, M. V. Very bright green fluorescent proteins from the Pontellid copepod Pontella mimocerami. PloS One 5, e11517, https://doi.org/10.1371/journal.pone.0011517 (2010).
Davis, A. D., Weatherby, T. M., Hartline, D. K. & Lenz, P. H. Myelin-like sheaths in copepod axons. Nature (London) 398, 571–571 (1999).
Lenz, P. H. & Hartline, D. K. in Trends in Copepod Studies - Distribution, Biology and Ecology (ed Uttieri, M.) Ch. 9, (Nova Science Publishers, Inc., 2017).
Weatherby, T. M. & Lenz, P. H. Mechanoreceptors in calanoid copepods: designed for high sensitivity. Arthropod Struct. Dev. 29, 275–288 (2000).
Esterly, C. O. Calanoid copepoda from the Bermuda Islands. Proc. Am. Acad. Arts Sci. 47, 219–226 (1911).
Lenz, P. H. et al. De novo assembly of a transcriptome for Calanus finmarchicus (Crustacea, Copepoda)–the dominant zooplankter of the North Atlantic Ocean. PloS One 9, e88589, https://doi.org/10.1371/journal.pone.0088589 (2014).
Roncalli, V., Cieslak, M. C., Sommer, S. A., Hopcroft, R. R. & Lenz, P. H. De novo transcriptome assembly of the calanoid copepod Neocalanus flemingeri: A new resource for emergence from diapause. Mar. Genomics 37, 114–119 (2018).
Lauritano, C., Roncalli, V., Ambrosino, L., Cieslak, M. C. & Ianora, A. First de novo transcriptome of the copepod Rhincalanus gigas from Antarctic waters. Biology 9, 410 (2020).
Roncalli, V., Cieslak, M. C. & Lenz, P. H. Transcriptomic responses of the calanoid copepod Calanus finmarchicus to the saxitoxin producing dinoflagellate Alexandrium fundyense. Sci. Rep. 6, 25708, https://doi.org/10.1038/srep25708 (2016).
Lenz, P. H., Lieberman, B., Cieslak, M. C., Roncalli, V. & Hartline, D. K. Transcriptomics and metatranscriptomics in zooplankton: Wave of the future? J. Plankton Res. 43, 3–9 (2021).
Bucklin, A. et al. in Population genomics: Marine organisms (eds Rajora, O. M. & Oleksiak, M.) 61–102 (Springer, 2018).
Jørgensen, T. S. et al. The genome and mRNA transcriptome of the cosmopolitan calanoid copepod Acartia tonsa Dana improve the understanding of copepod genome size evolution. Genome Biol. Evol. 11, 1440–1450 (2019).
Bron, J. E. et al. Observing copepods through a genomic lens. Front. Zool. 8, 1–15 (2011).
Coyle, K. O. & Pinchuk, A. I. Annual cycle of zooplankton abundance, biomass and production on the northern Gulf of Alaska shelf, October 1997 through October 2000. Fish. Oceanogr. 12, 227–251 (2003).
Blanco-Bercial, L., Bradford-Grieve, J. & Bucklin, A. Molecular phylogeny of the Calanoida (Crustacea: Copepoda). Mol. Phylogenet. Evol. 59, 103–113, https://doi.org/10.1016/j.ympev.2011.01.008 (2011).
Bradford-Grieve, J. M., Boxshall, G. A., Ahyong, S. T. & Ohtsuka, S. Cladistic analysis of the calanoid Copepoda. Invertebr. Syst. 24, 291–321, https://doi.org/10.1071/Is10007 (2010).
Andronov, V. N. Phylogenetic relations of large taxa within the suborder calanoida (Crustacea: Copepoda). Zool. Zhurnal 53, 1002–1012 (1974).
Baumgartner, M. F. & Tarrant, A. M. The physiology and ecology of diapause in marine copepods. Annu. Rev. Mar. Sci. 9, 387–411 (2017).
Hattori, H. Bimodal vertical distribution and diel migration of the copepods Metridia pacifica, M. okhotensis and Pleuromamma scutullata in the western North Pacific Ocean. Mar. Biol. 103, 39–50 (1989).
Koehl, M. A. R. & Strickler, J. R. Copepod feeding currents: food capture at low Reynolds number. Limnol. Oceanogr. 26, 1062–1073 (1981).
Andrews, S. FastQC: a quality control tool for high throughput sequence data Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512, https://doi.org/10.1038/Nprot.2013.084 (2013).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, https://doi.org/10.1186/Gb-2009-10-3-R25 (2009).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Consortium, T. U. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021).
Lenz, P. H. et al. Diapause vs. reproductive programs: transcriptional phenotypes in a keystone copepod. Commun. Biol. 4, 1–13 (2021).
Xu, L. et al. OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30, 772–780, https://doi.org/10.1093/molbev/mst010 (2013).
Katoh, K., Misawa, K., Kuma, K. I. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic biology 61, 539–542 (2012).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Whelan, S. & Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Molecular biology and evolution 18, 691–699 (2001).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP289633 (2020).
Lenz, P. H. Genbank https://identifiers.org/ncbi/insdc:GJRU00000000 (2020).
Lenz, P. H. Genbank https://identifiers.org/ncbi/insdc:GJRT01000000 (2020).
Lenz, P. H. Genbank https://identifiers.org/ncbi/insdc:GJSD01000000 (2020).
Lenz, P. H. Genbank https://identifiers.org/ncbi/insdc:GJRH00000000 (2020).
Lenz, P. H. Genbank https://identifiers.org/ncbi/insdc:GJRG00000000 (2020).
Lenz, P. H. Genbank https://identifiers.org/ncbi/insdc:GJAO00000000 (2020).
Lenz, P. H. Genbank https://identifiers.org/ncbi/insdc:GJRL00000000 (2020).
Lenz, P. H. Genbank https://identifiers.org/ncbi/insdc:GJRF00000000 (2020).
Lenz, P.H., Christie, A. E., Hartline, D. K., Hopcrofr, R. R. Collaborative Research: Molecular profiling of the ecophysiology of dormancy induction in calanid copepods of the Northern Gulf of Alaska LTER site. https://www.bco-dmo.org/project/720280 (march 2023).
Roncalli, V. et al. A deep transcriptomic resource for the copepod crustacean Labidocera madurae: A potential indicator species for assessing near shore ecosystem health. PloS One 12 (2017).
Plourde, S., Campbell, R. G., Ashjian, C. J. & Stockwell, D. A. Seasonal and regional patterns in egg production of Calanus glacialis/marshallae in the Chukchi and Beaufort Seas during spring and summer, 2002. Deep Sea Rs. Part II. Top. Stud. Oceanogr. 52, 3411–3426 (2005).
Ashjian, C. J., Campbell, R. G., Gelfman, C., Alatalo, P. & Elliott, S. M. Mesozooplankton abundance and distribution in association with hydrography on Hanna Shoal, NE Chukchi Sea, during August 2012 and 2013. Deep Sea Rs. Part II. Top. Stud. Oceanogr. 144, 21–36 (2017).
Lizano, A. M., Smolina, I., Choquet, M., Kopp, M. & Hoarau, G. Insights into the species evolution of Calanus copepods in the northern seas revealed by de novo transcriptome sequencing. Ecol. Evol. 12, e8606 (2022).
Tarrant, A. M., Nilsson, B. & Hansen, B. W. Molecular physiology of copepods-from biomarkers to transcriptomes and back again. Comp. Biochem. Phys. D 30, 230–247 (2019).
Ma, M. et al. Combining in silico transcriptome mining and biological mass spectrometry for neuropeptide discovery in the Pacific white shrimp Litopenaeus vannamei. Peptides 31, 27–43 (2010).
Gard, A. L., Lenz, P. H., Shaw, J. R. & Christie, A. E. Identification of putative peptide paracrines/hormones in the water flea Daphnia pulex (Crustacea; Branchiopoda; Cladocera) using transcriptomics and immunohistochemistry. Gen. Comp. Endocrinol. 160, 271–287, https://doi.org/10.1016/J.Ygcen.2008.12.014 (2009).
Roncalli, V., Uttieri, M., Capua, I. D., Lauritano, C. & Carotenuto, Y. Chemosensory-Related Genes in Marine Copepods. Mar. Drugs 20, 681 (2022).
Roncalli, V., Lauritano, C. & Carotenuto, Y. First report of OvoA gene in marine arthropods: A new candidate stress biomarker in copepods. Mar. Drugs 19, 647 (2021).
Porter, M. L., Steck, M., Roncalli, V. & Lenz, P. H. Molecular characterization of copepod photoreception. Biol. Bull. 233, 96–110 (2017).
Low, W. Y. et al. Molecular evolution of glutathione S-transferases in the genus. Drosophila. Genetics 177, 1363–1375 (2007).
Breugelmans, B., Simonet, G., van Hoef, V., Van Soest, S. & Broeck, J. V. Pacifastin-related peptides: structural and functional characteristics of a family of serine peptidase inhibitors. Peptides 30, 622–632 (2009).
Acknowledgements
We would like to thank Russell R. Hopcroft (University of Alaska Fairbanks) for assistance with zooplankton collections and species identification, Lynn Hata for administrative support from the University of Hawai’i at Mānoa, the science party and the crew of the USFWS R/V Tiĝlax̂ for at sea support. We are grateful for the constructive comments from two anonymous reviewers, which materially improved the quality of the MS, and in particular encouraged the inclusion of the cladograms of Figs. 3, 4. The work was supported by the National Science Foundation (NSF) OCE-1756767 (Lenz & Hartline); OCE-1756859 (Hopcroft) and LTER-1656070 (Hopcroft, Strom, Aguilar-Islas, Fiechter, Danielson). Additional support was provided by NCGAS under grants DBI-1062432 and ABI-1062432 to Indiana University. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation, the National Center for Genome Analysis Support, or Indiana University. This is the University of Hawai’i at Mānoa School of Ocean and Earth Science and Technology contribution number 11652.
Author information
Authors and Affiliations
Contributions
V.R., D.K.H. and P.H.L. conceived study design; V.R. and P.H.L. collected and identified specimens, M.C.C., B.L., A.M.C., V.R., D.K.H. analyzed the data and interpreted the statistical results; V.R., D.K.H. and P.H.L. wrote and reviewed the manuscript. All authors were involved in editing the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hartline, D.K., Cieslak, M.C., Castelfranco, A.M. et al. De novo transcriptomes of six calanoid copepods (Crustacea): a resource for the discovery of novel genes. Sci Data 10, 242 (2023). https://doi.org/10.1038/s41597-023-02130-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02130-1
