
Abstract
Gammarids are amphipods found worldwide distributed in fresh and marine waters. They play an important role in aquatic ecosystems and are well established sentinel species in ecotoxicology. In this study, we sequenced the transcriptomes of a male individual and a female individual for seven different taxonomic groups belonging to the two genera Gammarus and Echinogammarus: Gammarus fossarum A, G. fossarum B, G. fossarum C, Gammarus wautieri, Gammarus pulex, Echinogammarus berilloni, and Echinogammarus marinus. These taxa were chosen to explore the molecular diversity of transcribed genes of genotyped individuals from these groups. Transcriptomes were de novo assembled and annotated. High-quality assembly was confirmed by BUSCO comparison against the Arthropod dataset. The 14 RNA-Seq-derived protein sequence databases proposed here will be a significant resource for proteogenomics studies of these ecotoxicologically relevant non-model organisms. These transcriptomes represent reliable reference sequences for whole-transcriptome and proteome studies on other gammarids, for primer design to clone specific genes or monitor their specific expression, and for analyses of molecular differences between gammarid species.
Measurement(s) | transcription profiling assay |
Technology Type(s) | RNA sequencing |
Factor Type(s) | sex • species |
Sample Characteristic - Organism | Gammarus • Echinogammarus |
Sample Characteristic - Environment | habitat |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.9777905
Similar content being viewed by others
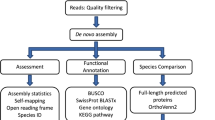


Background & Summary
Gammarid amphipods are animals that typically measure a few millimetres long and present in a wide range of aquatic habitats1. In freshwater ecosystems, they are often the most dominant macro-invertebrates, representing a significant proportion of the total biomass, and they also play a central role within food webs. Indeed, they are a prey for many species, but are also predators for many invertebrate species. Amphipods are also scavengers and shredders, and detritivores involved in leaf litter breakdown, playing a central role in the decomposition of organic matter in general. Thus, they modulate the composition of freshwater communities of invertebrates2. Thanks to these essential roles, they have been the subject of many recent studies investigating their sensitivity to pollutants3,4,5,6,7.
Marine and freshwater resources are of the utmost importance for Life. Human-made chemical contaminants released into aquatic environments compromise the quality of water bodies, threatening the resident biodiversity, and the utility of such ecosystems. The quality of these environments should be evaluated not only by measuring the concentrations of pollutants present, but also by monitoring how Life is affected by the bioavailable pollutants present and their synergistic/antagonist effects8. To do this, biomonitoring with caged representative sentinel species has proved to be a valuable tool for efficient ecotoxicological studies9,10,11,12,13. Specific traits such as moult delay, growth impairment, or reproductive defects can be monitored on sensitive animals exposed to toxic environments. These data can be then integrated into a quantitative water quality index that can be used by stakeholders in charge of the aquatic ecosystem and water resource management14. Because of their abundance and central ecological roles, invertebrates are commonly employed as test organisms in marine and ecotoxicological assessments. Specifically, gammarids have been successfully used as sentinel species for freshwater ecosystems following investigations of their physiological responses to toxicants15,16,17,18,19,20,21,22,23 and biomonitoring in caging systems9,12. Specific biomarkers have been proposed and can be monitored by innovative methods such as tandem mass spectrometry19,24,25,26. Next-generation proteomics contributed to improving our knowledge of the molecular responses of gammarids to toxicants, and led to the proposal of a broad panel of appropriate biomarkers27,28,29,30. This approach was successful after developing a protein sequence database from an RNA-Seq transcriptome translated in all the possible reading frames. This proteogenomics concept was used to establish an extensive catalogue of protein sequences comprising 1873 mass-spectrometry-certified proteins, thus representing a significant amphipod proteomic resource29.
Despite this progress, molecular resources relating to gammarids remain scarce31. No gammarid whole genome sequence was available until very recently, when a first-draft genome of Gammarus lacustris was released comprising 443,304 scaffolds32. The genomes of two related amphipods, Parhyale hawaiensis33 and Hyalella azteca34, have also been sequenced. RNA-Seq datasets are now available for P. hawaiensis35,36,37, Echinogammarus marinus38, Eogammarus possjeticus39, Gammarus fossarum29, Gammarus chevreuxi40, Gammarus pulex41, and Gammarus minus42. However, these datasets are not of equal quality in terms of mRNA sequence coverage, which is a crucial parameter for proteogenomics interpretation43. They are assembled from mRNAs extracted from a pool of several animals or from specific tissues, and in some cases are no longer accessible as it is the case for E. marinus because the repository used no longer exists44.
The data presented in this article consist of assembled transcriptome sequences for 14 different gammarids, seven males and seven females, namely Gammarus fossarum A (Müller type A), G. fossarum B (Müller type B), G. fossarum C (Müller type C), Gammarus wautieri, Gammarus pulex, Echinogammarus berilloni, and Echinogammarus marinus. These transcriptomes were assembled and translated using the same pipelines (full length whole-organism mRNAs), and thus are of equivalent sequencing depth and quality across the different taxa studied. Starting material was extracted from single animals to avoid sequence heterogeneity. The transcriptomes have been annotated to serve as reference protein sequence databases for proteogenomics studies involving these sentinel animals that will be soon conducted to gain more basic knowledge and thus improve how aquatic environmental risks are assessed. For these future studies, an interesting strategy could be to interpret MS/MS shotgun data first on the most appropriate specific single-organism database, and then perform a follow-up search on a multi-organism database. The transcriptomes presented here will also serve in comparative analyses to better define the molecular diversity amongst gammarids and will be a valuable sequence resource for future ecotoxicological studies.
Methods
Experimental design
Freshwater gammarids were collected in four geographically-distant French rivers (Table 1). One population of Gammarus fossarum was sampled in north-eastern France (Seebach river), which was previously shown to harbour the cryptic type A subspecies according to the three types defined in Müller et al.45, Westram et al.46, and Weiss et al.47. The second river (Pollon River) situated in the mid-eastern area of France, corresponding to a sympatric situation, supplied organisms belonging to Gammarus fossarum type B, type C, and Gammarus pulex species. Gammarus wautieri were collected in the Galaveyson river in the Dauphiné region, and Echinogammarus berilloni organisms from a fourth river in south-western France (Saucats river). These freshwater gammarids were all collected using a hand net following kick-sampling, and subsequently transported to the laboratory. After maintaining them for 1 week in the laboratory – at 12 °C with a constant aeration, under a 16/8-h light/dark photoperiod in buckets containing water sampled from their respective rivers of origin, and with conditioned alder leaves as food source – couples in amplexus were isolated for species determination before RNA extraction. Pairs where the females had well-developed ovaries were selected. Embryos were removed from the marsupial pouch of females for five of these couples. Based on the description of the reproductive cycle in Gammarus fossarum48, for RNA extraction, we were able to select one couple per species in the last stage of the reproductive cycle (pre-moulting stage for the female) by retaining pairs where the females were carrying embryos at the end of their embryonic development stage (stage 4 or 5). For the marine species, E. marinus were collected from beneath seaweed in the intertidal zone in Portsmouth, southern England. These species correspond to the same population as used in a previous study38. After maintaining them for 1 month in the laboratory – at 10 °C under a 12 h light/12 h dark photoperiod in buckets with filtered natural seawater and fed with fucoid seaweed - organisms were transported live in damp seaweed from United Kingdom to France (one-day travel). They were subsequently maintained for a few hours in aquaria containing reconstituted seawater (salinity 30‰) before organism selection. For this species, it was not possible to recover couples in amplexus. One free-swimming male and one free-swimming female were isolated from the batch of organisms available. Stage 1 embryos were recovered from the female marsupium, indicating that this female was in a post-moulting stage.
Species were first determined based on morphological criteria49. To distinguish between the three cryptic lineages, A, B, C, within the G. fossarum species, a molecular species assignment was carried out by amplifying the 5’ part of the mtDNA cytochrome c oxidase subunit I (COI) using universal primers (LCO1490 [GGT CAA ATC ATA AAG ATA TTG G] and HCO2198 [TAA ACT TCA GGG TGA CCA AAA AAT CA])50. Briefly, DNA was extracted from one or two pereopods (depending on individual size) cut from organisms before conditioning for RNA extraction. DNA was extracted using the Nucleospin tissue XS kit (Macherey-Nagel), and 10 ng of DNA for each organism was amplified. The PCR conditions consisted in 45 cycles of denaturation at 95 °C for 30 sec, annealing at 50 °C for 30 sec, and elongation at 72 °C for 1 min. PCR products were purified by ultrafiltration using the Nucleofast kit (Macherey-Nagel). Purified amplicons were prepared for sequencing using the BigDye Terminator v3.1 kit (ThermoFisher), and then sequenced on a DNA analyser ABI 3730XL (ThermoFisher). Sequencing data were analysed using the Sequencher 5.4.6 program (Genecodes). COI sequences (freely available from figshare, YC02_COI sequences and phylogenetic tree51) were aligned to build a phylogenetic tree including reference sequences from Weiss et al.47 and Lagrue et al.52. Using this phylogenetic tree (freely available from figshare, YC02_COI sequences and phylogenetic tree51) it is possible to position the COI sequences of the Gammarus organisms selected for RNA sequencing in relation to the published reference sequences (SeaView software53; BioNJ method based on J-C distance). The robustness of the different groupings was evaluated by a bootstrapping procedure (100 iterations). COI sequences were obtained for all Gammarus individuals, except for the female G. fossarum C as this individual was in precopulatory amplexus with the male COI-genotyped as G. fossarum C. However, in the same location (Pollon River), we also obtained the COI genotypes for 15 additional pairs, all of which were found to be non-heterospecific (4 G. fossarum B, 3 G. fossarum C, 8 G. pulex). Westraam et al.46 reported similar findings in the Glovelier river which harbours G. fossarum A and B, with only one heterospecific pair for a total of 64 genotyped pairs. Lagrue et al.52 also observed that mixed pairs are rare in the field for Gammarus lineages with a COI distance greater than 4%. Considering that the divergence between the COI-genotyped G. fossarum B and C specimens is about 17% in the Pollon River, it is very unlikely that this female does not belong to the G. fossarum C species.
Dataset generation
Gammarids were placed in RNAlater (Sigma) and stored at 4 °C overnight. The RNAlater was then removed, and the organisms were snap frozen in liquid nitrogen and stored at −80 °C until RNA was extracted. Organisms were first homogenized in lysis buffer using a bead homogenizer and then RNAs were extracted using the Qiagen fibrous tissue kit (Qiagen). RNA quantity, quality and integrity were assessed by Nanodrop (Thermo Fisher) and Bioanalyzer (Agilent) analysis. RNA-Seq libraries were generated using the TruSeq stranded mRNA Sample Prep kit (Illumina). mRNA was purified using poly-(T) beads from 2 µg of each total RNA sample, then cleaved in segments of 155 bp on average (120–210 bp range). Subsequently, cleaved RNA fragments were primed with random hexamers and reverse-transcribed into first-strand cDNA. A second strand of cDNA was consecutively synthetized, and double-stranded cDNA was purified on beads. The 3′ ends of the blunt fragments obtained were then adenylated. Indexed adapters were ligated to the PCR-enriched cDNA fragments (11 cycles). Libraries were purified and quality-assessed using a Fragment Analyzer (Advanced Analytical Technologies). The 16 libraries were quantified by qPCR using the Kapa Library Quantification Kit (Roche). Their concentrations were normalized, multiplexed in a single pool. Libraries were then sequenced on two lanes of Hiseq3000 (Illumina) using a paired-end read length of 2 × 150 bp with the HiSeq Reagent Kits (Illumina). The two HiSeq lanes produced an average of 40.0 ± 8 million read pairs per library. Quality control of reads was performed by FastQC version V0.11.2 (Babraham Bioinformatics). Detailed results are freely available from figshare (YC02_QC data51). The data records are stored in 14 folders, each containing four folders per transcriptome.
De novo assembly
For each sample, the forward or reverse reads were merged from two separate lanes. Data were filtered based on the mean Qphred score, with a threshold set at 16.99, and any remaining unpaired reads were removed using a homemade script. The numbers of reads for each sample before and after this filtering step are presented in Table 1. Trinity v2.454 was used to assemble reads for each sample considering pair-end and strand orientation (-SS_lib type RF); all other Trinity parameters were set to their default values, with k set to 25, and minimum contig length to 200 bp.
Assessing assembly quality
Transcriptome quality was assessed using Transrate v1.0.155, which generates standard metrics and remapping statistics. No reference protein sequences were used for the assessment with Transrate. The main metrics are shown in Table 2. To validate the quality of all the assemblies, BUSCO v2.056 was used. The database used for BUSCO analyses was Arthropoda_odb9 which contains 1066 orthologous genes at the nearest taxon level (i.e., Arthropods) available for Gammarus.
Annotation
For each sample, the transcripts were annotated using the Trinotate v3.1.1 annotation pipeline54. The Swissprot database was used as the main database, and amphipod proteins referenced on Uniref were used as a custom database. Similarity searches were performed with Blastx and Blastp, with an e-value cutoff set at 1e-2. Results from these searches were then used to generate the annotation report with the same e-value cutoff.
Data Records
Reads
Read sequences for each sample were deposited in the NCBI Sequence Reads Archive under accession Numbers SRR808972057, SRR8089722–SRR808972558,59,60,61, and SRR8089727–SRR808973562,63,64,65,66,67,68,69,70, as indicated in Table 3 alongside the corresponding Bioproject and Biosample codes. The FastQC results for the 14 samples are freely available from figshare (YC02 _QC data)51. The data records are stored as 14 folders, each of which contain four folders per transcriptome.
Transcriptomes
Transcriptome assemblies were deposited in the NCBI Transcriptome Shotgun Assembly Sequence Database. These data have been deposited in GenBank under identifiers GHCN0100000071, GHCP01000000-GHCR0100000072,73,74, GHCT01000000-GHCZ0100000075,76,77,78,79,80,81, GHDA01000000-GHDC0100000082,83,84, as indicated in Table 3 alongside the corresponding Bioproject and Biosample codes.
Proteogenomics databases
Translations of coding sequence regions were produced for each transcriptome from stop to stop codons by Transdecoder v3.0.154, analysing only the top strand. The 500 longest ORFs were used for training, retaining 600-bp ORFs and only proteins with a minimum length of 50 amino acids. The 14 translations are freely available for download as FASTA files from figshare (YC02_Transcriptome translated ORFs51).
Annotation
Annotations of each assembly are freely available for download as Excel files from figshare (YC02_Transcript annotations51). The folder contains 14 Excel files.
Technical Validation
Transrate
Transrate analyses showed good remapping of results, with more than 80% of reads remapped and most assemblies with more than 70% were classed as well mapped. Raw results from Transrate are freely available through figshare (YC02_ Transrate results)51.
BUSCO
A high level of single-copy ortholog retrieval was noted for the 14 assemblies, with at least a 75% ratio, as shown in Fig. 1. Furthermore, fewer than 8% of orthologs were missing in the worst case, and fewer than 5% were missing in 11 transcriptomes.

BUSCO assessment results for the 14 assembled transcriptomes.
Code Availability
Filtering before assembly was performed with an in-house Pythonv2.7 script, which is freely available (https://github.com/YannickCogne/Qfiltering). The script was automated with a bash script for each sample.
References
MacNeil, C., Dick, J. T. A. & Elwood, R. W. The trophic ecology of freshwater Gammarus Spp. (crustacea:amphipoda): problems and perspectives concerning the functional feeding group concept. Biological Reviews 72, 349–364 (1997).
Kelly, D. W., Dick, J. T. A. & Montgomery, W. I. The functional role of Gammarus (Crustacea, Amphipoda): shredders, predators, or both? Hydrobiologia 485, 199–203 (2002).
Arce-Funck, J. et al. High stoichiometric food quality increases moulting organism vulnerability to pollutant impacts: An experimental test with Gammarus fossarum (Crustacea: Amphipoda). Sci Total Environ 645, 1484–1495 (2018).
Ganser, B. et al. Wastewater alters feeding rate but not vitellogenin level of Gammarus fossarum (Amphipoda). Sci Total Environ 657, 1246–1252 (2019).
Konemann, S. et al. Combination of In Situ Feeding Rate Experiments and Chemical Body Burden Analysis to Assess the Influence of Micropollutants in Wastewater on Gammarus pulex. Int J Environ Res Public Health 16, 883 (2019).
Munz, N. A., Fu, Q., Stamm, C. & Hollender, J. Internal Concentrations in Gammarids Reveal Increased Risk of Organic Micropollutants in Wastewater-Impacted Streams. Environ Sci Technol 52, 10347–10358 (2018).
von Fumetti, S. & Blaurock, K. Effects of the herbicide Roundup(R) on the metabolic activity of Gammarus fossarum Koch, 1836 (Crustacea; Amphipoda). Ecotoxicology 27, 1249–1260 (2018).
Gouveia, D. et al. Ecotoxicoproteomics: A decade of progress in our understanding of anthropogenic impact on the environment. J Proteomics 198, 66–77 (2018).
Besse, J. P. et al. Caged Gammarus fossarum (Crustacea) as a robust tool for the characterization of bioavailable contamination levels in continental waters: towards the determination of threshold values. Water Res 47, 650–660 (2013).
Chaumot, A., Geffard, O., Armengaud, J. & Maltby, L. In Aquatic Ecotoxicology - Advancing tools for dealing with emerging risks (eds Amiard-Triquet, C., Amiard, J.-C. & Mouneyrac, C.) 253–280 (Academic Press, London, 2015).
Ciliberti, A. et al. Caged Gammarus as biomonitors identifying thresholds of toxic metal bioavailability that affect gammarid densities at the French national scale. Water Res 118, 131–140 (2017).
Lacaze, E. et al. DNA damage in caged Gammarus fossarum amphipods: a tool for freshwater genotoxicity assessment. Environ Pollut 159, 1682–1691 (2011).
Trapp, J., Armengaud, J., Salvador, A., Chaumot, A. & Geffard, O. Next-generation proteomics: toward customized biomarkers for environmental biomonitoring. Environ Sci Technol 48, 13560–13572 (2014).
Coulaud, R. et al. In situ feeding assay with Gammarus fossarum (Crustacea): Modelling the influence of confounding factors to improve water quality biomonitoring. Water Res 45, 6417–6429 (2011).
Barros, S. et al. Chronic effects of triclocarban in the amphipod Gammarus locusta: Behavioural and biochemical impairment. Ecotoxicol Environ Saf 135, 276–283 (2017).
Chaumot, A., Gos, P., Garric, J. & Geffard, O. Additive vs non-additive genetic components in lethal cadmium tolerance of Gammarus (Crustacea): novel light on the assessment of the potential for adaptation to contamination. Aquat Toxicol 94, 294–299 (2009).
Correia, A. D., Lima, G., Costa, M. H. & Livingstone, D. R. Studies on biomarkers of copper exposure and toxicity in the marine amphipod Gammarus locusta (Crustacea): I. Induction of metallothionein and lipid peroxidation. Biomarkers 7, 422–437 (2002).
Felten, V. et al. Physiological and behavioural responses of Gammarus pulex (Crustacea: Amphipoda) exposed to cadmium. Aquat Toxicol 86, 413–425 (2008).
Jubeaux, G. et al. Vitellogenin-like proteins in the freshwater amphipod Gammarus fossarum (Koch, 1835): functional characterization throughout reproductive process, potential for use as an indicator of oocyte quality and endocrine disruption biomarker in males. Aquat Toxicol 112–113, 72–82 (2012).
Kohler, S. A., Parker, M. O. & Ford, A. T. Species-specific behaviours in amphipods highlight the need for understanding baseline behaviours in ecotoxicology. Aquat Toxicol 202, 173–180 (2018).
Maltby, L. & Crane, M. Responses of Gammarus pulex (Amphipoda, Crustacea) to metalliferous effluents: identification of toxic components and the importance of interpopulation variation. Environ Pollut 84, 45–52 (1994).
Xuereb, B., Chaumot, A., Mons, R., Garric, J. & Geffard, O. Acetylcholinesterase activity in Gammarus fossarum (Crustacea Amphipoda) Intrinsic variability, reference levels, and a reliable tool for field surveys. Aquat Toxicol 93, 225–233 (2009).
Xuereb, B., Noury, P., Felten, V., Garric, J. & Geffard, O. Cholinesterase activity in Gammarus pulex (Crustacea Amphipoda): characterization and effects of chlorpyrifos. Toxicology 236, 178–189 (2007).
Gouveia, D. et al. Ecotoxico-Proteomics for Aquatic Environmental Monitoring: First in Situ Application of a New Proteomics-Based Multibiomarker Assay Using Caged Amphipods. Environ Sci Technol 51, 13417–13426 (2017).
Gouveia, D. et al. Assessing the relevance of a multiplexed methodology for proteomic biomarker measurement in the invertebrate species Gammarus fossarum: A physiological and ecotoxicological study. Aquat Toxicol 190, 199–209 (2017).
Simon, R. et al. Mass spectrometry assay as an alternative to the enzyme-linked immunosorbent assay test for biomarker quantitation in ecotoxicology: application to vitellogenin in Crustacea (Gammarus fossarum). J Chromatogr A 1217, 5109–5115 (2010).
Trapp, J. et al. High-throughput proteome dynamics for discovery of key proteins in sentinel species: Unsuspected vitellogenins diversity in the crustacean Gammarus fossarum. J Proteomics 146, 207–214 (2016).
Trapp, J. et al. Proteomic investigation of male Gammarus fossarum, a freshwater crustacean, in response to endocrine disruptors. J Proteome Res 14, 292–303 (2015).
Trapp, J. et al. Proteogenomics of Gammarus fossarum to document the reproductive system of amphipods. Mol Cell Proteomics 13, 3612–3625 (2014).
Trapp, J. et al. Digging Deeper Into the Pyriproxyfen-Response of the Amphipod Gammarus fossarum With a Next-Generation Ultra-High-Field Orbitrap Analyser: New Perspectives for Environmental Toxicoproteomics. Frontiers in Environmental Science 6, 54 (2018).
Armengaud, J. et al. Non-model organisms, a species endangered by proteogenomics. J Proteomics 105, 5–18 (2014).
Jin, S. et al. Identification of Candidate Genes for the Plateau Adaptation of a Tibetan Amphipod, Gammarus lacustris, Through Integration of Genome and Transcriptome Sequencing. Front Genet 10, 53 (2019).
Kao, D. et al. The genome of the crustacean Parhyale hawaiensis, a model for animal development, regeneration, immunity and lignocellulose digestion. Elife 5, e20062 (2016).
Poynton, H. C. et al. The Toxicogenome of Hyalella azteca: A Model for Sediment Ecotoxicology and Evolutionary Toxicology. Environ Sci Technol 52, 6009–6022 (2018).
Blythe, M. J. et al. High through-put sequencing of the Parhyale hawaiensis mRNAs and microRNAs to aid comparative developmental studies. PLoS One 7, e33784 (2012).
Nestorov, P., Battke, F., Levesque, M. P. & Gerberding, M. The maternal transcriptome of the crustacean Parhyale hawaiensis is inherited asymmetrically to invariant cell lineages of the ectoderm and mesoderm. PLoS One 8, e56049 (2013).
Zeng, V. et al. De novo assembly and characterization of a maternal and developmental transcriptome for the emerging model crustacean Parhyale hawaiensis. BMC Genomics 12, 581 (2011).
Short, S. et al. Crustacean intersexuality is feminization without demasculinization: implications for environmental toxicology. Environ Sci Technol 48, 13520–13529 (2014).
Chen, J., Liu, H., Cai, S. & Zhang, H. Comparative transcriptome analysis of Eogammarus possjeticus at different hydrostatic pressure and temperature exposures. Sci Rep 9, 3456 (2019).
Truebano, M., Tills, O. & Spicer, J. I. Embryonic transcriptome of the brackishwater amphipod Gammarus chevreuxi. Mar Genomics 28, 5–6 (2016).
Gismondi, E. & Thome, J. P. Transcriptome of the freshwater amphipod Gammarus pulex hepatopancreas. Genom Data 8, 91–92 (2016).
Carlini, D. B. & Fong, D. W. The transcriptomes of cave and surface populations of Gammarus minus (Crustacea: Amphipoda) provide evidence for positive selection on cave downregulated transcripts. PLoS One 12, e0186173 (2017).
Trapp, J. et al. Proteogenomic insights into the core-proteome of female reproductive tissues from crustacean amphipods. J Proteomics 135, 51–61 (2016).
Jones, M. & Blaxter, M. afterParty: turning raw transcriptomes into permanent resources. BMC Bioinformatics 14, 301 (2013).
Muller, J. Mitochondrial DNA variation and the evolutionary history of cryptic Gammarus fossarum types. Mol Phylogenet Evol 15, 260–268 (2000).
Westram, A. M., Jokela, J., Baumgartner, C. & Keller, I. Spatial distribution of cryptic species diversity in european freshwater amphipods (Gammarus fossarum) as revealed by pyrosequencing. PLoS One 6, e23879 (2011).
Weiss, M., Macher, J. N., Seefeldt, M. A. & Leese, F. Molecular evidence for further overlooked species within the Gammarus fossarum complex (Crustacea: Amphipoda). Hydrobiologia 721, 165–184 (2014).
Geffard, O. et al. Ovarian cycle and embryonic development in Gammarus fossarum: application for reproductive toxicity assessment. Environ Toxicol Chem 29, 2249–2259 (2010).
Piscart, C. & Bollache, L. Crustacés amphipodes de surface: gammares d’eau douce. (Association Française de Limnologie, 2012).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3, 294–299 (1994).
Cogne, Y. et al. YC02. figshare. https://doi.org/10.6084/m9.figshare.c.4568087.v1 (2019).
Lagrue, C. et al. Confrontation of cryptic diversity and mate discrimination within Gammarus pulex and Gammarus fossarum species complexes. Freshwater Biology 59, 2555–2570 (2014).
Gouy, M., Guindon, S. & Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27, 221–224 (2010).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652 (2011).
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. M. & Kelly, S. TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res 26, 1134–1144 (2016).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol 1962, 227–245 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089720 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089722 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089723 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089724 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089725 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089727 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089728 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089729 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089730 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089731 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089732 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089733 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089734 (2019).
NCBI Sequence Read Archive, http://identifiers.org/insdc.sra:SRR8089735 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCN01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCP01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCQ01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCR01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCT01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCU01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCV01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCW01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCX01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCY01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHCZ01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHDA01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHDB01000000 (2019).
GenBank, https://identifiers.org/ncbi/insdc:GHDC01000000 (2019).
Acknowledgements
We thank the Institut National de Recherche en Sciences et Technologies pour l’Environnement et l’Agriculture (France), the Commissariat à l’Energie Atomique et aux Energies Alternatives (France), and the Agence Nationale de la Recherche “ProteoGam” program (ANR-14-CE21-0006-02) for financial support.
Author information
Authors and Affiliations
Contributions
Y.C., A.F., O.G., J.A., A.C. and C.A. conceptualised the study. D.D.E., D.G., A.F., O.B., C.E., A.C. and C.A. performed the experimental work. Y.C., D.D.E., O.P., J.A., A.C. and C.A. analysed the data. Y.C. and J.A. wrote the manuscript with input from all co-authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Cogne, Y., Degli-Esposti, D., Pible, O. et al. De novo transcriptomes of 14 gammarid individuals for proteogenomic analysis of seven taxonomic groups. Sci Data 6, 184 (2019). https://doi.org/10.1038/s41597-019-0192-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0192-5
This article is cited by
-
Organ-oriented proteogenomics functional atlas of three aquatic invertebrate sentinel species
Scientific Data (2023)
-
The diversity of opsins in Lake Baikal amphipods (Amphipoda: Gammaridae)
BMC Ecology and Evolution (2021)
-
Species status evaluation of Lirceus usdagalan, L. culveri, and L. hargeri populations (Isopoda; Asellidae) based on a large scale next-generation sequence data set
Conservation Genetics (2021)
-
Continental-scale patterns of hyper-cryptic diversity within the freshwater model taxon Gammarus fossarum (Crustacea, Amphipoda)
Scientific Reports (2020)
