
Abstract
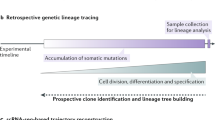
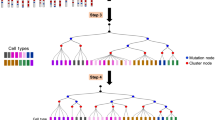
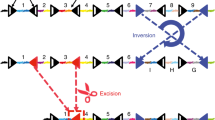
Classic approaches to mapping the developmental history of cells in vivo have relied on techniques that require complex interventions and often capture only a single trajectory or moment in time. We have previously described a developmental barcoding system to address these issues using synthetically induced mutations to record information about each cell’s lineage in its genome. This system uses MARC1 mouse lines, which have multiple homing guide RNAs that each generate hundreds of mutant alleles and combine to produce an exponential diversity of barcodes. Here, we detail two MARC1 lines that are available from a public repository. We describe strategies for using MARC1 mice and experimental design considerations. We provide a protocol for barcode retrieval and sequencing as well as the analysis of the sequencing data. This protocol generates barcodes based on synthetically induced mutations in mice to enable lineage analysis.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
Data availability
All sequencing data are available in the Sequence Read Archive under accession SRP155997; the parts of this dataset that correspond to the PB7 line were previously published7. Figs. 3–5 contain associated raw data for both PB3 and PB7 lines which are available in Supplementary Tables 1–4 and SRP155997.
Code availability
All code is available from our GitHub repository: https://github.com/Kalhor-Lab/MARC1-Pipeline. Code in this protocol has been peer reviewed.
References
Baron, C. S. & van Oudenaarden, A. Unravelling cellular relationships during development and regeneration using genetic lineage tracing. Nat. Rev. Mol. Cell Biol. 20, 753–765 (2019).
Wagner, D. E. & Klein, A. M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. https://doi.org/10.1038/s41576-020-0223-2 (2020).
Sulston, J. E., Schierenberg, E., White, J. G. & Thomson, J. N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119 (1983).
Frumkin, D., Wasserstrom, A., Kaplan, S., Feige, U. & Shapiro, E. Genomic variability within an organism exposes its cell lineage tree. PLoS Comput. Biol. 1, e50 (2005).
McKenna, A. et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016).
Alemany, A., Florescu, M., Baron, C. S., Peterson-Maduro, J. & van Oudenaarden, A. Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112 (2018).
Kalhor, R. et al. Developmental barcoding of whole mouse via homing CRISPR. Science 361, eaat9804 (2018).
Spanjaard, B. et al. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat. Biotechnol. 36, 469–473 (2018).
Frieda, K. L. et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111 (2017).
Chan, M. M. et al. Molecular recording of mammalian embryogenesis. Nature 570, 77–82 (2019).
Askary, A. et al. In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription. Nat. Biotechnol. 38, 66–75 (2020); correction 38, 245 (2020).
Bowling, S. et al. An engineered CRISPR-Cas9 mouse line for simultaneous readout of lineage histories and gene expression profiles in single cells. Cell https://doi.org/10.1016/j.cell.2020.04.048 (2020).
Kalhor, K. & Church, G. M. Single-cell CRISPR-based lineage tracing in mice. Biochemistry 58, 4775–4776 (2019).
Behjati, S. et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 513, 422–425 (2014).
Kalhor, R., Mali, P. & Church, G. M. Rapidly evolving homing CRISPR barcodes. Nat. Methods 14, 195–200 (2017).
Perli, S. D., Cui, C. H. & Lu, T. K. Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science 353, (2016).
Lieber, M. R. & Wilson, T. E. SnapShot: nonhomologous DNA end joining (NHEJ). Cell 142, 496–496.e1 (2010).
Allen, F. et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. https://doi.org/10.1038/nbt.4317 (2018).
Chen, W. et al. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 47, 7989–8003 (2019).
Shou, J., Li, J., Liu, Y. & Wu, Q. Precise and predictable CRISPR chromosomal rearrangements reveal principles of Cas9-mediated nucleotide insertion. Mol. Cell 71, 498–509.e4 (2018).
Guo, T. et al. Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 19, 170 (2018).
Shen, M. W. et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651 (2018).
Zuo, Z. & Liu, J. Cas9-catalyzed DNA cleavage generates staggered ends: evidence from molecular dynamics simulations. Sci. Rep. 5, 37584 (2016).
Lemos, B. R. et al. CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles. Proc. Natl Acad. Sci. USA 115, E2040–E2047 (2018).
Taheri-Ghahfarokhi, A. et al. Decoding non-random mutational signatures at Cas9 targeted sites. Nucleic Acids Res 46, 8417–8434 (2018).
Platt, R. J. et al. CRISPR–Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014).
Raj, B. et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 36, 442–450 (2018).
Lee, M. T., Bonneau, A. R. & Giraldez, A. J. Zygotic genome activation during the maternal-to-zygotic transition. Ann. Rev. Cell Dev. Biol. 30, 581–613 (2014).
Wilberg, E. W. What’s in an outgroup? The impact of outgroup choice on the phylogenetic position of Thalattosuchia (Crocodylomorpha) and the origin of Crocodyliformes. Syst. Biol. 64, 621–637 (2015).
Feng, J. et al. Estimation of cell lineage trees by maximum-likelihood phylogenetics. Preprint at BioRxiv https://doi.org/10.1101/595215 (2019).
Jones, M. G. et al. Inference of single-cell phylogenies from lineage tracing data using Cassiopeia. Genome Biol. 21, 92 (2020).
Beltman, J. B. et al. Reproducibility of Illumina platform deep sequencing errors allows accurate determination of DNA barcodes in cells. BMC Bioinformatics 17, 1–16 (2016).
Acknowledgements
The authors acknowledge J. Dapello for comments on the manuscript and L. Mejia for assistance in preparing sequencing libraries. The authors also acknowledge the team at MMRRC and UC-Davis Mouse Biology Program for making it possible to distribute MARC1 lines. This work was supported by Johns Hopkins Institutional Funds, grants from the Simons Foundation (SFARI 606178, R.K.), the NIH (MH103910, G.M.C. and R01GM123313, P.M.) and the Intelligence Advanced Research Projects Activity (IARPA) via the Department of Interior/Interior Business Center (DoI/IBC) contract number D16PC00008.
Author information
Authors and Affiliations
Contributions
R.K., P.M. and G.M.C. conceived the study; R.K. designed experiments; K.L. and R.K. carried out the experiments; K.L., K.K., and R.K. analyzed the data; A.G. supervised animal husbandry; A.V. assisted in animal husbandry and sample collection. R.K., K.L., and K.K. wrote the manuscript and prepared the analysis pipeline; and R.K. supervised the project.
Corresponding author
Ethics declarations
Competing interests
G.M.C.’s competing financial interests can be found at v.ht/PHNc. The remaining authors declare no competing financial interest.
Additional information
Peer review information Nature Protocols thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key reference using this protocol
Kalhor, R. et al. Science 361, eaat9804 (2018): https://doi.org/10.1126/science.aat9804
Supplementary information
Supplementary Information
Supplementary Figs. 1–4, Supplementary Table 2 and legends for Supplementary Tables 1 and 3–8.
Supplementary Table 1
List of all hgRNAs in the MARC1 founder males. For each hgRNA, its assigned number, ID sequence, spacer sequence, TSS to PAM length, spacer length, observed inheritance probability, chromosomal location, and confidence level of the chromosomal location are given. A “+” sign in the location column indicates that the hgRNA is transcribed in the same direction as the positive strand of the mm10 reference sequence.
Supplementary Table 3
Average observed mutation level of each hgRNA during different stages of development. Values indicate percent mutated while “NA” indicates values that were not measured. Functional category based on this mutation level is also shown. Barcodes with <2% maximum mutation rate in all samples were assigned as “inactive”, those with 2–50% maximum mutation rate in all samples were assigned as “slow”, those with >90% average mutation frequency at any point before E9 were assigned as “fast”, and the rest were assigned as “mid.”
Supplementary Table 4
For each mutant spacer allele of each hgRNA, its sequence, occurrence probability (fraction of mice with that hgRNA which contained the allele), occurrence (number of mice out of total with that hgRNA which contained the allele), and median abundance (median of its abundance fraction in all mice in which it was observed) are shown.
Supplementary Table 5
Example genotyping output of the analysis pipeline, corresponding to a mutated sample. A [sampleName]_genotypes.txt file is produced for each sample. It provides each identifier (# and ID) and the most commonly observed associated spacer sequence (SP), forming an identifier–spacer pair; the absolute count of observations of this pair (count); the percent of this pair relative to all observed pairs in a sample; and the percent of this pair relative to all pairs with that identifier.
Supplementary Table 6
Example filtered pairs output of the analysis pipeline on a mutated sample. A [sample]_filteredpairs.txt file, produced for each sample, contains information on mutation levels and all observed pairs. This table contains the observed identifier with a corresponding observed spacer, the total observation count for that pair, the percent of that pair within all observations of that identifier, and the percent of that pair within all sample observations.
Supplementary Table 7
Suggested primer sets for specific amplification of each hgRNA in non-barcoded mice. Each pair is expected to produce an amplicon of 141–155 bp in length. Using each primer pair in a standard PCR on genomic DNA of MARC1 mice can establish the presence of the targeted hgRNA if an amplicon of the correct size appears. Absence of the correct amplicon in the PCR product would indicate absence of the hgRNA. These primer sets are not experimentally validated and might fail to amplify mutated hgRNA sequences, as they depend on the spacer region to bind.
Supplementary Table 8
Table of observed mutant allele frequencies in each sample in Fig. 5. Rows correspond to samples and columns to each of the mutant spacer alleles that were observed. Alleles with <0.25% abundance in each sample are not included. parSP indicates spacer sequence observed in founder, mutSP indicates mutant spacer sequences observed in samples under analysis.
Supplementary Software
Data analysis pipeline.
Rights and permissions
About this article

Cite this article
Leeper, K., Kalhor, K., Vernet, A. et al. Lineage barcoding in mice with homing CRISPR. Nat Protoc 16, 2088–2108 (2021). https://doi.org/10.1038/s41596-020-00485-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-020-00485-y
This article is cited by
-
Genetic mosaicism in the human brain: from lineage tracing to neuropsychiatric disorders
Nature Reviews Neuroscience (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
