
Abstract
The CST–Polα/primase complex is essential for telomere maintenance and functions to counteract resection at double-strand breaks. We report a 4.6-Å resolution cryo-EM structure of human CST–Polα/primase, captured prior to catalysis in a recruitment state stabilized by chemical cross-linking. Our structure reveals an evolutionarily conserved interaction between the C-terminal domain of the catalytic POLA1 subunit and an N-terminal expansion in metazoan CTC1. Cross-linking mass spectrometry and negative-stain EM analysis provide insight into CST binding by the flexible POLA1 N-terminus. Finally, Coats plus syndrome disease mutations previously characterized to disrupt formation of the CST–Polα/primase complex map to protein–protein interfaces observed in the recruitment state. Together, our results shed light on the architecture and stoichiometry of the metazoan fill-in machinery.
Similar content being viewed by others


Main
Human telomeric DNA terminates in a 3′ overhang of the G-rich strand, which is required for t-loop formation and telomere protection1. The mature 3′ overhang must be regenerated during each cell cycle in a controlled manner. Following replication, nucleolytic resection of the 5′ strand can result in excessively long overhangs. The loss of sequences from the 5′ strand is counteracted through fill-in DNA synthesis by the CTC1–STN1–TEN1 (CST) complex and DNA polymerase α–primase (Polα/primase) (Fig. 1a), which are recruited to telomeres by the shelterin complex2,3,4,5,6,7. Dysfunctional fill-in, primarily driven by mutations in CST, causes the severe developmental disorder Coats plus syndrome (CP)8,9.

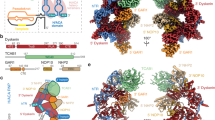
a, Domain schematics for proteins used in this study. Gray bars indicate regions modeled in c. NTD, N-terminal domain; CTD, C-terminal domain; OB, oligonucleotide/oligosaccharide binding fold; PDE, phosphodiesterase domain; wH, winged helix-turn-helix motif. b, Orthogonal views of the cryo-EM map of CST–PPΔN, segmented and colored by subunit as in a. c, Model of CST–PPΔN shown in cartoon representation.
In addition to its telomeric function, CST–Polα/primase performs an analogous fill-in reaction at resected double-strand breaks (DSBs). At DSBs, CST–Polα/primase is recruited by the 53BP1–RIF1–shieldin complex through a direct interaction between CST and shieldin. The fill-in reaction executed by CST–Polα/primase at DSBs is a main determinant of the lethality of PARP inhibitors in BRCA1-deficient cells10,11,12. Although a structure of decameric CST bound to a short oligonucleotide has been determined13, the molecular details of how CST interacts with Polα/primase are largely unknown14,15. Here, we present a combination of structural, biochemical, and biophysical data describing the molecular basis of their interaction. Our structure of the complex in a recruitment state reveals a novel interface between Polα/primase and CST that evolved in metazoans and informs on CP mutations found in the N-terminal oligonucleotide/oligosaccharide-binding (OB) folds of CTC1.
Results
Architecture of CST–Polα/primase in a pre-catalysis state
We sought to structurally characterize the complex using recombinant human CST and Polα/primase purified from insect cells. Although we could reconstitute a stable complex of CST–Polα/primase using size-exclusion chromatography (SEC), negative-stain EM images showed dissociation of CST and Polα/primase (Extended Data Fig. 1a,b). We added a (GGTTAG)3 substrate, as telomeric single-stranded DNA (ssDNA) has previously been shown to stabilize CST13, and used GraFix16 to cross-link the complex, resulting in a higher proportion of intact complexes in cryo-EM two-dimensional (2D) averages from a small dataset (no. 1; Extended Data Fig. 1c,d and Table 1). We collected an additional dataset from the same grid (no. 2) and could unambiguously identify CST and Polα/primase in the three-dimensional (3D) reconstructions. Although the mode of interaction between the two complexes appeared to be conserved in all maps, substantial conformational heterogeneity in peripheral regions limited the resolution to >16 Å (Extended Data Fig. 2 and Table 1).
The POLA1 subunit of Polα/primase contains a disordered N-terminal region (POLA1N, 1–335 aa) that is dispensable for catalysis and omitted in most structures of the enzyme17,18 (Fig. 1a). We purified Polα/primase lacking POLA1N (referred to hereafter as PPΔN), reconstituted a CST–PPΔN(–ssDNA) complex (Extended Data Fig. 3a) as for full-length Polα/primase (referred to hereafter as PPFL), and collected cryo-EM data (Extended Data Fig. 3b). The omission of POLA1N resulted in lower CST occupancy, so we introduced additional classification steps to select for particles containing intact CST (Extended Data Fig. 3c). We used 131,850 particles to generate the final map with a global resolution of 4.6 Å (Fig. 1b and Extended Data Fig. 3c,d). Local resolution estimates revealed lower resolution for the peripheral regions of CST and PPΔN (Extended Data Fig. 3e), likely due to flexibility, as suggested by the blurred-out regions in the 2D class averages (Extended Data Fig. 3d).
The crystal structure of apo PPΔN (PDB ID: 5EXR)17 and the cryo-EM structure of an ssDNA-bound CST monomer extracted from the decamer structure (PDB ID: 6W6W)13 could readily be docked into our density map. We then substituted the CTC1 structure with a model from the AlphaFold 2 database19,20, which provides information about the CTC1 N-terminus that was poorly resolved in the published CST cryo-EM map13. The ssDNA was included in the complex, as evidenced by the native gel of the GraFix fractions (Extended Data Fig. 3a), but no reliable density could be found for it owing to the low resolution of CST in the cryo-EM map. However, we observe low occupancy of the STN1 C-terminal half (STN1C, 184–398 aa) in that region (Fig. 1c) and, because ssDNA competes with STN1C for that binding site13, we retained the 4 nt of ssDNA from the previously determined CST structure13 in the model we docked into our map (Fig. 1c).
After initial rigid-body docking followed by flexible fitting and refinement, the overall conformations of the two subcomplexes did not show major changes from their structures in isolation (Fig. 1c and Extended Data Fig. 3f). In the complex, Polα/primase remains in the occluded state17, in which the POLA2 subunit is blocking entry of DNA into the active site of POLA1 (Fig. 1c). This finding is consistent with reported results showing that cross-linking preferentially stabilizes the more compact, occluded state compared with the flexible, extended state of the enzyme21,22. Thus, we conclude that our structure likely captures a recruitment state of the complex that forms prior to active RNA and DNA synthesis by Polα/primase.
Structural and evolutionary analysis of the CTC1–POLA1 interface
The primary interaction interface observed in our structure occurs between the C-terminal domain of POLA1 (POLA1CTD, 1,265–1,462 aa) and the N-terminal OB folds of CTC1 (Figs. 1c and 2a). The resolution is limiting for rigorous analysis of amino acid interactions, and analysis of surface electrostatic potential suggests that this interface is not driven by a dominant hydrophobic or charged interaction, but rather by shape complementarity of the two proteins, burying 1,250 Å2 of solvent-accessible surface area (Fig. 2a and Extended Data Fig. 4a,b).

a, Zoomed-in view of the interaction of POLA1CTD (cartoon) with CTC1 (cartoon and surface) superposed with POLA1CTD from the apo Polα/primase structure (PDB ID: 5EXR)17. The CTC1-recognition loop (CRL, 1400–1424 aa) is highlighted in cyan in the cartoon representation. The corresponding cleft in CTC1 is indicated with a cyan arc. b, Comparison of POLA1CTD models generated with AlphaFold 2 (refs. 19,20) from several species alongside domain comparisons of corresponding CTC1 orthologs. The metazoan CTC1 N-terminal expansion is indicated, and the binding pocket is represented by a cyan arc as in a or a dashed fuchsia arc in the non-metazoan species. c, Microscale thermophoresis measuring binding of POLA1CTD proteins to RED-tris-NTA-labeled His6-MBP-CTC1–STN1–TEN1. Error bars represent s.e.m. for each data point, calculated from three independent thermophoresis measurements (Extended Data Fig. 6e). d,e, Zoom-in of binding events 1 and 2 from c, respectively. KD values were calculated (N.D., not determined) with the MO Affinity Analysis (Nanotemper) software (split into two events for the CRLWT and generated with all data for the CRLGGSGGS and CRLS. pombe mutants). Fnorm, normalized fluorescence.
We identified a CTC1-recognition loop (CRL, 1400–1424 aa) in the POLA1CTD that is shifted relative to its position in the apo structure17 to contact CTC1 (Fig. 2a). Sequence conservation analysis of the interface revealed that both CTC1 and the interacting region on the POLA1CTD have low conservation at the primary sequence level (Extended Data Figs. 4c and 5a). However, the CRL is identifiable by an insertion of uniform length in metazoans, and we find that, when modeled using predictions from AlphaFold 2 (refs. 19,20), the CRL is structurally conserved in metazoans. Furthermore, we find that the presence of a CRL feature correlates with an expansion of metazoan CTC1 to contain the N-terminal OB folds that interact with the CRL. In unicellular eukaryotes, this loop diverges greatly between species and can be either shorter (for example, in Tetrahymena thermophila and Schizosaccharomyces pombe) or longer (for example, in Saccharomyces cerevisiae) and adopts a different predicted structure compared with that in the metazoan CRL (Fig. 2b and Extended Data Fig. 5b).
To assess the involvement of the CRL in CST binding, we generated human POLA1CTD constructs with the wild-type CRL (CRLWT, 1265–1462 aa), with the CRL swapped for a GGSGGS-linker (CRLGGSGGS, 1265–1402–GGSGGS–1423–1462 aa), or with the CRL swapped for the S. pombe short loop (CRLS. pombe, 1265–1399–QTTTGAT–1425–1462 aa) (Extended Data Fig. 6a–d). Although the POLA1CTD constructs ran as heterogeneously sized smears in SDS–PAGE, they compressed to single bands in native PAGE (Extended Data Fig. 6a,b). Sharp symmetric peaks in SEC elution profiles (Extended Data Fig. 6c) and spectrophotometric (Nanodrop) quantification indicated that the protein was pure and free from nucleic acid contamination, respectively. We measured the affinity of the interaction between the POLA1CTD constructs and fluorescently labeled CST using microscale thermophoresis (MST) (Fig. 2c). Two distinct binding events between the CRLWT protein and CST were observed: the first binding event (1; Fig. 2d) is higher affinity and characterized by a positive change in the normalized fluorescence (Fig. 2d), and the second binding event (2) is lower affinity and characterized by a negative change in the normalized fluorescence (Fig. 2e). We separated the data on the basis of the two inflection points23 and calculated dissociation constant (KD) values of ~3.3 μM and ~122 μM for binding events 1 and 2, respectively. In contrast, the CRLGGSGGS and CRLS. pombe mutant proteins displayed binding in only the second event (Fig. 2c,d). For binding event 2, the determined KD values for the CRLGGSGGS and CRLS. pombe proteins were ~96 μM and ~88 μM, respectively.
The CRL and one Zn2+-binding domain in POLA1CTD form the module that fits into a complementary cleft in CTC1. We speculate that the high-affinity binding mode (event 1; Fig. 2d) observed in the MST experiments involves both the CRL and Zn2+-binding domain, whereas the lower affinity binding mode (event 2; Fig. 2e) reflects the interaction of the Zn2+-binding domain with CTC1, which is CRL-independent. In this second binding mode, CST could rock about the Zn2+-binding domain, echoing the hinge-like flexible motion of CST about POLA1 suggested by the cryo-EM data (Extended Data Fig. 3d,e). CTC1OB-D, an elongated OB fold that shares no homology with known OB folds13, forms the major interaction with POLA1CTD (Fig. 2a and Extended Data Fig. 4). This finding is particularly interesting given the proposal that CST and Polα co-evolved in eukaryotes24. Our structure would then capture a metazoan-specific development in this trajectory.
Our structural model also places regions of CTC1 near POLA2 and PRIM2 (Fig. 1c). These two potential interaction sites are not well resolved in our cryo-EM map, but it is possible that these interactions are weak and/or more transient as CST flexes about the hinge generated by the POLA1CTD-CTC1 interaction.
CST–Polα/primase maintains a 1:1 stoichiometry
The CST–Polα/primase complex is sterically incompatible with the previously reported ssDNA-bound CST decamer13, as it would bind in the center of the ring and sterically clash with neighboring CST subunits (Fig. 3a). CST has also been shown to dimerize upon ssDNA binding13, but two additional lines of evidence suggest that active CST–Polα/primase has a 1:1 stoichiometry. First, we do not observe CST dimers in our 2D class averages (Extended Data Figs. 1 and 2). Second, we characterized the CST-POLA1N interaction to understand why CST–PPFL did not yield a high-resolution map although PPFL forms a more stable interaction with CST. We reconstituted a native complex of CST and MBP-tagged POLA1N and analyzed it by negative-stain EM and cross-linking mass spectrometry (CX-MS) (Extended Data Fig. 7). With MBP as a mass label, we localized the N-terminus of POLA1N to the primary CST dimerization interface (Fig. 3b). CX-MS analysis suggests that POLA1N binds in multiple modes to CST, which could partially explain the heterogeneity observed with CST–PPFL (Fig. 3c,d). It is possible that POLA1N binding is restrained in the presence of the full complex, but we observe similar cross-links between CTC1 and POLA1N in CX-MS analysis of CST–PPFL (Extended Data Fig. 7c). Thus, we conclude that POLA1N binds heterogeneously to CST in the region of the dimer interface, and the CST–Polα/primase fill-in machinery functions as a 1:1 complex.

a, CST–PPΔN structure (cartoon representation; colors as in Fig. 1) docked into the cryo-EM map13 of the CST decamer (transparent surface). b, Averages and cartoon representations of the six most populated RELION-3 2D classes of the MBP-POLA1N–CST complex. The MBP mass label is indicated with a white arrowhead when present. c, Cross-links between POLA1N and CST subunits identified by CX-MS. Blue lines, cross-links between CST and POLA1N. Dashed lines, inter-subunit cross-links in CST. d, Cartoon representation of the CST monomer in the same view as in b. Lysine residues cross-linked to POLA1N (c) are shown as blue spheres, and CP mutations previously characterized to disrupt Polα/primase binding14 are shown as red spheres. The white arrowhead indicates relative localization of the MBP mass label.
Coats plus mutations map to recruitment interfaces
Three CP point mutations (p.A227V, p.V259M, and p.V665G) in CTC1 were previously described to disrupt Polα/primase association with CST14,25 (Fig. 4a). We mapped these residues onto our structure to investigate the molecular basis of dysfunction caused by these mutations (Fig. 4b). V665 resides on a β-strand of CTC1OB-D, so it is plausible that a glycine substitution would destabilize the β-sheet and OB fold, disrupting the primary interaction. The mutations at A227 and V259 reside on CTC1OB-B, which does not contact Polα/primase in the CST–PPΔN structure (Fig. 4b). However, one major difference we observe between the cryo-EM maps of CST–PPΔN and CST–PPFL is the presence of connecting density between POLA2 and the CTC1 N-terminus, which we only observe when POLA1N is present (Fig. 4c). Because cooperative binding between POLA1N and POLA2NTD (1–78 aa, attached by a flexible linker and not visualized in our CST–PPΔN structure) in other settings has been described26, we speculate that this bridging density is a combination of these two termini. Although our resolution is limited, this connection could potentially explain the CP mutations occurring at the CTC1 N-terminus (Fig. 4a–c).

a, Summary of CP point mutations in CTC1. b, Mapping of mutations affecting Polα/primase binding on the CST–PPΔN structure. Colors as in a. Box, zoomed-in view of the primary CTC1OB-D-POLA1CTD interaction. c, Comparison of the CST–PPΔN and CST–PPFL (Extended Data Fig. 2a) cryo-EM density maps, segmented and colored according to docked CST and PPΔN models. d, Proposed model for the handoff of Polα/primase to shelterin-bound CST following telomere replication.
Discussion
In this study, we report a novel structure of the human CST–Polα/primase fill-in machinery. By chemically cross-linking the complex, we captured a conformational state that reveals how CST can recognize and bind the occluded state of Polα/primase through a newly uncovered interaction between CTC1 and POLA1CTD. We propose that this interaction is specific to metazoans and occurs during recruitment of Polα/primase to the telomere, prior to the start of RNA/DNA synthesis by the enzyme. Notably, this interface is formed by the N-terminal four OB folds of CTC1, a metazoan expansion of the subunit that further differentiates it from the paralogous RPA large subunit and from CTC1 homologs found in unicellular eukaryotes13,24. Our evolutionary conservation analysis identified a complementary species-specific loop in the POLA1CTD, termed the CRL, that appears to have co-evolved with the expansion in CTC1. Substitution of the CRL with a short loop, either a GGSGGS linker or the orthologous S. pombe sequence, abrogates binding in the high-affinity (KD = ~1–10 μM) binding mode between POLA1CTD and CST. MST experiments also captured an order-of-magnitude-lower affinity (KD = ~100 μM) binding mode that is CRL-independent. The two binding modes are consistent with our cryo-EM structure, in which POLA1CTD utilizes both the CRL and a Zn2+-binding motif to interact with CTC1.
Further work is needed to delineate the functional role of POLA1N in regulating the fill-in machinery. We uncovered an interaction between POLA1N and CST by using a combination of negative-stain EM, CX-MS, and cryo-EM with CST bound to POLA1N in isolation and additionally bound in the context of PPFL. The CST–PPFL complex associated more robustly than CST–PPΔN, as inferred from higher CST occupancy in cryo-EM 2D averages and stability during native SEC. However, we observed greater structural heterogeneity in the cryo-EM data for CST–PPFL. The addition of POLA1N may allow the complex to sample a greater conformational space, interfering with accurate alignment of the particles in the CST–PPFL complex. POLA1N is responsible for the flexible tethering of Polα/primase to the replisome via an interaction with AND-1 (refs. 27,28), and its extensive yet flexible interaction with CST suggests a potential spatiotemporal regulation of fill-in after telomere replication, where the replisome hands Polα/primase off to a shelterin-bound CST for C-strand synthesis (Fig. 4d).
Finally, our model informs on CP mutations (p.A227V, p.V259M, p.V665G) previously characterized to disrupt CST–Polα/primase association14,25. It is unlikely that these single point mutations result in complete loss of function, as such mutations would likely be lethal. Consistent with this framework of mild dysfunction, we observe that the three CP mutations map close to interaction interfaces but are not necessarily responsible for direct interaction with Polα/primase. For example, V665 resides on a β-strand in Ctc1OB-D that is not directly on the interface, but the glycine substitution could destabilize the OB fold and weaken the interaction. Similarly, the Ctc1OB-B mutations reside near the bridge observed in the CST–PPFL cryo-EM map. Since those mutants (p.A227V, p.V259M, p.V665G) were characterized14, three more CP mutations have been reported in CTC1OB-B (refs. 29,30). Presumably, these would be deleterious in a manner resembling that of p.A227V and p.V259M. Future genetic and functional studies are required to elucidate the precise mechanism by which these mutations cause CP, but our structural results provide a framework for understanding the molecular basis of human disease linked to CST–Polα/primase, its role in telomere maintenance, and its contribution to DSB repair.
Methods
DNA construct and baculovirus generation
DNA fragments encoding human CTC1, STN1, TEN1, POLA1 (POLA1N, 1–339 aa; POLA1FL, 1–1462 aa; POLA1ΔN, 335–1492 aa), POLA2, PRIM1, and PRIM2 were cloned using Gibson assembly into the biGBac vector system31 with affinity tags. The plasmids used in this study are pBIG1a(His6-MBP-PreSc-CTC1/STN1/TEN1), pBIG2ab((His6-MBP-PreSc-POLA1FL/POLA2)/(PRIM1/StrepII-TEV-PRIM2)), pBIG2ab((His6-MBP-PreSc-POLA1ΔN/POLA2)/(PRIM1/StrepII-TEV-PRIM2)), and pLIB(His6-MBP-PreSc-POLA1N). Recombinant bacmids were generated from these plasmids using MAX Efficiency DH10Bac competent cells (Gibco; cat. no. 10361012) and transfected into Sf9 insect cells (Gibco; cat. no. 11496015) with Cellfectin II Reagent (Gibco) to generate a P1 baculovirus stock. P1 baculovirus was amplified in adherent Sf9 insect cells to generate P2 and P3 stocks, and the P3 virus was used to infect Tni suspension insect cell culture (Expression Systems; cat. no. 94-002S) for protein expression.
Constructs for expression of POLA1CTD in E. coli (CRLWT: 1265–1462 aa; CRLGGSGGS: 1265–1402–GGSGGS–1423–1462 aa; CRLS. pombe: 1265–1399– QTTTGAT–1425–1462 aa) were cloned with an N-terminal His6-Smt3 tag into a pRSFDuet-1 vector in the first multiple cloning site. Mutants were generated by Gibson assembly and confirmed by Sanger sequencing.
Protein expression and purification
Fifty milliliters of P3 baculovirus were used per 500 mL of Tni culture, infected at a cell density of ~2 ×106 cells/mL. The infected cells were grown in spinner flasks at 150 r.p.m. for 72 h at 27 °C. Cells were collected by centrifugation at 1,000g and transferred to a syringe before droplets were flash frozen in liquid nitrogen. The frozen pellets were lysed by cryogenic milling (Retsch) and the cryo-milled powder was resuspended in a buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl, 15 mM 2-mercaptoethanol (β-ME), 20 mM imidazole, 10% (v/v) glycerol, and 1 mM phenylmethylsulfonyl fluoride (PMSF), supplemented with cOmplete EDTA-free protease inhibitor cocktail (Roche). The lysate was cleared by centrifugation at 4 °C and 40,000g for 1 h. Supernatants were incubated with end-over-end rotation for 1 h at 4 °C with Ni-NTA resin (Invitrogen) equilibrated with a buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl, 15 mM β-ME, 20 mM imidazole, and 5% glycerol and subsequently washed with 20–50 column volumes (CV) of the same buffer. Bound protein was eluted in a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP), 250 mM imidazole, and 5% glycerol.
After elution from the Ni-NTA resin, His6-MBP-PreSc-CTC1/STN1/TEN1 was incubated with rhinovirus 3C protease overnight at 4 °C to remove the His6-MBP tag. The cleaved protein was loaded onto a HiTrap Heparin HP column (GE Healthcare) equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM TCEP, and 5% glycerol and eluted with a linear gradient of NaCl concentration to 1 M. Protein-containing fractions were loaded onto a HiLoad Superdex 200 16/600 PG column (GE Healthcare) equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 300 mM NaCl, 0.5 mM TCEP, and 5% glycerol. For microscale thermophoresis experiments, the protease-cleavage step was omitted and the His6-MBP-PreSc-CTC1/STN1/TEN1 protein eluted from the Ni-NTA column was directly subjected to Heparin-affinity and size-exclusion chromatography as described above.
After elution from the Ni-NTA resin, His6-MBP-PreSc-POLA1FL/POLA2/PRIM1/StrepII-TEV-PRIM2 and His6-MBP-PreSc-POLA1ΔN/POLA2/PRIM1/StrepII-TEV-PRIM2 were loaded onto a RESOURCE Q column (Cytiva) equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM TCEP, and 5% glycerol, and were eluted with a linear gradient of NaCl concentration to 0.5 M. Protein-containing fractions were loaded onto a HiLoad Superdex 200 16/600 PG column equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM TCEP, and 5% glycerol.
After elution from the Ni-NTA resin, His6-MBP-PreSc-POLA1N was directly loaded onto a HiLoad Superdex 200 16/600 PG column equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM TCEP, and 5% glycerol.
His6-Smt3 tagged POLA1CTD proteins were expressed in E. coli Rosetta(DE3) cells (Novagen; cat. no. 70954-3) in Super Broth medium (Teknova) and induced at an OD600 of ~0.8 with 0.5 mM IPTG (Soltec). Cells were collected by centrifugation at 4,000g and were resuspended in a buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl, 4 mM β-ME, 20 mM imidazole, and 10% glycerol, supplemented with complete EDTA-free protease inhibitor cocktail (Roche) and flash frozen in liquid nitrogen. Cells were supplemented with 20 μg/mL DNase I (Sigma), 20 μg/mL RNase A (Sigma), 20 μg/mL lysozyme (Sigma), 1 mM CaCl2, and 1 mM MgCl2, lysed by sonication, and centrifuged at 4 °C and 40,000g for 1 h. Supernatants were filtered through a 0.45-μm syringe filter (Millipore), incubated with end-over-end rotation for 1 h at 4 °C with Ni-NTA resin (Invitrogen) equilibrated with a buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl, 4 mM β-ME, and 20 mM imidazole, and were subsequently washed with 20–50 CV of the same buffer. Bound protein was eluted with 250 mM imidazole in a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, and 0.1 mM TCEP. The His6-Smt3 tag was removed by cleavage with His6-Ulp1 protease concurrent with overnight dialysis into buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, and 0.1 mM TCEP. Dialyzed protein was incubated with end-over-end rotation for 1 h at 4 °C with Ni-NTA resin (Invitrogen) equilibrated with dialysis buffer to rebind the His6-Smt3, His6-Ulp1, and any uncleaved protein. The flowthrough (containing cleaved POLA1CTD protein) was collected, concentrated, and loaded onto a Superdex 200 10/300 GL column equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, and 0.1 mM TCEP.
Unless otherwise stated, all proteins were concentrated, flash frozen in liquid nitrogen, and stored in aliquots at -80 °C.
Reconstitution of native CST–PPFL and MBP-POLA1N–CST
Freshly purified PPFL and CST were mixed in equimolar amounts and incubated for 1 h at 4 °C prior to loading onto a HiLoad Superdex 200 16/600 PG column equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM TCEP, and 5% glycerol. The indicated fractions (Extended Data Fig. 1a) were pooled and concentrated. Purified CST was mixed with a twofold molar excess of His6-MBP-PreSc-POLA1N and incubated for 1 h at 4 °C prior to being loaded onto a HiLoad Superdex 200 16/600 PG column equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM TCEP, and 5% glycerol. The indicated fractions (Extended Data Fig. 5) were pooled and concentrated.
Negative-stain EM sample preparation, data collection, and image processing
Protein samples for negative-stain EM (3.5-μL drops, in a concentration range of 0.01–0.05 mg/mL) were adsorbed to glow-discharged carbon-coated copper grids with a collodion film, washed with three drops of deionized water, and stained with two drops of freshly prepared 0.7% w/v uranyl formate. Samples were imaged at room temperature using a Phillips CM10 electron microscope equipped with a tungsten filament and operated at an acceleration voltage of 80 kV. The magnification used corresponds to a calibrated pixel size of 2.8 Å. Particle coordinates were auto-picked using the Swarm picker in EMAN2 (ref. 32). Particle extraction and 2D classification were performed in RELION 3.0 or RELION 3.1, as indicated.
GraFix stabilization of CST–Polα/primase complexes
Purified CST and Polα/primase (PPFL or PPΔN) were first mixed in equimolar amounts (for a 1.5 μM final protein concentration of each component) and incubated on ice for 1 h before the addition of 5′-(GGTTAG)3-3′ ssDNA at a 2.25-fold molar excess. The protein–ssDNA mixture was incubated on ice for 2 h and then loaded on top of a 11-mL linear 10%–30% glycerol and 0–5 mM glutaraldehyde GraFix gradient in 20 mM HEPES, pH 7.5, 150 mM NaCl, 10 mM MgCl2, and 0.5 mM TCEP. Centrifugation was carried out at 288,000g (41,000 r.p.m.) in an SW41 Ti rotor (Beckman Coulter) for 18 h at 4 °C. The top 3 mL of the gradient was discarded and then 500-μL fractions were manually collected from the top of the gradient and quenched with Tris-HCl (pH 7.5) at a final concentration of 50 mM. Fractions containing CST–Polα/primase were concentrated and diluted three times in an Amicon Ultra concentrator (Millipore; 30-kDa cut-off) with glycerol- and glutaraldehyde-free buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP). The concentrated samples were checked first with negative-stain EM for homogeneity prior to cryo-EM specimen preparation.
Cryo-EM sample preparation and data collection
Four microliters of the GraFix-stabilized samples were applied to Quantifoil R 1.2/1.3 mesh Au400 holey carbon grids covered with a graphene oxide support layer (EMS), blotted for 1–1.5 sec, and plunge frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific) operated at 4 °C and 100% humidity.
Cryo-EM imaging was performed in the Cryo-EM Resource Center at the Rockefeller University using SerialEM33. Data-collection parameters are summarized in Table 1.
For CST–PPFL, one dataset (dataset 1) was collected on a 200-kV Talos Arctica electron microscope (Thermo Fisher Scientific) at a nominal magnification of ×28,000 (TEM nanoprobe), corresponding to a calibrated pixel size of 1.5 Å on the specimen level. Images were collected using a defocus range from −1.5 to −3 μm with a K2 Summit direct electron detector (Gatan) in super-resolution counting mode. Exposures of 10 sec were dose-fractionated into 50 frames (200 ms per frame) with a dose rate of 12 electrons/pixel/sec (approximately 1.07 electrons per Å2 per frame), resulting in a total dose of 53 electrons per Å2. The second dataset (dataset 2) was collected on a 300-kV Titan Krios electron microscope at a nominal magnification of ×53,000 (EFTEM nanoprobe), corresponding to a calibrated pixel size of 1.32 Å on the specimen level. Images were collected using a defocus range from -1 to -2.2 μm with a K3 direct electron detector (Gatan) in super-resolution counting mode. Exposures of 3 sec were dose-fractionated into 50 frames (60 ms per frame) with a dose rate of 33 electrons/pixel/sec (approximately 1.14 electrons per Å2 per frame), resulting in a total dose of 57 electrons per Å2.
For CST–PPΔN, data were collected on a 300-kV Titan Krios electron microscope at a nominal magnification of ×64,000, corresponding to a calibrated pixel size of 1.08 Å on the specimen level. Images were collected using a defocus range from –1 to –2.2 μm with a K3 direct electron detector (Gatan) in super-resolution counting mode. Exposures of 3 sec were dose-fractionated into 50 frames (60 ms per frame), with a dose rate of 30 electrons/pixel/sec (approximately 1.03 electrons per Å2 per frame), resulting in a total dose of 52 electrons per Å2.
Cryo-EM data processing
For all datasets, movie stacks were motion-corrected with the RELION-3 (ref. 34) implementation of MotionCor2 and motion-corrected micrographs were manually inspected and curated (graphene oxide coverage of grids were inconsistent) prior to CTF parameter estimation with CTFFIND4 (ref. 35) implemented in RELION-3. Particles were automatically picked with Gautomatch and extracted in RELION-3 for all further 2D and 3D processing steps. Auto-picked particles were examined by 2D classification, and particles in ‘bad’ classes corresponding to ice contamination or graphene oxide fold lines were discarded. The first reference model was generated using RELION-3’s 3D initial model job and subsequently improved as continued 3D classification produced better maps.
For the CST–PPFL complex, multiple processing strategies were pursued to generate higher-resolution maps but were unsuccessful owing to substantial heterogeneity among the particles. The reported standard image-processing pipeline was performed with twofold binned images (to speed up computation), as the resolution of the resulting map did not approach the Nyquist limit. Because the two datasets were collected on different microscopes, RELION 3.1 was used to combine the particle stacks (imported as two separate optics groups). The combined particles were used for further 3D classification and 3D refinement (Extended Data Fig. 2).
For the CST–PPΔN complex, a supervised 3D classification step in RELION-3 was introduced, using three references: CST–PPΔN, PPΔN alone, and a noise/junk ‘decoy’ class. After discarding particles assigned to the latter two classes, the remaining particles were subjected to 3D classification into four classes using a single reference. Because particles in the class with the best-defined features still showed low CST occupancy, a consensus 3D refinement was performed and the resulting map segmented into CST and PPΔN using UCSF Chimera36. Using a mask generated in RELION-3, the partial signal of PPΔN was subtracted and the signal-subtracted particles were subjected to focused 3D classification without alignment. The final stack contained 131,850 particles showing intact CST. The particles were reverted to include PPΔN, and used for three cycles of iterative 3D refinement, CTF refinement, and Bayesian polishing. The resulting density map was sharpened by post-processing, and Fourier shell correlation (FSC) curves and a local resolution map were calculated in RELION-3 (Extended Data Fig. 3).
Model building and refinement
An atomic model was built into the 4.6-Å resolution map of the CST–PPΔN complex. The crystal structure of the apo PPΔN (PDB ID: 5EXR) and cryo-EM structure of CST (PDB ID: 6W6W) were used for initial rigid-body docking into the map. The map showed only weak density corresponding to the C-terminal half of STN1 (STN1C, 184–368 aa), likely owing to the ssDNA charge preventing STN1C binding, as previously described13, and was therefore removed for model building. The geometry of PRIM1 in the PPΔN crystal structure is poor and it was substituted in our initial model with a crystal structure of PRIM1 determined at higher resolution (PDB ID: 6RB4, alternate conformers removed37). Although the geometries of the other PP subunits in the starting model (PDB ID: 5EXR) were also poor, we chose to continue with this model because it is the only experimentally determined model of apo POLA1/POLA2/PRIM2 in the occluded conformation and retains inter-subunit contact information that would not be captured by AlphaFold 2 models or other experimental structures of the subunits in isolation. The N-terminus of CTC1 was poorly resolved in the previously determined cryo-EM map and not modeled, so it was substituted with the AlphaFold 2 model of CTC1 (AF-Q2NKJ3-F1), which agrees with the experimentally determined structure of CTC1 (refs. 19,20). All subunits were docked into the cryo-EM map using the Chimera ‘Fit in Map’ tool, and the model was flexibly fitted using the ISOLDE plugin for ChimeraX38,39. After the model was improved by iterative cycles of refinement in Phenix (phenix.real_space_refine) and manual adjustment in Coot40, the geometry and map fit of the final model was validated (phenix.validation_cryoem)41.
Evolutionary conservation analysis
All sequence accession numbers used in this study are listed in Supplementary Table 1. Protein sequences obtained from BLAST (blastp suite) were analyzed using Jalview42,43 and multiple sequence comparison by log expectation (MUSCLE). Representative sequences for CTC1 and POLA1 were modeled in AlphaFold 2 (refs. 19,20) with the template database from 14 May 2020 and the casp14 preset. AlphaFold models were aligned in PyMOL (Schrödinger) and visualized in ChimeraX39.
Microscale thermophoresis
Microscale thermophoresis experiments were performed on a NanoTemper Monolith NT.115 machine. All samples were buffer exchanged in centrifugal concentrators (Amicon Ultra-0.5 mL) into buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM DTT, and 0.05% (v/v) Tween-20. His6-MBP-CST was labeled according to manufacturer instructions with RED-tris-NTA second-generation dye (NanoTemper Technologies). Fifty nanomolar labeled His6-MBP-CST was incubated with serial dilutions of unlabeled POLA1CTD constructs, and thermophoresis was measured at room temperature with an excitation power of 20% and an MST power of 20%. Titrations were performed in triplicate (experimental replicates), and capillary scans were performed in triplicate (technical replicates). Data were analyzed at the 10-sec time point with the MO Affinity Analysis Software (version 2.3, NanoTemper Technologies) using the KD fit option with outliers owing to aggregation automatically determined by the software. For CRLWT, the data were manually split to account for the presence of two binding events23. Reported KD values were calculated in the MO Affinity Analysis software and data were plotted with Prism 9 (GraphPad).
In vitro cross-linking
BS3 cross-linker (Proteochem, c1103) was dissolved in LC–MS-grade H2O (Proteochem, LC6330) at 50 mM. DSS cross-linker (Proteochem, c1105) was dissolved in oxygen-depleted DMSO (Millipore-Sigma, 900645) to 100 mM stock concentration. Cross-linker was added to the target complex (BS3: CST–POLA1N; DSS: CST–PPFL) prepared in NHS-ester non-reactive buffer (1 mg/mL) to the final concentration of 0.35–0.75 mM. Reactions were performed at 25 °C in disposable inert cuvettes (UVette, Eppendorf), and monitored by continuous looped dynamic light scattering measurements of polydispersity44 (Pd < 10%; DynaPro NanoStar, Wyatt). Cross-linking was quenched after 30 min of incubation by addition of Tris-HCl (pH 8.0) to a final concentration of 5 mM.
Mass spectrometry and data analysis
Samples were dialyzed against 100 mM ammonium bicarbonate, reduced with 50 mM TCEP at 60 °C for 10 min and alkylated with 50 mM iodoacetamide in the dark for 15 min at 37 °C. Digestion was carried out at 37 °C overnight with 125 ng/mL sequencing-grade modified trypsin (Fisher Scientific, PI90057) in 25 mM ammonium bicarbonate supplemented with ProteaseMax (Fisher Scientific, PRV2071). Reaction mix was supplemented with trifluoroacetic acid (TFA, Fisher Scientific, A116) to a final concentration of 0.1%. The resulting peptides were passed through C18 Spin Tips (Fisher Scientific, PI84850) before elution with 40 μL of 80% acetonitrile (ACN, Fisher Scientific, A9561) in 0.1% TFA. Eluted peptides were dried and resuspended in 20 μL 0.1% formic acid (FA; Fisher Scientific, A117) for MS analysis. Peptides were analyzed in an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) coupled to an EASY-nLC (Thermo Scientific) liquid chromatography system, with a 2 μm, 500 mm EASY-Spray column. The peptides were eluted over a 120-min linear gradient from 96% buffer A (0.1 % FA in water) to 40% buffer B (0.1 % FA in ACN), then continued to 98% buffer B over 20 min with a flow rate of 250 nL/min. Each full MS scan (R = 60,000) was followed by 20 data-dependent MS2 scans (R = 15,000) with high-energy collisional dissociation (HCD) and an isolation window of 2.0 m/z. Normalized collision energy was set to 35. Precursors of charge state ≤ 3 were collected for MS2 scans in enumerative mode, precursors of charge state 4–6 were collected for MS2 scans in cross-link discovery mode (both were performed for each sample); monoisotopic precursor selection was enabled and a dynamic exclusion window of 30.0 sec was set. Raw files obtained in enumerative mode were analyzed by pFind3 software45 in open search mode and protein modifications inferred by pFind3 and comprising >0.5% of total protein were included as the variable modifications in pLink2 (ref. 46) search parameters. pLink2 results were filtered for FDR (<5%), e-value (<1 × 10–3), score (<1 × 10–2), and abundance (PSMs ≥ 5). Cross-links were visualized using xiNET47.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Starting models used in this study can be found in the Protein Data Bank under the accession codes PDB 6W6W, PDB 5EXR, and PDB 6RB4 and in the AlphaFold Protein Structure Database under accession code AF-Q2NKJ3-F1. The cryo-EM maps generated in this study have been deposited at the Electron Microscopy Data Bank under accession codes EMD-26346 (CST–PPΔN) and EMD-26347 (CST–PPFL), and the CST–PPΔN coordinates have been deposited in the Protein Data Bank under accession code PDB 7U5C. Source data are provided with this paper.
References
de Lange, T. Shelterin-mediated telomere protection. Annu Rev. Genet 52, 223–247 (2018).
Chen, L. Y., Redon, S. & Lingner, J. The human CST complex is a terminator of telomerase activity. Nature 488, 540–544 (2012).
Wan, M., Qin, J., Songyang, Z. & Liu, D. OB fold-containing protein 1 (OBFC1), a human homolog of yeast Stn1, associates with TPP1 and is implicated in telomere length regulation. J. Biol. Chem. 284, 26725–26731 (2009).
Wu, P., Takai, H. & de Lange, T. Telomeric 3′ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 150, 39–52 (2012).
Takai, H. et al. A POT1 mutation implicates defective telomere end fill-in and telomere truncations in Coats plus. Genes Dev. 30, 812–826 (2016).
Stewart, J. A. et al. Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J. 31, 3537–3549 (2012).
Feng, X., Hsu, S. J., Kasbek, C., Chaiken, M. & Price, C. M. CTC1-mediated C-strand fill-in is an essential step in telomere length maintenance. Nucleic Acids Res 45, 4281–4293 (2017).
Anderson, B. H. et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat. Genet 44, 338–342 (2012).
Savage, S. A. Beginning at the ends: telomeres and human disease. F1000Res https://doi.org/10.12688/f1000research.14068.1 (2018).
Mirman, Z. et al. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 560, 112–116 (2018).
Mirman, Z. & de Lange, T. 53BP1: a DSB escort. Genes Dev. 34, 7–23 (2020).
Mirman, Z., Sasi, N. K., King, A., Chapman, J. R. & de Lange, T. 53BP1-shieldin-dependent DSB processing in BRCA1-deficient cells requires CST-Polα-primase fill-in synthesis. Nat. Cell Biol. 24, 51–61 (2022).
Lim, C. J. et al. The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science 368, 1081–1085 (2020).
Chen, L. Y., Majerská, J. & Lingner, J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev. 27, 2099–2108 (2013).
Ganduri, S. & Lue, N. F. STN1-POLA2 interaction provides a basis for primase-pol α stimulation by human STN1. Nucleic Acids Res 45, 9455–9466 (2017).
Kastner, B. et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5, 53–55 (2008).
Baranovskiy, A. G. et al. Mechanism of concerted RNA–DNA primer synthesis by the human primosome. J. Biol. Chem. 291, 10006–10020 (2016).
Coloma, J., Johnson, R. E., Prakash, L., Prakash, S. & Aggarwal, A. K. Human DNA polymerase α in binary complex with a DNA:DNA template-primer. Sci. Rep. 6, 23784 (2016).
Jumper, J. et al. Applying and improving AlphaFold at CASP14. Proteins 89, 1711–1721 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. gkab1061 (2021).
Kilkenny, M. L. et al. Structural basis for the interaction of SARS-CoV-2 virulence factor nsp1 with DNA polymerase α-primase. Protein Sci. 31, 333–344 (2022).
Núñez-Ramírez, R. et al. Flexible tethering of primase and DNA Pol α in the eukaryotic primosome. Nucleic Acids Res 39, 8187–8199 (2011).
Seidel, S. A. et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 (2013).
Lue, N. F. Evolving linear chromosomes and telomeres: a C-strand-centric view. Trends Biochem. Sci. 43, 314–326 (2018).
Gu, P. et al. CTC1-STN1 coordinates G- and C-strand synthesis to regulate telomere length. Aging Cell 17, e12783 (2018).
Huang, H. et al. Structure of a DNA polymerase alpha-primase domain that docks on the SV40 helicase and activates the viral primosome. J. Biol. Chem. 285, 17112–17122 (2010).
Villa, F. et al. Ctf4 is a hub in the eukaryotic replisome that links multiple CIP-box proteins to the CMG helicase. Mol. Cell 63, 385–396 (2016).
Kilkenny, M. L. et al. The human CTF4-orthologue AND-1 interacts with DNA polymerase α/primase via its unique C-terminal HMG box. Open Biol. 7, 170217 (2017).
Walne, A. J. et al. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica 98, 334–338 (2013).
Hidalgo-Sanz, J. et al. Infant with intracranial calcifications and retinopathy. Rev. Neurol. 69, 289–292 (2019).
Weissmann, F. et al. biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc. Natl Acad. Sci. USA 113, E2564–E2569 (2016).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Pettersen, E. F. et al. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
Holzer, S. et al. Structural basis for inhibition of human primase by arabinofuranosyl nucleoside analogues fludarabine and vidarabine. ACS Chem. Biol. 14, 1904–1912 (2019).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D. Struct. Biol. 74, 519–530 (2018).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr 66, 213–221 (2010).
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M. & Barton, G. J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Meyer, A. et al. Systematic analysis of protein-detergent complexes applying dynamic light scattering to optimize solutions for crystallization trials. Acta Crystallogr F. Struct. Biol. Commun. 71, 75–81 (2015).
Chi, H. et al. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat. Biotechnol. https://doi.org/10.1038/nbt.4236 (2018).
Chen, Z. L. et al. A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat. Commun. 10, 3404 (2019).
Combe, C. W., Fischer, L. & Rappsilber, J. xiNET: cross-link network maps with residue resolution. Mol. Cell Proteom. 14, 1137–1147 (2015).
Acknowledgements
We are grateful to H. Suzuki and Y. Zhang for guidance with data processing. We thank M. Ebrahim, J. Sotiris, and H. Ng at the Evelyn Gruss Lipper Cryo-EM Resource Center of The Rockefeller University for assistance with cryo-EM data collection. We thank I. Nudelman of the Fisher Drug Discovery Resource Center of The Rockefeller University for providing training and guidance on the microscale thermophoresis experiments. S.W.C. is supported by the David Rockefeller Graduate Program and a National Science Foundation Graduate Research Fellowship under grant no. 1946429. J.C.Z. was previously supported by an NIH cancer biology training grant (T32 CA009673) and is currently the Lorraine Egan Fellow of the Damon Runyon Cancer Research foundation (DRG 2337-19). This work is supported by grants from the NIH (5 R35 CA210036) and the Breast Cancer Research Foundation (BCRF-19-036) to T.d.L. and by funding from the Howard Hughes Medical Institute, NIH (5 R01 GM126891) and Blavatnik Family Foundation to E.N. and V.S.
Author information
Authors and Affiliations
Contributions
This study was conceived by T.d.L., T.W., J.C.Z., and S.W.C.S.W.C. purified and reconstituted protein complexes, performed EM studies, and carried out computational and biophysical analyses. J.C.Z. and S.W.C. designed and cloned expression constructs. CX-MS data were obtained by E.N. and V.S.M.W.B. assisted with negative-stain EM experiments. S.W.C. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
T.d.L. is a member of the SAB of Calico Life Sciences LLC, San Francisco. The other authors have no conflicts to declare.
Peer review
Peer review information
Nature Structural and Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Editor recognition statement Beth Moorefield was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Reconstitutions of CST–PPFL.
a, SEC-elution profile of CST–PPFL (top). The grey line indicates the fractions visualized on the SYPRO Ruby-stained SDS-PAGE gel (bottom). This reconstitution was independently reproduced at least three times with similar results. b, Negative-stain EM image-processing pipeline and averages of the 35 (of 50) most populated 2D classes of the reconstituted complex characterized in a. The size of the 128-px box corresponds to 358 Å. c, GraFix preparation of CST–PPFL–ssDNA. Top: Coomassie blue-stained SDS-PAGE gel (4–12% (w/v) Tris-Glycine; Invitrogen) of GraFix fractions, showing formation of cross-linked CST–PPFL species. Fractions marked with an asterisk (*) were pooled for analysis by cryo-EM. Bottom: SYBR Gold-stained native PAGE gel (4–20% (w/v) TBE; Invitrogen) of GraFix fractions, showing free and bound ssDNA. This reconstitution was independently reproduced at least three times with similar results. d, Cryo-EM image-processing pipeline and 2D-class averages obtained after vitrification and imaging of the GraFixed complex shown in c. See Table 1 for full data collection and processing details.
Extended Data Fig. 2 Cryo-EM image-processing pipeline of CST–PPFL.
a, Cryo-EM image-processing pipeline used for the CST–PPFL complex. Two datasets were combined in RELION 3.1 as separate optics groups, and particles were refined together with a reference generated from dataset no. 1. See Table 1 for full data collection and processing details. b, 2D-class averages from a cleanup step prior to 3D classification. Classes clearly representing ice contamination or graphene oxide edges were removed during cleanup.
Extended Data Fig. 3 GraFix preparation of CST–PPΔN complex and analysis by cryo-EM.
a, GraFix preparation of CST–PPΔN–ssDNA. Top: Coomassie blue-stained SDS-PAGE gel (4–12% Tris-Glycine; Invitrogen) of GraFix fractions, showing formation of cross-linked CST–PPΔN species. Fractions marked with an asterisk (*) were pooled for analysis by cryo-EM. Bottom: SYBR Gold-stained native PAGE gel (4–20% TBE; Invitrogen) of GraFix fractions, showing free and bound ssDNA. This reconstitution was independently reproduced at least three times with similar results. b, Selected high defocus motion-corrected cryo-EM micrograph of CST–PPΔN (representative of 9,282 curated micrographs). c, Cryo-EM image-processing pipeline used for the CST–PPΔN complex, including supervised 3D classification and focused 3D classification steps used to select particles with intact CST. Gold-standard FSC curves for the final map of CST–PPΔN indicate a global resolution of 4.6 Å (FSC = 0.143). See Table 1 for full data collection and processing details. d, 2D-class averages obtained with the particle stack (131,850 particles) used to generate the final CST–PPΔN map show blurring at the peripheral regions, indicating flexibility. e, Local resolution estimates of the CST–PPΔN map. f, Orthogonal views of the CST–PPΔN–ssDNA model fit into the cryo-EM map.
Extended Data Fig. 4 Analysis of the CTC1-POLA1CTD interface surfaces.
Surface representations of the CTC1-POLA1CTD interface shown in two orientations related by a 180° rotation and views CTC1 and POLA1CTD after separation to reveal the interface surfaces (outlined in black). a, Surface overlaid with cartoon representation of CTC1–POLA1CTD. Identification of CTC1 and POLA1CTD binding surfaces, colored in dark blue. b, Surface representation colored according to electrostatic potential. c, Surface representation colored according to sequence conservation for metazoans using an alignment containing all species listed in Supplementary Table 1.
Extended Data Fig. 5 Conservation analysis of the CTC1-POLA1CTD interface.
a, Multiple sequence alignment showing the CRL region in POLA1. Residues are colored according to the color scheme used by Clustal X43. Accession codes for sequences are listed in Supplementary Table 1. b, Sequences for POLA1CTD and CTC1 (when present) were modeled in AlphaFold 219,20 and aligned in PyMOL (Schrödinger) (Supplementary Table 1). The CRL in metazoans is colored cyan, and the corresponding insertion region in unicellular eukaryotes is colored in fuchsia. For CTC1, the elongated OB-D is colored yellow and the rest of the N-terminus (OB-A, OB-B, OB-C, three-helix bundle) is colored cyan.
Extended Data Fig. 6 Purification and microscale thermophoresis analysis of POLA1CTD CRL mutants.
a, Coomassie blue-stained SDS-PAGE gels (4–12% (w/v) Bis-Tris, run in MES-SDS; Invitrogen) showing proteins used for MST analyses. POLA1CTD constructs run as heterogeneously sized smears on SDS-PAGE. Gels are representative of three independent protein preparations. b, Native PAGE (4–16% Bis-Tris; Invitrogen) gel showing POLA1CTD constructs and a 1 μg BSA loading control next to a NativeMark unstained protein standard. Gels are representative of three independent protein preparations. c, SEC-elution profiles of the POLA1CTD constructs (estimated MW ~20 kDa) superposed with gel-filtration standards (BioRad). d, Models generated with AlphaFold 219,20 of the human POLA1CTD constructs encoding CRLWT (cyan), CRLGGSGGS (red), or CRLS. pombe (fuchsia), demonstrating the accommodation of shortened linkers. e, Independent microscale thermophoresis experiments with POLA1CTD constructs and RED-tris-NTA-labeled His6-MBP-CTC1–STN1–TEN1. Dissociation constant (KD) values were calculated with the MO Affinity Analysis (Nanotemper) software. Error bars represent the s.e.m. for each data point calculated from three capillary scans. The combined data are shown in Fig. 2c.
Extended Data Fig. 7 Reconstitution and analysis of the CST-POLA1N interaction.
a, SEC-elution profile of reconstituted His6-MBP-POLA1N–CST (top). The grey line indicates the fractions visualized on the SYPRO Ruby-stained SDS-PAGE gel (4–12% Bis-Tris, run in MOPS-SDS; Invitrogen) (bottom). The black bar indicates the fractions pooled for negative-stain EM and CX-MS analysis (see Fig. 3). This reconstitution was independently reproduced at least three times with similar results. b, Negative-stain EM image-processing pipeline and averages of the 35 (of 50) most populated classes of the reconstituted complex characterized in a. The size of the 128-px box corresponds to 358 Å2. c, Cross-links between POLA1N and CST subunits identified by CX-MS analysis of the CST–PPFL complex (Extended Data Fig. 1a). Blue solid lines: cross-links between CST and POLA1N shared with the His6-MBP-POLA1N–CST dataset (Fig. 3c). Blue dashed lines: cross-links between CST and POLA1N not shared with the His6-MBP-POLA1N–CST dataset (Fig. 2c). Black dashed lines: all other inter-subunit cross-links.
Supplementary information
Supplementary Information
Supplementary Table 1
Source data
Source Data Fig. 2
Statistical Source Data
Source Data Fig. 3
Statistical Source Data
Source Data Extended Data Fig. 1
Unprocessed Gels
Source Data Extended Data Fig. 3
Unprocessed Gels, Uncropped Micrograph
Source Data Extended Data Fig. 6
Unprocessed Gels
Source Data Extended Data Fig. 6
Statistical Source Data
Source Data Extended Data Fig. 7
Unprocessed Gels
Source Data Extended Data Fig. 7
Statistical Source Data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cai, S.W., Zinder, J.C., Svetlov, V. et al. Cryo-EM structure of the human CST–Polα/primase complex in a recruitment state. Nat Struct Mol Biol 29, 813–819 (2022). https://doi.org/10.1038/s41594-022-00766-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-022-00766-y
This article is cited by
-
A mechanistic model of primer synthesis from catalytic structures of DNA polymerase α–primase
Nature Structural & Molecular Biology (2024)
-
CST–polymerase α-primase solves a second telomere end-replication problem
Nature (2024)
-
The evolving genetic landscape of telomere biology disorder dyskeratosis congenita
EMBO Molecular Medicine (2024)
-
Orchestrating nucleic acid–protein interactions at chromosome ends: telomerase mechanisms come into focus
Nature Structural & Molecular Biology (2023)
-
CaMKK2 and CHK1 phosphorylate human STN1 in response to replication stress to protect stalled forks from aberrant resection
Nature Communications (2023)
