
Abstract
Blindsnakes of infraoder Scolecophidia (order Squamata) are the most basal group of extant snakes, comprising of more than 450 species with ecological and morphological features highly specialized to underground living. The Brahminy blindsnake, Indotyphlops braminus, is the only known obligate parthenogenetic species of snakes. Although the origin of I. braminus is thought to be South Asia, this snake has attracted worldwide attention as an alien species, as it has been introduced to all continents except Antarctica. In this study, we present the first draft genome assembly and annotation of I. braminus. We generated approximately 480 Gbp of sequencing data and produced a draft genome with a total length of 1.86 Gbp and N50 scaffold size of 1.25 Mbp containing 89.3% of orthologs conserved in Sauropsida. We also identified 0.98 Gbp (52.82%) of repetitive genome sequences and a total of 23,560 protein-coding genes. The first draft genome of I. braminus will facilitate further study of snake evolution as well as help to understand the emergence mechanism of parthenogenetic vertebrates.
Measurement(s) | Whole genome sequence |
Technology Type(s) | Next generation DNA sequencing |
Factor Type(s) | Whole genome of Indotyphlops braminus |
Sample Characteristic - Organism | Indotyphlops braminus |
Sample Characteristic - Environment | Soil and rotten wood |
Sample Characteristic - Location | Japan and India |
Similar content being viewed by others


Background & Summary
The Infraorder Scolecophidia (blindsnakes) is the most basal lineage of extant snakes1. All constituent species are subterranean and are found mainly in the southern hemisphere and on tropical islands. They can range from 10 cm to nearly 1 m in length2, and they have highly specialized morphologies, including a vestigial organ form of eyes that can only perceive light. Although 462 species in five families have been described in Scolecophidia3, the true species diversity is thought to be greatly underestimated due to their cryptic ecology4,5.
As of April 2022, there are 32 available genome assemblies for snakes. Among the three major groups that comprise Serpentes (Caenophidia, Henophidia, and Scolecophidia), genomic data have been accumulated in Caenophidia, mainly for poisonous snakes belonging to the families Elapidae and Viperidae6 and in Henophidia, which includes the families Boidae and Pythonidae, for which the genome of Python molurus bivittatus has been reported7. However, there are currently no datasets for draft genome assemblies or annotations for snakes in the Scolecophidia group, despite the evolutionarily importance of this group, with the exception of low-quality assembly data (N50 < 2kbp)8.
The Brahminy blindsnake, or Indotyphlops braminus, is one of the most well-known species in Scolecophidia (Fig. 1). No male I. braminus have been found, and this species of snake is the only known obligate parthenogenesis snake9,10. Further, I. braminus is an allotriploid (triploid) species11,12,13 and is considered to have emerged via inter-species hybridization, as has occurred with other parthenogenetic reptiles14,15. The geographic origin of this species is thought to be in South Asia based on the distribution of congeneric species16,17. However, due to their small size and fossorial and parthenogenetic nature, they have been transported around the world, hidden in the rotting woods and soils of ornamental plants. Consequently, I. braminus has now been colonized artificially and unintentionally in all continents except Antarctica18,19. Because I. braminus can be found globally, various studies regarding their osteology20,21, anatomy22, neurology23, and ethology24,25 have been conducted worldwide. For these reasons, I. braminus has the potential to serve as a useful snake model organism and is a suitable species in which to investigate the emergence mechanism of parthenogenesis in vertebrates.

Live specimen of Indotyphlops braminus.
In this study, we present the first draft genome of I. braminus. We extracted genomic DNA from liver and muscle tissues, constructed three pair-end (PE) libraries, and sequenced libraries using the Illumina Hiseq2500 platform. In addition, we conducted long-read sequencing of four libraries using Oxford Nanopore MinION and performed hybrid de novo assembly. The draft genome was assembled into 4,851 scaffolds (N50 = 1.25 Mbp) with a total size of 1.86 Gbp, comparable to the estimated genome size (1.50 Gbp) in k-mer analysis. Our BUSCO assessment indicated that 89.3% of orthologs conserved in Sauropsida were present in the genome assembly. Structural annotation of the genome identified 23,560 protein-coding genes. In the future, this highly-quality scolecophidian genome will be a crucial reference for further understanding of both snake evolution and the emergence mechanism of parthenogenetic species.
Methods
Sample Collection and DNA Extraction
We used two I. braminus specimens collected from India (Ooty: 11°24′26″ N, 76°41′27″ E) and Japan (Okinawajima Island: 26°15′09″N, 127°45′55″E), since I. braminus individuals are parthenogenetic clones, and the worldwide colonization of this blindsnake is thought to have occurred recently26. Indeed, the partial sequence of the mitochondrial cytochrome b gene of I. braminus from Japan (obtained by methods described previously in Smíd et al.27) matched perfectly with the corresponding region of the India specimen constructed by short-read data using NOVOPlasty v3.228. The specimens used were picked up from under stones, euthanized, and dissected to isolate the liver and muscle tissues for DNA extraction. These experiments were performed under permissions received from the Ethics Committees for Animal Experiments by Dr. Babasaheb Ambedkar Marathwada University (permit No. A01) and Nagahama Institute of Bio-Science (permit No. 085).
For genome sequencing using Illumina, the I. braminus specimen from India was used, and DNA was extracted using the Wizard® Genomic DNA Purification Kit (Promega Corporation, WH, Madison, WI, USA). For Oxford Nanopore long-read sequencing, the specimen from Japan was used, and DNA extraction was performed using the Blood & Cell Culture DNA Midi Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Purified precipitates were dissolved in TE buffer (pH 8.0) and stored at −30 °C until further processing.
Library preparation and sequencing
Short-read sequencing libraries were prepared using the Illumina trueseq LT kit (Illumina, San Diego, CA, USA). Three PE libraries were prepared with an insert size of 550 bp and sequenced by Hiseq2500. Raw sequencing data were converted to fastq format using bcl2fastq2 v2.20. A total of 422 Gbp of sequences were obtained (Table 1), which were approximately 226.9 x coverage of I. braminus genome (1.86 Gbp, see below).
For long-read sequencing using MinION (Oxford Nanopore Technology, Oxford, UK), the extracted genomic DNA was fragmented to ~20 kbp using Covaris g-TUBE (Covaris, Woburn, MA, USA). After purification using 0.4 x AMPure XP beads (Beckman Coulter, Brea, CA, USA), library preparation was performed using the SQK-LSK109 Ligation Sequencing kit (Oxford Nanopore Technologies) based on the manufacturer’s protocol. Four libraries were prepared and loaded onto R9.4.1 chemistry flowcell (FLO-MIN106) and sequenced using MinKNOW v 19.06.7. After sequencing, Guppy v3.2.2 was used for basecalling. A total of 57.9 Gbp of long-read data were obtained (Table 1), which were 31.1 x coverage of I. braminus genome. The raw reads were checked using LongQC v1.2.0c29, and quality filtered using Filtlong v0.2.1 (https://github.com/rrwick/Filtlong) with a minimum QV of 10 and a minimum read length of 1 Kbp.
Genome assembly
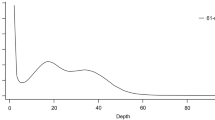
We estimated the overall characteristics of the I. braminus genome, including its genome size, heterozygosity, and repeat content, by k-mer frequencies calculated from Illumina short-reads. KMC v3.1.130 was used to obtain a 21-mer count histogram (Fig. 2). GenomeScope v2.031 estimated a genome size of 1.50 Gbp, which was comparable with that of our draft genome (1.86 Gbp). The genome size of I. braminus fell within the range of other snake species whose genomes have been reported previously (1.13–2.03 Gbp).

The k-mer distribution (k = 21) of Indotyphlops braminus. The 21-mer distribution was calculated by GenomeScope based on 422 Gbp Illumina short-reads data. K-mer coverages (x axis) were plotted against the value of coverage multiplying frequency (y axis).
We applied a hybrid de novo assembly approach based on Illumina short-reads and Nanopore long-reads. Short- and long-reads were assembled to contigs using MaSuRCA v4.0.532. For gap-closing, assembled contigs were scaffolded into the draft genome using HaploMerger2 v2018060333. The resultant draft genome had a total length of 1.86 Gbp, scaffold number of 4,851, N50 of 1.25 Mbp and the longest scaffold of length 7.0 Mbp, as calculated by QUAST v5.0.234 (Table 2). We evaluated the gene completeness of our draft genome using BUSCO v5.2.235,36. BUSCO assessment showed that 89.3% of orthologs conserved in Sauropsida were present in this genome assembly (sum of the percentages of single-copy and duplicate), suggesting that our draft genome possessed a sufficient gene repertoire from I. braminus (Table 2).
Repeat analysis
Repetitive regions of I. braminus were identified using a combination of de novo and homology-based approaches. For homology-based prediction, known repetitive elements were identified using RepeatMasker v4.1.1 (http://www.repeatmasker.org) to search against published RepBase sequences. For de novo prediction, RepeatModeler v2.0.1 was executed on the I. braminus assembly to build a de novo repeat library for this species. Then, RepeatMasker was used to annotate repetitive elements using this library. The estimated repeat regions of total length 0.98 Gbp accounted for 52.82% of the genome. Long interspersed nuclear elements were the most abundant elements and accounted for 20% of the genome. A summary of the annotation is shown in Table 3.
Gene prediction and annotation
A BLAST search with the known mitochondrial DNA sequence of I. braminus (Accession number: NC_010196) identified a contig showing 99.9% homology. This was a mitochondrial DNA excluded from the assembly data. We also masked repeat regions and conducted gene prediction using Augustus v3.4.037 trained with the assessment result of BUSCO with respect to the genome assembly. In total, 23,560 protein-coding genes were annotated in the I. braminus genome (Table 4). Next, we investigated the closest protein homolog of each entry in the gene model of I. braminus using diamond v2.0.1338, and visualized results by Krona39 (Fig. 3). Approximately 91% of the closest protein homolog of each gene of the gene model belonged to Sauropsida. Of the proteins detected in Sauropsida, approximately 76% were derived from Serpentes, indicating that the gene model is quite consistent with the systematic position of I. braminus.

Krona chart representing taxonomic composition of Indotyphlops braminus gene model. Taxonomy charts, which consist of all taxa (left) and Sauropsida (right), are shown.
The BUSCO analysis with Sauropsida conserved genes databases found 72.9% completeness in our annotation dataset (Table 4), which was lower than that estimated in the genome assembly (89.3%: Table 2). Since the completeness of predicted genes was evaluated based on the codon reading frame, it is likely that there were low-quality genes exhibiting premature termination. In this analysis, we applied a hybrid assembly with short-reads (accuracy >99.9%) and long-reads (<85%), which may have resulted in a lower base accuracy for the assembled regions with only long-reads and in low BUSCO value. To improve the assembly of the I. braminus genome, it would be necessary to obtain novel transcriptome data or perform further high accuracy short- and long-read sequencing.
Technical Validation
Quality assessment of the genome assembly
The total assembly length is 1.86 Gbp, which is almost comparable with the estimated genome size (1.50 Gbp). The scaffold N50 is 1.25 Mbp (Table 2). BUSCO analysis was performed with Sauropsida conserved genes databases to assess the completeness of the genome assembly, resulting in a BUSCO value of 89.3%.
Gene prediction and annotation validation
Gene models in the assembly were predicted using Augustus trained with the BUSCO assessment result. The final gene set consisted of 23,560 genes (Table 4). The BUSCO value was 72.9%, which was lower than that in the genome assembly, probably due to the insufficient reliability of the regions assembled using only long-reads data.
Code availability
All analyses were conducted on Linux systems. The version and code and parameters of the main software tools are described below.
(1) LongQC, version 1.2.0c, parameters used: default.
(2) Filtlong, version 0.2.1, parameters used: min_length 1000, keep_percent 90, split 100, mean_q_weight 10.
(3) KMC, version 3.1.1, parameters used: k21, ci1, cs10000.
(4) GenomeScope, version 2.0, parameters used: ploidy 3, kmer_length 21.
(5) MaSuRCA, version 4.0.5, parameters used: LIMIT_JUMP_COVERAGE = 300, CA_PARAMETERS = cgwErrorRate = 0.15, FLYE_ASSEMBLY = 0.
(6) HaploMerger2, version 20180603, parameters used: default; hm.batchA and hm.batchB.
(7) QUAST, version 5.0.2, parameters used: default.
(8) BUSCO, version 5.2.2, parameters used: lineage_dataset sauropsida_odb10.
(9) RepeatMasker, version 4.1.1, parameters used: engine ncbi, xsmall, Database: Dfam with RBRM.
(10) RepeatModeler, version 2.0.1, parameters used: default, Database: The scaffolds assembled with MaSuRCA and HaploMerger2.
(11) Augustus, version 3.4.0, parameters used: species = Database trained with BUSCO, alternatives-from-evidence = true, hintsfile = Output of RepeatMasker.
(12) Diamond, version 2.0.13, parameters used: more-sensitive, max-target-seqs. 1, evalue 1e-5.
References
Pyron, R. A., Burbrink, F. T. & Wiens, J. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol. Biol. 13, 93 (2013).
O’Shea, M. The Book of Snakes: A Life-Size Guide to Six Hundred Species from around the World (Ivy Press, 2018).
Uetz, P., Freed, P., Aguilar, R. & Hošek, J. The Reptile Database http://www.reptile-database.org (2022).
Marin, J. et al. Hidden species diversity of Australian burrowing snakes (Ramphotyphlops). Biol. J. Linn. Soc. 110, 427–441 (2013).
Hedges, S. B., Marion, A. B., Lipp, K. M., Marin, J. & Vidal, N. A taxonomic framework for typhlopid snakes from the Caribbean and other regions (Reptilia, Squamata). Caribb. Herpetol. 49, 1–61 (2014).
Kerkkamp, H. M. I. et al. Snake genome sequencing: Results and future prospects. Toxins 8, 360.
Castoe, T. A. et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. USA 110, 20645–20650 (2013).
Gower, D. J. et al. Eye-transcriptome and genome-wide sequencing for Scolecophidia: Implications for inferring the visual system of the ancestral snake. Genome Biol. Evol. 13, evab253 (2021).
McDowell, S. B. A catalogue of the snakes of New Guinea and the Solomons, with special reference to those in the Bernice P. Bishop Museum, Part I. Scolecophidia. J. Herpetol. 8, 1–57 (1974).
Nussbaum, R. A. The Brahminy blind snake (Ramphotyphlops braminus) in the Seychelles Archipelago: distribution, variation, and further evidence for parthenogenesis. Herpetologica 36, 215–221 (1980).
Wynn, A., Cole, C. J. & Gardner, A. L. Apparent triploidy in the unisexual Brahminy blind snake, Ramphotyphlops braminus. American Museum Novitates 2868, 1–7 (1987).
Ota, H., Hikida, T., Matsui, M., Mori, A. & Wynn, A. H. Morphological variation, karyotype and reproduction of the parthenogenetic blind snake, Ramphotyphlops braminus, from the insular region of East Asia and Saipan. Amphibia-Reptilia 12, 181–193 (1991).
Matsubara, K., Kumazawa, Y., Ota, H., Nishida, C. & Matsuda, Y. Karyotype analysis of four blind snake species (Reptilia: Squamata: Scolecophidia) and karyotypic changes in Serpentes. Cytogenet. Genome Res. 157, 98–106 (2019).
Ryskov, A. P. et al. The origin of multiple clones in the parthenogenetic lizard species Darevskia rostombekowi. PLoS One 12, e0185161 (2017).
Spangenberg, V. et al. Cytogenetic mechanisms of unisexuality in rock lizards. Sci. Rep. 10, 8697 (2020).
Wallach, V. Ramphotyphlops braminus (Daudin): a synopsis of morphology, taxonomy, nomenclature and distribution (Serpentes: Typhlopidae). Hamadryad 34, 34–61 (2009).
Pyron, R. A. & Wallach, V. Systematics of the blindsnakes (Serpentes: Scolecophidia: Typhlopoidea) based on molecular and morphological evidence. Zootaxa 3829, 1–81 (2014).
Kuraus, F. Alien reptiles and amphibians: a scientific compendium and analysis (Springer, 2008).
Wallach, V. First appearance of the Brahminy Blindsnake, Virgotyphlops braminus (Daudin 1803) (Squamata: Typhlopidae), in North America, with reference to the states of Mexico and the USA. IRCF Reptiles & Amphibians 27, 326–330 (2020).
Mahendra, B. C. Contributions to the osteology of the Ophidia. I. The endoskeleton of the so-called ‘Blind-snake’, Typhlops braminus Daud. Proceedings of the Indian Academy of Sciences 3, 128–142 (1936).
List, J. C. Comparative osteology of the snake families Typhlopidae and Leptotyphlopidae (University of Illinois Press, 1966).
Abdeen, A., Mostafa, N. A., Abo-Eleneen, R. E. & Elsadany, D. A. Anatomical studies on the alimentary tract of the Egyptian typhlopid snake Rhamphotyphlops braminus. J. Am. Sci. 9, 504–517 (2013).
Dakrory, A. I., Ali, H. M., Ali, R. S., Abdel-Kader, T. G. & Mahgoub, A. F. Innervation of the olfactory apparatus of the brahaminy blind snake, Ramphotyphlops braminus (Daudin, 1803)-(the nervi terminalis, vomeronasalis and olfactorius) (Reptilia-Squamata- Ophidia- Typhlopidae). Jokull Journal 68, 70–92 (2018).
Mizuno, T. & Kojima, Y. A blindsnake that decapitates its termite prey. J. Zool. 297, 220–224 (2015).
O’Shea, M., Kathriner, A., Mecke, S., Sánchez, C. & Kaiser, H. ‘Fantastic voyage’: a live blindsnake (Ramphotyphlops braminus) journeys through the gastrointestinal system of a toad (Duttaphrynus melanostictus). Herpetology Notes 6, 467–470 (2013).
Ineich, I., Wynn, A., Giraud, C. & Wallach, V. Indotyphlops braminus (Daudin, 1803): distribution and oldest record of collection dates in Oceania, with report of a newly established population in French Polynesia (Tahiti Island, Society Archipelago). Micronesica 2017-01, 1–13 (2017).
Smíd, J. et al. Out of Arabia: a complex biogeographic history of multiple vicariance and dispersal events in the gecko genus Hemidactylus (Reptilia: Gekkonidae). PLoS One 8, e64018 (2013).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18 (2017).
Fukasawa, Y., Ermini, L., Wang, H., Carty, K. & Cheung, M.-S. LongQC: A quality control tool for third generation sequencing long read data. G3: Genes, Genomes, Genetics 10, 1193–1196 (2020).
Kokot, M., Dlugosz, M. & Deorowicz, S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761 (2017).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Zimin, A. V. et al. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 27, 787–792 (2017).
Huang, S., Kang, M. & Xu, A. HaploMerger2: rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33, 2577–2579 (2017).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Waterhouse, R. M. et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Ondov, B. D., Bergman, N. H. & Phillippy, A. M. Interactive metagenomic visualization in a web browser. BMC Bioinform. 12, 384 (2011).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR374853 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR374854 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR374855 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR374856 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR374857 (2022).
NCBI Se quence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR374858 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR374859 (2022).
Acknowledgements
We would like to thank Enago (www.enago.jp) for the English language review. We would like to thank Rashtriya Uchchatar Shiksha Abhiyan (RUSA), Maharashtra, India, (Grant no. PD/RUSA/Order/2018/127 dated February 14th, 2018) for financial support to G.K. This study was also financially supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Numbers 20J22729 and 18H02497 to C.K. and A.K., respectively.
Author information
Authors and Affiliations
Contributions
G.K. and C.K. contributed equally to this work. G.K., A.O., A.K. conceived the study. G.K., C.K., H.T. contributed to the sample collection and sequence data acquisition. H.T., I.T., R.M., A.O. analyzed the genomic data. C.K., A.O., A.K. wrote the manuscript with contributions from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khedkar, G., Kambayashi, C., Tabata, H. et al. The draft genome sequence of the Brahminy blindsnake Indotyphlops braminus. Sci Data 9, 410 (2022). https://doi.org/10.1038/s41597-022-01530-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01530-z
This article is cited by
-
Polyploidization of Indotyphlops braminus: evidence from isoform-sequencing
BMC Genomic Data (2024)
