
Abstract
Lead optimization is a crucial step in the drug discovery process, which aims to design potential drug candidates from biologically active hits. During lead optimization, active hits undergo modifications to improve their absorption, distribution, metabolism, excretion and toxicity (ADMET) profiles. Medicinal chemists face key questions regarding which compound(s) should be synthesized next and how to balance multiple ADMET properties. Reliable transformation rules from multiple experimental analyses are critical to improve this decision-making process. We developed OptADMET (https://cadd.nscc-tj.cn/deploy/optadmet/), an integrated web-based platform that provides chemical transformation rules for 32 ADMET properties and leverages prior experimental data for lead optimization. The multiproperty transformation rule database contains a total of 41,779 validated transformation rules generated from the analysis of 177,191 reliable experimental datasets. Additionally, 146,450 rules were generated by analyzing 239,194 molecular data predictions. OptADMET provides the ADMET profiles of all optimized molecules from the queried molecule and enables the prediction of desirable substructure transformations and subsequent validation of drug candidates. OptADMET is based on matched molecular pairs analysis derived from synthetic chemistry, thus providing improved practicality over other methods. OptADMET is designed for use by both experimental and computational scientists.
Key points
-
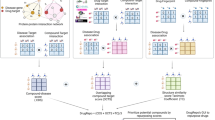
OptADMET is an integrated web-based tool that uses the data-driven chemical transformation rules for 32 absorption, distribution, metabolism, excretion and toxicity properties. The database contains 41,779 validated transformation rules generated from the analysis of 177,191 reliable experimental datasets.
-
OptADMET provides the absorption, distribution, metabolism, excretion, and toxicity profiles of optimized molecules from a queried lead candidate and informs on the subsequent validation of drug candidates.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
The output files can be downloaded from https://cadd.nscc-tj.cn/deploy/optadmet/.
Code availability
The software is available from https://cadd.nscc-tj.cn/deploy/optadmet/. Code for deployment of OptADMET is in the GitHub repository (https://github.com/antwiser/OptADMET)
References
Vogt, M., Yonchev, D. & Bajorath, J. Computational method to evaluate progress in lead optimization. J. Med. Chem. 61, 10895–10900 (2018).
Segall, M. Advances in multiparameter optimization methods for de novo drug design. Expert Opin. Drug Discov. 9, 803–817 (2014).
Sutherland, J. J., Raymond, J. W., Stevens, J. L., Baker, T. K. & Watson, D. E. Relating molecular properties and in vitro assay results to in vivo drug disposition and toxicity outcomes. J. Med. Chem. 55, 6455–6466 (2012).
Campbell, I. B., Macdonald, S. J. F. & Procopiou, P. A. Medicinal chemistry in drug discovery in big pharma: past, present and future. Drug Discov. Today 23, 219–234 (2018).
Khanna, I. Drug discovery in pharmaceutical industry: productivity challenges and trends. Drug Discov. Today 17, 1088–1102 (2012).
Gaulton, A. et al. The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954 (2017).
Kim, S. et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 49, D1388–D1395 (2021).
Sterling, T. & Irwin, J. J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Panteleev, J., Gao, H. & Jia, L. Recent applications of machine learning in medicinal chemistry. Bioorg. Med. Chem. Lett. 28, 2807–2815 (2018).
Xu, Y., Yao, H. & Lin, K. An overview of neural networks for drug discovery and the inputs used. Expert Opin. Drug Discov. 13, 1091–1102 (2018).
Winter, R., Montanari, F., Noé, F. & Clevert, D.-A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 10, 1692–1701 (2019).
Bell, D. R. et al. Dynamics-based peptide-MHC binding optimization by a convolutional variational autoencoder: a use-case model for CASTELO. J. Chem. Theory Comput. 17, 7962–7971 (2021).
Long, T. Z. et al. Structural analysis and prediction of hematotoxicity using deep learning approaches. J. Chem. Inf. Model. 63, 111–125 (2023).
Abbasi, M. et al. Designing optimized drug candidates with generative adversarial network. J. Cheminf. 14, 40 (2022).
Sheridan, R. P., Hunt, P. & Culberson, J. C. Molecular transformations as a way of finding and exploiting consistent local QSAR. J. Chem. Inf. Model. 46, 180–192 (2006).
Leach, A. G. et al. Matched molecular pairs as a guide in the optimization of pharmaceutical properties; a study of aqueous solubility, plasma protein binding and oral exposure. J. Med. Chem. 49, 6672–6682 (2006).
Tyrchan, C. & Evertsson, E. Matched molecular pair analysis in short: algorithms, applications and limitations. Comput. Struct. Biotechnol. J. 15, 86–90 (2017).
Bush, J. T. et al. A turing test for molecular generators. J. Med. Chem. 63, 11964–11971 (2020).
Griffen, E., Leach, A. G., Robb, G. R. & Warner, D. J. Matched molecular pairs as a medicinal chemistry tool: miniperspective. J. Med. Chem. 54, 7739–7750 (2011).
Birch, A. M., Kenny, P. W., Simpson, I. & Whittamore, P. R. Matched molecular pair analysis of activity and properties of glycogen phosphorylase inhibitors. Bioorg. Med. Chem. Lett. 19, 850–853 (2009).
Gleeson, P., Bravi, G., Modi, S. & Lowe, D. ADMET rules of thumb II: a comparison of the effects of common substituents on a range of ADMET parameters. Bioorg. Med. Chem. 17, 5906–5919 (2009).
Sushko, Y. et al. Prediction-driven matched molecular pairs to interpret QSARs and aid the molecular optimization process. J. Cheminf. 6, 1–18 (2014).
Lumley, J. A., Desai, P., Wang, J., Cahya, S. & Zhang, H. The derivation of a matched molecular pairs based ADME/Tox knowledge base for compound optimization. J. Chem. Inf. Model. 60, 4757–4771 (2020).
Kofler, M. in The Definitive Guide to MySQL 3–19 (Springer, 2004).
Xiong, G. et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 49, W5–W14 (2021).
Yang, Z.-Y. et al. Semi-automated workflow for molecular pair analysis and QSAR-assisted transformation space expansion. J. Cheminf. 13, 86 (2021).
Fu, L. et al. QSAR-assisted-MMPA to expand chemical transformation space for lead optimization. Brief. Bioinform. 22, bbaa374 (2021).
Hussain, J. & Rea, C. Computationally efficient algorithm to identify matched molecular pairs (MMPs) in large data sets. J. Chem. Inf. Model. 50, 339–348 (2010).
Papadatos, G. et al. Lead optimization using matched molecular pairs: inclusion of contextual information for enhanced prediction of HERG inhibition, solubility, and lipophilicity. J. Chem. Inf. Model. 50, 1872–1886 (2010).
Wei, Y., Li, S., Li, Z., Wan, Z. & Lin, J. Interpretable-ADMET: a web service for ADMET prediction and optimization based on deep neural representation. Bioinformatics 38, 2863–2871 (2022).
Yang, H. et al. ADMETopt: a web server for ADMET optimization in drug design via scaffold hopping. J. Chem. Inf. Model 58, 2051–2056 (2018).
Yang, H. et al. admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 35, 1067–1069 (2019).
Shan, J. & Ji, C. MolOpt: a web server for drug design using bioisosteric transformation. Curr. Comput. Aided Drug Des. 16, 460–466 (2020).
Keefer, C. E., Chang, G. & Kauffman, G. W. Extraction of tacit knowledge from large ADME data sets via pairwise analysis. Bioorgan. Med. Chem. 19, 3739–3749 (2011).
Kanetaka, H. et al. Discovery of InhA inhibitors with anti-mycobacterial activity through a matched molecular pair approach. Eur. J. Med. Chem. 94, 378–385 (2015).
Ertl, P. & Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminf. 1, 8 (2009).
Nogawa, H. & Kawai, T. hERG trafficking inhibition in drug-induced lethal cardiac arrhythmia. Eur. J. Pharmacol. 741, 336–339 (2014).
Jamieson, C., Moir, E. M., Rankovic, Z. & Wishart, G. Medicinal chemistry of hERG optimizations: highlights and hang-ups. J. Med. Chem. 49, 5029–5046 (2006).
Das, N. et al. Mitigating hERG liability of toll-like receptor 9 and 7 antagonists through structure-based design. ChemMedChem 18, e202300069 (2023).
Acknowledgements
We thank the user community for using OptADMET and providing us with valuable suggestions regarding further improvement of this tool. We also acknowledge H. Xu, and the High-Performance Computing Center of Central South University for support. The study was approved by the university’s review board. This work was supported by National Key Research and Development Program of China (2021YFF1201400), National Natural Science Foundation of China (22173118, 22220102001), Hunan Provincial Science Fund for Distinguished Young Scholars (2021JJ10068), the science and technology innovation Program of Hunan Province (2021RC4011), the Natural Science Foundation of Hunan Province (2022JJ80104), and the 2020 Guangdong Provincial Science and Technology Innovation Strategy Special Fund (2020B1212030006, Guangdong-Hong Kong-Macau Joint Lab).
Author information
Authors and Affiliations
Contributions
T.H. and D.C. conceived and supervised the project. J.Y. and Z.Y. designed and developed the web server. S.S., L.F. and J.Y. wrote the manuscript. P.N., A.L., C.W., Y.D., C.H. and X.Z. co-supervised the project and helped with troubleshooting. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Xiong, G. et al. Nucleic Acids Res. 49, W5–W14 (2021): https://doi.org/10.1093/nar/gkab255
Yang, Z.-Y. et al. J. Cheminform. 13, 86 (2021): https://doi.org/10.1186/s13321-021-00564-6
Fu, L. et al. Brief. Bioinform. 22, bbaa374 (2021): https://doi.org/10.1093/bib/bbaa374
Supplementary information
Supplementary Information
Supplementary Figs. 1–5 and Tables 1–5.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yi, J., Shi, S., Fu, L. et al. OptADMET: a web-based tool for substructure modifications to improve ADMET properties of lead compounds. Nat Protoc 19, 1105–1121 (2024). https://doi.org/10.1038/s41596-023-00942-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-023-00942-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
