
Abstract
Liquid chromatography coupled with high-resolution mass spectrometry (LC–HRMS) has become a workhorse in global metabolomics studies with growing applications across biomedical and environmental sciences. However, outstanding bioinformatics challenges in terms of data processing, statistical analysis and functional interpretation remain critical barriers to the wider adoption of this technology. To help the user community overcome these barriers, we have made major updates to the well-established MetaboAnalyst platform (www.metaboanalyst.ca). This protocol extends the previous 2011 Nature Protocol by providing stepwise instructions on how to use MetaboAnalyst 5.0 to: optimize parameters for LC–HRMS spectra processing; obtain functional insights from peak list data; integrate metabolomics data with transcriptomics data or combine multiple metabolomics datasets; conduct exploratory statistical analysis with complex metadata. Parameter optimization may take ~2 h to complete depending on the server load, and the remaining three stages may be executed in ~60 min.
Similar content being viewed by others
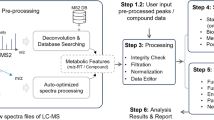


Introduction
The goal of metabolomics is to comprehensively study all metabolites in biological samples. For research concerning predefined lists of compounds (targeted metabolomics), various protocols have been established and an increasing number of commercial kits are becoming available. However, unbiased comprehensive metabolome profiling (global metabolomics) remains a critical bottleneck owing to several complex analytical and bioinformatics challenges1,2,3. Developing high-throughput global metabolomics technologies has become a high-priority task in metabolomics4 as well as the burgeoning fields of exposomics5 and precision medicine6. Among different technologies available, high-resolution mass spectrometry (HRMS) has shown great promise3,7. HRMS instruments such as Orbitrap or time-of-flight systems coupled with gas chromatography (GC) or liquid chromatography (LC) can simultaneously measure a wide range of endogenous and exogenous compounds to characterize an individual’s metabolic phenotype, environmental exposures and associated biological responses. LC–HRMS is arguably the most widely used platform as it can measure a broad range of compounds with minimal sample preparation. However, the associated data processing and analysis remain particularly challenging to most researchers.
The computational workflow for LC–HRMS-based global metabolomics can be summarized in three general steps: (1) spectra processing to convert raw spectra into a peak intensity table; (2) peak annotation to characterize peak relationships and to assign putative compound identities (ID); (3) data analysis to identify important features, patterns of variation and their functional interpretations. Although these steps are conceptually similar to the analysis of other omics data such as gene expression or microbiome data analysis8,9, the inherent characteristics of the data generated in LC–HRMS-based global metabolomics present unique challenges especially for those without statistical programming skills and/or a deep understanding of the analytical instrumentation. To address this gap, we have recently made major updates to the widely used MetaboAnalyst platform to support LC–HRMS-based global metabolomics data analysis and interpretation10. This protocol provides an overview of these new features followed by stepwise instructions through several example datasets using MetaboAnalyst 5.0.
MetaboAnalyst and other web-based tools
MetaboAnalyst was launched in 2009, and since then there have been major update releases every 3 years10,11,12,13,14. MetaboAnalyst versions 1.0–3.0 were mainly designed for general statistical and functional analysis of targeted metabolomics data. Starting with v4.0, the development of MetaboAnalyst has gradually shifted toward addressing more complex bioinformatics and statistical needs arising from global metabolomics data, including raw spectra processing, functional analysis and integration with other omics data. According to Google Analytics, the public platform (www.metaboanalyst.ca) is currently being accessed by ~2,000 users worldwide on a daily basis.
Many software tools have been developed in the past decade for processing and analyzing metabolomics data15,16,17. Most of these tools need to be locally installed by the users. For web-based platforms, there are several popular options, including MetaboAnalyst, XCMS online18, W4M19 and NOREVA20. These web-based tools have been compared with MetaboAnalyst 5.0 in our recent publication10. In summary, MetaboAnalyst offers the most features for statistical and functional analysis; XCMS online and W4M provide more comprehensive support for raw data processing; while NOREVA is dedicated for metabolomics data normalization and quality assessment. A hallmark of MetaboAnalyst is its ease of use, which enables researchers from diverse backgrounds to perform various complex tasks of data analysis. For instance, most tools require users to manually adjust multiple parameters to achieve satisfactory performance for raw LC–MS spectra processing. MetaboAnalyst 5.0 provides a largely automated workflow through efficient optimization of parameters from default settings for common LC–MS instruments. In addition, the results of spectra processing can be directly transferred to other compatible modules within MetaboAnalyst for streamlined statistical and functional analysis. The overall workflow of MetaboAnalyst is depicted in Fig. 1.

Version 5.0 focuses on comprehensive support for LC–MS-based global metabolomics including spectral processing, functional interpretation, statistical analysis with complex metadata, and multi-omics integration. ROI, regions of interest; DoE, design of experiments.
Limitations of this protocol and software
Owing to the proprietary formats and large file sizes generated from LC–HRMS instruments, MetaboAnalyst currently does not accept raw spectra uploaded in vendor-specific formats. Thus, raw data generated from different MS instruments need to be first converted into an open data format using either a vendor-provided conversion tool or a free tool such as ProteoWizard21. More details surrounding data preparation are provided in ‘Materials’ and ‘Procedure’ (Steps 1 and 2).
To ensure fast turnover, the public platform of MetaboAnalyst currently limits the processing of raw spectra up to 200 samples per job, which, in our experience, is usually sufficient for common metabolomic studies. For large-scale projects, users are encouraged to perform spectra processing locally using the OptiLCMS package developed by our team (https://github.com/xia-lab/OptiLCMS). Please refer to Box 1 for more details on this package.
MetaboAnalyst currently does not support processing spectra from GC–MS or MS/MS, which are also commonly employed in global metabolomics. Users are advised to explore other powerful tools, including MZmine22, MS-DIAL23, MS-Finder24 and OpenMS25, for dealing with these data types.
In addition, functional analysis and multi-omics integration in MetaboAnalyst mainly focuses on biological samples, while environmental and industrial samples are not well supported owing to lack of well-established conceptual frameworks and knowledgebases required for these types of analysis26.
For statistical analysis involving complex metadata, the covariate adjustment in MetaboAnalyst is based on the widely used linear regression approach27 to model the level of individual features with the response variable and uncontrolled covariates. The current interface does not permit users to include complex interaction terms; the algorithm assumes that the response variable will impact the feature in the same way across covariate values.
Experimental design
Driven by user feedback, we have published several comprehensive tutorials and protocols for each major release of MetaboAnalyst28,29,30,31,32,33. These tutorials provide detailed instructions for tasks related to data processing, filtering, normalization, statistical analysis of datasets with single experimental factors, and functional analysis for targeted metabolomics. Our 2019 protocol32 is still up to date on these topics. Accordingly, this current protocol provides detailed instructions for four major updates that have been made to MetaboAnalyst 5.0:
-
1
Optimized processing of LC–HRMS spectra (Steps 1–18)
-
2
Deriving functional insights from LC–HRMS peaks (Steps 19–31)
-
3
Meta-analysis and integration with transcriptomics data (Steps 32–45)
-
4
Statistical analysis and exploration with complex metadata (Steps 46–65)
Spectra processing
This is the first task in global metabolomics. The continuum/profile data of a single raw spectrum obtained from an LC–HRMS instrument (such as Q-Exactive) is typically 1–2 GB in mzXML/mzML format. A common practice is to first perform peak centroiding to condense the Gaussian-shaped mass peak to a single mass centroid. This step can substantially reduce the file size to ~100 MB.
The next step is to detect peaks (also known as peak picking) from the centroid data. Multiple algorithms have been developed to identify peaks in different dimensions such as retention time34,35,36. Please see Table 1 for more details. Among them, the centWave algorithm34 implemented in XCMS has been shown to perform well in processing LC–HRMS spectra. However, a practical difficulty associated with using default XCMS is to decide appropriate values for several key parameters, which requires a relatively deep understanding of both MS instrumentation and the peak picking algorithm. To address this challenge, we have developed OptiLCMS to enable automated parameter specification for XCMS in MetaboAnalyst37. After peak detection, peak alignment is performed to address retention time variations across spectra. These aligned peaks form a peak intensity table with varying proportions of missing values. These missing values indicate that either peak detection failed or the corresponding feature is absent from the respective sample. Therefore, the final step is ‘gap filling’ by reperforming direct peak extraction on corresponding regions in the raw spectra.
Peak annotation
A typical LC–HRMS spectrum of common biofluids (such as blood or urine samples) can often produce >10,000 peaks. However, this number is not equivalent to the number of compounds detected. The correspondence between the number of peaks and the number of actual metabolites remains elusive38. Multiple peaks can be derived from the same compounds. These are real, biologically relevant peaks and might result from the formation of adducts, incorporation of isotopes, or fragmentation during sample preparation or LC–MS analysis. The remaining peaks are now considered to be largely from background noise (artifacts or noise peaks)39. Therefore, the first step in peak annotation aims to identify real peaks, and to clarify the relationships among them. Many empirical and statistical rules have been developed to address this problem, including CAMERA40 and CliqueMS41, which are two popular R packages. Peak annotation in MetaboAnalyst 5.0 is currently based on CAMERA.
The next step is to assign putative compound IDs to those peaks. This is a challenging task even with high-resolution instruments, as a single peak can potentially match multiple compounds42. To facilitate peak annotation for global metabolomics, it is highly recommended to acquire MS2-level data, whenever possible. It is important to keep in mind that, unlike targeted metabolomics, the primary goal of global metabolomics is to understand the overall patterns and to identify promising features to inform the design of follow-up studies for more targeted analysis. As discussed in the sections below, accurate peak annotation, although important, is not an absolute prerequisite for functional interpretation of global metabolomics data.
Functional interpretation of global metabolomics data
Identifying a list of genes or compounds of interest, and performing enrichment analysis against predefined libraries of gene sets or metabolite sets using gene set enrichment analysis (GSEA)43 or metabolite set enrichment analysis (MSEA)44 is well established for transcriptomics or targeted metabolomics data. When functions are defined in the form of pathways with structure information available, it is sensible to consider the positions of perturbed genes/metabolites in these pathways45. Common topology measures such as degree and betweenness values are often employed to prioritize pathways involving more changes at ‘hub’ or ‘bottleneck’ positions in functional analysis46. Please refer to many excellent tutorials as well as our previous published protocols28,32. For discovery of novel pathways or metabolic functions, users are encouraged to define their own metabolite sets or signatures and upload them as customized libraries during enrichment analysis using MetaboAnalyst.
For global metabolomics, since the mapping of peaks to compounds is often inaccurate, researchers wonder if it is still possible to identify meaningful functional changes by performing enrichment analysis based on those ‘fuzzy’ annotations. This question has been answered by the ‘mummichog’ algorithm47, which has clearly demonstrated that the collective behavior (i.e., changes across multiple compounds involved in a pathway) are robust to random errors introduced during individual compound assignments. In other words, enrichment analysis of putatively annotated peaks is a valid approach to identify changes at pathway or network level, as long as the annotation errors are random. MetaboAnalyst 5.0 offers both mummichog and GSEA-based approaches to predict pathway activities from LC–MS peaks. In general, high-resolution MS peaks are preferred as they provide better coverage of pathways with reduced error rates in putative annotation, compared with using peaks from low-resolution MS instruments42.
Multi-omics integration
Integrating multiple omics datasets from the same study or integrating the same type of omics datasets across multiple studies can help reduce false positives and derive a more holistic understanding. Data integration approaches have been relatively well established in other omics fields, including targeted metabolomics where the same compound IDs can be mapped consistently across different datasets48,49. However, this is not the case in global metabolomics where features are generally not comparable across different studies. This is because LC–MS peaks, characterized by their mass-to-charge ratio (m/z) and retention time (rt), are highly susceptible to experimental parameters and analytical batch effects. Since we can compute functional activities from such data, integrating data at a higher level (i.e., pathways) becomes conceptually clear and practical. As the same set of metabolic pathways are defined for both transcriptomics and metabolomics, such an approach also permits integrative analysis with transcriptomics data as well as across multiple global metabolomics datasets. Given the exploratory nature of such analysis, statistical integration should always be combined with intuitive data visualization to gain a more comprehensive data understanding.
Statistical analysis with complex metadata
Metadata describes the data, and contains details on the experimental conditions, sample sources (i.e., species, tissue), sample collection (i.e., location, time) and other factors. Such metadata are critical for data interpretation, because they allow researchers to account for the biological and environmental context when they analyze the data, and facilitate data reuse by allowing other researchers to search for, and meaningfully compare and potentially integrate, results from across diverse studies. Details on the context and sample source are becoming increasingly important as observational studies that collect omics data from human populations or animals outside laboratory settings are becoming more common50. Epidemiologic studies in biomedical or environmental sciences generally involve a primary variable of interest, such as presence/absence of a certain disease or exposure to a specific chemical, as well as variables such as age, sex or other potential factors that covary with the primary metadata. Statistical analyses that take these covariates into account can lead to substantial increases in power and draw more robust conclusions about the relationships between the primary variable and the omics data51.
Transparent and reproducible analysis
Exploratory analysis of omics data often involves employing a diverse set of approaches coupled with versatile visualization techniques. Reproducing these results after a few months have elapsed can be a daunting task, because it is often very difficult to remember what choices were made and the order in which steps were executed. MetaboAnalyst addresses this issue through three complementary ways:
-
Transparent stepwise analysis through proper interface design to facilitate procedural reproducibility
-
A comprehensive PDF analysis report documenting all major steps and associated results
-
A detailed R command history together with the underlying MetaboAnalystR package (https://github.com/xia-lab/MetaboAnalystR)37,52,53 to allow batch execution of the workflow
These main features are illustrated in Fig. 2.

Several key features of MetaboAnalyst are illustrated here: (1) the navigation tree allowing users to access different pages, (2) the results (files and images) generated during analysis, (3) the R Command History containing the underlying R commands executed, (4) the analysis report documenting all steps with results embedded, and (5) the module switching panel for users to continue analysis in other compatible modules.
Materials
Equipment
Computer requirements
-
Browser requirements: MetaboAnalyst 5.0 runs on all modern web browsers. For the best results, we recommend Google Chrome 92+, Firefox 92+, Safari 12+ and Microsoft Edge v93+. JavaScript must be enabled in your browser
-
Internet connection requirements: a fast connection is highly recommended. At least 1 MB per second is required for uploading raw spectra
-
Hardware requirements: >4 GB of RAM and a screen resolution of at least 1,200 × 800 is preferred. At least 8 GB available hard drive space is needed to store the raw spectra files
Data files
-
Raw MS spectra. MetaboAnalyst accepts centroid LC–MS spectra in various open data formats: mzXML, mzData, mzML and netCDF. The first three are XML-based data formats. Among them, mzML and mzXML are the preferred formats for MS spectra, while mzData is phasing out. If your spectra files are not centroided, please do it locally with ProteoWizard21. This will substantially reduce the file size and increase data upload speed. All spectra need to be compressed into individual *.zip files and uploaded together with a metadata file (.txt). Quality control (QC) and blank samples need to be specified in the metadata file as ‘QC’ and ‘BLANK’, respectively. If no metadata file is provided when uploading spectral data, QC files should start with ‘QC_’ and blank samples should start with ‘BLANK_’
-
LC–MS peak list. Peak lists can contain up to five columns - ‘m.z’, ‘r.t’, ‘p.value’, ‘t.score’ and ‘mode’ separated by a space or tab. The required column is ‘m.z’, which is a list of m/z values for all MS peaks. The others are optional but are required for specific analyses. The ‘r.t’ column is required if users would like to perform functional analysis with mummichog v2. The ‘p.value’ is highly recommended for the mummichog algorithm, otherwise the single ‘m.z’ column must be preranked. The ‘t.score’ is required for the GSEA algorithm. The ‘mode’ column is used to specify the ion mode if the data contain peaks from different ion modes
-
Gene/protein/compound list. The list must contain gene, protein or compound IDs in the first column. The optional second column should contain fold change (FC) values that are used for visualization
-
Generic feature table. A generic table with features in rows and samples in columns (or its transposed format) is the most common format for statistical analysis in MetaboAnalyst. The first row or column of the table should contain unique sample/feature names using a combination of English letters, numbers, underscore or hyphen. When users are not asked to provide a separate metadata file, the sample group information must be coded in the same feature table, immediately following the sample names with at least three replicates per group
-
Metadata table. To accommodate complex data structure or study design, two modules in MetaboAnalyst (‘LC-MS Spectra Processing’ and ‘Statistical Analysis [metadata table]’) now ask users to provide a separate metadata file. The first column of the metadata table must be the same sample names used by the data file. The second column contains the primary condition of interest. Other metadata (e.g., sex, age or batch) can be included in the remaining columns. Both categorical and continuous variables are acceptable. MetaboAnalyst currently cannot deal with missing values (NA or empty) in the metadata file. Users will be provided an interface to manually fix this issue if missing values are detected during the metadata integrity check stage
-
Example datasets. Multiple built-in example datasets are offered in each MetaboAnalyst module. Users can simply choose an example dataset and click the ‘Submit’ button to start exploring the tool. Six datasets are used in this protocol:
-
1
Raw spectra data (malaria_raw.zip) consisting of 12 plasma samples from naive and malaria semi-immune subjects along with 3 QCs54.
-
2
A feature table (malaria_feature_table.csv) generated from processing the above raw spectra for functional analysis.
-
3
A gene list (integ_genes_1.txt) and a compound list (integ_cmpds.txt) to demonstrate the integration of targeted metabolomics data with transcriptomics data.
-
4
A gene list (integ_genes_2.txt) and a peak list (integ_peaks.txt) to demonstrate the integration of global metabolomics data with transcriptomics data.
-
5
Three peak intensity tables (A1_pos.csv, B1_pos.csv, C1_pos.csv) from global metabolomics studies of serum samples from patients with coronavirus disease 2019 (COVID-19) to demonstrate the integration of multiple global metabolomics datasets55.
-
6
A feature table (TCE_feature_table.csv) and its associated metadata table (TCE_metadata.csv) from an exposomics study on metabolic changes associated with occupational exposure to trichloroethylene (TCE) to demonstrate statistical analysis with complex metadata and covariate adjustment56.
-
1
Equipment setup
(Optional) Download the example datasets
Go to the MetaboAnalyst home page (https://www.metaboanalyst.ca/), and click the ‘Data Formats’ link from the left panel. Users can download all the example datasets used in the protocol. These datasets are also available as built-in examples in each corresponding module. Users can directly select those datasets and follow the protocols without downloading and uploading them.
Procedure
Stage 1: LC–HRMS raw spectra processing
Timing 1.5–2 h
-
1
Starting up. Go to the MetaboAnalyst home page (https://www.metaboanalyst.ca/), and click the ‘Click here to start’ button in the middle of the page. The module page displays all modules as a pyramid organized into four tiers. The top tier contains one button ‘LC-MS Spectra Processing’. Click the button to enter the raw spectra processing module.
Critical step
Almost all modern browsers support multiple tabs. Please keep MetaboAnalyst open in only one tab. If MetaboAnalyst is accessed from multiple tabs, they could interfere with one another, leading to unpredictable results.
-
2
Data uploading.
-
Unzip the dataset #1 (malaria_raw.zip) into individual files
-
Click the ‘Select’ button to open a File Chooser dialog
-
Locate the and select all spectra files (.zip) and the metadata file (.txt)
-
Click the ‘Upload’ button to start uploading. At any time, users can cancel the upload by clicking the ‘Reset’ button below the uploading progress bars
-
-
3
Once the upload has finished, click the ‘Proceed’ button. Alternatively, users can use the built-in example dataset without uploading spectra. To do this, locate the table under ‘Try our example data’, select the second option and click ‘Submit’.
-
4
Data integrity check. This ‘data integrity check’ page summarizes key information from the user uploaded spectra. The file names, sizes, centroid status and group information are displayed in the first four columns. Only centroid data are supported for further processing. MetaboAnalyst evaluates whether a given spectrum is in centroid format based on MSnbase57. If the data do not conform to this format, click the arrowhead icon to centroid the data on the fly using MSnbase. By default, all samples passing the data integrity check will be processed. Users can also choose to exclude certain samples by deselecting the corresponding checkboxes in the ‘Include’ column. Click the ‘Next’ button.
-
5
Customize profiling parameters. The ‘LC-MS Spectra Processing’ page displays all important parameters for processing raw spectra. Several platform-specific parameters are provided by default. Three algorithms are available for peaks picking and two algorithms for peak alignment.
-
Select an LC–MS platform. For this dataset, select ‘UPLC-Q/E’
-
There are two options for parameter setting: ‘Default/manual’ and ‘Auto-optimized’. Here we select ‘Auto-optimized’ option for spectra processing. Please refer to Box 1 for more details on how parameter optimization is achieved in MetaboAnalyst
-
Advanced users can manually configure the parameters using the ‘Default/manual’ option. Please refer to Table S1 for detailed explanations of these parameters.
Critical step
The ‘Auto-optimized’ procedure has been developed for XCMS centWave algorithm, which generally performs well for HRMS spectra. For low-resolution MS spectra, users may want to use MatchedFilter algorithm. For users interested in detecting low-intensity peaks, Massifquant could perform better58. Implementation of different algorithms for peak picking and alignment within MetaboAnalyst are summarized in Table 1.
-
-
6
Customize annotation parameters. The peak annotation parameters (including ion mode and potential adducts) must be specified manually on the basis of the experimental conditions. Make sure the ‘Polarity’ option is set to ‘positive’, and keep other options as default.
-
7
(Optional) Spectral inspection for potential contaminants.
-
If the ‘Auto-optimized’ option is selected for peak profiling, the ‘Contaminants’ option will be enabled. This option allows users to exclude strong instrumental noise or potential contaminants present during the chromatographic run for parameters’ optimization. The option is selected by default
-
Visually inspect the spectral data by clicking the ‘View’ link to bring up the ‘Spectral Inspection’ dialog (Supplementary Fig. 1A). By default, a random QC sample (if provided) is displayed in 3D style
-
Switch to other spectra files or different spectral regions based on m/z and rt ranges (Supplementary Fig. 1B)
-
Alternatively, use 2D heatmaps to view the same information (Supplementary Fig. 1C). In this example, we could see multiple peaks that persist over half of the chromatogram; therefore, we keep the ‘Remove’ option checked
Critical step
In addition to removal of contaminant peaks, spectra files with mean signal intensity below 5% of the average signal intensity calculated on the basis of all uploaded spectra files will be excluded as these outliers will cause error during peak alignment.
-
-
8
Job submission. Once the parameter configuration is done, click ‘Submit Job’ and ‘Confirm’ to proceed. Once the job is submitted, users cannot come back to this page and modify parameters until the job is complete or cancelled.
Tip: The optimized parameters can be downloaded in a text file as an experimental record or for further reuse. Users can manually enter these parameters using the ‘Default/manual’ option the next time they process the same or similar data to achieve faster analysis.
-
9
Create a job URL. Raw spectra processing could take hours to finish depending on the server load. After the job is submitted, users will be directed to the ‘Job Status View’ page. A ‘Job ID’ will be assigned to this job automatically. Users can view the running status of the submitted job from the ‘Current Status’. The status will show ‘Pending’ if the job is still in queue. After the job starts running, the actual processing takes ~75 min for this data using the ‘Auto-optimized’ mode, or ~10 min using the ‘Default/manual’ mode.
-
Create a URL bookmark, and save the link in a text file. Then click ‘Exit’ from the navigation tree of the left panel. Users will then be able to return to this page via the bookmark URL at any time to check the status of their jobs
-
-
10
Job progress and status. The job execution process is displayed as a progress bar in the middle of the ‘Job Status’ box. The data processing details are displayed in the ‘Text Output’ box. The job status is refreshed every 3 s.
-
Click ‘Cancel Job’ to cancel the current job at any time
-
Once the job is complete, click ‘Proceed’ to view the results
-
-
11
Result visualization. Explore the processed results; these are summarized using several graphics (e.g., principal component analysis (PCA), total ion chromatogram (TIC), base peak ion chromatogram (BPI)) in separate tabs (Fig. 3a).
-
Rotate the PCA 3D plot to view data from different angles or zoom in and out with mouse scroll wheel
-
By default, all features are shown in the loading plot. Drag the ‘PCA Loadings’ slider to keep the top features based on their contributions to the sample separation patterns in the score plot. The PCA score plot can be displayed on the basis of all peaks or on the peaks that you have included in the loading plot
Fig. 3: Results from raw spectra processing. 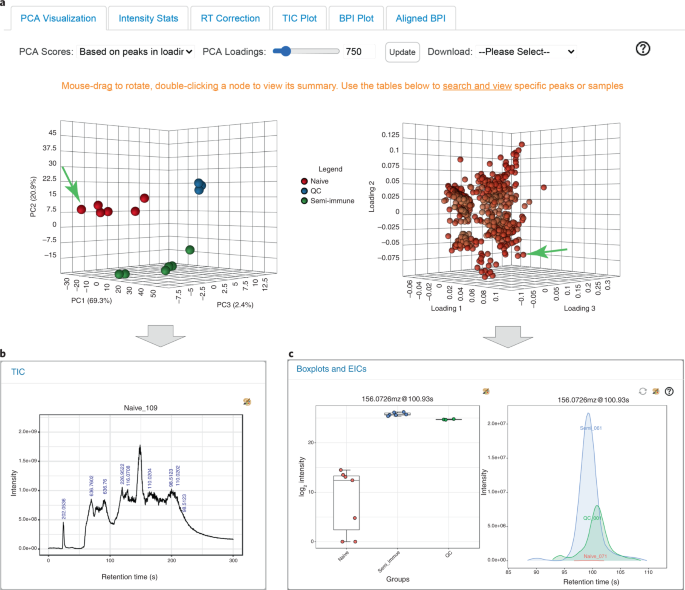
a, Overview of metabolic profiles based on 3D PCA visualization. PCA score plot (left) shows the clustering pattern of all samples, while the PCA loading plot (right) shows the corresponding features. The PCA score and loading plots can be rotated synchronously. b, A TIC plot after clicking a sample point (indicated by the green arrow) in the PCA score plot. c, Box plots (left) after clicking a feature point (indicated by the green arrow) in the PCA loading plot; EIC plots (right panel) after clicking three sample points on the box plots.
Tip: PCA summarizes the main patterns of variation of the data. The top 25–50% features can usually reveal the same pattern without overcrowding the loading plot.
-
-
12
Exploring samples and features using PCA.
-
Double click a data point in the PCA score plot to generate a TIC plot of the corresponding sample (Fig. 3b)
-
Double-clicking a data point in the PCA loading plot will display a dialog box with two panels (Fig. 3c). The left panel shows box plots summarizing the intensity distributions of the selected feature across sample groups defined by the metadata file uploaded in Step 2
-
Double-clicking a data point on the box plot will generate its corresponding extracted ion chromatogram (EIC) plot. Clicking more data points will overlay their EIC plots. The function allows users to create and visually compare typical EICs in different groups
-
Click the reset icon on the top-right corner to reset the process
-
-
13
Result tables. The bottom half of the result page consists of two tables. ‘Spectra/Sample Table’ summarizes peak detection results from different samples, while the ‘Feature/Peak Table’ displays all peaks (m/z, retention time) and their annotations (e.g., isotopes, adducts, formula, potential compounds). Use these two tables to search for specific samples or features for more detailed inspection and click the ‘View’ button in the last column for more details.
Tip: The ‘Annotation’ column in the ‘Feature/Peak Table’ contains annotation formula calculated from CAMERA40, in which the character ‘M’ represents the precursor ion. The ‘Putative IDs’ column contains their potential chemical formulas and matched compounds based on HMDB59. Empty cell means the corresponding feature is either a precursor ion or there are no annotations available.
-
14
Analysis report generation and downloading results. Click the ‘Download Page’ button to enter the corresponding page. All processed results are shown in a table on the ‘Results Download’ tab. Click the ‘Generate Report’ to create a PDF report of the current analysis.
-
15
(Optional) Users can compare their results with those obtained using the default parameter settings.
-
Click the ‘Spectra Processing’ node from the navigation tree to return to the ‘Parameter Setting’ page
-
Select ‘UPLC-Q/E’ platform, make sure the ‘Default/manual’ mode is selected, set ‘Polarity’ as ‘positive’ and submit the job
Table 2 presents the results based on the ‘Optimized’ and the ‘Default’ options, in which optimized parameters produced greater number of peaks and higher proportions of peaks with putative annotations. In addition, the top two PCs capture more variations of the data. These results clearly validate the effectiveness of the optimization strategies described in Box 1.
Table 2 Comparison of results obtained using the optimized and the default pipelines from the example raw spectra (n = 15) -
-
16
After finishing the download, click ‘Logout’ to end the analysis. Users can also select the ‘Start New Journey’ tab to explore results in other compatible modules.
Tip: By default, the complete raw peak table will be transferred to other modules for analysis. It is highly recommended to perform appropriate data filtering, normalization, transformation and scaling in these modules to get optimal results.
-
17
(Optional) Create a user account. Users can set up an account to better manage their jobs. Click ‘Log in’ button from the left panel of the data uploading page (Step 2). This page also allows users to create a new account by clicking the ‘Create account’ button or clicking the ‘Forgot password?’ button to set a new password.
-
18
(Optional) Manage projects and accounts. After logging into their accounts, all projects are displayed on the Project page. A maximum of ten projects are allowed per account. Each project will be saved for at most 90 days. At any time, users can click the ‘Delete Account’ button from the projects management page to completely remove their accounts from MetaboAnalyst.

Stage 2: functional analysis of LC–HRMS peaks
Timing ~20 min
-
19
Starting up. Go to the MetaboAnalyst homepage, and select the ‘Click here to start’ button to enter the modules overview page. Locate the ‘Functional Analysis’ button (on the second tier). Click the button to enter this module.
-
20
Data uploading. Users can upload two types of data: a peak list or a complete peak intensity table (see ‘Materials’ for more details). In this protocol, we will use the ‘A peak intensity table’ tab for malaria_feature_table.csv.
-
In this tab, enter the following parameters: Ion Mode: ‘Positive Mode’; Mass Tolerance (ppm): 4.3 (based on the optimized value from the Raw Spectra Processing module); Retention time: ‘Yes - Seconds’
-
Click the ‘Choose’ button to select the data #2 (malaria_feature_table.csv)
-
Click the ‘Submit’ button
Alternatively, users can simply select the third example, ‘Malaria’, which is the same data, and click ‘Submit’ at the bottom of the page.
Critical step
Functional enrichment analysis of global metabolomics data requires permutation tests based on the complete features in order to compute empirical P values to evaluate the significance of the pathways. Users should provide a peak list or a peak intensity table containing the complete features detected from the raw spectra processing step, instead of just significant features. The feature list or table can also be generated by other tools, such as MZmine or MS-DIAL. Make sure their formats are compatible with MetaboAnalyst. Please refer to the Box 2 for more details on functional enrichment analysis.
-
-
21
Perform data integrity check. The data integrity check summarizes the key information of this data. Note that features containing all zero values or a single non-zero values will be excluded automatically. This is not uncommon for global metabolomics data with a small number of samples. Click the ‘Proceed’ button.
-
22
Data filtering. The data filtering step is used to remove the noninformative variables. Select ‘None’ and click the ‘Submit’ button to keep all features for this dataset. Click the ‘Proceed’ button to the next page.
-
23
Perform data normalization. MetaboAnalyst offers comprehensive options for data normalization. The choice of option depends on the purpose of the data normalization steps; the implementation principals are introduced in the previous protocol of MetaboAnalyst32. For the example dataset, select ‘Log transformation’ and click ‘Normalize’ to perform normalization. Then, click ‘View Result’ to inspect the normalization results. Click the ‘Proceed’ button to continue.
-
24
(Optional) Group removal. Functional analysis is mainly designed to detect functional changes between two experimental groups. If there are three or more groups (e.g., healthy, disease and QC) in the data table, the QC group should be excluded for analysis. To do this, click the ‘Data Editor’ from the navigation tree. Click the ‘Edit Groups’ tab. Select ‘QC’ from the ‘Available’ box, and move it into the ‘Exclude’ box by clicking the left arrow. Click ‘Submit’ to finish editing.
Critical step
Users need to perform the normalization again after editing data.
-
25
Parameter setting. The page is organized into two sections: algorithm parameters and library selection. Two algorithms are available for functional analysis: mummichog (default) and GSEA.
-
For mummichog, users could manually specify the P-value cutoff and the version of the algorithm. The default P-value cutoff is to keep the top 10% peaks, and using mummichog 2.0. GSEA is a cutoff-free method
-
The results can be viewed in a scatter plot or heatmap (available for peak intensity tables only)
-
The library selection is based on the organism under study
-
The parameter at the bottom of the page is for excluding small pathways—if the number of compounds is very small, there is usually not sufficient information to allow reliable identification of the underlying pathways
Here we keep the default options and click ‘Submit’. See Box 2 for more details on the concepts of functional enrichment analysis.
-
-
26
Pathway activity prediction. The results from the mummichog algorithm are displayed in the ‘Mummichog Pathway Activity Profile’ page. The top half page is an interactive scatter plot with ‘Enrichment Factor’ as x axis and ‘−log10(P)’ as y axis (Fig. 4a).
Fig. 4: Functional analysis of ranked peak lists. 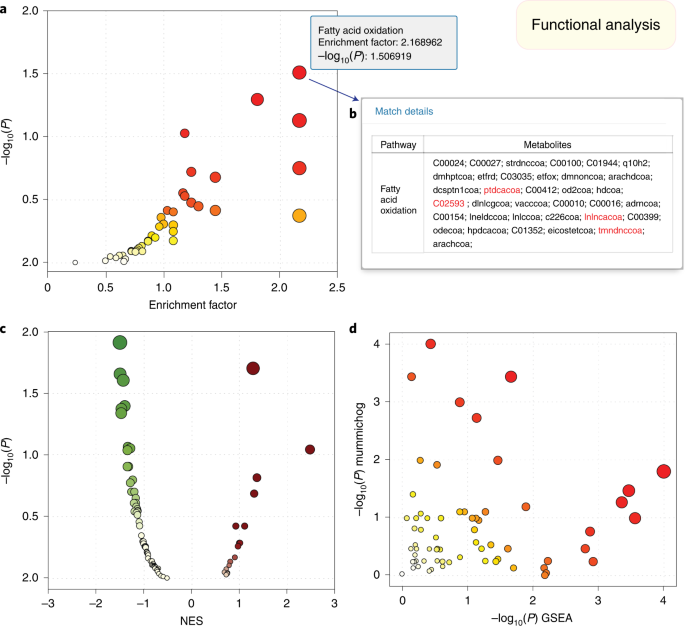
a, The enriched pathways predicted from mummichog algorithm. b, A dialog showing the compound matches of the corresponding pathway with significant compounds in red. c, The result from the GSEA algorithm. d, The joint result from the mummichog and GSEA methods. The sizes of the data points are based on their x values, while the colors correspond to their y values.
-
Hover the mouse over a point to find out the pathway name and the corresponding statistics
-
Click on a point to display the compounds matched to that pathway (Fig. 4b)
In this example, mummichog has identified several fatty-acid-metabolism-related pathways that are consistent with the literature on the roles of fatty acid metabolism in the pathogenesis of malaria60,61. User can download the ‘Pathway Hits’ and ‘Compound Hits’ files directly. The pathway results are displayed at the bottom of this page. Click the ‘View’ link from the last column of the table to view all potentially matched compounds (Fig. 4b).
-
-
27
(Optional) GSEA analysis. Use the navigation tree to go back to the ‘Set Parameters’ page. Uncheck the mummichog option and select GSEA algorithm, click ‘Submit’ for further exploration. Different from mummichog results, GSEA returns a volcano plot with normalized enrichment score (NES) as the x axis and ‘−log(P)’ as the y axis for scatter plot visualization (Fig. 4c).
-
28
(Optional) Joint analysis with mummichog and GSEA. To combine results from both algorithms, select both options from the ‘Set parameters’ page. The results would be a joint scatter plot with GSEA plotted on the x axis and mummichog on the y axis (Fig. 4d). The P values from both algorithms are merged with Fisher’s method.
-
29
(Optional) Network visualization. From the ‘Pathway Activity Profile’ page, click the ‘Network Explorer’ button, and explore the enriched pathways within the KEGG global metabolic network.
-
30
Functional analysis of metabolic patterns. Click ‘Set parameters’ from the navigation tree to go back to the parameter configuration page, select ‘Heatmaps’ as the visual analytics option and click ‘Submit’. As shown in Fig. 5, the page is divided into three vertical sections: Overview, Focus View and Enrichment Analysis. Overview allows researchers to use the mouse to drag and select peaks of interest from the Overview to the Focus View and perform enrichment analysis based on the selected peaks. By default, Overview displays all peaks ranked by their P values and Focus View shows the significant peaks. Performing enrichment analysis on those significant peaks is equivalent to the analysis as described in Steps 25 and 26.
-
Here we show how to gain functional insights into peaks showing similar patterns of variations, similar to the gene co-expression analysis in transcriptomics. First explore different cluster algorithms (from ‘Cluster peaks’ menu) to perform cluster analysis; visually identify an area with interesting patterns of variation and select the region by performing drag-select on the Overview heatmaps for enrichment analysis (Fig. 5)
-
Watch the second part video demo of No. 2 (www.metaboanalyst.ca/MetaboAnalyst/resources/data/8_Video_Tutorial.pdf) to learn how to manually select multiple regions of interest to compile a composite pattern for enrichment analysis
-
All patterns and results can be downloaded easily from the ‘Download’ menu
Critical step
The interactive heatmaps run on users’ browser rather than on the MetaboAnalyst server. Please directly download the images using the ‘Download’ menu at the current page, as the final Download page will not be able to capture these heatmap images.
Fig. 5: Functional analysis of manually selected metabolic patterns. 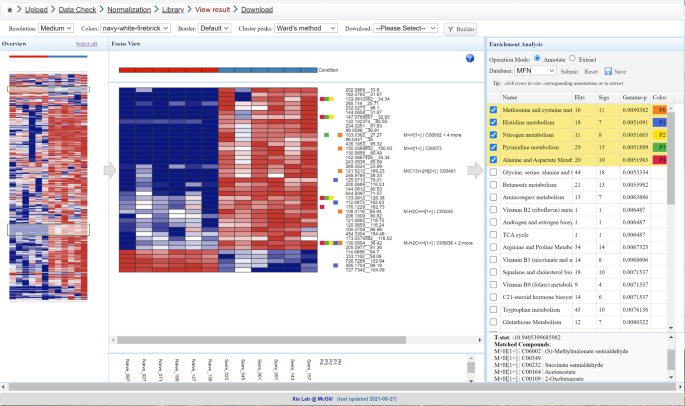
This is a screenshot of the heatmap-based functional analysis of user-selected peak clusters. Users can use a mouse to drag-select one or more metabolic patterns of interest from the Overview (left) to the Focus View (middle). The functional analysis will be performed on the basis of the selected metabolic patterns. Enriched pathways are displayed (right) with potential compounds annotated beside the metabolic patterns in the Focus View.
-
-
31
Result downloading. Click the ‘Download’ node from the navigation tree at the left panel. At the download page, generate the analysis report, download all results and exit.


Stage 3: pathway-level integration of multiple datasets
Timing 20–35 min
Critical
MetaboAnalyst provides two approaches to integrate multiple datasets. The first one is the integration of datasets from different omics layers, and the other method is the integration of multiple metabolomic datasets. These two approaches are introduced sequentially in this section.
Integration of transcriptomics and targeted metabolomics datasets
-
32
Starting up. From the MetaboAnalyst home page, select ‘Click here to start’ to access the module selection page and click the ‘Joint-Pathway Analysis’ to enter the module.
-
33
Select species. A total of 25 species are currently supported for this analysis. Select ‘Homo sapiens (Human)’ because the data we are going to use are from a study on human subjects.
-
34
Data uploading. This module accepts two lists from transcriptomics/proteomics (genes or proteins) and metabolomics (metabolites or peaks). Please refer to ‘Materials’ for more details.
-
Open the data (integ_genes_1.txt) with a text editor and copy-paste the text into the gene/protein list box. Set the ID type as ‘Official Gene Symbol’
-
For metabolomics data, the compound list (integ_cmpds.txt) can be entered in the same way as uploading a gene list, with the ‘Metabolomics Type’ set to ‘Targeted (compound list)’ and the ID type as ‘Compound Name’.
For demonstration purpose, we will use an example dataset to demonstrate the integration of transcriptomics data with targeted metabolomics data. Click the ‘Try our example’ link at the bottom of the page, select the first example, which contains the two files (integ_genes_1.txt and integ_cmpds.txt) described above. Click the ‘Submit’ button.
-
-
35
Data integrity check. The genes and compounds uploaded from the previous step are matched to the MetaboAnalyst knowledgebase. The unrecognized genes or compounds will be highlighted. Delete or correct any unmatched items. Click the ‘Proceed’ button when finished.
-
36
Parameter setting 1. The ‘Parameter Selection’ page allows users to choose a pathway library and an algorithm to perform enrichment analysis. Different pathway libraries are offered to allow users to compare results from integrative analysis with those obtained on the basis of genes or metabolites alone.
-
Select and configure the algorithms from the list of options. These include enrichment analysis algorithms, topology measurements and integration methods. Here we select the ‘Combined p values (overall)’ as the integration method. Leave other parameters as default
-
Click the ‘Submit’ button to perform integration analysis
Critical step
MetaboAnalyst provides four options for integration: (1) combine queries, (2) combine P values (unweighted), (3) combine P values (overall) and (4) combine P values (pathway level). The first two options treat transcriptomics and metabolomics equally, even though the number of genes detected for any given pathway is usually much higher than the number of metabolites detected. The last two options aim to mitigate this issue by introducing weights to genes/metabolites based on their proportions at the global level or individual pathway level. Please refer to Box 3 for more guidance on different integration methods.
-
-
37
Explore results from pathway view. The results from the pathway analysis are summarized as an interactive scatter plot on the left (Fig. 6a). The y axis shows negative log transformed P values from enrichment analysis results. Pathways are further differentiated along the x axis on the basis of their topological impact values, which are the sums of the normalized degree values (or betweenness values based on the topology measure specified in Step 36) over all perturbed genes/metabolites in each pathway.
-
Click a data point on the scatter plot; the corresponding pathway will be displayed on the right panel. Genes/proteins are represented as rectangles, and compounds as circles. The upregulated features (positive logFC values) are in red, while the downregulated features (negative logFC values) are in green. The numerical details of the pathway results are provided in the table at the bottom of the page
-
Click the ‘Results Table’ button to download the statistics of all pathways
-
Click the ‘Matched Features Table’ button to download the matched genes/proteins and compounds of all pathways
Fig. 6: Results of joint-pathway analysis. 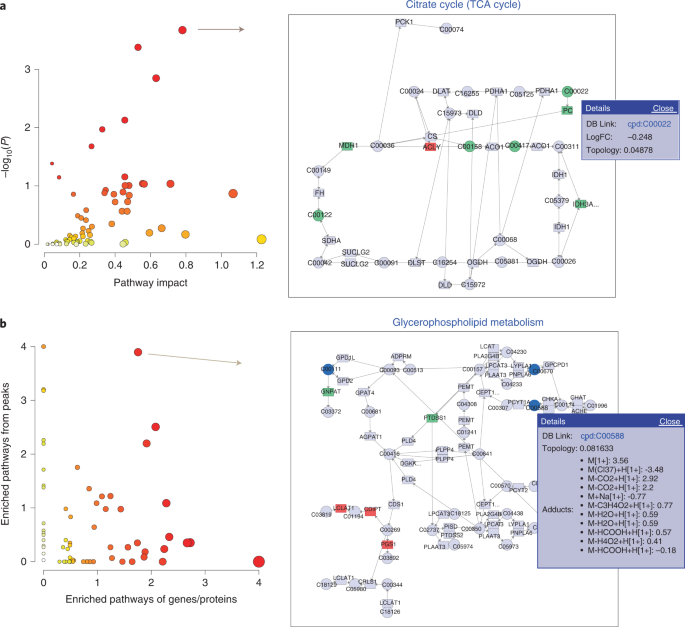
a, The result from the integration of transcriptomics (gene list) and targeted metabolomics data (compound list). The pathways are displayed as a scatter plot (left). The x axis shows pathway impact scores, which summarize normalized topology measures of those perturbed genes/metabolites in each pathway. The y axis shows −log10(P) values of the enrichment analysis results. The sizes of the data points are correlated with their x values, and the color gradients correspond to their y values. By clicking a node in the left panel, one can view the corresponding pathway in the right panel. Genes are shown as rectangles, and compounds as circles. The matched nodes are colored on the basis of their logFC, with green for negative values and red for positive values. b, The result from the integration of transcriptomics (gene list) and untargeted metabolomics data (peak list). Both axes show −log10(P) values of the pathway enrichment analysis based on transcriptomics (x axis) and metabolomics (y axis), respectively. The size and color of the data points are based on their merged −log10(P) values. Clicking a node in the left panel will show the corresponding pathway in the right panel. Clicking a compound (blue circle node) in the pathway will show all the peaks assigned to this compound.
-
-
38
(Optional) Explore results from network view. Click the ‘Network Explorer’ button from the pathway view page to explore the integration results from a network view (Supplementary Fig. 2). All pathways in the left panel can be highlighted on the network. The network view can be exported as PNG or SVG images.

Integration of transcriptomics and untargeted metabolomics datasets
-
39
Integration of genes and peaks. Return to the Data Upload page, and select the second example for integration. This example utilizes the original datasets of the study54. Click ‘Submit’ to submit your data. Alternatively, users can upload the downloaded gene lists (integ_genes_2.txt) similar as described in Step 34. For the metabolomics data (integ_peaks.txt), choose the ‘Untargeted (peak list)’ option; an interface similar to the data upload page in Step 20 will appear. Specify the parameters, and then click the File Chooser to upload the peak list file.
-
40
Data integrity check. The sanity check page summarizes the results of the uploaded genes and peaks. Click ‘Proceed’ to continue.
-
41
Parameter setting 2. This page provides an introduction on the mechanism of integration as well as a parameter setting panel.
-
Choose which of the two pathway libraries to use—metabolic pathways or all pathways. The latter option also includes regulatory pathways containing only genes/proteins
-
Choose a method to merge P values. There are two options (Fisher’s and Stouffer’s methods); the difference is that Stouffer’s method attributes different weights when combining p values and is not as sensitive as Fisher to very small P values
-
Set the ‘Sig. peaks cutoff’ as 0.001, and leave the other parameters as default.
-
Click the ‘Submit’ button to get the results
-
-
42
Results display. The results are shown in Fig. 6b.
-
Explore the pathways using the scatter plot on the left. Pathways that are supported by both genes and metabolite peaks will be located on the top right area of the plot, while those supported mainly by one type of omics data will be distributed along their corresponding axes
-
Click a data point on the scatter plot; the corresponding pathway will be displayed on the right panel. Genes/proteins in the pathway are rectangles, while compounds/peaks are circles. The upregulated genes (positive logFC values) are in red, while the downregulated genes (negative logFC values) are in green. The potential compounds predicted on the basis of matched peaks are highlighted in dark blue. Clicking a compound node will show all its matched peaks as shown in Fig. 6b (right). All figures can be re-generated in higher resolutions for download by clicking the palette icon located at the top-right corner
-
(Optional) Explore the pathways from the KEGG network view as described in Step 38
-
Integration of multiple untargeted metabolomic datasets
-
43
Uploading and processing multiple global metabolomics datasets.
-
Go to MetaboAnalyst module selection page, and click ‘Functional Meta-analysis’ to enter the module for integrating multiple metabolomics datasets
-
Upload and process each dataset (A1_pos.csv, B1_pos.csv, C1_pos.csv) separately using the table provided
-
Click the button of each step to perform data visualization, normalization and identification of significant features through the corresponding dialogs. The meaning of all parameters here are consistent with those described in the ‘Functional Analysis’
-
For demonstration purpose, we will click the ‘Try Examples’ button and select the first COVID-19 example dataset
-
Then, click the ‘Proceed’ button to enter the parameter setting page for functional meta-analysis. Global metabolomics datasets can be integrated at pathway level by combining P values from individual pathway analysis results, or at feature level by pooling peaks
-
Since the datasets are from different studies, click the ‘Submit’ button in the ‘Pathway-level Integration’ box to continue
Critical step
Pooling peaks should be used only when the peaks are generated from identical or very similar instruments. A typical use case is to combine peaks generated from both positive and negative modes by the same LC–HRMS to increase the metabolome coverage.
-
-
44
Integration at pathway level. The integration results are shown as a bubble plot (Fig. 7a). Pathways from all datasets are organized on the basis of merged P values. The bubble sizes are correlated with their enrichment ratios. The intersections of significant pathways identified across different datasets can be explored through an interactive Upset plot, which can easily show the overlap of a large number of sets by arranging them as bar charts of their frequency. An example output is shown in Fig. 7b using a P-value cutoff of 0.35. Click on a bar to view the corresponding pathway names in the right panel.
Fig. 7: Graphical results of functional meta-analysis. 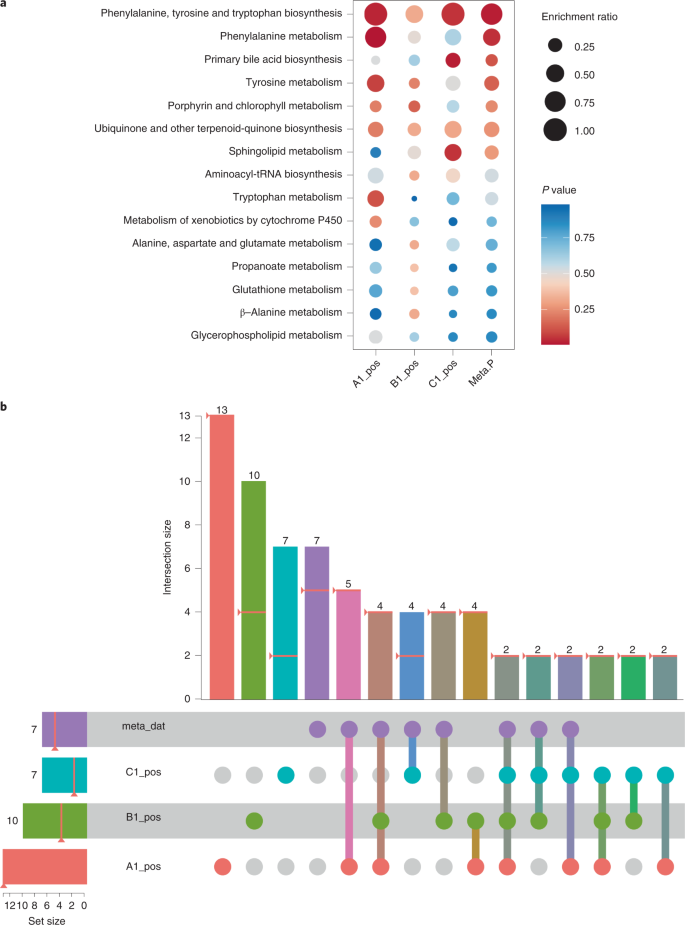
a,b, Results from the three global metabolomics datasets together with the meta-analysis results visualized as a bubble plot (a) and an Upset plot (b). The rightmost column (Meta.P) in the bubble plot (a) consists of merged P values of all corresponding rows of the left columns. The rows of the bubble plot are sorted on the basis of their merged P values. The Upset plot (b) in MetaboAnalyst is an interactive graph. The heights of the vertical bars at the top half of the image correspond to intersection size—the number of significant pathways that are shared among the corresponding datasets (as highlighted with different colors at the bottom half of the image). For example, the first vertical red bar shows that dataset ‘A1_pos’ contains 13 significant pathways, while the tenth vertical bar indicates that two significant pathways are shared across all four datasets. Users can directly click any bar to see the corresponding pathways displayed on the right panel of the page.
-
45
Result Downloading. Click the ‘Download’ node from the navigation tree to finalize the analysis and download the results.

Stage 4: analyzing metabolomics data with complex metadata
Timing 20–30 min
-
46
Starting up. Go to the MetaboAnalyst home page, and select the ‘Click here to start’ button to enter the Modules Overview page.
-
47
Locate the ‘Statistical Analysis [metadata table]’ module at the bottom row. Click the module name to enter the data upload page.
-
48
Data upload. Metabolomics data and its associated metadata should be uploaded as two separate files. This protocol uses LC–HRMC untargeted metabolomics data measured in blood plasma collected from both TCE-exposed and unexposed workers55. TCE exposure has been linked to multiple types of cancer. The metadata includes the TCE concentration (‘TCE_Exp_Conc’) and the corresponding exposure level (‘TCE_Exp_Category’), age, sex, alcohol and smoking status, body mass index (BMI) and analytical batch. Users can upload the files (TCE_feature_table.csv and TCE_metadata.csv). They are also available as the second example dataset. In this case, select this example and click ‘Submit’.
-
49
Missing value estimation.
-
On ‘Data Integrity Check’, click ‘Missing Values’ at the middle bottom
-
This dataset has lots of missing values, so uncheck the top box to avoid filtering out many of the metabolites
-
For ‘Step 2. Estimate the remaining missing values’, use ‘Replace by LoDs’ as default. Then click ‘Process’.
-
-
50
Metadata checking. The ‘Meta-data check’ page is essential for downstream statistical analysis. MetaboAnalyst estimates whether metadata are continuous or categorical based on the presence of strings and the number of replicates for each value. The metadata table shows that MetaboAnalyst has correctly classified all variables as categorical except for exposure concentration, age and BMI. The ‘Status’ column shows that all variables pass the integrity check.
Critical step
Carefully review the automatic metadata classifications to verify whether MetaboAnalyst has assigned the correct type. Proceeding with incorrect metadata types can greatly impact the downstream steps.
-
51
By default, the groups will be ordered alphabetically. This may not be ideal. For instance, ‘Low’, ‘Moderate’ and ‘High’ will be shown as ‘High’, ‘Low’ and ‘Moderate’ by default. To address this issue, we have added support to allow users to manually specify the order for categorical metadata. Click the ‘Edit’ link for ‘TCE_Exp_Category’ and, in the ‘Order (factor-level)’ tab, change the order to be ‘Low’, ‘Moderate’ and ‘High’. Click ‘Proceed’.
Critical step
It is highly recommended that ‘order’ be manually assigned to variables that have an inherent order (i.e., low, medium, high) using the ‘Edit’ link. This could lead to more accurate and interpretable results.
-
52
For data filtering, leave the default selection. Click ‘Submit’ and then ‘Proceed’.
-
53
On the normalization page, select ‘Normalization by median’, ‘Log transformation’ and ‘Auto scaling’ according to the practice of the original publication56. Click ‘Normalize’ and then ‘Proceed’.
Tip: Users are advised to first check if data are already normalized by visualization. For unnormalized data, start with simple methods (such as log transformation) and gradually apply more advanced ones if necessary. Although there are no hard rules in deciding the optimal combination, the data can often give some hints—including clustering patterns of QC samples, presence of batch effects, etc.
-
54
Metadata visualization. Click the ‘Metadata Visualization’ link to compute relationships between metadata variables and visualize them in a heatmap. From the heatmap automatically generated with default parameters (Fig. 8a), we can see that the two TCE exposure variables (‘TCE_Exp_Category’ and ‘TCE_Exp_Conc’) are clustered together and appear to have very similar distributions. This makes sense since one is a derivative of the other. ‘Sex’, ‘Smoking_Status’ and ‘Alcohol_Use’ are clustered together, as are ‘Age’ and ‘BMI’.
Fig. 8: Metadata correlations and associations with metabolomics data. 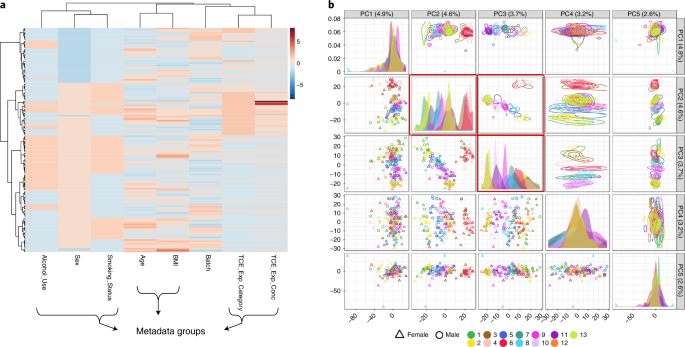
a, Heatmap showing similarities and patterns between different metadata. For continuous and ordered categorical variables, higher and lower values are colored red and blue, respectively. b, Pairwise plot of the top five principal components (PCs). Colors are based on ‘Batch’ variable (13 batches in total), and shapes in scatter plots are according to ‘Sex’. The diagonal panels describe the distribution patterns of current metadata groups (e.g., batches) in the specific PC. In this example, PC2 and PC3 show very distinctive patterns based on batches.
Critical step
Understanding the metadata structure is important for downstream analysis, as including multiple, correlated variables in a multivariate regression model can lead to parameter instability. This is something to be aware of before constructing the model and interpreting the results.
-
55
Principal components analysis (PCA). Next, we will use PCA to investigate general patterns between the metabolomics data and selected metadata of interest.
-
Click the ‘iPCA’ link in the left navigation panel, underneath the ‘Multi-factors’ heading. By default, the scores plot will be annotated with the first two metadata
-
Examine the separation of samples with respect to each metadata by repeatedly changing the ‘Color based on’ dropdown and clicking ‘Update’. Of all the metadata, the ‘Batch’ metadata correspond to clear and obvious sample separation along the PC2 and PC3 (Fig. 8b). The 3D PCA is also updated simultaneously in the ‘Synchronized 3D Plots’ tab
-
Rotate or zoom to discover patterns in the score plot (left)
-
Double click a feature variable in the loading plot (right) to display a summary of its distribution with regard to different metadata of interest
Tip: PCA can effectively summarize the overall patterns of variations. PCA combined with metadata highlighting can quickly help identify main patterns if present in the data. Heatmap is another effective visualization method that provides detailed feature-level variations. Heatmap with clustering can complement PCA to provide a more holistic data overview.
-
-
56
Differential analysis with covariate adjustment.
-
Click the ‘Linear Model’ node in the left navigation panel
-
Choose the primary metadata variable. In this case, we are most interested in metabolites associated with TCE exposure and choose the continuous concentration version (‘TCE_Exp_Conc’) rather than the derived categorical version (‘TCE_Exp_Category’)
Critical step
It is very important to leave the other TCE variable out of the model. As these two variables are highly correlated, including both will add little new information and will lead to unstable model coefficients that are difficult to interpret.
-
-
57
Next, we decide which covariates to adjust for by including them in the model. Ideally, if we had an extremely large sample size, we would add all additional covariates to the model. However, as the number of variables increases, the statistical power decreases.
-
In this example, add ‘Sex’, ‘Age’ and ‘Batch’ to the model while leaving out the variables that they are correlated with (‘Alcohol_Use’, ‘Smoking_Status’ and ‘BMI’). If we had a much lower sample size (i.e., n = 30) and therefore were choosing a single covariate to adjust for, we would pick ‘Batch’, since this variable had the most noticeable association with the metabolomics data in the PCA plot (see Box 4 for more details on covariate adjustment)
-
-
58
Keep the rest of the parameters as default, and click ‘Submit’. Note that there is no reference group because our primary variable is continuous.
-
59
Explore the covariate adjustment results. The pop-up message tells us that, after covariate adjustment, there are 333 metabolites with P < 0.05. As shown in Fig. 9, adjusting for the covariates improved the significance of the TCE coefficient for many features, ultimately increasing the number past the threshold from 216 to 333.
Fig. 9: Results of covariate adjustment. 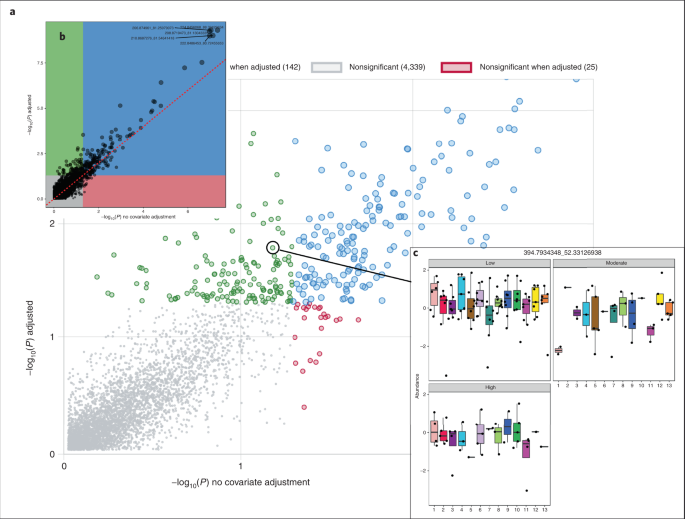
a, Screenshot of the interactive figure, zoomed in to the area of interest. b, High-resolution static image available for download. Panels a and b show P values for association of each metabolite with TCE concentration with covariate adjustment (y axis) and without (x axis). c, Example boxplots generated after clicking a point in the interactive plot. The boxplots show the abundance distributions of the current feature with respect to the covariate of interest (13 batches) within each category of the primary metadata (Low, Moderate and High).
-
60
Click on individual features to see their values across all samples, annotated by the metadata of choice. The PCA plot (Fig. 8b) showed that the main covariate associated with metabolite variation is Batch, which has 13 levels. With so many groups, it is difficult to visually identify how adjusting for the covariates changes the P values for individual features. This would be easier to visualize where a covariate like sex had more influence.
-
61
Click on the table icon at the top right of the plot to enter the ‘Feature Details View’ page. The first column (after the ‘Name’ column) shows the coefficient(s) in the linear model associated with the primary metadata. In the case of a continuous variable, the sign of the coefficient indicates positive or negative association between the primary metadata and that feature, and the magnitude indicates effect size. For categorial metadata, the sign shows whether that metabolite has a lower or higher level in the given metadata group, compared with the reference group. Click the ‘View’ button to visualize their distributions regarding different metadata.
-
62
Classification using Random Forest.
-
Select ‘Random Forest’ under the ‘Multi-factors’ heading on the left navigation panel. We will try to build a model to predict whether a worker is exposed or not using the Random Forest method
-
Select the ‘TCE_Exp_Category’ from the primary metadata dropdown, and click ‘Update’. The result shows that the algorithm can accurately predict the ‘Low’ category (94/95, error rate 0.01), followed by ‘High’ (error rate 0.29) and ‘Moderate’ (error rate 0.69). The error rates here might be slightly different owing to the random nature of the algorithm. The result is not surprising as ‘Low’ and ‘High’ are two extremes with clear phenotypes, while the ‘Moderate’ tends to be moderated by individual differences in metabolism
-
-
63
In exposomics studies, a common objective is to distinguish exposed from nonexposed. We would like to test the performance for such binary classification task. To do this:
-
Click the ‘Metadata check’ on the navigation tree to return to the corresponding page
-
Click the ‘Edit’ link for ‘TCE_Exp_Category’
-
On the dialog, select ‘Edit (factor-level)’ tab. Update the labels: ‘Not_exposed’ for ‘Low’, and ‘Exposed’ for both ‘High’ and ‘Moderate’. Click ‘Update’. A new metadata will be created (‘TCE_Exp_Category.1’) appearing at the bottom of the metadata table. This is not very meaningful. Directly click on this name to enter the editing mode. Update the name to ‘TCE_Exp_Binary’. Click anywhere outside the editing box to save the changes
-
Click ‘Skip to Analysis’ to the analysis page
-
Select ‘Random Forest’ to enter the previous page. In this case, choose the new metadata ‘TCE_Exp_Binary’ and click ‘Update’
-
-
64
View the results. The model is able to correctly classify most workers this time! Note that these are only the training results, and the model would need to be tested with additional data to properly validate it. Click the ‘Var. Importance’ tab to see the top features in the model. In addition, users can explore whether including some metadata such as age or sex could improve the prediction.
-
65
Result download. Click the ‘Download’ node from the navigation tree. On the download page, click ‘Generate Report’ to create a PDF report summarizing the analysis. Download the results and analysis report. Click ‘Exit’ to finalize the analysis.


Troubleshooting
Troubleshooting advice can be found in Table 3.
Timing
-
The duration required to execute all steps detailed in this protocol, especially the raw spectra processing, highly depends on the server load. The expected time for the steps is summarized below.
-
Steps 1–18, LC–HRMS raw spectra processing: ~2 h, depending on the sample size and server load
-
Steps 19–31, functional analysis of LC–HRMS peaks: ~20 min
-
Steps 32–45, pathway-level integration of multiple datasets: ~20 min
-
Steps 46–65, analyzing metabolomics data with complex metadata: ~20 min
Anticipated results
In this protocol, we show results for several example datasets used in the four main tasks covered in this protocol. The major outputs generated during the sequential analyses are shown in Figs. 3–9, Table 2 and Supplementary Figs. 1 and 2. Other results generated during the process are summarized separately as below.
LC-HRMS raw spectra processing
Raw spectra processing (Step 13) produces a peak intensity table (metaboanalyst_input.csv) and a peak annotation table (annotated_peaklist.csv). A total of 5,113 peaks have been found in dataset 1 with ‘Auto-optimized’ pipeline, and of these, 2,214 peaks (43.3% of the total) were annotated as isotopes or adducts. The peak intensity table is directly used in the downstream statistical and functional analysis to identify significant features and pathways. For researchers who are familiar with analytical chemistry, the annotation table can be used to narrow down the candidates and design targeted experiments for tandem MS. BPI, TIC and specific EIC are all generated and downloadable to show the chromatographical results.
Functional analysis of LC–HRMS peaks
On the basis of the mummichog algorithm, the top significantly perturbed pathways are related with fatty acid metabolism (Step 26), while the GSEA algorithm also reports some amino-acid-metabolism-related pathways (Step 27). The combined approach of GSEA and mummichog confirms two significant pathways (‘fatty acid oxidation’ and ‘methionine and cysteine metabolism’, Step 28). All intermediate normalized data, matching compound list and predicted pathways are available as different tables from the result download page.
Integration of multi-omics data and multiple metabolomics data
Joint pathway analysis of transcriptomics and targeted metabolomics data (the first example, Step 37) reports seven significant pathways (P < 0.05, y axis). Many of them are located in the top-right area (Fig. 6a), indicating that many underlying perturbed genes or metabolites are at important positions with high pathway impact values (x axis). For joint analysis of transcriptomics and untargeted metabolomics data (the second example, Step 42), 23 significant pathways are reported (merged P < 0.05). The gene/compounds matching results and mummichog prediction results are available from the result download page. Functional meta-analysis integrates three untargeted metabolomics data tables. Two amino-acid-metabolism-associated pathways are reported as significant (merged P < 0.05).
Analyzing metabolomics data with complex metadata
Covariate adjustment results in 333 significant features after adjusting for sex, age and batch (Step 59). The main figure showing the impact of covariate adjustment can be downloaded in high resolution by clicking the paint icon to the top right of the interactive scatterplot. Statistical results of the covariate adjustment are saved in the ‘covariate_result.csv’ file. The Random Forest method correctly classifies ~99% of nonexposed workers and ~88% of exposed workers (results will vary slightly each time owing to use of random numbers, Step 64). More detailed results are in the ‘randomforests_sigfeatures.csv’ file.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All example datasets used in the protocol are integrated as example datasets in their respective modules and are also available for download from the ‘Format’ page of MetaboAnalyst (https://www.metaboanalyst.ca/MetaboAnalyst/docs/Format.xhtml). There are no restrictions on their use.
Code availability
MetaboAnalyst is freely accessible as a web-based application. The underlying R code is freely available at GitHub as the MetaboAnalystR (https://github.com/xia-lab/MetaboAnalystR) and OptiLCMS (https://github.com/xia-lab/OptiLCMS) packages under the GNU General Public License version 2 or later.
References
Alseekh, S. et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat. Methods 18, 747–756 (2021).
Alseekh, S. & Fernie, A. R. Metabolomics 20 years on: what have we learned and what hurdles remain? Plant J. 94, 933–942 (2018).
Doerr, A. Global metabolomics. Nat. Methods 14, 32–32 (2017).
Cajka, T. & Fiehn, O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal. Chem. 88, 524–545 (2016).
Vermeulen, R., Schymanski, E. L., Barabasi, A. L. & Miller, G. W. The exposome and health: where chemistry meets biology. Science 367, 392–396 (2020).
Beger, R. D. et al. Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metabolomics 12, 149 (2016).
Want, E. J. et al. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 8, 17–32 (2013).
Zhou, G. et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 47, W234–W241 (2019).
Chong, J., Liu, P., Zhou, G. & Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821 (2020).
Pang, Z. et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396 (2021).
Xia, J., Psychogios, N., Young, N. & Wishart, D. S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 37, W652–W660 (2009).
Xia, J., Mandal, R., Sinelnikov, I. V., Broadhurst, D. & Wishart, D. S. MetaboAnalyst 2.0—a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 40, W127–W133 (2012).
Xia, J., Sinelnikov, I. V., Han, B. & Wishart, D. S. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 43, W251–W257 (2015).
Chong, J. et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486–W494 (2018).
Stanstrup, J. et al. The metaRbolomics Toolbox in Bioconductor and beyond. Metabolites https://doi.org/10.3390/metabo9100200 (2019).
Gardinassi, L. G., Xia, J., Safo, S. E. & Li, S. Bioinformatics tools for the interpretation of metabolomics data. Curr. Pharmacol. Rep. 3, 374–383 (2017).
Chang, H. Y. et al. A practical guide to metabolomics software development. Anal. Chem. 93, 1912–1923 (2021).
Tautenhahn, R., Patti, G. J., Rinehart, D. & Siuzdak, G. XCMS Online: a web-based platform to process untargeted metabolomic data. Anal. Chem. 84, 5035–5039 (2012).
Giacomoni, F. et al. Workflow4Metabolomics: a collaborative research infrastructure for computational metabolomics. Bioinformatics 31, 1493–1495 (2015).
Yang, Q. et al. NOREVA: enhanced normalization and evaluation of time-course and multi-class metabolomic data. Nucleic Acids Res. 48, W436–W448 (2020).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Du, X. X., Smirnov, A., Pluskal, T., Jia, W. & Sumner, S. Metabolomics data preprocessing using ADAP and MZmine 2. Computational Methods Data Anal. Metabolomics 2104, 25–48 (2020).
Tsugawa, H. et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 38, 1159–1163 (2020).
Tsugawa, H. et al. Hydrogen rearrangement rules: computational MS/MS fragmentation and structure elucidation using MS-FINDER software. Anal. Chem. 88, 7946–7958 (2016).
Rost, H. L. et al. OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 13, 741–748 (2016).
Danczak, R. E. et al. Using metacommunity ecology to understand environmental metabolomes. Nat. Commun. 11, 6369 (2020).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Xia, J. & Wishart, D. S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 6, 743–760 (2011).
Xia, J. & Wishart, D. S. Metabolomic data processing, analysis, and interpretation using MetaboAnalyst. Curr. Protoc. Bioinformatics https://doi.org/10.1002/0471250953.bi1410s34 (2011).
Xia, J. & Wishart, D. S. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinforma. 55, 14 10 11–14 10 91 (2016).
Xia, J., Broadhurst, D. I., Wilson, M. & Wishart, D. S. Translational biomarker discovery in clinical metabolomics: an introductory tutorial. Metabolomics 9, 280–299 (2013).
Chong, J., Wishart, D. S. & Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinforma. 68, e86 (2019).
Chong, J. & Xia, J. Using MetaboAnalyst 4.0 for metabolomics data analysis, interpretation, and integration with other omics data. Methods Mol. Biol. 2104, 337–360 (2020).
Tautenhahn, R., Bottcher, C. & Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinforma. 9, 504 (2008).
Yu, T., Park, Y., Johnson, J. M. & Jones, D. P. apLCMS—adaptive processing of high-resolution LC/MS data. Bioinformatics 25, 1930–1936 (2009).
Kenar, E. et al. Automated label-free quantification of metabolites from liquid chromatography-mass spectrometry data. Mol. Cell Proteom. 13, 348–359 (2014).
Pang, Z., Chong, J., Li, S. & Xia, J. MetaboAnalystR 3.0: toward an optimized workflow for global metabolomics. Metabolites https://doi.org/10.3390/metabo10050186 (2020).
Chaleckis, R., Meister, I., Zhang, P. & Wheelock, C. E. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr. Opin. Biotechnol. 55, 44–50 (2019).
Sindelar, M. & Patti, G. J. Chemical discovery in the era of metabolomics. J. Am. Chem. Soc. 142, 9097–9105 (2020).
Kuhl, C., Tautenhahn, R., Bottcher, C., Larson, T. R. & Neumann, S. CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 84, 283–289 (2012).
Senan, O. et al. CliqueMS: a computational tool for annotating in-source metabolite ions from LC-MS untargeted metabolomics data based on a coelution similarity network. Bioinformatics 35, 4089–4097 (2019).
Kind, T. & Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics https://doi.org/10.1186/1471-2105-8-105 (2007).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Xia, J. & Wishart, D. S. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 38, W71–W77 (2010).
Tarca, A. L. et al. A novel signaling pathway impact analysis. Bioinformatics 25, 75–82 (2009).
Xia, J. & Wishart, D. S. MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 26, 2342–2344 (2010).
Li, S. et al. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 9, e1003123 (2013).
Xia, J. et al. INMEX—a web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 41, W63–W70 (2013).
Zhou, G. & Xia, J. OmicsNet: a web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 46, W514–W522 (2018).
Schriml, L. M. et al. COVID-19 pandemic reveals the peril of ignoring metadata standards. Sci. Data 7, 188 (2020).
Kahan, B. C., Jairath, V., Dore, C. J. & Morris, T. P. The risks and rewards of covariate adjustment in randomized trials: an assessment of 12 outcomes from 8 studies. Trials 15, 139 (2014).
Chong, J. & Xia, J. MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 34, 4313–4314 (2018).
Chong, J., Yamamoto, M. & Xia, J. MetaboAnalystR 2.0: from raw spectra to biological insights. Metabolites https://doi.org/10.3390/metabo9030057 (2019).
Gardinassi, L. G. et al. Integrative metabolomics and transcriptomics signatures of clinical tolerance to Plasmodium vivax reveal activation of innate cell immunity and T cell signaling. Redox Biol. 17, 158–170 (2018).
Pang, Z., Zhou, G., Chong, J. & Xia, J. Comprehensive meta-analysis of COVID-19 global metabolomics datasets. Metabolites https://doi.org/10.3390/metabo11010044 (2021).
Walker, D. I. et al. High-resolution metabolomics of occupational exposure to trichloroethylene. Int. J. Epidemiol. 45, 1517–1527 (2016).
Gatto, L., Gibb, S. & Rainer, J. MSnbase, efficient and elegant R-based processing and visualization of raw mass spectrometry data. J. Proteome Res. 20, 1063–1069 (2021).
Conley, C. J. et al. Massifquant: open-source Kalman filter-based XC-MS isotope trace feature detection. Bioinformatics 30, 2636–2643 (2014).
Wishart, D. S. et al. HMDB 5.0: the Human Metabolome Database for 2022. Nucleic Acids Res. 50, D622–D631 (2022).
Vaughan, A. M. et al. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell Microbiol. 11, 506–520 (2009).
Cumnock, K. et al. Host energy source is important for disease tolerance to malaria. Curr. Biol. 28, 1635–1642 e1633 (2018).
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R. & Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 (2006).
Prince, J. T. & Marcotte, E. M. Chromatographic alignment of ESI–LC–MS proteomics data sets by ordered bijective interpolated warping. Anal. Chem. 78, 6140–6152 (2006).
Libiseller, G. et al. IPO: a tool for automated optimization of XCMS parameters. BMC Bioinforma. 16, 118 (2015).
McLean, C. & Kujawinski, E. B. AutoTuner: high fidelity and robust parameter selection for metabolomics data processing. Anal. Chem. 92, 5724–5732 (2020).
Zhou, G., Ewald, J. & Xia, J. OmicsAnalyst: a comprehensive web-based platform for visual analytics of multi-omics data. Nucleic Acids Res. 49, W476–W482 (2021).
Rappoport, N. & Shamir, R. Multi-omic and multi-view clustering algorithms: review and cancer benchmark. Nucleic Acids Res. 47, 1044 (2019).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Acknowledgements
We thank Genome Canada, Génome Québec, US National Institutes of Health (U01 CA235493), Natural Sciences and Engineering Research Council of Canada (NSERC) and Canada Research Chairs (CRC) Program for funding support.
Author information
Authors and Affiliations
Contributions
Z.P., J.E., N.B. and J.X. prepared the manuscript. Z.P., G.Z., J.E., L.C., O.H. and J.X. contributed to the development and testing of the MetaboAnalyst. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks Julia Kuligowski, Zhenzhen Xu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations
Related links
Key references using this protocol
Pang, Z. et al. Metabolites 10, 186 (2020): https://doi.org/10.3390/metabo10050186
Pang, Z. et al. Metabolites 11, 44 (2021): https://doi.org/10.3390/metabo11010044
Pang, Z. et al. Nucleic Acids Res. 49, W388–W396 (2021): https://doi.org/10.1093/nar/gkab382
Key data used in this protocol
Gardinassi, L. G. et al. Redox Biol. 17, 158–170 (2018): https://doi.org/10.1016/j.redox.2018.04.011
Walker, D. I. et al. Int. J. Epidemiol. 45, 1517–1527 (2016): https://doi.org/10.1093/ije/dyw218
This protocol is an extension to: Nat. Protoc. 6, 743–760 (2011): https://doi.org/10.1038/nprot.2011.319.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2 and Supplementary Table 1.
Rights and permissions
About this article

Cite this article
Pang, Z., Zhou, G., Ewald, J. et al. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc 17, 1735–1761 (2022). https://doi.org/10.1038/s41596-022-00710-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-022-00710-w
This article is cited by
-
Interactions between Balantidium ctenopharyngodoni and microbiota reveal its low pathogenicity in the hindgut of grass carp
BMC Microbiology (2024)
-
Acute venous thromboembolism plasma and red blood cell metabolomic profiling reveals potential new early diagnostic biomarkers: observational clinical study
Journal of Translational Medicine (2024)
-
Lipid alterations play a role in the integration of PD-1/PD-L1 inhibitors and anlotinib for the treatment of advanced non–small-cell lung cancer
Lipids in Health and Disease (2024)
-
The causal relationship of human blood metabolites with the components of Sarcopenia: a two-sample Mendelian randomization analysis
BMC Geriatrics (2024)
-
Untargeted metabolomic profiling reveals molecular signatures associated with type 2 diabetes in Nigerians
Genome Medicine (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
