
Abstract
A high number of viral metagenomes have revealed countless genomes of putative bacteriophages that have not yet been identified due to limitations in bacteriophage cultures. However, most virome studies have been focused on marine or gut environments, thereby leaving the viral community structure of freshwater lakes unclear. Because the lakes located around the globe have independent ecosystems with unique characteristics, viral community structures are also distinctive but comparable. Here, we present data on viral metagenomes that were seasonally collected at a depth of 1 m from Lake Soyang, the largest freshwater reservoir in South Korea. Through shotgun metagenome sequencing using the Illumina MiSeq platform, 3.08 to 5.54-Gbps of reads per virome were obtained. To predict the viral genome sequences within Lake Soyang, contigs were constructed and 648 to 1,004 putative viral contigs were obtained per sample. We expect that both viral metagenome reads and viral contigs would contribute in comparing and understanding of viral communities among different freshwater lakes depending on seasonal changes.
Measurement(s) | Metagenome • DNA viral genome |
Technology Type(s) | whole genome sequencing |
Factor Type(s) | season |
Sample Characteristic - Organism | unclassified bacterial viruses |
Sample Characteristic - Environment | oligotrophic lake • freshwater lake biome |
Sample Characteristic - Location | South Korea |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.12924506
Similar content being viewed by others
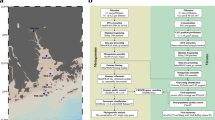

Background & Summary
Bacteriophages—viruses that infect bacteria—are the smallest but the most abundantly found biological entities on earth with approximately 1031 particles1. Despite their large population, only about 2,500 bacteriophages (phage) genomes have been announced so far (as of April 2020, RefSeq release 99, www.vogdb.org). A low number of phage isolates have been reported owing to difficulties in isolating and culturing phages in laboratory settings. As obligate parasites of bacteria, phage cultivation requires the preceding culture of bacterial hosts. However, most of the bacterial population remains uncultured2 despite the development of various culturing techniques. Consequently, phages are left as a large black box of the biosphere. Advances in viral metagenome (virome) studies have facilitated access to a vast amount of phage genomes without the need for phage cultivation. Although most of the virome sequences remain uninterpretable in terms of viral taxonomy and their host information due to a dearth of reference phage genomes across public databases, viral community structures within diverse environments, including ocean3, freshwater4, soil5, and gut6, have been accessed and predicted. The virome sequences are valuable for quantifying the abundance of specific phage genomes within the environment in silico3. The virome data can also be used to predict putative host-phage systems by matching virome sequences to the CRISPR array sequences of bacterial CRISPR-Cas systems7,8 or bacterial signature genes9,10. Virome sequences are also very useful for the discovery of novel phage genes, including antibiotic resistance genes, that are carried by uncultured phages11,12.
Most of the virome studies have been focused on marine and gut environments, leaving freshwater viral communities in question1,8. As one of the major biospheres of earth, freshwater systems encompass diverse organisms, including methylotrophic, nitrifying, and sulfur-oxidizing bacteria, that contribute to biogeochemical cycles. Hence, numerous ecological and genomic studies on freshwater bacterial communities have been performed, whereas a lesser number of studies have been conducted on phages. Lake Soyang is the largest artificial freshwater reservoir in South Korea that is represented in GLEON (Global Lake Ecological Observatory Network13). As a temperate monomictic lake with seasonal physicochemical turnovers and phytoplankton blooms14, Lake Soyang is rich in microbial diversity. From Lake Soyang, numerous novel bacterial strains and phages have been isolated and cultured15,16,17,18. Particularly, phage P19250A, which is the most abundantly found freshwater phage that infects the methylotrophic bacterial strain “Candidatus Methylopumilus planktonicus,” was originally isolated and cultured from Lake Soyang19. Novel phages, P2621816 and P26059A, and B15 infecting heterotrophic bacterial strains isolated from Lake Soyang such as Rhodoferax lacus17 and a bacterial strain belonging to the Comamonadaceae family, respectively, have also been isolated from the Lake Soyang. Bacterial strains from the acI group of the Actinobacteria phylum, which is a ubiquitous and the most frequently found freshwater bacterial group, has been recently isolated and successfully cultured from Lake Soyang for the very first time20.
Here, we present six viral metagenomes collected from the surface of Lake Soyang from October 2014 to May 2016. Each virome represents different seasons and is expected to show a seasonal shift in viral community structures as late-autumn turnover takes place in the monomictic lake14. The collected water samples were enriched for virus particles using a combination of filtration, precipitation, and CsCl purification targeting double-stranded DNA phages (see Fig. 1 & Methods). The viromes were sequenced with Illumina MiSeq platform, and each virome yielded between approximately 3.08-Gbps to 5.54-Gbps of raw sequencing reads (Table 1). The proportion of bacterial rRNA sequences and marker genes was low, and viral gene enrichment scores were approximately 3 to 8-fold higher compared with non-viral metagenome data, showing that the possibility of bacterial contamination in the virome reads was negligible (Table 1). The virome reads were subjected for taxonomic prediction via MG-RAST server (mg-rast.org)21, with 3.1 to 17.3% of the reads taxonomically classified. Of those classified reads, 8.4 to 26.0% were predicted to be of viral origin (Fig. 2a) and most of the reads assigned to viruses belonged to the order Caudovirales (Fig. 2b). The virome contigs were assembled from the virome reads, resulting in a total of 809,964 contigs with minimum length of 128 bp; 6,480 of these contigs with a sequence length longer than 10-kb were used for further analysis (Table 2). Among those, 5,203 contigs were predicted as either viral or prophage genomes (Table 2), according to VirSorter22. A low proportion of bacterial sequences and a high proportion of viral contigs in the virome data of Lake Soyang indicate that the viral community sequences were well sampled. Therefore, we anticipate that the virome data of Lake Soyang would prove to be a useful resource for facilitating the discovery of novel phage genomes and the study of seasonal changes in viral community structure.

A map depicting the sampling site in Lake Soyang and an overview of the metagenome preparation. The red dot represents the sampling site.

Taxonomic annotation of virome reads collected from Lake Soyang. The taxonomic prediction of virome reads is shown in the domain level (a). Only the virome reads that were able to be taxonomically classified by MG-RAST using the NCBI RefSeq database are shown here. The “others” shown here means reads that had a significant hit in RefSeq database but could not be assigned to a specific taxon. The reads that were annotated as viruses in (a) were further shown in family levels in (b).
Methods
Environmental Sampling and metagenome sequencing
From October 2014 to May 2016, 20 L of water samples were collected six times at a depth of 1 m from the Dam station of Lake Soyang, located in Gangwon province, South Korea (37.947421 N, 127.818872 E, Fig. 1). Physicochemical parameters such as temperature, concentration of dissolved oxygen (DO), and pH were measured on site using the YSI Multi-parameter water quality meter, 556 MPS model (Table 3, YSI Incorporated, Yellow Springs, OH, USA). The other physicochemical parameters were measured using the HACH spectrophotometer (HACH DR-28000, Loveland, CA, USA) or QuAAtro microflow analyzer (SEAL analytical, Mequon, Wisconsin, USA). Using the HACH spectrophotometer, 14’ Oct. and ’15 Jan. samples were analyzed for ammonia (HACH method 8155), nitrite (HACH method 8507), nitrate (HACH method 8171), phosphorous (HACH method 8048), and silica (HACH method 8186), according to the manufacturer’s instructions. The collected water samples were maintained at 4 °C and brought to the laboratory. Upon arrival at the lab, 5 L of each water sample was filtered through a 142 mm 0.2-μm Supor® PES Membrane filter (Pall Corporation, New York, USA) using a polycarbonate filter holder (Geotech, Denver, CO, USA) to remove bacteria-like particles. Five milligrams of FeCl3·6H2O was added to 5 L of filtered water samples for flocculating viral particles within the samples23. The samples were incubated at room temperature for 1 hour, with intermittent vigorous shaking to promote flocculation of viral particles. The flocculated viral particles were then collected on a 0.8-μm Isopore polycarbonate filter (Merck Millipore, Darmstadt, Germany). The polycarbonate filters were placed in a conical tube and stored at 4 °C under dark conditions with moist until further treatment24.
The polycarbonate filters were inoculated in 5 ml of 0.1 M EDTA-0.2 M MgCl2-0.2 M ascorbate acid buffer (pH 6) to resuspend flocculated viral particles. Then, the resuspended viral concentrate was treated with DNase I and RNase A at final concentrations of 10 U/ml and 1 U/ml (Sigma-Aldrich, St. Louis, MO, USA), respectively, to remove external nucleic acids. After incubating for 1 hour of incubation with both enzymes at 20 °C, DNase and RNase were deactivated by adding EDTA and EGTA25 at final concentrations of 100 mM. The nuclease-treated viral particles were purified via cesium chloride (CsCl) step-gradient ultracentrifugation26. Different densities of CsCl were stacked from bottom to top layers in a centrifuge tube in the following order: 1.7, 1.5, 1.35, and 1.2 g/cm3; above the top layer, approximately 15 ml of viral particles was added. The samples were centrifuged at 24,000 rpm for 4 hours at 4 °C in a Beckman Coulter L-90K ultracentrifuge with an SW32 Ti swing bucket. After centrifugation, the density fraction ranging between 1.5 and 1.35 g/cm3, corresponding to the density of double-stranded DNA phages, especially of Caudovirales, was retrieved using a syringe. The CsCl remnants in the sample were removed through washing with SM buffer (50 mM Tris-HCl, pH 7.5; 100 mM NaCl; 10 mM MgSO4·7H2O; 0.01% gelatin). The samples were loaded onto the 30 kDa Centrifugal Device (Pall Corporation) and centrifuged at 3,000 rpm until the supernatants were flowed through. Then 10 ml of SM buffer was added to resuspend the sample and was centrifuged again. This process was repeated three times to wash out the CsCl. To remove any remaining bacterial-size contaminants, the samples were filtered through a 0.2-μm pore size Acrodisc® Syringe filter with Supor® membrane (Pall Corporation). Viral DNA was extracted from the filtrates using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instruction with a slight modification24. To 70 μl of the sample, 300 μl of ATL buffer, 30 μl of Proteinase K, and 6 μl of RNase A were added to lyse capsid proteins, followed by addition of 300 μl of 99% ethanol and AL buffer. Then DNA was washed and eluted using the spin column. The extracted viral DNA (211–702 ng per each sample) was subsequently used for constructing a TruSeq library without any amplification. Sequencing was performed at ChunLab Inc. (Seoul, South Korea) using Illumina MiSeq platform, with 2 × 300-bp paired-end reads and no sequencing controls was used. The overall schematic for viral metagenome preparation is shown in Fig. 1.
Quality trimming, assembly, and analysis of viral metagenome reads and contigs
Using the CLC Genomics Workbench (Qiagen), the raw metagenome sequencing data were mapped to the phiX174 genome to remove technical sequencing control reads. The virome data were uploaded in the MG-RAST server21. Taxonomic assignment of the virome reads were performed with the analysis tools provided by the MG-RAST, using the RefSeq as a reference database with default parameters (Fig. 2). The MG-RAST pipeline predicts potential protein encoding genes from each read and compare these translated sequences against reference databases for taxonomic and functional assignment21.
The virome reads with phiX174 adapters removed were trimmed of low-quality reads using Trimmomatic program27 for further analysis. The degree of viral enrichment and non-viral contamination were investigated using the ViromeQC program28 with -w environmental option. Within quality trimmed metagenome reads, the enrichment scores were computed by dividing the abundance of bacterial 16 S small subunit ribosomal RNA gene (SSU rRNA) and 23 S large subunit rRNA gene (LSU rRNA), as well as single-copy universal bacterial and archaeal marker gene sequences by those found within viromes.
The trimmed reads were assembled using SPAdes version 3.5.0 (for ’14 Oct. and ’15 Jan. samples) and 3.8.2 (for all the other samples)29 with k-mer values of 27, 47, 67, 87, 107, and 127 and–careful option. Of all the constructed contigs, only those that were 10-kb in length or longer were selected for VirSorter analysis (Table 2). All the selected contigs from Lake Soyang were used as an input to VirSorter algorithm22 with the virome decontamination option using the virome database to screen for contigs that are of the viral or prophage origin (http://de.cyverse.org/de/). The VirSorter identified viral or prophage contigs by searching for viral proteins within the submitted contigs. Based on the number of viral protein-coding genes found, the submitted contigs were classified into three categories, “pretty sure,” “quite sure,” and “not so sure.” For further analysis, only the contigs that were classified as “pretty sure” and “quite sure” categories were accepted (Table 2)24.
Data Records
The raw data of Lake Soyang viromes are available on the European Nucleotide Archive (ENA) under the accession number of PRJEB15535 (ERP017347)30. The virome reads from which PhiX174 and adapters had been removed were uploaded in the MG-RAST server for basic analysis under the accession numbers of mgm4632933.3 (’14 Oct.), mgm4632937.3 (’15 Jan.), mgm4694059.3 (’15 Sept.), mgm4709782.3 (’15 Nov.), mgm4709783.3 (’16 Feb.), and mgm4709863.3 (’16 May)31. The virome contigs that were 10-kb in length or longer were selected and deposited in the JGI IMG/MER database with accession numbers of IMG3300007735 (’14 Oct.), IMG3300007734 (’15 Jan.), IMG3300011113 (’15 Sept.), IMG3300011116 (’15 Nov.), IMG3300011114 (’16 Feb.), and IMG3300011115 (’16 May)32.
Technical Validation
The virome reads were evaluated for their sequencing qualities using the fastp program33, using default parameters. The Q scores for the raw virome reads were calculated and showed that 68.35 to 73.78% of reads scored Q30 or higher (Table 4), indicating that most of the virome reads were constructed with low error rates. To evaluate how well the purification protocol employed in this study enriched virus-like particles (VLPs) and reduced contamination by bacteria, ViromeQC program was used. For each virome, ViromeQC first calculates the proportion of virome reads that are aligned to SSU and LSU rRNA gene sequences obtained from the Silva database, or matched to 31 conserved bacterial marker proteins database. Then, this program calculates enrichment scores by dividing the median proportions calculated from >2,000 non-enriched (i.e. non-viral) metagenomes by the proportions calculated form each virome, with the underlying premise that better enrichment of VLPs would lead to the decrease of aligned or matched reads, resulting in the increase of enrichment score. Among the three enrichment scores (SSU rRNA, LSU rRNA, and marker proteins), the minimum one is regarded as a comprehensive enrichment score. The enrichment score of Lake Soyang viromes ranged from 2.95 to 8.29 (median: 6.56), which indicates that the purification protocol of this study worked well compared to the scores of ~2,000 viromes calculated by ViromeQC where ~50% of viromes showed enrichment scores of ≤328.
Code availability
The options used for the generation and processing of the virome data are as follows: Trimmomatic (v. 0.33): ILLUMINACLIP: TruSeq. 3-PE-2.fa:2:30:10 LEADING:10 TRAILING:10 SLIDINGWINDOW:4:16 MINLEN:100 SPAdes (v. 3.5.0 and v. 3.8.2): -k 27, 47, 67, 87, 107, 127--careful
References
Dion, M. B., Oechslin, F. & Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 18, 125–138 (2020).
Steen, A. D. et al. High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 13, 3126–3130 (2019).
Roux, S. et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature. 537, 689–693 (2016).
Okazaki, Y., Nishimura, Y., Yoshida, T., Ogata, H. & Nakano, S.-i Genome-resolved viral and cellular metagenomes revealed potential key virus-host interactions in a deep freshwater lake. Environ. Microbiol. 21, 4740–4754 (2019).
Williamson, K. E., Fuhrmann, J. J., Wommack, K. E. & Radosevich, M. Viruses in soil ecosystems: an unknown quantity within an unexplored territory. Annu.Rev. Virol. 4, 201–219 (2017).
Yutin, N. et al. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 3, 38–46 (2018).
Davison, M., Treangen, T. J., Koren, S., Pop, M. & Bhaya, D. Diversity in a polymicrobial community revealed by analysis of viromes, endolysins and CRISPR spacers. PLoS One. 11, e0160574 (2016).
Paez-Espino, D. et al. Uncovering earth’s virome. Nature. 536, 425–430 (2016).
Ghai, R., Mehrshad, M., Mizuno, C. M. & Rodriguez-Valera, F. Metagenomic recovery of phage genomes of uncultured freshwater actinobacteria. ISME J. 11, 304–308 (2017).
Kavagutti, V. S., Andrei, A.-Ş., Mehrshad, M., Salcher, M. M. & Ghai, R. Phage-centric ecological interactions in aquatic ecosystems revealed through ultra-deep metagenomics. Microbiome. 7, 135 (2019).
Balcazar, J. L. Bacteriophages as vehicles for antibiotic resistance genes in the environment. PLoS Pathog. 10, e1004219 (2014).
Moon, K. et al. Freshwater viral metagenome reveals novel and functional phage-borne antibiotic resistance genes. Microbiome. 8, 75 (2020).
Weathers, K. C. et al. The global lake ecological observatory network (GLEON): the evolution of grassroots network science. Limnol. Oceanogr. Bull. 22, 71–73 (2013).
Kim, B., Choi, K., Kim, C., Lee, U.-H. & Kim, Y.-H. Effects of the summer monsoon on the distribution and loading of organic carbon in a deep reservoir, Lake Soyang, Korea. Water Res. 34, 3495–3504 (2000).
Moon, K., Kang, I., Kim, S., Kim, S.-J. & Cho, J.-C. Genomic and ecological study of two distinctive freshwater bacteriophages infecting a Comamonadaceae bacterium. Sci. Rep. 8, 7989 (2018).
Moon, K., Kang, I., Kim, S., Cho, J.-C. & Kim, S.-J. Complete genome sequence of bacteriophage P26218 infecting Rhodoferax sp. strain IMCC26218. Stand. Genomic. Sci. 10, 111 (2015).
Park, M., Song, J., Nam, G. G. & Cho, J.-C. Rhodoferax lacus sp. nov., isolated from a large freshwater lake. Int. J. Syst. Evol. Microbiol. 69, 3135–3140 (2019).
Joung, Y. et al. Lacihabitans soyangensis gen. nov., sp. nov., a new member of the family Cytophagaceae, isolated from a freshwater reservoir. Int. J. Syst. Evol. Microbiol. 64, 3188–3194 (2014).
Moon, K., Kang, I., Kim, S., Kim, S.-J. & Cho, J.-C. Genome characteristics and environmental distribution of the first phage that infects the LD28 clade, a freshwater methylotrophic bacterial group. Environ. Microbiol. 19, 4714–4727 (2017).
Kim, S., Kang, I., Seo, J.-H. & Cho, J.-C. Culturing the ubiquitous freshwater actinobacterial acI lineage by supplying a biochemical ‘helper’ catalase. ISME J. 13, 2252–2263 (2019).
Meyer, F. et al. The metagenomic RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386 (2008).
Roux, S., Enault, F., Hurwitz, B. L. & Sullivan, M. B. VirSorter: mining viral signal from microbial genomic data. PeerJ. 3, e985 (2015).
John, S. G. et al. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 3, 195–202 (2011).
Moon, K. Ecological and genomic study on freshwater bacteriophages. (Seoul National University, 2018).
Hurwitz, B. L., Deng, L., Poulos, B. T. & Sullivan, M. B. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ. Microbiol. 15, 1428–1440 (2013).
Thurber, R. V., Haynes, M., Breitbart, M., Wegley, L. & Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 4, 470–483 (2009).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014).
Zolfo, M. et al. Detecting contamination in viromes using ViromeQC. Nat. Biotechnol. 37, 1408–1412 (2019).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Moon, K., Kang, I. & Cho, J.-C. Viral metagenome of Lake Soyang. European Nucleotide Archieve https://identifiers.org/ncbi/bioproject:PRJEB15535 (2018).
Moon, K., Kang, I. & Cho, J.-C. Viral metagenome of Lake Soyang. MG-RAST http://www.mg-rast.org/linkin.cgi?project=mgp13279 (2020).
Moon, K., Kang, I. & Cho, J.-C. Freshwater viral communities from Lake Soyang, Gangwon-do, South Korea. Joint Genome Institute IMG/MER https://gold.jgi.doe.gov/study?id=Gs0118096 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Acknowledgements
This study was supported by the Mid-Career Research Program through the National Research Foundation (NRF) funded by the Ministry of sciences and ICT (NRF-2019R1A2B5B02070538 to J-CC), Science Research Center grant of the NRF (NRF-2018R1A5A1025077 to J-CC), and Research Staff Program (NRF-2019R1I1A1A01063401 to IK and NRF-2019R1I1A1A01062072 to KM). Field sampling on Lake Soyang was conducted using the research boat belonging to the Korea Water Resources Corporation (K-water) with help of captain Do Gyeom Lee.
Author information
Authors and Affiliations
Contributions
K.M. constructed virome data. K.M. and I.K. performed metagenome analyses. S.K. performed ion measurements of samples. I.K. and J.-C.C. supervised the study. K.M., I.K., and J.-C.C. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Moon, K., Kim, S., Kang, I. et al. Viral metagenomes of Lake Soyang, the largest freshwater lake in South Korea. Sci Data 7, 349 (2020). https://doi.org/10.1038/s41597-020-00695-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-00695-9
This article is cited by
-
Metagenome sequencing and recovery of 444 metagenome-assembled genomes from the biofloc aquaculture system
Scientific Data (2023)
-
Metaviromics coupled with phage-host identification to open the viral ‘black box’
Journal of Microbiology (2021)
-
Occurrence and diversity of viruses associated with cyanobacterial communities in a Brazilian freshwater reservoir
Brazilian Journal of Microbiology (2021)
