
Abstract
Gestational diabetes (GD) is one of the most prevalent metabolic diseases in pregnant women worldwide. GD is a risk factor for adverse pregnancy outcomes, including macrosomia and preeclampsia. Given the multifactorial etiology and the complexity of its pathogenesis, GD requires advanced omics analyses to expand our understanding of the disease. Next generation RNA sequencing (RNA-seq) was used to evaluate the transcriptomic profile of subcutaneous and omental adipose tissues (AT) collected from patients with gestational diabetes and matched controls. Samples were harvested during cesarean delivery. Results show differences based on anatomical location and provide whole-transcriptome data for further exploration of gene expression patterns unique to GD patients.
Measurement(s) | RNA sequencing |
Technology Type(s) | Paired-End Sequencing |
Factor Type(s) | Group • Adipose Tissue Site |
Sample Characteristic - Organism | Homo sapiens |
Sample Characteristic - Environment | city |
Sample Characteristic - Location | Bogota, Colombia |
Similar content being viewed by others

Background & Summary
Approximately 7% of all pregnant women develop gestational diabetes (GD) worldwide1. GD prevalence in the US is higher, with 9.2% of the pregnancies diagnosed every year2. Short and long-term maternal and child health complications prevalent in GD patients include macrosomia, preeclampsia, and type 2 diabetes3. Macrosomia in GD patients appears to be associated with peripheral insulin resistance and lipolysis dysregulation in adipose tissues (AT)4. The latter is defined as enhanced and protracted lipolysis that is not responsive to insulin’s anti-lipolytic actions leading to increased and sustained levels of free fatty acids in the blood5,6. In fact, circulating free fatty acids can better predict macrosomia in cases of GD and in those pregnancies complicated by obesity6,7. As a consequence, even in GD patients with adequate glucose control, the incidence of large for gestational age babies is high, reaching 30–50%8,9. Elucidating early triggers of AT insulin resistance and lipolysis dysregulation will minimize the incidence of maternal and neonatal complications in GD.
Using next generation RNA sequencing (RNA-seq), this study evaluated the whole transcriptome of subcutaneous (SC) and omental (OM) AT from patients with gestational diabetes (GD) and healthy matching controls collected during cesarean delivery (C-section). The inclusion of SC and OM supports the evaluation of AT site variations considering depot-specific differences in inflammatory and immune responses and insulin sensitivity10,11. Results show a strong separation of the transcriptomic profiles based on anatomical location and reveal specific RNA expression patterns unique to GD patients.
Methods
Ethics statement
This experiment was approved by Michigan State University and Hospital Universitario Clinica San Rafael institutional review boards (IRB). All patients provided written informed consent following the guidelines established by the ethics committee of Hospital Universitario Clinica San Rafael. Prior to sample preparation, all samples were anonymized by assigning a patient ID number.
Patients and adipose tissue sampling
GD patients (n = 5) were recruited during the third trimester of gestation. Inclusion criteria were age = 18–45; gestational age at the moment of C-section = 37–41weeks; programmed C-section with fasting of at least 8 h; GD diagnosis during the second trimester of gestation based on blood biomarkers of dysfunctional carbohydrate metabolism. Matching controls (n = 5) had the same inclusion criteria except for GD diagnosis. Patients with multiple pregnancy, diabetes (Type I or II) diagnosis prior to pregnancy, hypertension, hypo or hyperthyroidism, autoimmune diseases, chronic diseases, and active tuberculosis were excluded. Table 1 presents a descriptive summary of demographics and blood biomarkers of the GD patients and controls.
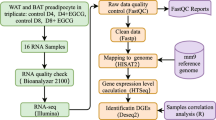
AT samples from the SC and OM depots were collected during the C-Section. In brief, the SC samples were harvested from the incision area using a surgical scalpel. OM samples were collected from the surgical area using scissors and ligature at the omentum majus level. Both AT samples were flash-frozen and stored in liquid nitrogen until processing. Then, total RNA was extracted from OM and SC samples using Trizol and the Quick RNA MiniPrep kit (R1054; Zymo Research, Irving, CA, USA) that includes a DNase step to remove genomic DNA according to the manufacturer’s protocol.
Data Records
Raw FASTQ data is available in the NCBI Gene Expression Omnibus (GEO) NCBI GSE18879912. Raw read count matrix was also deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE18879912. Processed read count matrix and DEGs found in patients with gestational diabetes are available in (Supplemental Table 113).
Technical Validation
Purity, concentration, and integrity of mRNA were checked using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and an Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). All samples had a 260:280 nm ratio between 1.9 and 2.1 and RNA integrity number ≥ 7 (Table 2). At least 1 µg of each sample was used for NGS.
RNA sequencing
All RNA-seq was performed at the Beijing Genomics Institute [BGI, Shenzhen/Hong Kong, China (www.genomics.cn)] and paired-end sequencing (100 bp) was performed on the DNBSEQ platform. BGI’s process includes filtration and exclusion of reads with excessively high levels of unknown base N, adaptor contamination and low-quality reads with a score below 15. On average, 4.5 million adapter sequences were filtered, and the average size of clean reads was 4.46 Gb per sample (range 4.43–4.48 Gb). The ratio of clean reads was 93,7% (Table 3). RNA raw sequencing data was obtained in fastq-files from BGI and subsequent data processing and quality control was performed with FastQC v0.11.814 (www.bioinformatics.babraham.ac.uk/projects/fastqc/) by the authors.
Quality assessment of total RNA and RNA-Seq data
Data quality of the raw RNA-seq reads from FastQC was compiled using MultiQC15. Basic quality assessments included: Phred scores, per sequence and per base quality score, GC contents, overrepresented k-mers, duplicated reads and presence of adaptors were re-checked. To identify global tendencies in the quality metrics output from MultiQC shows the quality across SC and OM samples (Fig. 1).

Evaluation of sequence quality scores in raw FASTQ data. The quality of FASTQ files was estimated using FastQC and summary plots for different samples were mapped on MultiQC. All 40 FASTQ files were assessed, and plots for GC content, mean quality per-base and per-sequence quality in terms of Phred score are presented. In (A) Results for Omental samples and (B) results for Subcutaneous samples.
Reads mapping and counts
After quality check, reads were mapped to the Homo sapiens reference genome (GRCh37/hg19) using HISAT 2.1.016. BAM files obtained were sorted using SAMtools17 in the High Performance Computing at the Institute for Cyber-Enabled Research (ICER), Michigan State University. Mapping results are summarized in (Fig. 2A). The average mapping ratio with the reference genome was 91.8%. Next, featureCounts v.2.0.118 was used to summarize the number of raw reads (Fig. 2B). On average 35,9 millions of reads (73,8%) were assigned to coding genes.

Mapping summary and counts assignment for each omental and subcutaneous adipose tissue sample collected from patients with gestational diabetes and matching controls. (A) Percentage of reads mapped to the (GRCh37/hg19). (B) Percentage of read counts assigned for Subcutaneous samples (left panel) and Omental (right panel).
Differential expression analysis in tissue-specific profiles
For differential expression analysis purposes, data counts were normalized through DESEQ. 2.0 negative binomial distribution model19. Sample variance was established using principal component analysis (PCA) plotting and hierarchical clustering (complete linkage method) using the Euclidean distances between samples (Fig. 3A). Samples from the same anatomic region clustered together indicating their expression profile is highly specific in both tissues (Fig. 3B).

Tissue expression profile summary (A) Euclidean sample-to-sample distances. Samples were clustered using hierarchical clustering analysis, and dendrograms represent the clustering results. The heatmap illustrates the pairwise distances between the indicated samples. (B) PCA illustrates the cluster between subcutaneous (SC) and omental (OM) samples.
Differential Expressed Genes DEGs were determined by paired comparison between controls (OMC-SCC) and patients (OMG-SCG) in each specific tissue as follow OMG-OMC and SCG- SCC. For each comparison, the variance and gene expression changes between patients and control were established by DESEQ. 2. Genes with fold changes > 1 and FDR < 0.05 were defined as DEGs and captured for analysis (Fig. 4A-B), list of DEGs is available in (Supplemental Table 220).

Transcriptomic expression profile in subcutaneous (SC) and omental (OM) adipose tissues from patients with gestational diabetes and matching controls. (A) PCA illustrates the cluster between OM samples from gestational diabetes patients (OMG) and thoses collected from matching controls (OMC). The MA plot shows the changes in gene expression of OM adipose tissue in gestational diabetes patients. (B) PCA illustrates the cluster between SC samples from gestational diabetes patients (SCG)and matching controls (SCC). MA plot shows the changes in gene expression of SC adipose tissue in gestational diabetes patients.
Enrichment Analysis GSEA
After differential expression, Gene Set Enrichment Analysis (GSEA) was performed using the fgsea library in-house implementation in R studio (1000 permutations, term size of 15, and maximum term size of 500) to assess enrichment signatures in the expression profiles21. The entire gene lists were pre-ranked based on the mean fold change and significance (p-value) of each gene. The analysis included the gene set from the Molecular Signatures Database (MSigDB) pathways “Hallmarks”. The significance of enrichment was set by Benjamini-Hochberg false-discovery rate (FDR p‐value < 0.05) (Fig. 5).

Signature enrichment in Omental and Subcutaneous tissue from Diabetes gestational patients (A) Omental (B) Subcutaneous. GSEA shows in the y axis 50 hallmark categories according to the Molecular signature Database and its enrichment in the profile expression of both tissues, in x axis Normalized Enriched Score (NES). Processes in red are not differentially enriched while those in green are differentially enriched at a p-adjusted value > 0.05.
Code availability
This study was supported in part through computational resources provided by the Institute for Cyber-Enabled Research at Michigan State University (ICER). The following software was used to perform quality and expression analyses of the dataset:
1. FastQC v0.72 https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
2. MultiQC v1.9 https://multiqc.info/.
3. HISAT2 v2.2.1 http://daehwankimlab.github.io/hisat2/.
4. SAMtools v1.9 http://www.htslib.org/.
5. featureCounts v2.0.1 https://www.rdocumentation.org/packages/Rsubread/versions/1.22.2/topics/featureCounts.
6. DESEQ v2.11.40.6 https://bioconductor.org/packages/release/bioc/html/DESeq2.html.
7. R v3.6.3.
References
American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 34(Suppl 1), S62–9 (2011).
DeSisto, C. L., Kim, S. Y. & Sharma, A. J. Prevalence estimates of gestational diabetes mellitus in the United States, Pregnancy Risk Assessment Monitoring System (PRAMS), 2007-2010. Prev Chronic Dis 11, E104 (2014).
Damm, P. et al. Gestational diabetes mellitus and long-term consequences for mother and offspring: a view from Denmark. Diabetologia 59(7), 1396–9 (2016).
Diderholm, B. et al. Increased lipolysis in non-obese pregnant women studied in the third trimester. BJOG 112(6), 713–8 (2005).
Schaefer-Graf, U. M. et al. Maternal lipids as strong determinants of fetal environment and growth in pregnancies with gestational diabetes mellitus. Diabetes Care 31(9), 1858–63 (2008).
Diderholm, B. et al. Maternal rates of lipolysis and glucose production in late pregnancy are independently related to foetal weight. Clin Endocrinol (Oxf) 87(3), 272–278 (2017).
Cade, W. T. et al. Maternal Glucose and Fatty Acid Kinetics and Infant Birth Weight in Obese Women With Type 2 Diabetes. Diabetes 65(4), 893–901 (2016).
Combs, C. A. et al. Relationship of fetal macrosomia to maternal postprandial glucose control during pregnancy. Diabetes Care 15(10), 1251–7 (1992).
Evers, I. M. et al. Macrosomia despite good glycaemic control in Type I diabetic pregnancy; results of a nationwide study in The Netherlands. Diabetologia 45(11), 1484–9 (2002).
Harlev, A. et al. Macrophage infiltration and stress-signaling in omental and subcutaneous adipose tissue in diabetic pregnancies. J Matern Fetal Neonatal Med 27(12), 1189–94 (2014).
Mazaki-Tovi, S. et al. Characterization of Visceral and Subcutaneous Adipose Tissue Transcriptome and Biological Pathways in Pregnant and Non-Pregnant Women: Evidence for Pregnancy-Related Regional-Specific Differences in Adipose Tissue. PloS one 10(12), e0143779–e0143779 (2015).
Salcedo-Tacuma, D. et al. Transcriptome dataset of two different adipose tissues in gestational diabetes patients. NCBI Gene Expression Omnibus (GEO) https://identifiers.org/geo/GSE188799 (2021).
Salcedo-Tacuma, D. et al. Transcriptome dataset of omental and subcutaneous adipose tissues from gestational diabetes patients. Suplemental Table 2. FigShare https://doi.org/10.6084/m9.figshare.17185469 (2022).
Babraham, B. FastQC v0.11.8. A quality control tool for high throughput sequence data. 2021, Available from: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
MultiQC. MultiQC v1.9. 2021; Available from: https://multiqc.info/.
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37(8), 907–915 (2019).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16), 2078–2079 (2009).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7), 923–930 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biology 15(12), 550 (2014).
Salcedo-Tacuma, D. et al. Transcriptome dataset of omental and subcutaneous adipose tissues from gestational diabetes patients. Suplemental Table 1. FigShare https://doi.org/10.6084/m9.figshare.19093601 (2022).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 102(43), 15545–15550 (2005).
Acknowledgements
All authors acknowledge the technical resources and support provided by at the Institute for Cyber-Enabled Research (ICER), Michigan State University. The authors thank all the clinical personal at the gynecology and obstretics section of the Hospital Universitario Clinica San Rafael, Bogota, Colombia. The authors also thank Diana Milena Parra Olaya for logistics and executive assistance.
Author information
Authors and Affiliations
Contributions
G.A.C., D.S.T., M.C.G.M., and L.B developed the idea. D.S.T. performed the bioinformatics analyses. M.C.G.M., L.B. coordinated patient recruitment. M.C.G,N., L.B., J.E.N.G., and S.M.S.G harvested tissue samples. M.C. processed all samples and performed quality control. D.S.T and G.A.C. drafted the frst version of the manuscript and all authors contributed signifcantly to the revision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salcedo-Tacuma, D., Bonilla, L., Montes, M.C.G. et al. Transcriptome dataset of omental and subcutaneous adipose tissues from gestational diabetes patients. Sci Data 9, 344 (2022). https://doi.org/10.1038/s41597-022-01457-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01457-5
