
Abstract
Aging is the strongest risk factor for Alzheimer’s disease (AD), although the underlying mechanisms remain unclear. The chromatin state, in particular through the mark H4K16ac, has been implicated in aging and thus may play a pivotal role in age-associated neurodegeneration. Here we compare the genome-wide enrichment of H4K16ac in the lateral temporal lobe of AD individuals against both younger and elderly cognitively normal controls. We found that while normal aging leads to H4K16ac enrichment, AD entails dramatic losses of H4K16ac in the proximity of genes linked to aging and AD. Our analysis highlights the presence of three classes of AD-related changes with distinctive functional roles. Furthermore, we discovered an association between the genomic locations of significant H4K16ac changes with genetic variants identified in prior AD genome-wide association studies and with expression quantitative trait loci. Our results establish the basis for an epigenetic link between aging and AD.
Similar content being viewed by others


Main
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. Accumulation of intercellular β-amyloid plaques and intracellular neurofibrillary tangles are two hallmarks of AD that may drive neuronal death and the corresponding dramatic loss of cognitive abilities. A complex interaction between genetic and environmental factors likely contributes to the molecular processes that drive AD. Although genetic variation in specific genes increases the risk of AD1, age is the strongest known risk factor2. How molecular processes of aging predispose to AD, or become deregulated in AD, remains to be understood.
Studies in model organisms such as yeast and Caenorhabditis elegans show that epigenetic factors that integrate environmental stimuli into structural changes in the chromatin are major determinants of whole organism aging, mean lifespan and health span3. In mouse, epigenetic marks such as histone acetylation are associated with learning and age-related memory decline4,5. Histone acetylation is reduced at memory genes in mouse models for AD, and treatments with nonselective histone deacetylase inhibitors aiming to reverse loss of acetylation have shown promising results in restoring synaptic and cognitive plasticity in mouse models of AD5.
The power and unbiased nature of genome-wide studies can reveal mechanisms previously unknown to contribute to disease pathogenesis. However, their application to the study of human brain has been limited by the availability of postmortem tissue and the stability of nuclear molecules. Nonetheless, several studies have examined the stability of the chromatin, including histone H3 acetylation and methylation, under different conditions of postmortem interval, tissue pH, tissue storage (frozen versus fixed) and chromatin preparation (native versus cross-linked); these studies have shown that histone modifications can be stably detected within a wide range of postmortem interval (5–72 h) and pH (6.0–6.8)6,7,8,9,10. The past ~5 years have seen several interrogations of the human brain epigenome through chromatin immunoprecipitation sequencing (ChIP-seq) studies showing chromatin changes, for example H3K4me3, during development and substance abuse11,12.
Among the histone acetylation marks, H4K16ac is a key modification because it regulates chromatin compaction, gene expression, stress responses and DNA damage repair13,14,15,16. In model organisms, modulators of H4K16ac play a role in whole organism aging and cellular senescence17,18. Also, senescent cells display H4K16ac enrichment over promoter regions of expressed genes19. Therefore, we considered that epigenetic regulation by H4K16ac may be involved in aging of the human brain and perhaps in the progression of AD. Here we compare the genome-wide profiles of H4K16ac in the brain tissue of AD patients with age-matched and younger individuals without dementia, to elucidate key mechanisms that drive AD. In particular, our findings indicate that the normal course of age-related, and perhaps protective, changes in brain H4K16ac is perturbed in AD. Our findings provide insights into epigenetic alterations that underlie AD pathology and provide a foundation for investigating pharmacological treatments targeting chromatin modifiers that could ameliorate the progression of AD.
Results
H4K16ac is redistributed during normal aging and AD
To begin to elucidate the role of H4K16ac in aging and AD, we profiled the genome-wide enrichment of H4K16ac by ChIP-seq in the lateral temporal lobe (one of the regions affected early in AD) of postmortem brain tissue from either cognitively normal elder individuals (hereafter ‘Old’, n = 10, mean age = 68), AD subjects (n = 12, mean age = 68), or younger cognitively normal subjects (hereafter ‘Young’, n = 9, mean age = 52; Fig. 1a and Supplementary Table 1). All selected AD subjects had high levels of AD neuropathological changes while the Young and Old controls had no change or minimal changes. In addition, to reduce the number of explanatory variables to a minimum, we controlled for gender (mainly male subjects), comorbidity (excluding cases with other neuropathologies) and neuronal loss (excluding cases with severe loss; see Methods section “Brain tissue samples” for full description).

a, Coronal section of human brain indicating the lateral temporal lobe (red circle) used in this study. b, Bar plot of total number of H4K16ac peaks. c, UCSC Genome browser track view of H4K16ac peak at the SLC35D1 gene promoter in Young, Old and AD subjects. d, Venn diagram of peak overlap among Young, Old and AD subjects. e–g, Meta-profile of H4K16ac enrichment at (e) TSSs (±1 kb) of constitutive peaks; (f) TSS (±1 kb) where no peak is detected; and (g) intergenic constitutive peaks (peaks shared across Young, Old and AD subjects).
We performed H4K16ac ChIP-seq in individual brain samples, marking each sequencing library with a unique bar code, and subsequently pooled sequencing reads across samples of the same group to improve coverage and sensitivity of peak detection (Supplementary Table 2). H4K16ac peaks were detected in each group using the MACS2 peak calling method (false discovery rate < 1 × 10−3), and differential peak enrichment was statistically assessed by considering the enrichment of the corresponding region in individual ChIP-seq samples.
Because neuronal loss could potentially account for some of the H4K16ac changes observed in AD, we additionally quantified neuron percentages in the samples through NeuN (a neuron-specific mark) immunostaining of temporal lobe sections (Supplementary Fig. 1a). This showed a mild but not significant trend in neuronal reduction in both normal aging and AD (Supplementary Fig. 1b; P = 0.087, one-way ANOVA). Despite this mild trend, we additionally assessed whether there was any correlation between neuronal proportions across all samples and H4K16ac peaks detected in the combined data analysis. To improve accuracy, neuron proportions for this analysis were measured by flow cytometry in NeuN-stained nuclei isolated from the same brain region used for ChIP-seq (see Methods section “Neuron quantification by flow cytometry” and Supplementary Table 1). Using principal component analysis of the top 10,000 peaks by standard deviation (s.d.) we measured the Spearman’s correlation coefficient for the first two principal components, PC1 and PC2, which revealed no correlation between neuronal fractions and H4K16ac (Spearman’s ρ PC1 = –0.006; Spearman’s ρ PC2 = 0.076), highlighting a lack of contribution from any neuronal losses. Furthermore, to reduce the risk of this potentially confounding variable to a minimum, we masked from the analysis the top 50,000 peaks associated with neuronal proportion (10% of such peaks) by Spearman’s ρ (see Methods section “ChIP-seq analysis” for details) and then processed the data downstream.
Using this method to call peaks in each study group, we detected ~239,000 peaks in Young, ~349,000 peaks in Old and ~323,000 peaks in AD subjects (Fig. 1b), indicating an overall increase in the total number of H4K16ac peaks with age but not with AD. Representative peaks at the SLC35D1 gene, which codes for a nucleotide sugar transporter, provided a clear example of the higher accumulation around the transcription start site (TSS) in Old compared to Young or AD subjects (Fig. 1c); comparison of the individual samples showed similar accumulations of higher levels in Old compared to Young or AD subjects (Supplementary Fig. 2). The lower number of peaks in AD subjects compared to Old could reflect either loss or lack of complete H4K16ac upregulation with age in AD subjects. However, when comparing peaks across the three study groups, both gains and losses were evident, despite the overall higher number of peaks in Old (Fig. 1d). Comparison of the constitutive peaks (~114,000 peaks common to Young, Old and AD subjects) with the remaining peaks in each group showed that at least 50% of peaks in each group were redistributed, thus suggesting that Young, Old and AD subjects had different chromatin states (Fig. 1d).
Examination of the enrichment profile of the constitutive H4K16ac peaks showed a bimodal distribution around the TSS (Fig. 1e) compared to TSSs where no H4K16ac peaks were called (Fig. 1f). Similarly to the trend observed in overall peak number (Fig. 1b), the constitutive TSS peaks showed a higher level of H4K16ac in Old compared to similar levels in Young and AD subjects (Fig. 1e). In addition to the constitutive TSS peaks, we detected smaller intergenic peaks corresponding to regulatory elements, such as enhancers (Fig. 1g). Thus, both the total number of H4K16ac peaks and the level of acetylation at the TSS of constitutive peaks were higher in Old compared to Young or AD subjects.
We examined the genome-wide locations of H4K16ac accumulation in our data relative to previous observations. Because no H4K16ac ChIP-seq data are available in the brain, we compared our results with genome-wide H4K16ac data from mouse20 (Supplementary Fig. 3a) and human cells15 (Supplementary Fig. 3b). The comparisons revealed a high degree of similarity in peak location and genomic compartmentalization, with 68% of human fibroblast peaks (IMR90 cells) being detected in the constitutive brain peaks, confirming the reliability of our data. Furthermore, tissue enrichment analysis showed brain as the top enriched category (Supplementary Fig. 3c) in the constitutive peaks, providing further confidence to proceed with the analysis.
To gain insight into the dynamics of the H4K16ac changes, we quantified the number of peaks that were gained or lost in each pairwise situation. Comparison of Young and Old revealed a substantially higher number of peaks gained in Old (~196,000) than lost in Old (~86,000; Fig. 2a). Comparison between Old and AD subjects indicated a higher number of peaks lost in AD subjects (~166,000) than gained in AD subjects (~140,000; Fig. 2b). Comparison of Young to AD subjects showed a higher number of peaks gained with AD than lost (~177,000 peaks gained versus ~92,000 peaks lost; Fig. 2c). This analysis underscored that the redistribution in H4K16ac peaks was remarkably different during normal aging compared to AD: during aging, H4K16ac trends toward gains, whereas in AD it trends toward losses.

a–c, Venn diagram of H4K16ac peak overlap between Young and Old (a), Old and AD subjects (b), and Young and AD subjects (c). d–f, Scatter plot of H4K16ac fold-change vs. peak size average (measured as area under the curve or AUC) for (d) Young vs. Old, (e) Old vs. AD subjects and (f) Young vs. AD subjects comparisons for peaks called in Young, Old or AD subjects. Blue dots represent peaks with significant changes (P < 0.05, Welch’s t test, two-sided) in H4K16ac enrichment. For graphical representation, 1,000 randomly chosen points are shown in each panel. g–i, Histogram of H4K16ac fold-change vs. frequency for peaks with significant (P < 0.05, Welch’s t test, two-sided) H4K16ac changes (blue dots in d–f) for (g) Young vs. Old, (h) Old vs. AD subjects and (i) Young vs. AD subjects comparisons. j–l, Boxplot of H4K16ac fold-changed based on the distance of the peak from the closest TSS ordered into quintiles for peaks with significant (P < 0.05, Welch’s t test, two-sided) H4K16ac changes (blue dots in d–f) for (j) Young to Old, (k) Old to AD subjects and (l) Young to AD subjects comparisons. Boxplots show minimum, first quartile, median (center line), third quartile and maximum.
Given the overall increase in H4K16ac peaks with aging, we wanted to gain further insight into its dynamics. We therefore expanded our analysis to quantitative measurements of H4K16ac enrichment. This would also ensure that the observed trends were statistically significant, since patient heterogeneity could in principle contribute to variable peaks. For each peak detected in Young, Old or AD subjects, we measured the corresponding area under the curve in each patient and compared it across the three study groups. When comparing Young to Old, we detected ~20,000 peaks with significant increase in H4K16ac and ~7,000 peaks with significant loss in H4K16ac with age (P < 0.05, Welch’s t test; Fig. 2d,g). In contrast, comparison of Old to AD subjects showed a reversed pattern in H4K16ac gains and losses, with ~25,000 peaks with H4K16ac losses and ~9,000 peaks with H4K16ac gains in AD subjects (P < 0.05, Welch’s t test; Fig. 2e,h). The number of H4K16ac peaks gained or lost in the Young-to-AD subjects comparison was similar, with ~11,000 peaks lost versus ~13,000 peaks gained in AD subjects (P < 0.05, Welch’s t test; Fig. 2f,i).
To assess the genomic locations of peaks with significant H4K16ac changes relative to TSSs, we first divided these peaks into quintiles based on distance to the nearest TSS and measured the change in enrichment. In the Young-to-Old comparison (Fig. 2j), we found that the variance in H4K16ac fold-change was smaller in the quintile closest to the TSS, despite the fact that the median values were invariant across all quintiles. This was not the case for the comparison of Old to AD subjects or for Young to AD subjects (Fig. 2k,l), where there were no differences in variance across the quintiles. The smaller variance near the TSS for gains in Old (Fig. 2j) may point to a functional impact of H4K16ac on the proximal gene, which is possibly lost in AD. Thus, changes in H4K16ac associated with age, and with disease in AD subjects, appear to preferentially affect the regulatory regions most likely to impact gene expression.
To specifically address whether H4K16ac changes affect nearby gene expression, we performed RNA-seq in individual patient samples from the same brain region (Supplementary Table 3). Overall, we found a positive linear correlation between the enrichment at the closest H4K16ac peak (relative to the TSS) and gene expression in Young, Old and AD subjects (Supplementary Fig. 4a–c). In addition, a mild correlation was evident between the magnitude of differential gene expression and differential enrichment of the nearest H4K16ac peak for the significantly (P < 0.05, false discovery rate < 0.05) differentially expressed genes (P values of correlation ranging between 1 × 10−1 and 4 × 10−29; Supplementary Fig. 4d–f). We also observed agreement between published microarray datasets of gene expression from hippocampal sections21 and our RNA-seq dataset (Supplementary Fig. 5). Taken together, these data indicate that the changes in H4K16ac associated with age and AD correlated with nearby gene expression.
H4K16ac changes during normal aging are negatively correlated with changes in AD
We next asked whether the direction of H4K16ac changes is correlated with the processes of aging and disease, as how these processes interrelate is an important and outstanding question in the neurodegeneration field. To do this, we made pairwise comparisons of H4K16ac fold-changes for all peaks among the three processes: Young-to-Old representing aging; Old-to-AD subjects representing disease; and Young-to-AD subjects representing components of aging mixed with components of disease. The relationships between these three sets of H4K16ac changes are represented in three-dimensional space, where each comparison is represented as a projection onto two-dimensional space (Fig. 3a). This analysis revealed a positive linear correlation between aging and aging mixed with disease (Young-to-Old versus Young-to-AD subjects; Fig. 3b), thus demonstrating a component of normal aging in AD. Also, a positive linear correlation was detected between aging mixed with disease and disease alone (Young-to-AD subjects versus Old-to-AD subjects; Fig. 3c), suggesting a strong, age-independent disease component. In clear contrast, a remarkable and robust negative linear correlation was observed between aging and disease (Young-to-Old versus Old-to-AD subjects; Fig. 3d). This latter finding indicates that aspects of normal aging fail to occur or are dysregulated in AD and is consistent with the observations above of an opposite trend in H4K16ac enrichments during normal aging and AD, with predominant gains in normal aging and predominant losses in AD (Fig. 2). Indeed, as discussed below, the negative correlation between aging and disease clarifies an important question in the field, that is, whether AD is a simple exacerbation of aging or rather a dysregulation of aging. Our results reveal the more complex latter scenario, where there is a clear component of dysregulation of aging in the pathology of AD.

a, 3D scatter plot showing the correlations between H4K16ac changes during aging (Young-to-Old), disease (Old-to-AD subjects) and aging mixed with disease (Young-to-AD subjects) for all H4K16ac peaks detected in the three groups (black dots). Projections on the 2D orthogonal subspaces represent pairwise comparison of the three processes (blue, red and green dots), which are enlarged in panels b–d. b–d, Scatter plot shows correlation between H4K16ac changes in (b) Aging and Disease + aging (positive correlation), (c) Disease + aging and Disease (positive correlation) and (d) Aging and Disease (negative correlation). For graphical representation, 500 randomly chosen points are shown in each case. Pearson correlation coefficient for the entire dataset is indicated. Black dots in b–d represent centroids of underlying ovals.
Three classes of H4K16ac changes detected in AD: age-regulated, age-dysregulated and disease-specific
Having established an overall pattern of H4K16ac changes in aging and disease, we focused on identification of functional pathways. For gene ontology (GO) analysis, we considered all significant H4K16ac changes (P < 0.05, Welch’s t test; Fig. 4a,d) up to 10 kb from TSSs to include regulatory elements such as enhancers. Categories of genes showing significantly increased or decreased H4K16ac (P < 0.05, Welch's t test) during aging included terms related to response to oxygen levels, insulin stimulus, aging, inflammatory response, defense response, phosphorylation, actin filaments, etc., the majority of which have been shown to be altered in the aging brain and in cellular senescence (Supplementary Fig. 6a,b and Supplementary Table 4)22,23,24,25. Gene sets with H4K16ac gains or losses in AD included GO terms related to myeloid differentiation, cell death, and Wnt and Ras signal transduction (Supplementary Fig. 6c,d). These functional categories are in agreement with published reports of aging and AD-specific pathways. For example, immunity is known to be involved in the pathology of AD26, and the Wnt signaling pathway, required for synaptic transmission and plasticity, is downregulated by β-amyloid in AD27,28. Also, the Aβ42 oligomers have been shown to enhance the Ras–ERK signaling pathways, inducing tau hyperphosphorylation in AD29,30.

a,d, Scatter plot showing H4K16ac peak enrichment (measured as AUC) between (a) Young and Old and (d) Old and AD subjects for peaks detected in each of the three groups. Blue dots represent peaks with significant H4K16ac changes (P < 0.05, Welch’s t test, two-sided). For graphical representation, 1,000 randomly chosen points are shown in each case. b,c,e,f, Top DNA motifs from SeqPos analysis are shown for peak regions with significant (P < 0.05, Welch’s t test, two-sided) H4K16ac (b) gains or (c) losses in aging (Young-to-Old comparison) and H4K16ac (e) gains or (f) losses in AD (Old-to-AD subjects comparison; within top 6 DNA motifs by significance) within 1 kb from TSS.
To gain additional insight into the regulation of these genes, we analyzed the DNA sequence under the H4K16ac peaks near the TSSs of these genes (within 1 kb) for occurrence of transcription factor binding sites using SeqPos in the Cistrome site31. Binding sites for REST, a repressor of neuronal genes in nonbrain tissue and neuroprotective to aging brain32,33, were enriched in genes that had loss of H4K16ac with age (Fig. 4c). On the other hand, CEBPA (a regulator of proliferation and myeloid differentiation) sites were more enriched in genes with upregulated H4K16ac in AD (Fig. 4e). CEBPA expression has been correlated to clinical scores of incipient AD and is induced in microglia activated upon hypoxic stress34,35. It is therefore striking that our analyses revealed regulatory elements under H4K16ac peaks that control both stress response (REST) and immunity (CEBPA). Most notably, we detected enrichment for binding sites for the transcription factor HIC1, involved in p53-mediated DNA-damage response36 and Wnt signaling pathways37, at both classes of genes exhibiting increased H4K16ac with aging and decreased H4K16ac in AD (Fig. 4b,f).
Given the finding of HIC1-motif enrichment in genes with H4K16ac peaks displaying opposing gains in aging as compared to losses with AD, we wanted to determine more globally whether the H4K16ac changes were occurring at the same peak locations. A three-way comparison of H4K16ac enrichments comparing Young, Old and AD subjects simultaneously was performed to determine how the changes in AD were related to aging. This analysis allowed detection of three major classes of H4K16ac changes that we defined in relation to AD: age-regulated, age-dysregulated and disease-specific (Fig. 5a–f). Because the patients were collected and sequenced in two replication sets, their similarities were assessed by clustering over the three classes of peaks, revealing that they separated primarily by study group (Supplementary Fig. 7). Age-regulated changes were defined as changes that are established with normal aging (either gains or losses) and are maintained in AD (Fig. 5a,d). Age-dysregulated changes are those that are established with age (either gains or losses) and either fail to be established or fail to be maintained in AD (Fig. 5b,e). Disease-specific changes are gains or losses specific to AD and not seen with normal aging (Fig. 5c,f). In each of the three classes, the number of significant gains and losses (P < 0.05, one-way ANOVA) were similarly represented, except for the age-dysregulated class, in which the losses in AD were more pronounced (Fig. 5b,e; ~2,000 gains versus ~10,000 losses); this was anticipated given the trends seen in Fig. 2.

a–c, Peak schematic of the three classes of H4K16ac changes in AD: (a) age-regulated, (b) age-dysregulated and (c) disease-specific. Each class is further separated into two subclasses based on H4K16ac gains or losses in AD. The number of significant gains or losses (P < 0.05, one-way ANOVA) in each defined subclass is reported below the schematic. d–f, Box-plots of H4K16ac enrichment in each subclass reported in a–c. Boxplots show minimum, first quartile, median (center line), third quartile and maximum. Outliers are represented as black dots. g–i, Representative UCSC Genome browser track views of H4K16ac changes defined above. Chr, chromosome. j–l, Bar plot of top eight GO terms (Biological Process and Cellular component; DAVID Bioinformatics Resources v6.7) in each of the three classes of H4K16ac changes with at least 20 genes per term and false discovery rate (FDR) < 10%.
A functional analysis of genes showing H4K16ac changes in each of the three major classes was then performed. We considered all H4K16ac peaks within 10 kb from the TSS of the closest gene to include regulatory elements (Fig. 5g–l and Supplementary Table 5). Compared to the two-way analysis, the three-way analysis traced the enrichment changes of a peak across aging and disease, thereby specifying the exact functional pathway dysregulated. For example, categories related to neuron and synapses were found in both age-regulated and disease-specific classes of changes, pointing at neuroplasticity as a known feature of brain aging and early stages of dementia38,39. On the other hand, categories related to immunity and stress response, such as to hypoxia, were found in age-regulated and age-dysregulated classes of changes. It is known that immunity and stress responses are induced in aging22,23 and that excessive glia activation is a feature of AD40; this points at age-dysregulation of immunity as a possible mechanism in AD. Regulation of cell death was present as a top category in age-dysregulated changes, reminiscent of REST-mediated stress response in aging, and in AD33. Notably, a category related to chromatin modifications was present among the age-regulated GO terms, pointing at a role for epigenetics in aging and disease. This opens the question of how genetic risk factors for AD relate to epigenetic changes; this relationship has only recently been explored in the context of human tissue aging and age-related diseases.
Regions of H4K16ac changes are enriched for AD single-nucleotide polymorphisms and regulatory expression quantitative trait loci
Genome-wide association studies (GWAS) of single nucleotide polymorphisms (SNPs) identify genetic variants associated with specific traits and complex diseases. Often these disease-associated SNPs are located outside of gene bodies and may coincide with genetic elements that are subject to epigenetic regulation, such as enhancers and promoters affecting gene expression. Since H4K16ac is known to mark both active enhancers and promoters20, we considered that there may be a significant overlap between the H4K16ac changes that we defined in AD and the AD SNPs that have emerged from GWAS. To examine this, we used a curated list of disease-associated SNPs (GWAS association P < 1 × 10−5) passing two stages of clinical testing in the International Genomics of Alzheimer’s Project meta-analysis study, which includes four different GWAS datasets41, and applied INRICH, an interval-based GWAS analysis tool, to infer their overlap with regions of H4K16ac changes.
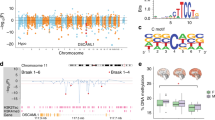
SNPs that are in linkage disequilibrium were merged into one region (using PLINK, a whole-genome association analysis tool), ultimately yielding a total of 260 merged SNP regions. We then examined these merged SNP regions for overlap with H4K16ac alterations in each of the three major classes described above (Fig. 6a–c). Notably, we found significant associations between the AD SNPs and both the age-regulated and disease-specific changes (P = 0.0018 and P = 0.0118, respectively; Fig. 6d), but not the age-dysregulated changes (P = 0.4071; Fig. 6d; see Fig. 6e for an example genomic view of disease-specific associated SNPs).

a–c, Manhattan plots showing the 260 AD SNP regions (vertical lines) in all chromosomes overlapped with H4K16ac changes (color-coded circles) for each of the three classes in Fig. 5a–c. The y axis indicates the –log10(P) of the SNP with the strongest AD association within each SNP region (P values are from the International Genomics of Alzheimer’s project (IGAP)41). d, Bar plot showing the significance (–log10(P)) of the association between the AD SNP regions and each of the three classes of H4K16ac changes assessed by INRICH. Black dotted horizontal line denotes the threshold of significance (P < 0.05). e, Representative UCSC Genome browser track view showing a cluster of AD SNPs (top) within a SNP region associated with disease-specific class of H4K16ac change (bottom; highlighted in pink) at the NME8 locus. f, Heatmap of Bonferroni adjusted P values for sampling-based analysis of H4K16ac peak overlap (three classes of changes) with temporal cortex (TX) eQTLs from Zou et al.43 eQTLs are split into those from AD cases (TX_AD), non-AD but with other brain pathologies (TX_CTL), and combined conditions (TX_ALL). g, Overlap analysis with TX eQTLs from Zou et al.43 using GREGOR.
To further assess the extent to which H4K16ac peaks mark regulatory elements involved in AD, we overlapped them with expression quantitative trait loci (eQTLs) detected in AD studies. eQTLs are genetic variants that have a substantial effect on the expression level of an mRNA transcript and therefore tend to mark transcriptional regulatory elements42. Because no meta-analysis has yet been performed with AD eQTLs, we chose one dataset43 with relatively high numbers of eQTLs and used a bootstrapping method followed by a Bonferroni correction to test the significance of association with each of the three classes of H4K16ac changes.
We used a dataset of eQTLs in temporal cortex from subjects with AD (n = 202) and subjects with non-AD pathologies (n = 197; other brain pathologies)43. This dataset contains significant eQTLs from the AD cases (85,359 SNP transcript pairs), eQTLs from the non-AD cases (68,337 SNP transcript pairs) and eQTLs from the combined set of AD and non-AD cases (156,134 SNP transcript pairs), and it was highly powered due to sample size as well as an imputation scheme (HapMap2) that allowed more SNPs to be analyzed for eQTL activity. In performing this analysis we found significant enrichments for all combinations of peak and eQTL conditions (Bonferroni P values ranging from 9 × 10−4 to 3.96 × 10−1; Fig. 6f and Supplementary Table 6), suggesting that all classes of H4K16ac peaks harbor regulatory elements involved in AD pathology as well as other neurodegenerative processes. Additional analysis on the same dataset using GREGOR44, a tool for assessing the enrichment between genetic variants and genomic elements, confirmed an association between AD eQTLs and the three classes of H4K16ac peaks (P values ranging from 4.69 × 10−50 to 4.46 × 10−14; Fig. 6g).
To assess the specificity of these enrichment results to AD eQTLs and not to any other unrelated but highly powered eQTLs, we examined the enrichment in our peaks with eQTL datasets from the GTEx (Genotype–Tissue Expression) project45 that include eQTL analyses of normal human tissues (including blood and nonbrain). Across the 44 datasets tested, we found significant enrichment in two datasets only: ‘Cells_Transformed_fibroblasts’ for age-regulated and age-dysregulated peaks and ‘Thyroid’ for age-dysregulated only (P = 0.0132); we found none for disease-specific peaks (Supplementary Fig. 8). Because only 2 of 44 datasets showed significant enrichments in our classes of peaks, and none for the disease-specific peaks, as expected for normal tissue eQTLs, these findings accentuate the significance of the association with AD eQTLs selectively.
Overall, these data underscore the significant association of AD GWAS SNPs and AD eQTLs with H4K16ac changes defined by the analysis of Young, Old and AD subject brains. This relationship emphasizes the biological relevance of chromatin changes to the genetic factors impacting AD.
Discussion
We report the first genome-wide profile of a histone modification in human brains affected with AD. Given that age is the number one risk factor for late-onset AD, we carefully designed our study to take into account epigenetic changes associated with aging by including brain samples from younger and older adults, to reveal how aging affects the epigenetic profile of AD. To our knowledge, such a comparison has not been performed previously, as most studies have used mouse models, which do not naturally develop AD with age and are artificially induced to develop plaques and tangles and which therefore can only be used to study the downstream consequences of these pathologies. In contrast, our study traces the natural changes in AD with age in human brain tissue.
We studied the acetylation of H4K16ac due to its ties with aging in model organisms and senescence in mammalian cell culture17,18,19,46,47,48. Comparison of Young and Old samples revealed a redistribution of H4K16ac with age characterized by a greater number of gains than losses (Fig. 2a,d). This finding is in general agreement with studies in yeast and mammalian senescent cells, where H4K16ac is observed to increase at specific genomic loci with age17,18,19. In contrast to normal aging, comparison of Old and AD subjects revealed a redistribution of H4K16ac in AD subjects, with more losses than gains (Fig. 2b,e). These data are congruent with analyses of histone acetylation in mouse models of AD, in which loss of acetylation (H2BK5ac, H3K14ac, H4K5ac and H4K12ac) occurs at neuronal genes49. Additionally, a targeted proteomics approach in human brains showed reduction of H3K18ac and H3K23ac in AD50. Our comparison of H4K16ac changes between aging and AD revealed that changes during aging and changes during disease are negatively correlated.
These analyses point to a model wherein Alzheimer’s disease is not simply an advanced state of normal aging, but rather dysregulated aging that may induce disease-specific chromatin structural changes and/or transcription programs. Indeed, the three-way comparison of Young, Old and AD subjects revealed a specific class of H4K16ac changes in AD subjects that were opposite to normal age-established changes (Fig. 5). Hence this suggests that certain normal aging changes could guard against AD and thus, when dysregulated, predispose to AD (Fig. 5b). A similar trend of age-dysregulation in AD has been observed for the transcriptional co-repressor REST, which increases with age but decreases in AD and plays a neuroprotective role in aging through modulation of H3K9ac33. However, no genome-wide assessment of REST has been performed in the human brain. For H4K16ac changes that are age-regulated and maintained in AD, these changes could predispose, be protective or simply correlate with aging with no effect on disease. In addition to the two classes of H4K16ac changes that are age-dependent, we observed a third class of changes that we defined as disease-specific. These changes (for example, affecting neuronal function) could be secondary to the age-associated changes, but contribute to the pathogenesis of the disease.
Finally, by assessing the relationship between AD eQTLs with the H4K16ac changes, we found significant association with the three classes of H4K16ac changes, indicating that our analysis can pinpoint regulatory mechanisms discovered through SNP analysis of AD patients. Further, the significant overlap of the AD GWAS SNPs with age-regulated and disease-specific peaks, but not age-dysregulated peaks, highlights the discovery of additional regulatory mechanisms through our epigenomic analysis and supports the inclusion of epigenomic GWAS in understanding complex diseases.
Our study proposes a mechanism to explain how age is a risk factor for AD: a particular histone modification, whose accumulation is strongly associated with aging, is dysregulated in AD. These findings and their replication in future work using patients from other biobanks open the possibility that prevention of age-dysregulation at the chromatin level may be a therapeutic avenue for AD.
Methods
Brain tissue samples
Postmortem human brain samples from lateral temporal lobe (Brodmann area 21 or 20) were obtained from the Center for Neurodegenerative Disease Research (CNDR) brain bank at the University of Pennsylvania (Penn). Informed consent for autopsy was obtained for all patients and the study was approved by the Penn Institutional Review Board (Penn IRB). The CNDR autopsy brain bank protocols were exempted from full human research (research on tissue derived from an autopsy is not considered human research; see https://humansubjects.nih.gov/human-specimens-cell-lines-data). A detailed description of the brain bank standard operating procedures has been reviewed elsewhere51. A neuropathological diagnosis of AD was established based on the presence of plaques and tangles using the CERAD scores and Braak stages, respectively52,53. The CERAD plaque score assesses the burden of neuritic plaques (0 and A–C in order of increasing frequency) in the neocortex. Braak staging is based on the progression of neurofibrillary tangles from the transentorhinal cortex (stage I) to widespread neocortical pathology including primary visual cortex (stage VI). The tissue samples were selected based on the presence of plaques and neurofibrillary tangles using the CERAD scores and Braak stages, respectively52,53. All selected AD cases had high levels of AD neuropathological changes (Braak = V/VI and CERAD = C; Supplementary Table 1). The Young and Old control brains had no or minimal neuritic amyloid plaques (CERAD = 0) or neurofibrillary tangles (CERAD = 0). None of the AD cases had other coincident neurodegenerative diseases. Control subjects had no deposits consistent with a frontotemporal lobar degeneration– or Lewy body–related pathology diagnosis. AD cases with severe neuronal loss were not included. The neuronal loss was originally assessed through semiquantitative measurements by hematoxylin and eosin (H&E) staining by board-certified neuropathologists of the CNDR. The H&E scoring for neuronal loss ranges from 0–3, where 0 signifies no neuronal loss and 3 is severe neuronal loss. Only cases with neuronal loss of 1 or 2 (mild or moderate) were included.
Quantification of neuron abundance by IF
Neural percentages in the samples were also quantified by NeuN immunofluorescence staining, as described54. Briefly, formalin-fixed, paraffin-embedded (FFPE) temporal lobe tissue sections (5 µm thick) were placed on glass slides at the CNDR (University of Pennsylvania). Slides were deparaffinized and hydrated by serial washes in xylene followed by 100%, 90% and 70% ethanol and ddH2O. Antigen retrieval was performed by keeping the slides in 10 mM citrate, pH 6.0, for 25 min in a chamber exposed to boiling water. Slides were blocked with subsequent incubations in 1 mg/mL sodium borohydride and 5% goat serum in PBS with 0.25% Triton X-100 / 0.1% BSA. Slides were incubated with 1:500 dilution of anti-NeuN antibody (MAB377, EMD Millipore55) overnight at 4 °C, washed with PBS / 0.1% BSA / 0.1% Triton X-100, and incubated 90 min at room temperature (20–25 °C) with Oregon Green 488 anti-mouse antibody (Life Technologies). Slides were subsequently incubated with 1 ug/mL DAPI for 10 min to visualize nuclei, and autofluorescence was blocked by incubation with 0.1% Sudan Black in 70% ethanol. Cover glasses were mounted on the slides using Fluoromount-G mounting medium (Southern Biotech) and slides were visualized on an Olympus BX60 Widefield Fluorescence Microscope using a Hamamatsu ORCA-ER CCD camera running Slidebook 5.5 software. For each slide we visualized 20–30 fields from random locations in each of the gray and white matter. NeuN+ cells were quantified by marking manually in a blinded fashion in Microsoft Paint and subsequently counting on Cell Profiler (Broad Institute). Total cell numbers were obtained by automated counting of DAPI+ objects in Cell Profiler. Tissue from each of the Young (n = 9), Old (n = 10) and AD groups (n = 12), also used for H4K16ac ChIP-seq, were stained and quantified. One slide was analyzed per patient. For the combined white and gray matter percentages, the counts per field of white and gray matter were averaged by weighting the gray matter count by 2.7 and white matter count by 1, to reflect the composition of the human temporal cortex56.
ChIP-seq
ChIP-seq was performed as previously described17 with modifications for brain preparation. Briefly, 200 mg brain tissue from each patient was minced on ice and nuclei were prepared by dounce homogenization in nuclei isolation buffer (50 mM Tris-HCl at pH 7.5, 25 mM KCl, 5 mM MgCl2, 0.25 M sucrose) with freshly added protease inhibitors and sodium butyrate, followed by ultracentrifugation on a 1.8-M sucrose cushion. Nuclei pellet was resuspended in 2 mL PBS and cross-linked in 1% formaldehyde for 10 min at room temperature. Crosslinking reactions were quenched with addition of glycine to 125 mM for 5 min followed by two washes in cold PBS. We then lysed 2 × 106 nuclei in nuclei lysis buffer (10 mM Tris-HCl at pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium-deoxycholate, 0.5% N-lauroylsarcosine) with freshly added protease inhibitors and sodium butyrate, and chromatin was sheared using a Covaris S220 sonicator to ~250 bp. Equal aliquots of sonicated chromatin were used per immunoprecipitation reaction with 5 µL H4K16ac antibody (Millipore, #07-32915,19) preconjugated to Protein G Dynabeads (Life Technologies), and 10% of the amount was saved as input. ChIP reactions were incubated overnight at 4 °C with rotation and washed three times in wash buffer. Immunoprecipitated DNA was eluted from the washed beads, purified and used to construct sequencing libraries with 5 ng of DNA (ChIP or input) using the NEBNext Ultra DNA library prep kit for Illumina (New England Biolabs, NEB). Libraries were multiplexed using NEBNext Multiplex Oligos for Illumina (dual index primers) and single-ended sequenced (75 bp) on the NextSeq 500 platform (Illumina) in accordance with the manufacturer’s protocol.
ChIP-seq analysis
ChIP-seq tags generated with the NextSeq 500 platform were de-multiplexed with the bcl2fastq utility and aligned to the human reference genome (assembly NCBI37/hg19) using Bowtie v1.1.157, allowing up to two mismatches per sequencing tag (parameters: -m 1–best). Peaks were detected using MACS258 (tag size = 75 bp; FDR <1 × 10−3) from pooled H4K16ac tags of patients belonging to the same study group (Young, Old or AD subjects) along with treatment-matched input tags as control. Within each pooled sample, peaks whose termini were within 150 bp were merged into one peak. The MTL method59 was then used to compare H4K16ac enrichment across the three study groups. A ‘region of analysis across the three study groups’ was defined by having at least one peak called in Young, Old or AD subjects. Furthermore, if peaks across the three study groups had their centers within 200 bp distance, the entire area including these peaks (from peak to peak termini) was considered one unique region of analysis. H4K16ac enrichment was then calculated by summing the H4K16ac tags overlapping this unique region of analysis and adjusting them by a per-patient reads-per-million (RPM) scalar coefficient and by the size of the region of analysis (in kb). Adjusted tag counts were averaged over all patients belonging to the same study group and input subtracted, resulting in an H4K16ac enrichment value, or AUC (area under the curve). AUC values were then transformed in log2(AUC + 1) for downstream analysis. Statistical significance of differential H4K16ac enrichments was assessed by performing a Welch’s t test for two-way comparisons (i.e., Young vs. Old) or one-way ANOVA for three-way comparisons (Young vs. Old vs. AD subjects). Scatter plots, histograms and box plots of ChIP-seq data were visualized using Python package Seaborn (v0.7.1.) or Matplotlib (v 1.5.1.).
Removal of confounding factors
A principal component analysis (PCA) was performed in R using the top 10,000 H4K16ac peaks by s.d. across all patients. The first two principal components (PC1 and PC2) were examined for rank correlation with neuronal proportions measured by flow cytometry, yielding Spearman’s ρ PC1 = 0.006; Spearman’s ρ PC2 = 0.076. The PCA was also performed on the ~30,000 differentially enriched H4K16ac peaks in the three classes (age-regulated, age-dysregulated, disease-specific) combined, and rank correlation was re-assessed, yielding Spearman’s ρ PC1 = –0.261 and Spearman’s ρ PC2 = 0.375. To correct for the mild correlation between neuronal proportion and the two PCs, all peaks were assessed for correlation between H4K16ac enrichment and neuronal proportion on a per-patient basis, and the top 50,000 peaks by correlation were masked. PCA was then redone on the differentially enriched H4K16ac peaks, and the correlation analysis yielded Spearman’s ρ PC1 = 0.008 and Spearman’s ρ PC2 = 0.133. Peak masking was done using custom python scripts while R was used for the PCA and correlation analyses.
Neuron quantification by flow cytometry
To remove the contribution of neuronal loss to the H4K16ac peak analysis, we measured neuronal proportions by NeuN staining and flow cytometry analysis in nuclei isolated from the same tissue regions used for ChIP-seq (values reported in Supplementary Table 1). Isolated nuclei (prepared as in the ChIP-seq protocol) were stained with an anti-NeuN antibody (Millipore # MAB 377×60; Alexa Fluor-488 conjugated) in presence of 5% goat serum and incubated in the dark for 1 h. NeuN-stained nuclei were analyzed on a BD LSR II flow cytometer (at the UPenn FACS core facility) with gates set according to nuclei size, NeuN intensity and an IgG control.
Genome browser tracks
Generation and visualization of ChIP-seq tracks was conducted as follows. BED files of each aligned dataset were converted into coverage maps using the BEDtools utility genomeCoverageBed. Resulting bedGraphs were scaled by using the RPM (reads per million) coefficient, a measure of the millions of tags sequenced per sample to correct for sequencing efficiency biases, and subsequently normalized by subtracting an input coverage map. Finally, BigWig files were generated and uploaded on the UCSC (University of California Santa Cruz) Genome Browser.
Meta-profiles
Meta-profiles of H4K16ac enrichment at TSSs were generated by taking a 2-kb window around the TSS of all RefSeq genes associated with an H4K16ac peak in Young, Old and AD subjects (or genes associated with no H4K16ac peak) and tabulating the average of H4K16ac enrichment (AUC) in 20-bp intervals. A meta-profile of intergenic peaks was generated similarly by selecting a 2-kb window around the center of H4K16ac peaks detected in each of Young, Old and AD subjects and not overlapping with gene bodies or 1-kb upstream promoter regions.
Functional analysis
Downstream functional analysis of genes targeted by H4K16ac changes was performed by associating each RefSeq transcript to its nearest peak. Gene ontology (GO) enrichment analysis of genes associated with significant H4K16ac changes was performed using DAVID (David Bioinformatics Resources v6.7)61. For representation of GO terms in the text figures, terms with shared genes were collapsed to a single representative term. Also, if one GO term was a subset of another GO term, that GO term was dropped in favor of the other (see Supplementary Tables 4 and 5 for a complete list of biological process (BP), cellular component (CC), molecular function (MF) and tissue at FDR < 10%; FDR <10% represents the threshold of significance in DAVID). DNA motif analysis was performed using SeqPos in the Cistrome site31 with default parameters and DNA motif scanning window = 1.2 kb.
RNA-seq
Total RNA was isolated from 20 mg frozen brain tissue using the RNAeasy Mini kit (Qiagen) coupled to an RNase-free DNase step (Qiagen). Ribosomal RNA was removed using the rRNA Depletion kit (NEB) and the resulting RNA was used to construct sequencing libraries using the NEBNext Ultra Directional RNA library Prep Kit for Illumina (NEB). Libraries were multiplexed using NEBNext Multiplex Oligos for Illumina (dual index primers) and single-ended sequenced (75 bp) on the NextSeq 500 platform (Illumina) in accordance with the manufacturer’s protocol.
RNA-seq tags reads were aligned to the human reference genome (assembly GRCh37.75/hg19) using STAR with default parameters. Alignments with a mapping score <10 were discarded using SAMtools and alignments mapped to mitochondria and chrUn (contigs that cannot be confidently placed on a specific chromosome) were removed using BEDtools. FeatureCounts was used to generate a matrix of mapped fragments per RefSeq annotated gene, from which genes annotated by RefSeq as rRNA were discarded. Analysis for differential gene expression was performed using the DESeq2 R package with FDR <0.05. For comparison of our RNA-seq data to published microarray data in the hippocampus of AD and control patients21, the published data were downloaded from NCBI’s GEO (accession GSE28146) and requantified using Limma. Transcripts were then organized into deciles by overall expression in control or AD subjects and compared to old or AD subjects RNA-seq respectively.
Association between AD SNPs and H4K16ac changes
To curate a list of Alzheimer’s-associated SNPs, a set of 2,371 SNPs passing stage I and stage II GWAS meta-analysis with P ≤ 1 × 10−5 were downloaded from the International Genomics of Alzheimer’s Project (IGAP)41. INRICH62 was used to infer the relationship between H4K16ac changes and PLINK-joined63 AD GWAS SNP intervals (linkage due to HapMap release 23) using standard parameters. The set of all H4K16ac changed peaks, filtered for a one-way ANOVA P < 0.05, was the background for the experiment.
IGAP is a large two-stage study based on GWAS of individuals of European ancestry. In stage 1, IGAP used genotyped and imputed data on 7,055,881 single nucleotide polymorphisms (SNPs) to meta-analyze four previously-published GWAS datasets consisting of 17,008 Alzheimer’s disease cases and 37,154 controls (The European Alzheimer’s Disease Initiative, EADI; the Alzheimer Disease Genetics Consortium, ADGC; the Cohorts for Heart and Aging Research in Genomic Epidemiology consortium, CHARGE; the Genetic and Environmental Risk in AD Consortium, GERAD). In stage 2, 11,632 SNPs were genotyped and tested for association in an independent set of 8,572 Alzheimer’s disease cases and 11,312 controls. Finally, a meta-analysis was performed combining results from stages 1 and 2.
eQTL data processing and sampling analysis
For the Zou et al. data43, eQTL data tables were downloaded from the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site at the University of Pennsylvania, funded by the National Institute on Aging (grant U24-AG041689-01). The original paper analyzed samples from cerebellum in addition to temporal cortex, but we only used the temporal cortex data due to the cortical origin of our H4K16ac measurements and because regulatory elements are variable across brain regions64. Custom awk-based bash scripts, available by request, were used to convert eQTL data tables into BED format using the liftOver utility from the UCSC Genome Browser65 to convert annotations from the hg18 genome build to hg19 to overlap with the H4K16ac peaks. Twelve AD, 10 non-AD, and 18 combined-condition eQTLs were unmapped by liftOver. We then used the intersect tool from the bedtools suite66 to overlap our H4K16ac peaks with the eQTL bed files.
For the sampling analysis, the shuffle tool from bedtools was used to generate 10,000 sets of matched control intervals, where unmappable regions, as defined by the DAC blacklisted regions, were downloaded from the UCSC genome browser and ENCODE67. For each dataset, custom scripts, also available by request, were used to summarize the overlap counts in easily parse files that were then read into the R programming language, which was used to perform the empirical enrichment analyses.
GREGOR enrichment analysis
The GREGOR tool requires LD-pruned sets of variants as input, so the sets of significant eQTLs for each target gene in each condition were pruned using PLINK v1.90b2i 64-bit68 with a cutoff of R2 ≥ 0.7 to define the LD blocks and using data from the phase 3 version 1 (11 May, 2011) European population of the 1,000 Genomes Project69. Then, using the matching reference data, the GREGOR tool was run on each set of pruned eQTLs against the H4K16ac BED format files, using an R2 threshold of 0.7, an LD window size of 1,000,000 bp and a minimum of 500 control SNPs for each index eQTL.
Statistical analysis
Statistical analysis of ChIP-seq data was performed with Welch’s t test (two-sided) or one-way ANOVA (one-sided). Differences were considered statistically significant for P < 0.05 (uncorrected for multiple hypothesis testing). Statistical analysis of RNA-seq data was performed using DESeq (Wald test) and differences were considered statistically significant for P < 0.05 (FDR < 0.05, controlled by Benjamini–Hochberg). For all figures derived by the analysis of ChIP-seq data (all figures except Supplementary Fig. 4), sample sizes were Young = 9; Old = 10; AD = 12 (independent brain samples). For RNA-seq analysis (Supplementary Fig. 4), the sample size was Young = 8; Old = 10; AD = 12 (independent brain samples, from the same subjects as those used for the ChIP-seq experiments). No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous studies in the field70,71. Data distribution was assumed to be normal, but this was not formally tested. Data collection and analysis were not performed blind to the conditions of the experiments, except for quantitative analysis of IF staining. Samples were not subject to randomization, but were assigned to experimental group based on their age and disease status (Young, Old and AD subjects). No data points were excluded from the analyses.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability
The data that support the findings of this study are available through the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE84618.
Code availability
Code and pipeline for the analyses performed in this study are available at http://165.123.66.72/btracks/sulfa/Nativio.11112017.
Change history
19 March 2018
In the version of this article initially published online, the fifth author’s name was given as Alexander Amlie-Wolf. The correct name is Alexandre Amlie-Wolf. The error has been corrected in the print, PDF and HTML versions of this article.
References
Toledo, J. B. et al. Alzheimer’s disease cerebrospinal fluid biomarker in cognitively normal subjects. Brain 138, 2701–2715 (2015).
Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 11, 332–384 (2015).
Sen, P., Shah, P. P., Nativio, R. & Berger, S. L. Epigenetic mechanisms of longevity and aging. Cell 166, 822–839 (2016).
Kawahara, T. L. et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 62–74 (2009).
Gräff, J. & Tsai, L. H. Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111 (2013).
Huang, H. S., Matevossian, A., Jiang, Y. & Akbarian, S. Chromatin immunoprecipitation in postmortem brain. J. Neurosci. Methods 156, 284–292 (2006).
Stadler, F. et al. Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J. Neurochem. 94, 324–336 (2005).
Nagy, C. et al. Effects of postmortem interval on biomolecule integrity in the brain. J. Neuropathol. Exp. Neurol. 74, 459–469 (2015).
Matevossian, A. & Akbarian, S. A chromatin assay for human brain tissue. J. Vis. Exp. 13, 717 (2008).
Bennett, D. A. et al. Epigenomics of Alzheimer’s disease. Transl. Res. 165, 200–220 (2015).
Cheung, I. et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl Acad. Sci. USA 107, 8824–8829 (2010).
Zhou, Z., Yuan, Q., Mash, D. C. & Goldman, D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc. Natl Acad. Sci. USA 108, 6626–6631 (2011).
Shogren-Knaak, M. et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847 (2006).
Akhtar, A. & Becker, P. B. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell 5, 367–375 (2000).
Sammons, M. A., Zhu, J., Drake, A. M. & Berger, S. L. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 25, 179–188 (2015).
Sharma, G. G. et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell. Biol. 30, 3582–3595 (2010).
Dang, W. et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459, 802–807 (2009).
Kozak, M. L. et al. Inactivation of the Sas2 histone acetyltransferase delays senescence driven by telomere dysfunction. EMBO J. 29, 158–170 (2010).
Rai, T. S. et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 28, 2712–2725 (2014).
Taylor, G. C., Eskeland, R., Hekimoglu-Balkan, B., Pradeepa, M. M. & Bickmore, W. A. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 23, 2053–2065 (2013).
Blalock, E. M., Buechel, H. M., Popovic, J., Geddes, J. W. & Landfield, P. W. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer’s disease. J. Chem. Neuroanat. 42, 118–126 (2011).
Lu, T. et al. Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891 (2004).
Lee, C. K., Weindruch, R. & Prolla, T. A. Gene-expression profile of the ageing brain in mice. Nat. Genet. 25, 294–297 (2000).
Chen, Q. & Ames, B. N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl Acad. Sci. USA 91, 4130–4134 (1994).
von Zglinicki, T., Saretzki, G., Ladhoff, J., d’Adda di Fagagna, F. & Jackson, S. P. Human cell senescence as a DNA damage response. Mech. Ageing Dev. 126, 111–117 (2005).
Heppner, F. L., Ransohoff, R. M. & Becher, B. Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372 (2015).
Inestrosa, N. C. & Varela-Nallar, L. Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 6, 64–74 (2014).
Wan, W., Xia, S., Kalionis, B., Liu, L. & Li, Y. The role of Wnt signaling in the development of Alzheimer’s disease: a potential therapeutic target? BioMed Res. Int. 2014, 301575 (2014).
Kirouac, L., Rajic, A. J., Cribbs, D. H. & Padmanabhan, J. Activation of Ras-ERK signaling and GSK-3 by amyloid precursor protein and amyloid beta facilitates neurodegeneration in Alzheimer’s disease. eNeuro https://doi.org/10.1523/ENEURO.0149-16.2017 (2017).
Jiang, S. et al. Trafficking regulation of proteins in Alzheimer’s disease. Mol. Neurodegener. 9, 6 (2014).
Liu, T. et al Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12, R83 (2011).
Chong, J. A. et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 80, 949–957 (1995).
Lu, T. et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454 (2014).
Blalock, E. M. et al. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl Acad. Sci. USA 101, 2173–2178 (2004).
Walton, M. et al. CCAAT-enhancer binding protein alpha is expressed in activated microglial cells after brain injury. Brain Res. Mol. Brain Res. 61, 11–22 (1998).
Chen, W. Y. et al. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123, 437–448 (2005).
Valenta, T., Lukas, J., Doubravska, L., Fafilek, B. & Korinek, V. HIC1 attenuates Wnt signaling by recruitment of TCF-4 and beta-catenin to the nuclear bodies. EMBO J. 25, 2326–2337 (2006).
Gutchess, A. Plasticity of the aging brain: new directions in cognitive neuroscience. Science 346, 579–582 (2014).
Hill, N. L., Kolanowski, A. M. & Gill, D. J. Plasticity in early Alzheimer’s disease: an opportunity for intervention. Top. Geriatr. Rehabil. 27, 257–267 (2011).
Meda, L., Baron, P. & Scarlato, G. Glial activation in Alzheimer’s disease: the role of Abeta and its associated proteins. Neurobiol. Aging 22, 885–893 (2001).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Nica, A. C. & Dermitzakis, E. T. Expression quantitative trait loci: present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. https://doi.org/10.1098/rstb.2012.0362 (2013).
Zou, F. et al. Brain expression genome-wide association study (eGWAS) identifies human disease-associated variants. PLoS Genet. 8, e1002707 (2012).
Schmidt, E. M. et al. GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 31, 2601–2606 (2015).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Contrepois, K., Thuret, J. Y., Courbeyrette, R., Fenaille, F. & Mann, C. Deacetylation of H4-K16Ac and heterochromatin assembly in senescence. Epigenetics Chromatin 5, 15 (2012).
Wan, Y. Z. et al. SIRT1-mediated epigenetic downregulation of plasminogen activator inhibitor-1 prevents vascular endothelial replicative senescence. Aging Cell 13, 890–899 (2014).
Krishnan, V. et al. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl Acad. Sci. USA 108, 12325–12330 (2011).
Gräff, J. et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226 (2012).
Zhang, K. et al. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 12, 1261–1268 (2012).
Toledo, J. B. et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimer’s Dement. 10, 477–484 (2014).
Mirra, S. S. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol. Aging 18 Suppl., S91–S94 (1997).
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H. & Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404 (2006).
Bhat, R. et al. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 7, e45069 (2012).
García-Moreno, F. & Molnár, Z. Subset of early radial glial progenitors that contribute to the development of callosal neurons is absent from avian brain. Proc. Natl Acad. Sci. USA 112, E5058–E5067 (2015).
Schenker, N. M., Desgouttes, A. M. & Semendeferi, K. Neural connectivity and cortical substrates of cognition in hominoids. J. Hum. Evol. 49, 547–569 (2005).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq aMACSa. Genome Biol 9, R137 (2008).
Chen, X. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 (2008).
Kerimoglu, C. et al. KMT2A and KMT2B mediate memory function by affecting distinct genomic regions. Cell Rep. 20, 538–548 (2017).
Huang, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Lee, P. H., O'Dushlaine, C., Thomas, B. & Purcell, S. M. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics 28, 1797–1799 (2012).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Kang, H. J. et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Vermunt, M. W. et al. Epigenomic annotation of gene regulatory alterations during evolution of the primate brain. Nat. Neurosci. 19, 494–503 (2016).
Flavahan, W. A. et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016).
Acknowledgements
We thank T. Schuck and G. Gibbons for assistance with brain tissue selection and retrieval; the Penn Flow Cytometry core for technical assistance; P. Sen and M. Sammons for edits and comments to the manuscript and P. Ortega for insightful scientific discussions. This work was funded by NIH grant R01-NS078283 (to S.L.B., N.M.B. and F.B.J.). J.Q.T. is supported by AG10124 and AG17586.
Author information
Authors and Affiliations
Contributions
R.N., F.B.J., N.M.B. and S.L.B. conceived the project. R.N. performed most of the experiments. R.N., G.D., A.B., Y.L., A.A.W., F.T., J.B.T., S.J.G., B.D.G., C.T., J.Q.T., L.S.W., F.B.J., N.M.B. and S.L.B. contributed to methodology and resources. R.N., G.D. and Y.L. analyzed ChIP-seq and RNA-seq data. G.D. performed the AD SNP enrichment analysis. A.A.W. performed the AD eQTL enrichment analysis. F.T. performed IF staining and analysis. R.N., F.B.J., N.M.B. and S.L.B. wrote the manuscript. All authors reviewed the manuscript and discussed the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Quantification of neurons in temporal cortex of Young, Old and AD subjects.
(A) Representative IF images showing total nuclei (DAPI-blue, left column), NeuN positive nuclei (green, central column) and merged images (right column) for Young (top row), Old (middle row), AD (bottom row). (B) Barplot (with data points) representing mean ± SEM of neuron percentages quantified by IF (no Young = 9; no Old = 10; no AD = 12). Sample size (no) represents individual brain samples. Differences are not significant (P=0.087, 1-way ANOVA; F (2,28)=2.669).
Supplementary Figure 2 H4K16ac enrichment in individual and pooled samples.
UCSC Genome browser track views of H4K16ac enrichment at the promoter of SLC35D1 gene in individual Young (blue), Old (orange), AD (gray) samples and corresponding pooled samples. The numbers on the right of each track refer to the sample ID as reported in Table S1.
Supplementary Figure 3 Comparison between H4K16ac peaks detected in the brain and other cell types.
(A) Genomic compartment analysis showing % of H4K16ac shared peaks (in common between Young, Old and AD) is similar to Taylor et al. [20] in mouse ESC and NPC cells. (B) Heatmap showing H4K16ac enrichment in Young, Old and AD over previously mapped H4K16ac peaks in human IMR90 cells [15]. Peaks are sorted in decreasing order of Young brain enrichment. (C). Barplot showing % of tissue-specific expressed genes targeted by H4K16ac (top 10% of Young peaks and up to 1 kb from TSS) for H4K16ac shared peaks (UniProt tissue expression database, DAVID). Tissue terms were filtered by gene count > 200 and FDR < 1%.
Supplementary Figure 4 Correlation between H4K16ac and gene expression in Young, Old and AD subjects.
(a-c) Scatter plot of H4K16ac peak enrichment (log2 AUC +1) vs gene expression (log2 DESeq-score +1) of genes expressed in (a) Young, (b) Old and (C) AD considering the closest peak from the TSS. For graphical representation, 3000 randomly chosen points are shown in each panel. (d-f) Absolute H4K16ac fold-changes (closest peak from the TSS) vs absolute gene expression changes for the significantly (P < 0.05, FDR< 0.05) differentially expressed genes for the comparisons (d) Young to Old, (e) Old to AD and (f) Young to AD. Linear regression trendlines and Pearson correlation coefficients are indicated.
Supplementary Figure 5 Comparison between RNA-seq and published microarray data (Blalock et al. 2011) in Old and AD subjects.
Boxplot showing RNA-seq gene expression (DEseq-scores) in Old (a) or AD (b) for genes identified in a published AD microarray study [21] from human hippocampus. All expressed genes from the published microarray data (control Old and AD) were sorted into ten groups of increasing expression and compared to Old (a) or AD (b) in our RNA-seq data.
Supplementary Figure 6 GO analysis of genes with H4K16ac changes in aging and AD subjects.
(A-D) Bar plot for top eight most significantly enriched GO terms (BP; DAVID) for genes with significant (P < 0.05, Welch's t-test, two-sided) H4K16ac (A) gains or (B) losses during aging (Young-Old comparison) and H4K16ac (C) gains or (D) losses in AD (Old-AD comparison). Reported GO terms contain > 20 genes and FDR < 10%.
Supplementary Figure 7 Correlation across patients at the three classes of H4K16ac changes.
Dendrogram of Spearman Rank Correlation across patients (rep1 and rep2) for the three classes of peaks (Age-regulate, Age-dysregulated and Disease-specific; P < 0.05, 1-way Anova) showing patients clustering according to study group (Young, Old and AD). The numbers for each patient refer to the sample ID as reported in Table S1.
Supplementary Figure 8 H4K16ac overlap analysis with tissue eQTLs from the GTEx project.
Heatmap of Bonferroni adjusted p-values for sampling-based analysis of H4K16ac overlap (the three classes of changes) with tissue eQTLs from the GTEx project.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–8
Supplementary Table 1 - Tissue donor information
Tissue donors were categorized by gender, age at death, postmortem interval (PMI), Cerad scores and Braak stages, RNA integrity number (RIN) of RNA extracted from the same brain region, neuronal fractions (measured by flow cytometry) and cause of death. Four control brains had a small degree of neurofibrillary tangles (Braak I/II) limited to the medial temporal lobe; low level pathology is commonly present in aged subjects and is not associated with cognitive changes in the absence of other brain pathologies (Montine et al., Acta Neuropathol, 2012. 123(1): p. 1-11).
Supplementary Table 2 - Sequencing and alignment statistics for H4K16ac ChIP-seq experiment
For each H4K16ac ChIP-seq sample the number of total tags, uniquely aligned tags and % uniquely aligned tags are reported.
Supplementary Table 3 - Sequencing and alignment statistics for RNA-seq experiment
For each RNA-seq sample the number of total tags, uniquely aligned tags and % uniquely aligned tags are reported. No RNA-seq is available for sample 1 as there was no tissue left for this experiment.
Supplementary Table 4
Functional analysis of genes with significant H4K16ac changes (P < 0.05, Welch's t-test, two-sided) in the 2-way comparison (Aging and Disease).
Supplementary Table 5
Functional analysis of genes with significant H4K16ac changes (P < 0.05, 1-way Anova) in the three-way comparison (Age-regulated, Age-dysregulated, Disease-specific).
Supplementary Table 6
Supplementary Table 6.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nativio, R., Donahue, G., Berson, A. et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat Neurosci 21, 497–505 (2018). https://doi.org/10.1038/s41593-018-0101-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41593-018-0101-9
This article is cited by
-
Novel histone post-translational modifications in Alzheimer’s disease: current advances and implications
Clinical Epigenetics (2024)
-
Advanced patient-specific microglia cell models for pre-clinical studies in Alzheimer’s disease
Journal of Neuroinflammation (2024)
-
Histone acetylation in an Alzheimer’s disease cell model promotes homeostatic amyloid-reducing pathways
Acta Neuropathologica Communications (2024)
-
Alzheimer’s Disease-Related Epigenetic Changes: Novel Therapeutic Targets
Molecular Neurobiology (2024)
-
ACSS2-dependent histone acetylation improves cognition in mouse model of Alzheimer’s disease
Molecular Neurodegeneration (2023)
