
Abstract
Accumulation of damaged mitochondria is a hallmark of aging and age-related neurodegeneration, including Alzheimer’s disease (AD). The molecular mechanisms of impaired mitochondrial homeostasis in AD are being investigated. Here we provide evidence that mitophagy is impaired in the hippocampus of AD patients, in induced pluripotent stem cell-derived human AD neurons, and in animal AD models. In both amyloid-β (Aβ) and tau Caenorhabditis elegans models of AD, mitophagy stimulation (through NAD+ supplementation, urolithin A, and actinonin) reverses memory impairment through PINK-1 (PTEN-induced kinase-1)-, PDR-1 (Parkinson’s disease-related-1; parkin)-, or DCT-1 (DAF-16/FOXO-controlled germline-tumor affecting-1)-dependent pathways. Mitophagy diminishes insoluble Aβ1–42 and Aβ1–40 and prevents cognitive impairment in an APP/PS1 mouse model through microglial phagocytosis of extracellular Aβ plaques and suppression of neuroinflammation. Mitophagy enhancement abolishes AD-related tau hyperphosphorylation in human neuronal cells and reverses memory impairment in transgenic tau nematodes and mice. Our findings suggest that impaired removal of defective mitochondria is a pivotal event in AD pathogenesis and that mitophagy represents a potential therapeutic intervention.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Code availability
ANY-maze (version 4.99) was used for behavioral tracking in the mouse studies. The ZEN (blue edition) software was used for taking the images. FCS Express (version 4) was used for flow cytometry. Microsoft Excel 2016 and Prism 7 (GraphPad Software) were used for the data analysis. ImageJ with the plug-in ObjectJ (https://sils.fnwi.uva.nl/bcb/objectj/download/) was used for the morphological quantification of mitochondria.
Data availability
The microarray GEO accession number for the data reported in this paper is GSE111737. All data are available from the corresponding author upon reasonable request.
References
Scheibye-Knudsen, M., Fang, E. F., Croteau, D. L., Wilson, D. M.3rd & Bohr, V. A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 25, 158–170 (2015).
Kerr, J. S. et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 40, 151–166 (2017).
Lustbader, J. W. et al. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452 (2004).
Sorrentino, V. et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193 (2017).
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G. & Anandatheerthavarada, H. K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068 (2006).
Mairet-Coello, G. et al. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron 78, 94–108 (2013).
Vossel, K. A. et al. Tau reduction prevents Aβ-induced defects in axonal transport. Science 330, 198 (2010).
Mandelkow, E. M., Stamer, K., Vogel, R., Thies, E. & Mandelkow, E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 24, 1079–1085 (2003).
Dixit, R., Ross, J. L., Goldman, Y. E. & Holzbaur, E. L. Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089 (2008).
Palikaras, K., Lionaki, E. & Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528 (2015).
Fang, E. F. et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD+/SIRT1 reduction. Cell 157, 882–896 (2014).
Fang, E. F. et al. NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581 (2016).
Cantó, C. et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 (2009).
Toyama, E. Q. et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281 (2016).
Egan, D. F. et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 (2011).
Lazarou, M. et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 (2015).
Handel, A. E. et al. Assessing similarity to primary tissue and cortical layer identity in induced pluripotent stem cell-derived cortical neurons through single-cell transcriptomics. Hum. Mol. Genet. 25, 989–1000 (2016).
Shi, Y., Kirwan, P. & Livesey, F. J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836–1846 (2012).
Menzies, F. M., Fleming, A. & Rubinsztein, D. C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357 (2015).
Kimura, S., Noda, T. & Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 (2007).
Fivenson, E. M. et al. Mitophagy in neurodegeneration and aging. Neurochem. Int. 109, 202–209 (2017).
Zhang, J. H., Chung, T. D. & Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 (1999).
Fang, E. F. et al. In vitro and in vivo detection of mitophagy in human cells, C. elegans, and mice. J. Vis. Exp. https://doi.org/10.3791/56301 (2017).
Aman, Y., Qiu, Y., Tao, J. & Fang, E. F. Therapeutic potential of boosting NAD+ in aging and age-related diseases. Transl. Med. Aging 2, 30–37 (2018).
Fang, E. F. et al. NAD+ in aging: molecular mechanisms and translational implications. Trends Mol. Med. 23, 899–916 (2017).
Ryu, D. et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22, 879–888 (2016).
Sun, N. et al. Measuring in vivo mitophagy. Mol. Cell 60, 685–696 (2015).
Fatouros, C. et al. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum. Mol. Genet. 21, 3587–3603 (2012).
Lublin, A. L. & Link, C. D. Alzheimer’s disease drug discovery: in vivo screening using C. elegans as a model for β-amyloid peptide-induced toxicity. Drug Discov. Today Technol. 10, e115–e119 (2013).
Iaccarino, H. F. et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235 (2016).
Voglis, G. & Tavernarakis, N. A synaptic DEG/ENaC ion channel mediates learning in C. elegans by facilitating dopamine signalling. EMBO J. 27, 3288–3299 (2008).
Selkoe, D. J. Alzheimer’s disease is a synaptic failure. Science 298, 789–791 (2002).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017).
Babcock, A. A. et al. Cytokine-producing microglia have an altered beta-amyloid load in aged APP/PS1 Tg mice. Brain Behav. Immun. 48, 86–101 (2015).
Ip, W. K. E., Hoshi, N., Shouval, D. S., Snapper, S. & Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519 (2017).
Heneka, M. T. et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 (2013).
Spillantini, M. G. & Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622 (2013).
Hanger, D. P. et al. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biol. Chem. 282, 23645–23654 (2007).
Hou, Y. et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl Acad. Sci. USA 115, E1876–E1885 (2018).
Mazure, C. M. & Swendsen, J. Sex differences in Alzheimer’s disease and other dementias. Lancet Neurol. 15, 451–452 (2016).
Swerdlow, R. H., Burns, J. M. & Khan, S. M. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231 (2014).
Martín-Maestro, P. et al. Mitophagy failure in fibroblasts and iPSC-derived neurons of Alzheimer’s disease-associated presenilin 1 mutation. Front. Mol. Neurosci. 10, 291 (2017).
Ye, X., Sun, X., Starovoytov, V. & Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 24, 2938–2951 (2015).
Thornton, C., Bright, N. J., Sastre, M., Muckett, P. J. & Carling, D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid β-peptide exposure. Biochem. J. 434, 503–512 (2011).
Yoon, S. O. et al. JNK3 perpetuates metabolic stress induced by Aβ peptides. Neuron 75, 824–837 (2012).
Sarlus, H. & Heneka, M. T. Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249 (2017).
Venegas, C. et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361 (2017).
Du, F. et al. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 140, 3233–3251 (2017).
Khandelwal, P. J., Herman, A. M., Hoe, H. S., Rebeck, G. W. & Moussa, C. E. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Aβ in AD models. Hum. Mol. Genet. 20, 2091–2102 (2011).
Bu, X. L. et al. Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol. Psychiatry 23, 1–9 (2018).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Samara, C., Syntichaki, P. & Tavernarakis, N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 15, 105–112 (2008).
Bargmann, C. I. & Horvitz, H. R. Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron 7, 729–742 (1991).
Palikaras, K. & Tavernarakis, N. Assessing mitochondrial selective autophagy in the nematode Caenorhabditis elegans. Methods Mol. Biol. 1567, 349–361 (2017).
Oddo, S. et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 39, 409–421 (2003).
Vorhees, C. V. & Williams, M. T. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 1, 848–858 (2006).
Ghosal, K. et al. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl Acad. Sci. USA 106, 18367–18372 (2009).
Scheibye-Knudsen, M. et al. A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855 (2014).
Hou, Y. et al. Smart Soup, a traditional Chinese medicine formula, ameliorates amyloid pathology and related cognitive deficits. PLoS One 9, e111215 (2014).
Koopman, M. et al. A screening-based platform for the assessment of cellular respiration in Caenorhabditis elegans. Nat. Protoc. 11, 1798–1816 (2016).
Fang, E. F. et al. Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci. Rep. 7, 46208 (2017).
Brewer, G. J. & Torricelli, J. R. Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2, 1490–1498 (2007).
Forester, C. M. et al. Revealing nascent proteomics in signaling pathways and cell differentiation. Proc. Natl Acad. Sci. USA 115, 2353–2358 (2018).
Skamagki, M. et al. ZSCAN10 expression corrects the genomic instability of iPSCs from aged donors. Nat. Cell Biol. 19, 1037–1048 (2017).
Yuan, H. et al. Heat-induced radiolabeling and fluorescence labeling of Feraheme nanoparticles for PET/SPECT imaging and flow cytometry. Nat. Protoc. 13, 392–412 (2018).
Acknowledgements
We thank R. Legouis (Institut de Biologie Intégrative de la Cellule) for the C. elegans strain expressing GFP::LGG-2. Some of the nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Center for Research Resources of the National Institutes of Health, and S. Mitani (National Bioresource Project) in Japan. We thank A. Fire for the plasmid vectors. We thank S. Cordonnier, H. Kassahun, J. Tian, W.B. Iser, M.A. Wilson, D. Figueroa, E. Fivenson, and Q.P. Lu for the experiments involving C. elegans and mice; K. Marosi and T.G. Demarest for the Seahorse studies; M. Kang, Y.Q. Zhang, E. Lehrmann, and K. Becker for the array data analysis; and A. Pasparaki for the technical support with the experiments. We thank N.F. Borhan for reading the paper. This research was supported by the Intramural Research Program of the NIH; the NIA (V.A.B.); the Helse Sør-Øst RHF (E.F.F., grant no. 2017056 and H.N., grant no. 275911); The Research Council of Norway (E.F.F., grant nos. 262175 and 277813); Center for Healthy Aging, University of Copenhagen; the European Research Council (ERC—GA695190—MANNA, ERC—GA737599 —NeuronAgeScreen); the European Commission Framework Programmes; the Greek Ministry of Education (N.T.); and the Olav Thon Foundation (V.A.B.). K.P. is supported by an AXA Research Fund postdoctoral long-term fellowship.
Author information
Authors and Affiliations
Contributions
E.F.F., Y.H., K.P., and V.A.B. designed the experiments. E.F.F. performed the microarray and D.L.C. analyzed the data. E.F.F., Y.H., D.C., and J.S.K. performed the western blot. E.F.F., J.S.K., and M.M.H.-O. performed the Seahorse experiments. Y.H., J.S.K., B.Y., and S. L. performed the animal treatment, histology, IHC, and ELISA experiments. K.P., N.T., E.F.F., J.S.K., and X.D. performed the C. elegans experiments. B.A.A., P.R., and M.Z.C. performed the stem cell experiment. E.F.F., Y.H., K.P., D.L.C., M.A., N.H.G., T.F., H.N., M.P.M., and V.A.B. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
E.F.F., V.A.B., and H.N. have Cooperative Research and Development Agreement (CRADA) arrangements with ChromaDex. V.A.B. also has a CRADA with Elysium Health.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Characterization of postmortem hippocampal samples and iPSC-derived neurons from AD patients and neurologically normal controls.
a, Quantification of Western blot data of the indicated proteins. Data were shown in mean ± s.e.m. (n = 7 biologically independent samples from 7 AD patients or 7 healthy controls; *p < 0.05; Two-sided Student’s t-test). b, Levels of proteins related to mitochondrial homeostasis in postmortem hippocampal samples from AD patients and age-matched controls. The right panel shows quantified data between normal and AD groups. Experiments were repeated independently twice with similar results. c, Representative IHC images showing colocalization of TOMM20 (mitochondrial outer membrane protein) and LAMP2 (lysosomal protein) in postmortem hippocampal regions of AD patients and age-matched healthy controls (n = 3 samples/group). Experiments were repeated independently twice with similar results. d, (upper) Levels of neuronal markers in two AD patient iPSC-derived neuronal lines and a sex- and age-matched control. (lower) Western blotting showing levels of mitophagy-related proteins in a PSEN1 AD patient iPSC-derived neuronal lines and matched control. Experiments were performed once. e and f, Levels of proteins involved in mitochondrial function and dynamics, metabolism, DNA repair, and mitophagy in two AD patient iPSC-derived neuronal lines and a shared sex- and age-matched control. g, Quantification of Western blot data of the indicated proteins. Data are shown in mean ± s.e.m. (n = n = 3 independent experiments; n.s., p>0.05 and *p < 0.05, **p < 0.01, ***p < 0.001; One-way ANOVA). Experiments were performed three times. h, ATP levels in postmortem hippocampal samples from AD patients and age-matched controls. Data are shown in mean ± s.e.m. (n = 7 biologically independent samples; ***p < 0.001; Two-sided Student’s t-test). i, Quantification of autophagosomes and autolysosomes in the APP and control cells with the ptfLC3 plasmid, with data shown in mean ± s.e.m. (n = 20 neurons from 3 independent experiments; ***p < 0.001; Two-sided Student’s t-test). Full scans of all the blots are in Supplementary Note.
Supplementary Figure 2 Monitoring of neuronal mitophagy in vivo.
Transgenic nematodes expressing mtRosella biosensor in neuronal cells treated with NMN, UA, and AC. Decreased GFP/DsRed ratio of mtRosella indicates neuronal mitophagy stimulation. DCT-1, PDR-1 and PINK-1 were required for neuronal mitophagy induction in response to UA, NMN and AC treatment. Scale bars, 20 μm. Quantitative data shown in Fig. 2d. Experiments were repeated independently three times with similar results.
Supplementary Figure 3 Molecular mechanisms of mitophagy induction by UA and AC and defective mitophagy in the Aβ1–42 (CL2241) nematodes.
a, Effects of UA and AC on the protein levels of a list of mitophagy-related proteins. The human SH-SY5Y cells were treated with UA (10–100 µM) and AC (10–100 µM) for 24 h, followed by detection of protein expression through western blotting. b, UA induces PINK1 expression in the APP/PS1 mice. The APP/PS1 mice were treated with UA (200 mg/kg/day) by daily gavage for 2 months starting from 6 months of age, and then the hippocampal tissues were subjected to western blotting analysis. c, Representative images showing changes of neuronal mitophagy in Aβ1–42 (CL2241) nematodes under normal and oxidative stress conditions (paraquat/para. 8 mM). Colocalization between the mitophagy receptor DCT-1 fused with GFP and the autophagosomal protein LGG-1 fused with DsRed depicts mitophagy events. Scale bars, 2 μm. For (a), experiments were repeated independently twice with similar results. For (b), one repeat. For (c), experiments were repeated independently twice with similar results. Full scans of all the blots are in Supplementary Note.
Supplementary Figure 4 The optimized doses of NMN, UA, and AC do not induce general macro-autophagy in C. elegans.
a-b, Supplementation with NMN (5 mM), UA (0.1 mM), and AC (1 mM) does not alter general autophagy levels in C. elegans neurons. Representative images showing transgenic nematodes expressing the autophagosomal marker LGG-1 fused with GFP (a) or DsRed (b) (n = 30 neuronal cells/group; ***p < 0.001; one-way ANOVA followed by Sidak’s multiple comparisons test). Scale bars, 5 μm. c-h, C. elegans transgenic animals expressing full-length plgg-1GFP::LGG-1, plgg-2GFP::LGG-2 and patg-18ATG-18::GFP fusion proteins indicative of autophagic activity treated with NMN (5 mM), UA (0.1 mM), and AC (1 mM) (n = 30 nematodes/group; ***p < 0.001; one-way ANOVA followed by Sidak’s multiple comparisons test). Several tissues were evaluated, including intestines (c-d; Scale bars, 40 μm), embryos (e; Scale bars, 10 μm), muscles (f; Scale bars, 20 μm), and neurons (g; Scale bars, 5 μm). Starvation-induced autophagy used as an internal positive control. Error bars, ± s.e.m.
Supplementary Figure 5 Mitophagy induction protects against memory impairment in C. elegans AD models.
We evaluated memory defects in the AD nematodes using an aversive olfactory learning paradigm. To determine whether memory retention is dependent on mitophagy in the Aβ1–42 (CL2355) nematodes, we generated three mitophagy mutant strains, pink-1(tm1779);CL2355, pdr-1(gk488);CL2355, and dct-1(tm376);CL2355. a-c, NMN-, UA-, and AC-induced memory improvement depends differentially on PINK-1, PDR-1 and DCT-1 activity (n = 400 nematodes/group; n.s., p > 0.05, ****p < 0.0001; Two-way ANOVA followed by Tukey’s multiple comparisons test). d, Beneficial effects of NMN, UA, and AC on memory of Aβ1–42 expressing nematodes (CL2355) differentially depend on mitophagy genes. For all experiments, nematodes were treated with NMN (5 mM) and AC (1 mM) for 2 generations, and with UA (0.1 mM) from eggs to the day of experiment for one generation. Adult Day 1 nematodes were used for the memory assay. For all worm experiments, 2 to 4 independent experiments were performed. For all ‘dot-plot’ figures, center value represents mean and error bars represent s.e.m.
Supplementary Figure 6 Mitophagy induction improves mitochondrial homeostasis and mitochondrial function in APP/PS1 AD mice and APOE4/E4 iPSC-derived neurons.
APP/PS1 AD mice were treated with UA (200 mg/kg/day) or AC (30 mg/kg/day) by daily gavage for 2 months starting at 6 months of age, and subsequently tested in behavioral studies. Thereafter, mice were sacrificed for tissue collection. a, Representative EM images of neuronal mitochondria (n = 3 mice/group). b, Summary of mitochondrial parameters and mitophagy events in hippocampus tissues upon EM analysis. Data were shown in mean ± s.e.m. (n = 3 mice/group; ***p < 0.001 compared with WT (veh), ###p < 0.001 compared with APP/PS1 (veh); One-way ANOVA). c-e, Measurement of mitophagy levels (c), mitochondrial content (d) and mitochondrial ROS (e) in freshly isolated neurons from hippocampal tissues of treated and untreated animals. Center value represents mean and error bars represent s.e.m. (n = 3 mice/group; *p < 0.05, **p < 0.01, and ***p < 0.001; One-way ANOVA). f-h, Measurement of mitophagy (f), mitochondrial content (g), and mitochondrial ROS (h) in isolated neurons from prefrontal cortex (PFC) tissues of treated and untreated animals. Center value represents mean and error bars represent s.e.m. (n = 3 mice/group; *p < 0.05, **p < 0.01, and ***p < 0.001; One-way ANOVA). i, UA improves mitochondrial function in APOE/E4 iPSC-derived neurons. The APOE/E4 iPSC-derived neurons and WT controls were treated with UA (50 µM) for 24 h, followed by the evaluation of OCR using a seahorse machine. Data were shown in mean ± s.e.m. (n = 6 technical repeats; One-way ANOVA with ***p < 0.001). J-k, UA increases OCR in the Aβ1–42 (CL2355) nematodes. Experiments for (j-k) were performed together with Fig. 2e, f and, thus share the same data for the vehicle groups (n = 3 independent experiments; **p < 0.01, and ***p < 0.001; One-way ANOVA). l, UA treatment reduced mitochondrial ROS levels in WT, Aβ1–42 (CL2355), and tau (BR5270) nematodes. UA (0.1 mM) was fed from L4 to adult day 3, followed by isolation of fresh mitochondria, stained with DFCA, and the ROS signals were quantified through FACS. Data were shown in mean ± s.e.m. (n = 3 independent experiments; ***p < 0.001 compared with WT veh.; ##p < 0.01 or ###p < 0.001 compared with each own respective vehicle control; One-way ANOVA).
Supplementary Figure 7 Mitophagy induction increases memory, diminishes soluble Aβ in PFC, and improves the quality of microglial mitochondria in the APP/PS1 AD mice.
APP/PS1 AD mice were treated with UA (200 mg/kg/day) or AC (30 mg/kg/day) by daily gavage for 2 months starting from 6 months to 8 months, tested in behavioral assays, and then sacrificed for tissue collection. a, Representative images of the latency to a hidden platform in the Morris water maze test (n = 13 mice in the WT veh. group, or 11 mice in all the other groups). b, There was no difference in swimming speed between groups in Morris water maze test. Center value represents mean and error bars represent s.e.m. (n = 13 mice in the WT veh. group, or 11 mice in all the other groups). c, Mitophagy stimulation improves spatial memory in the Y-Maze. Center value represents mean and error bars represent s.e.m. (n = 13, 12, 14, 12 mice for each group). d, Quantification of GFAP using histological tissues. Center value represents mean and error bars represent s.e.m. (n = 30, 30, 25, 11 sections, from 3 mice, for each group). e-f, Analysis of soluble and insoluble Aβ1–42 and Aβ1–40 levels in prefrontal cortex (PFC) tissues using an ELISA method. Center value represents mean and error bars represent s.e.m. (n = 9 mice in the AD UA. group, or 8 mice in all the other groups; *p < 0.05, **p < 0.01; One-way ANOVA). g, Quantification of Western blot data of the indicated proteins from 3 mice/group (associated with Fig. 4b). Data are shown in mean ± s.e.m. (n.s., p>0.05 and *p < 0.05, **p < 0.01, ***p < 0.001; One-way ANOVA). h, Protein levels of APP intermediates in hippocampal samples in response to UA and AC supplementation (n = 3 in the veh. group and the UA group, n = 4 in the AC group). Quantification values are shown in mean ± s.e.m. (**p < 0.01 with p =0.029; One-way ANOVA). i, Representative EM images of mitochondria in microglial cells (n = 3 mice/group). Error bars, ± s.e.m. j, Quantification of a list of parameters related to microglial phagocytosis, numbers and morphology (activation). Supplementation of UA and AC resulted in enhanced Aβ plaque sequestration by microglia in hippocampus (n = 5 mice/group; ***p < 0.001; One-way ANOVA). UA and AC treatment influences the microglia population, the number of processes and the process length of microglia in hippocampus. Data are shown in mean ± s.e.m. (n = 5 mice/group; ***p < 0.001; One-way ANOVA). Full scans of all the blots are in Supplementary Note.
Supplementary Figure 8 Bioinformatic analysis indicates that mitophagy induction improves neuronal function in APP/PS1 AD mice.
APP/PS1 AD mice were treated with UA (200 mg/kg/day) by daily gavage for 2 months starting from 6 months of age. Hippocampal tissue was collected for microarray analysis. a, Effects of UA treatment on mRNA levels of designated neuron-specific genes. Right side, neuronal functions of designated proteins. b, Changes of GO pathways among designated groups. n = 5, 6, 4 mice for AD (veh.), AD (UA), and WT (veh.), respectively.
Supplementary Figure 9 Rescue of learning and memory deficits by NMN, UA, and AC is dependent on mitophagy.
a, A summary of the effects of UA on pTau levels (associated to Fig. 5a). Conclusions were based on two independent experimental repeats and with similar results. b-e, Using an aversive olfactory learning paradigm, we investigated whether memory retention is dependent on mitophagy in the Tau (BR5270) nematodes. We generated three mitophagy mutant Tau strains, pink-1(tm1779);BR5270, pdr-1(gk488);BR5270, and dct-1(tm376);BR5270. b-d, PINK-1, PDR-1 and DCT-1 were differentially required to mediate memory improvement in transgenic animals expressing Tau (BR5270) upon NMN, UA and AC supplementation. Center value represents mean and error bars represent s.e.m. (n = 400 nematodes/group; n.s., p > 0.05, ****p < 0.0001; Two-way ANOVA followed by Tukey’s multiple comparisons test). For all experiments, nematodes were treated with NMN (5 mM) and AC (1 mM) for 2 generations, or with UA (0.1 mM) from eggs to the day of experiment. Adult Day 1 nematodes were used for the memory assay. For all worm experiments, 2 to 4 independent experiments were performed. e, Beneficial effects of NMN, UA, and AC on memory of Tau (BR5270) nematodes depend differentially on mitophagy genes. Conclusions were based on two independent experimental repeats and with similar results.
Supplementary Figure 10 Effects of sex difference on mitophagy-induction-induced benefits in two AD mouse models.
a-b, The APP/PS1 mice were treated with UA (200 mg/kg/day) or AC (30 mg/kg/day) by daily gavage for 2 months starting from 6 months of age, and then the Aβ1–42 and Aβ1–40 levels in the hippocampal region were detected using standard ELISA techniques. Data shown in changes of hippocampal Aβ1–42 (a) or Aβ1–40 (b) levels in male and female mice. For the mouse numbers in (a) and (b), n = 8 (4 males + 4 females) in WT (veh.), n = 8 (4 males + 4 females) in AD (veh.), n = 9 (5 males + 4 females) in AD (UA), and n = 8 (5 males + 3 females) in AD (AC). Center value represents mean and error bars represent s.e.m. (*p < 0.05, **p < 0.01, ***p < 0.001; One-way ANOVA). Sex difference of the data shown in Fig. 3f, g were reanalyzed here. c-f, effects of one-month UA treatment on memory performance in 3xTgAD mice. Thirteen-month old 3xTgAD mice were treated with UA (200 mg/kg/day) by daily gavage for 1 month. To investigate any sex difference, the data show in Fig 5i–l were further analyzed here. Contextual and cued fear conditioning test (c, d), object recognition test (e), and Y-maze test (f) were performed. For the mouse numbers using in c-f, n = n = 7 (5 males + 2 females) in WT (veh.), n = 7 (3 males + 4 females) in 3xTgAD (veh.), n = 7 (3 males + 4 females) in 3xTgAD (UA). Center value represents mean and error bars represent s.e.m. (*p < 0.05, **p < 0.01, ***p < 0.001; Two-sided Student’s t-test was used for the comparison between 2 groups, while One-way ANOVA was used to compare three groups.).
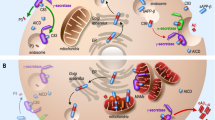
Supplementary Figure 11 Working model.
Upper panel: The roles of mitophagy were evaluated in AD pathology utilizing postmortem human AD brain samples, AD iPSC-derived neurons, and transgenic animal models of AD, including C. elegans and mice. Lower panel: We propose the synergistic roles and relationships between Aβ, p-Tau, and defective mitophagy in AD progression. In the red panel: The underlying cause in most AD cases is complex, likely reflecting risk associated with aging, multiple genetic factors as well as non-genetic (for example, environmental, lifestyle/behavioral and metabolic) factors. These factors can directly/indirectly cause mitophagy defects, leading to accumulation of damaged mitochondria, a major feature in both familial and sporadic AD patients. Defective mitophagy, damaged mitochondria, and Tau tangles/Aβ plaques, exacerbate one another (dashed arrow, further work necessary), causing neurodegeneration and impaired phagocytosis by microglia, and the gradual development of AD pathology in brain. Evidence of Tau/Aβ-induced reduction of mitochondrial motility were from (Ram D. et al., Science 2008; Tammineni P et a., Autophagy 2017). In the blue panel: Mitophagy induction maintains a healthy mitochondrial pool through efficient clearance of dysfunctional organelles. Healthy mitochondria augment neuronal function and survival as well as promote the clearance of extracellular Aβ plaques by microglia. Proficient mitophagy maintains a healthy brain.
Rights and permissions
About this article

Cite this article
Fang, E.F., Hou, Y., Palikaras, K. et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22, 401–412 (2019). https://doi.org/10.1038/s41593-018-0332-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41593-018-0332-9
This article is cited by
-
Mutual interaction of neurons and astrocytes derived from iPSCs with APP V717L mutation developed the astrocytic phenotypes of Alzheimer’s disease
Inflammation and Regeneration (2024)
-
Bioinformatic identification and experiment validation reveal 6 hub genes, promising diagnostic and therapeutic targets for Alzheimer’s disease
BMC Medical Genomics (2024)
-
Pediococcus acidilactici reduces tau pathology and ameliorates behavioral deficits in models of neurodegenerative disorders
Cell Communication and Signaling (2024)
-
Mitochondrial metabolism in neural stem cells and implications for neurodevelopmental and neurodegenerative diseases
Journal of Translational Medicine (2024)
-
A review on traditional Chinese medicine natural products and acupuncture intervention for Alzheimer’s disease based on the neuroinflammatory
Chinese Medicine (2024)
