
Abstract
We introduce the TRUST4 open-source algorithm for reconstruction of immune receptor repertoires in αβ/γδ T cells and B cells from RNA-sequencing (RNA-seq) data. Compared with competing methods, TRUST4 supports both FASTQ and BAM format and is faster and more sensitive in assembling longer—even full-length—receptor repertoires. TRUST4 can also call repertoire sequences from single-cell RNA-seq (scRNA-seq) data without V(D)J enrichment, and is compatible with both SMART-seq and 5′ 10x Genomics platforms.
Similar content being viewed by others

Main
Both T and B cells can generate diverse receptor (TCR and BCR, respectively) repertoires, through somatic V(D)J recombination, to recognize various external antigens or tumor neoantigens. Following antigen recognition, BCRs also undergo somatic hypermutations (SHMs) to further improve antigen-binding affinity. Repertoire sequencing has been increasingly adopted in infectious disease1, allergy2, autoimmune3, tumor immuology4 and cancer immunotherapy5 studies, but it is an expensive assay and consumes valuable tissue samples. Alternatively, RNA-seq data contain expressed TCR and BCR sequences in tissues or peripheral blood mononuclear cells (PBMC). However, because repertoire sequences from V(D)J recombination and SHM are different from the germline, they are often eliminated in the read-mapping step.
Previously we developed the TRUST algorithm6,7,8, utilized to de novo assemble immune receptor repertories directly from tissue or blood RNA-seq data. When applied to The Cancer Genome Atlas (TCGA) tumor RNA-seq data, TRUST revealed profound biological insights into the repertoires of tumor-infiltrating T cells6 and B cells8, as well as their associated tumor immunity. Although less sensitive than TCR-seq and BCR-seq, TRUST is able to identify the abundantly expressed and potentially more clonally expanded TCRs/BCRs in the RNA-seq data that are more likely to be involved in antigen binding9. Recent years have also seen other computational methods introduced for immune repertoire construction from RNA-seq data, including V’DJer10, MiXCR11, CATT12 and ImRep13. These methods focus on reconstruction of complementary-determining region 3 (CDR3), with limited ability to assemble full-length V(D)J receptor sequences, although CDR1 and CDR2 on the V sequence still contribute considerably to antigen recognition and binding. For example, five out of six mutations predicted in a recent study to influence antibody affinity in the acidic tumor environment are located in CDR1 and CDR2 (ref. 14), and four out of nine positions contributing most to 4A8 antibody binding to the SARS-CoV-2 spike protein are in CDR1 and CDR2 (ref. 15). Therefore, algorithms that can infer full-length immune receptor repertoires can facilitate better receptor–antigen interaction modeling.
With the advance of scRNA-seq technologies, researchers can study immune cell gene expression and receptor repertoire sequences simultaneously. Several algorithms, including MiXCR11, BALDR16, BASIC17 and VDJPuzzle18, have been developed to construct full-length paired TCRs or BCRs from the SMART-seq scRNA-seq platform19. In contrast to SMART-seq, droplet-based scRNA-seq platforms such as 10x Genomics20, while yielding sparser transcript coverage per cell, can process orders of magnitude more cells at lower cost. To analyze immune repertoires using the 10x Genomics platform, researchers currently need to prepare extra libraries to amplify TCR/BCR sequences.
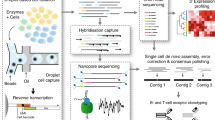
In this study, we redesigned the TRUST algorithm to TRUST4 with substantially enhanced features and improved performance for immune repertoire reconstruction (Fig. 1a). First, TRUST4 supports fast extraction of TCR/BCR candidate reads from either FASTQ or BAM files. Second, TRUST4 prioritizes candidate read assembly by abundance and assembles all candidate reads with partial overlaps against contigs, thus increasing algorithm speed. Third, TRUST4 explicitly represents highly similar reads in the contig consensus, thus accommodating somatic hypermutations and improving memory efficiency (Methods). Fourth, TRUST4 can assemble full-length V(D)J sequences on TCRs and BCRs. Finally, TRUST4 supports repertoire reconstruction from scRNA-seq platforms without requiring the extra 10x V(D)J amplification steps.

a–d, TRUST4 applied to bulk RNA-seq data. a, Overview of TRUST4. b, Number of TRB CDR3s reported by MiXCR, CATT, TRUST3 and TRUST4 from in silico RNA-seq data. c, Precision–recall of six bulk RNA-seq samples using BCR-seq results as the gold standard. d, Evaluation of full-length V, CDR3 and J sequences assembled by TRUST4 and MiXCR on pseudobulk RNA-seq by grouping SMART-seq data. Each row represents whether the cell’s sequences were recovered in the pseudobulk data.
We evaluated the performance of TRUST4 on TCR/BCR reconstruction from bulk RNA-seq using three different approaches. First, for TCR evaluation we used in silico RNA-seq datasets with known TRB sequences from an earlier study11. On average, TRUST4 called 281% more CDR3s than MiXCR, 22.9% more than CATT, 57.8% more than TRUST3 and maintained a zero false-positive rate across different read lengths (Fig. 1b; further parameter settings are given in Supplementary Fig. 1a). Second, for BCR evaluation, we used six tumor RNA-seq samples of ~100 million pairs of 150 base pair (bp) reads with corresponding immunoglobulin heavy-chain (IGH) BCR-seq as the gold standard8. Since BCRs also have somatic hypermutation and isotype switching during clonal expansion, we required the algorithm call to match CDR3 and V, J and C (isotype) gene assignments as BCR-seq. TRUST4 showed better precision (>18%) and sensitivity (>74%) than MiXCR in five out of six samples (Fig. 1c; further parameter settings are given in Supplementary Fig. 2a). On the sixth sample, TRUST4 lost only 6% precision with twice the sensitivity compared to MiXCR (FZ-97). We note that BCR-seq and RNA-seq were conducted on different slices of the same tumor. Even two technical replicates of repertoire sequencing on the same DNA/RNA could not achieve 100% precision and sensitivity, so the performance metrics are likely to be underestimations. TRUST4 consistently assembled more IGHs across different abundance ranges reported in BCR-seq (Supplementary Fig. 2b), and found twice as many IGHs with a single RNA copy than MiXCR. In addition, TRUST4 required only 20–25% of the time, on average, needed by MiXCR to process these samples (Supplementary Table 1), at <6 GB of memory usage on an eight-thread processor. Furthermore, TRUST4 run directly on FASTQ files was notably faster than read mapping used to generate BAM files, followed by TRUST4 run on BAM files. Third, for base-level, full-length assembly evaluation, we created pseudobulk RNA-seq data by randomly selecting 25 million read pairs from 137 SMART-seq B cells as a test case. To establish a gold standard for BCR calls, we used the 128 IGH assemblies consistently called by BALDR and BASIC at the single-cell level (Supplementary Fig. 3a). TRUST4 and MiXCR correctly identified all 128 CDR3s and TRUST4 reconstructed 93 full-length IGH sequences, while MiXCR found only 39 (Fig. 1d). TRUST4 was able to call some BCRs with only 5,000 randomly sampled read pairs in the SMART-seq dataset (18 read pairs per chain), and showed higher sensitivity than MiXCR across all abundance ranges (Supplementary Fig. 3b). The high efficiency of TRUST4 allowed us to characterize the immune repertoire in tumor samples, and we identified an association of IgA1 B-cell clonal expansion with poor prognosis in colon adenocarcinoma (COAD) from TCGA RNA-seq samples (Methods and Supplementary Fig. 4). We note that IGHA1 overexpression is not associated with survival, suggesting that immune repertoire analysis provides additional insights into tumor immunity.
Next, we evaluated TRUST4 performance on 5′ 10x Genomics scRNA-seq data on PBMC. For this dataset, the two separately processed T- and B-cell 10x V(D)J libraries served as the gold standard. When considering single cells that passed the Seurat21 cell-level quality control, TRUST4 made 5,091 T- and 1,318 B-cell calls (Fig. 2a and Supplementary Fig. 5a). Among the CDR3s reported by 10x V(D)J, TRUST4 recovered 48.1% (6,035/12,558) of TCR CDR3s and 78.0% (1,946/2,494) of BCR CDR3s. The higher sensitivity of TRUST4 on BCR is due to the higher expression level of BCR in B cells (Supplementary Fig. 5b). For precision, 94.6% of TCR CDR3s and 98.2% of BCR CDR3s from TRUST4 were identical to 10x V(D)J (Fig. 2b). Although CellRanger_VDJ was designed for 10x V(D)J data, we tested it on 5′ 10x scRNA-seq data, which have the same format. TRUST4 found 78% more TCR CDR3s and 16% more BCR CDR3s in cells that passed quality control (Supplementary Fig. 5c). In addition, TRUST4 was over ten times faster and over twice more memory efficient than CellRanger_VDJ. Furthermore, TRUST4 also reported 83 γδT cells, for which 10x V(D)J currently does not have a kit to profile. In these data, Seurat did not annotate any γδT cells but rather called 71 out of 83 TRUST4-annotated γδT cells as CD8 T cells. Close examination of gene expression in these 83 cells revealed that they had higher δV and δC gene expression but lower CD8A or CD8B expression (Supplementary Fig. 5d), supporting TRUST4’s annotation of these cells as γδT cells.

a–c, Application of TRUST4 to 5′ 10x Genomics scRNA-seq data. a, Uniform manifold approximation and projection (UMAP) of 5′ 10x Genomics PBMC data. b, Number of CDR3s matched with 10x Genomics V(D)J-enriched library from cells annotated by Seurat. c, Comparison of TRUST4-assembled V gene similarities and reference germline V gene sequences from paired, full-length IGH and IGK/IGL assemblies of 5′ 10x Genomics NSCLC data. Each dot represents one cell. Treg, regulatory T cells; RestNKs, resting natural killer cells; RestDCs, resting dendritic cells; cor, Pearson correlation.
We further tested TRUST4 on a 10x Genomics non-small cell lung cancer (NSCLC) dataset. In this case, TRUST4 called 1,241 T cells and 2,478 B cells (Supplementary Fig. 6). TRUST4 assembled 142 IGH CDR3s out of the 144 Seurat-annotated plasma B cells while 10x V(D)J found only 131. For these plasma B cells, TRUST4 also reconstructed full-length paired BCRs for 104 cells in which we observed a high correlation for SHM rate between IGHs and IGK/IGLs (Fig. 2d; Pearson r = 0.67, P = 8 × 10–15), suggesting coordinated SHMs on two chains during B-cell division. Furthermore, TRUST4 found more somatic hypermutations on IGH than on IGK/IGL (P < 1 × 10–10, two-sided Wilcoxon signed-rank test), supporting the more important role of antibody heavy chain in antigen-binding affinity.
In summary, TRUST4 is an effective method to infer TCR and BCR repertoires from bulk RNA-seq or scRNA-seq data. TRUST4 not only has high efficiency, sensitivity and precision in reconstruction of CDR3s, but can also assemble full-length immune receptor sequences from bulk RNA-seq data. Furthermore, TRUST4 can reconstruct immune receptor sequences at the single-cell level, including γδT cells, directly from 5′ 10x Genomics scRNA-seq data without specific 10x V(D)J enrichment libraries. Our results support the advantage of the 5′ 10x Genomics scRNA-seq platform, which not only provides gene expression information but also enables computational calling of immune repertoires. TRUST4 is available open source at https://github.com/liulab-dfci/TRUST4, and could be an important method for tumor immunity and immunotherapy studies.
Methods
Algorithm overview
TRUST4 reconstructs the immune repertoire in three stages: candidate reads extraction, de novo assembly and annotation (Fig. 1a).
Candidate reads extraction
TRUST4 can find candidate TCR and BCR reads from either raw sequence files or the alignment file produced by aligners such as STAR22 and HISAT23. When input is an alignment file, if a read or its mate aligns on the V, J or C locus, this read is added to the candidate read set. If a read is unmapped and is not a candidate based on mate information, TRUST4 will test whether this read has a significant overlap with V, J and C genes. If so, this read and its mate are also candidate reads. When input is raw sequence files, TRUST4 applies the significant overlap criterion to every read or read pair to find candidate reads. To identify whether a read has significant overlap with one of the V, J or C genes, TRUST4 first locates the receptor gene with the highest number of k-mer hits (default, k = 9) from the read. TRUST4 then computes the longest chain from these k-mers to filter incompatible hits. Last, if the union bases of the k-mers in the longest chain reach threshold, TRUST4 will claim that the read has a significant overlap with the gene. The threshold is maximum(21, read_length/5 + 1), so data with shorter reads have less stringent criteria. Since TRUST4 avoids alignment in the candidate reads extraction algorithm, this stage is fast even if input data are raw sequence files.
If the data have barcode information, such as 10x Genomics scRNA-seq data, TRUST4 also corrects the barcode, if erroneous, for each candidate read when given the whitelist. TRUST4 first builds barcode usage distribution from the first two million reads before correcting. Then, for each input barcode that is not in the whitelist, TRUST4 finds all the neighbor barcode within one hamming distance in the whitelist (at most, fourfold barcode length) and reports the one that is the most frequent barcode in usage distribution. If there are multiple valid neighbor barcodes with the same frequency in usage distribution, TRUST4 will correct on the base with the lowest FASTQ quality.
De novo assembly
When assembling candidate reads into immune receptor sequences, TRUST4 adopts the read overlap scheme. Cells such as plasma B cells can generate thousands of reads for each recombined receptor gene, so comparison of every pair of reads to construct the overlap graph, as in previous versions of TRUST, is inefficient. TRUST4 implements a greedy extension approach by aligning the candidate read to existing contigs, one by one. To perform alignment, TRUST4 builds an index for all k-mers in the contigs and applies the seed-extension paradigm to identify alignments. TRUST4 deems that a read overlaps with a contig if they have a highly similar (90% for BCR, 95% for TCR) alignment block containing at least 31-bp exact matches and the unaligned bases of the read are outside of the contig. Based on overlaps, TRUST4 will update contigs with the following rules. (1) If a read partially overlaps one contig, TRUST4 extends this contig; (2) if a read partially overlaps several contigs, TRUST4 merges corresponding contigs; and (3) if a read does not overlap any existing contigs, TRUST4 creates a new contig with this read’s sequence. When processing reads, TRUST4 prioritizes those derived from highly expressed TCRs/BCRs. To achieve this, TRUST4 first counts the frequency of k-mers (21-mer by default) across all candidate reads. If a read comes from a highly expressed receptor sequence, all of its k-mers would be of high frequency in the data. Therefore, the minimum frequency of a read’s k-mers is a rough indicatation of gene abundance. TRUST4 then sorts the read based on the minimum k-mer frequency rule. The ordering of reads is equivalent to picking the most frequent k-mer as the starting point in the de Bruijn-graph-based transcriptome assembler, Trinity24.
TRUST4 clusters reads with somatic hypermutations into the same contig by representing a contig as the consensus of assembled reads. Each position in the contig records four weights according to the number of reads with the corresponding nucleotide for that position. The consensus means that the nucleotide for a specific position is that with the highest weight, and the index for alignments stores the k-mers of the consensuses. Read alignment takes the weights into account to tolerate the somatic hypermutations in BCRs. For example, for a particular position, if nucleotides A and T on the contig have high weights, it is a match if the read has nucleotide A or T. Therefore, reads with different somatic hypermutations can align to the same contig, which avoids the creation of redundant contigs.
If input data are paired end, TRUST4 will use mate-pair information to extend the contigs. In the first round of contig assembly, due to read sorting and greedy extension, a contig for the abundant recombined gene attracts all reads from the same V, J and C genes even though these reads come from different recombinations. The mate-pair information fixes this issue by reassigning reads to the appropriate contigs. Reassignment will extend the contigs and update position weights in the affected consensus. When input data are SMART-seq, since there is no need to perfect assemblies for low-abundant sequences in a cell, TRUST4 can skip the extension to reduce running time.
When input contains barcode information, TRUST4 will assign the read barcode to the contig when creating a new contig, and a read can align to the contigs only with that read’s barcode. As a result, two identical reads with different barcodes will change different sets of contigs. Furthermore, the read–contig overlap criterion is relaxed and requires 17- rather than 31-bp exact matches in the alignment.
Annotation
TRUST4 aligns the assembled contigs to sequences from the international ImmunoGeneTics (IMGT) database25 to identify V, J and C genes. The IMGT database curates the sequences for V, D, J and constant genes and is widely used to annotate BCR and TCR sequences, such as in previous TRUST versions and MiXCR. Besides the sequences, IMGT also annotates the start position of CDR3 in the V gene (104th amino acids of the V gene in IMGT coordination). IMGT also defines the end position of CDR3 as amino acid W/F in the amino acid motif W/FGxG in the J gene. TRUST4 determines the CDR3 coordinate based on these IMGT conventions after identification of V and J genes. If the contig is too short to identify the V gene, TRUST4 locates the CDR3 start position as amino acid C in the motif YYC by testing all open reading frames.
In the final step of annotation, TRUST4 retrieves somatic hypermutated CDR3s and estimates CDR3 abundances. If a read fully covers the CDR3 on a contig and the CDR3 sequence from the read is different from the consensus, TRUST4 will report the CDR3 from the read. If there is no such read, TRUST4 directly reports the consensus CDR3 sequence. In abundance estimation, if reads partially overlap with CDR3, each could be compatible with several different complete CDR3 sequences. Therefore, TRUST4 applies the expectation-maximization algorithm26, similar to that in RSEM27, to distribute read counts iteratively to their compatible CDR3s. For TCR, TRUST4 filters CDR3s with abundance <5% of the most abundant CDR3 from the same contig, to avoid sequencing errors.
For CDR3s that have only start or end positions determined in the contigs, TRUST4 reports these as partial CDR3s and tries to extend partial TCR CDR3s as in MiXCR. As an example of a missing start position, the extendable partial CDR3 must overlap with the identified V gene in the contig but cannot reach the start position. This scenario could happen when the V gene is identified through mate-pair information. TRUST4 then fills the missing sequences with germline sequences of the V gene to complete the partial CDR3. In scRNA-seq, TRUST4 also utilizes information across all cells to extend partial TCR CDR3s. For two cells, A and B with the same V and J genes on both chains, cell B can extend its partial CDR3 if B has a complete CDR3 identical to A’s corresponding complete CDR3, and B’s partial CDR3 is a substring of A’s corresponding complete CDR3.
Sequence data
We tested TRUST4 on both in silico and real data. In silico bulk RNA-seq data for evaluation of TCRs were generated using scripts from MiXCR11, where repseqio (https://github.com/repseqio/repseqio) and ART28 generated the simulated TRB and RNA-seq data. As a result, each of the in silico RNA-seq samples contained 1,000 read fragments randomly derived from 1,000 recombined TRBs. To evaluate BCR reconstruction, we used six sets of lung cancer RNA-seq data and their pairing BCR-seq data from our previous study8. iRepertoire processed the BCR-seq data, and results were the gold standard for evaluation. For SMART-seq evaluation we used three SMART-seq datasets from BALDR: AW1, 1620PV (AW2-AW3) and VH_CD19pos. For pseudobulk RNA-seq data we first added a pseudomate for 1620PV single-end data with a sequence of one nucleotide N. We then randomly selected 25 million read pairs across all the cells of these three samples to create the pseudobulk RNA-seq. Finally, 56, 33 and 11% of the pseudobulk RNA-seq data were derived from AW1, 1620PV and VH_CD19pos, respectively. We applied the same procedure to generate psuedobulk samples with fewer read pairs (12 million, 6 million, …, 2,500, 1,000). The 10x Genomics scRNA-seq data and 10x V(D)J data were downloaded from the 10x Genomics website.
Performance evaluation
All methods utilized were tested with their default parameters without explicit clarification. BAM files as input for TRUST4 were generated by STAR v.2.5.3a. In this study we used MiXCR v.3.0.12, CATT with GitHub commit ID 0e7b462, TRUST3 v.3.0.3, BALDR with GitHub commit ID e865b45 and BASIC v.1.5.1. All evaluations in this work were at the nucleotide level: for example, the match of CDR3s of TRUST4 and BCR-seq gold standard meant that their nucleotide sequences were identical. TRUST4 can report both partial and complete CDR3s, but we considered only complete CDR3s in evaluations.
In the TCR evaluation with in silico RNA-seq data, evaluation criteria were based on scripts from MiXCR’s manuscript11. We added read length 150 bp and ran MiXCR with default parameters. In MiXCR’s original manuscript, the authors used the option ‘--badQualityThreshold 0' for higher sensitivity (MiXCR_0), and TRUST4 still found about 8% more CDR3s than MiXCR_0 on average (Supplementary Fig. 1a). Furthermore, TRUST4 with input from FASTQ and BAM files showed almost identical results, which demonstrated the efficiency of the candidate extraction method. TRUST4 was also the most, or among the most, sensitive method in assembly of CDR3s for TRB chains with varying numbers of reads (Supplementary Fig. 1b).
For bulk RNA-seq data we mainly evaluated the performance of reconstructing BCR heavy chains, including V, J and C gene assignments and CDR3 sequences. We considered gene assignments in addition to CDR3 sequences in the evaluation because IGHs had different C genes as isotypes, such as IgM, IgG1 and IgA1, and were critical in determining antibody functions. Since CATT could not report the C gene and TRUST3 focused only on CDR3 assembly, we omitted CATT and TRUST3 in this evaluation. For the match of V and J genes we ignored allele ID. For example, if the V gene was annotated as IGHV1-18*01 we regarded it as IGHV1-18. In the evaluation, we excluded assemblies missing the V, J or C gene from TRUST4 and MiXCR. The IGH abundances reported by TRUST4 had a better correlation with the corresponding abundances in BCR-seq than MiXCR (Pearson r = 0.57 versus 0.53 on average; Supplementary Fig. 2a). We further checked the precision–recall curve by ranking inferred IGHs by abundance (top 100, 500, 1,000, …), and TRUST4 consistently outperformed MiXCR across different thresholds (Supplementary Fig. 2a). On these real data, MiXCR_0 did not outperform MiXCR as in the in silico data, suggesting that the parameter is not effective with real data. TRUST4 with FASTQ and BAM input still showed identical performance across six samples in this real-data evaluation. We further evaluated the performance on CDR3 sequences only, which included results from TRUST3 and CATT. TRUST4 showed the highest sensitivity consistently across all six samples, and reported 11% more correct CDR3s than MiXCR_0, the second most sensitive method, with similar precision on average.
The evaluations with SMART-seq data focused on whether the methods could reconstruct all nucleotides in the variable domain. If the assembled V and J sequences were shorter than gene lengths in the IMGT database, we regarded that as unreconstructed. The match of V or J sequences means that nucleotide bases were the same for regions annotated as V or J genes. In other words, we ignored bases before the V or after the J gene. In addition to the pseudobulk RNA-seq data from the three samples, we ran TRUST4 on original cell-level data and compared it with BALDR and BASIC on all three samples. We selected the top abundant heavy chain and light chain from TRUST4, and these were identical to either BALDR and BASIC on 272 out of 274 chains (Supplementary Fig. 3). The comparison result indicated that TRUST4 can effectively reconstruct the immune repertoire from SMART-seq scRNA-seq data.
For evaluation with 10x Genomics data, we used TCR library and IG library results from 10x Genomics Immune profiling (10x V(D)J) as the gold standard. Since the computational software CellRanger_VDJ can report multiple CDR3s for a cell, we regarded the most abundant CDR3 as the true CDR3 for a chain, and the less abundant CDR3s as secondary. TRUST4 took the BAM file generated by CellRanger as input, which included the barcode information in the field “CB”. TRUST4 also took FASTQ files as input, and corrected the erroneous barcodes based on the whitelist provided in the CellRanger package. TRUST4 with FASTQ input reported almost identical results to that with BAM input (Supplementary Fig. 5c). Even though CellRanger_VDJ (v.3.1.0) was designed for 10x V(D)J data, we ran it on the 10x 5′ scRNA-seq data using IMGT sequences as reference with eight cores. In our analysis of full-length assemblies, somatic hypermutation rate was represented by the proportion of matched bases (similarity) between the assembled V genes and germline sequences (Fig. 2c). When there are many somatic hypermutations, the similarity will be low. Besides the 5′ scRNA-seq data, we also evaluated TRUST4 on the 3′ 10x Genomics PBMC data, with only 335 cells having reconstructed CDR3s (Supplementary Fig. 7). We used LM22 marker genes from CIBERSORT29 to determine cell types.
Application of TRUST4 on TCGA COAD RNA-seq samples
We explored immune repertoire features on 466 COAD RNA-seq samples in TCGA cohorts. To reduce the effects of somatic hypermutated CDR3s, we first clustered highly similar CDR3 nucleotide sequences of the same length and with the same V and J gene assignments reported from TRUST4. We selected the similarity cutoff as 0.8 by comparison of similarity distribution among pairs of CDR3s within (intra-patient) and between (inter-patient) samples, where inter-patient distribution can be regarded as background random CDR3 pair similarity (Supplementary Fig. 4a). Therefore, we defined the clonotype for TCR as the CDR3 sequence and that for BCR as the cluster with the same V and J gene assignments and similar CDR3 sequences. Although TRB and IGH clonalities were positively correlated with their respective expression (Spearman r = 0.346 for TRB, r = 0.085 for IGH), they contained additional information on TCR and BCR clonal expansion (Supplementary Fig. 4b). The expression for a chain is computed by the sum of transcripts per million (TPM) obtained from TCGA cohorts on the constant genes of a chain. We defined clonality as 1 – (normalized Shannon entropy) based on the clonotype definition above.
We identified that IgA1 antibody clonal expansion was related to patient survival in COAD. Unlike in melanoma, where IgG1 and IgA expression and abundance fractions were respectively positively and negatively associated with survival time11, we did not observe such association of survival time in COAD (Supplementary Fig. 4c). However, higher clonality of IgA1 B cells was correlated with significantly shorter survival time (P = 8.1 × 10–5, hazard ratio = 9.14 by Cox proportional hazards regression corrected by age), supporting the immunosuppressive property of IgA antibodies30. We hypothesize that the clonal expansion of IgA1 B cells could be related to gut microbiota31, and future work is needed to elucidate the mechanisms involved.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The original scripts for generation and evaluation of in silico RNA-seq data are available at https://github.com/milaboratory/mixcr-rna-seq-paper.
The six bulk RNA-seq samples for BCR evaluation are available in the SRA repository, accession code PRJNA492301, and their matched iRepertoire data are available at https://bitbucket.org/liulab/ng-bcr-validate/src/master/iRep. SMART-seq data are available in the SRA repository, accession code SRP126429. 10x Genomics scRNA-seq data are available at https://support.10xgenomics.com/single-cell-vdj/datasets/3.1.0/vdj_nextgem_hs_pbmc3, https://support.10xgenomics.com/single-cell-vdj/datasets/2.2.0/vdj_v1_hs_nsclc_5gex and https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.1.0/5k_pbmc_protein_v3_nextgem.
Code availability
TRUST4 source code is available at https://github.com/liulab-dfci/TRUST4. Evaluation code for this work is available at https://github.com/liulab-dfci/TRUST4_manuscript_evaluation.
References
Lee, J. et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat. Med. 22, 1456–1464 (2016).
Kiyotani, K. et al. Characterization of the B-cell receptor repertoires in peanut allergic subjects undergoing oral immunotherapy. J. Hum. Genet. 63, 239–248 (2018).
Liu, S. et al. Direct measurement of B-cell receptor repertoire’s composition and variation in systemic lupus erythematosus. Genes Immun. 18, 22–27 (2017).
Kurtz, D. M. et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood 125, 3679–3687 (2015).
Riaz, N. et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949 (2017).
Li, B. et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet. 48, 725–732 (2016).
Li, B. et al. Ultrasensitive detection of TCR hypervariable-region sequences in solid-tissue RNA-seq data. Nat. Genet. 49, 482–483 (2017).
Hu, X. et al. Landscape of B cell immunity and related immune evasion in human cancers. Nat. Genet. 51, 560–567 (2019).
Cao, Y. et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182, 73–84 (2020).
Mose, L. E. et al. Assembly-based inference of B-cell receptor repertoires from short read RNA sequencing data with V’DJer. Bioinformatics 32, 3729–3734 (2016).
Bolotin, D. A. et al. Antigen receptor repertoire profiling from RNA-seq data. Nat. Biotechnol. 35, 908–911 (2017).
Chen, S.-Y., Liu, C.-J., Zhang, Q. & Guo, A.-Y. An ultrasensitive T-cell receptor detection method for TCR-seq and RNA-seq data. Bioinformatics 36, 4255–4262 (2020).
Mandric, I. et al. Profiling immunoglobulin repertoires across multiple human tissues using RNA sequencing. Nat. Commun. 11, 3126 (2020).
Sulea, T. et al. Structure-based engineering of pH-dependent antibody binding for selective targeting of solid-tumor microenvironment. mAbs 12, 1682866 (2020).
Chi, X. et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 369, 650–655 (2020).
Upadhyay, A. A. et al. BALDR: a computational pipeline for paired heavy and light chain immunoglobulin reconstruction in single-cell RNA-seq data. Genome Med. 10, 20 (2018).
Canzar, S., Neu, K. E., Tang, Q., Wilson, P. C. & Khan, A. A. BASIC: BCR assembly from single cells. Bioinformatics 33, 425–427 (2017).
Rizzetto, S. et al. B-cell receptor reconstruction from single-cell RNA-seq with VDJPuzzle. Bioinformatics 34, 2846–2847 (2018).
Hagemann-Jensen, M. et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 38, 708–714 (2020).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Stuart, T. et al. Comprehensive Integration of single-cell data. Cell 177, 1888–1902 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Lefranc, M.-P. IMGT, the international ImMunoGeneTics information system. Cold Spring Harb. Protoc. 2011, 595–603 (2011).
Dempster, A. P., Laird, N. M. & Rubin, D. B. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Series B Stat. Methodol. 39, 1–22 (1977).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Huang, W., Li, L., Myers, J. R. & Marth, G. T. ART: a next-generation sequencing read simulator. Bioinformatics 28, 593–594 (2012).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Sharonov, G. V., Serebrovskaya, E. O., Yuzhakova, D. V., Britanova, O. V. & Chudakov, D. M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 20, 294–307 (2020).
Bunker, J. J. & Bendelac, A. IgA responses to microbiota. Immunity 49, 211–224 (2018).
Acknowledgements
We thank B. Li and C. Wang for the helpful discussions. We also acknowledge funding from NIH (grant U01CA226196) and China Scholarship Council (Z.O. and Y.C.) to support this work. The study used data generated by the TCGA Research Network that are not otherwise cited: https://www.cancer.gov/tcga
Author information
Authors and Affiliations
Contributions
L.S., X.H. and X.S.L conceived the project. L.S. designed and implemented the methods. L.S., D.C., Z.O., Y.C., X.H. and X.S.L. evaluated the methods and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
X.S.L. is a cofounder, scientific advisory board (SAB) member and consultant of GV20 Oncotherapy and its subsidiaries, SAB memner of 3DMedCare, consultant for Genentech, stockholder of AMGN, JNJ, MRK and PFE and receives sponsored research funding from Takeda and Sanofi. X.H. conducted the work while a postdoctorate fellow at DFCI, and is currently a full-time employee of GV20 Oncotherapy.
Additional information
Peer review information Nature Methods thanks Aly Azeem Khan, Gur Yaari and the other, anonymous reviewer(s) for their contribution to the peer review of this work. Lin Tang was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Table 1 and Figs. 1–7.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Song, L., Cohen, D., Ouyang, Z. et al. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat Methods 18, 627–630 (2021). https://doi.org/10.1038/s41592-021-01142-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-021-01142-2
This article is cited by
-
B cell receptor repertoire analysis in primary Sjogren’s syndrome salivary glands identifies repertoire features associated with clinical activity
Arthritis Research & Therapy (2024)
-
Blood transcriptomics analysis offers insights into variant-specific immune response to SARS-CoV-2
Scientific Reports (2024)
-
Predictability of B cell clonal persistence and immunosurveillance in breast cancer
Nature Immunology (2024)
-
IGH repertoire analysis at scale: deciphering the complexity of B cell infiltration and migration in esophageal squamous cell carcinoma
Cancer Gene Therapy (2024)
-
The HLA-DRB1*09:01-DQB1*03:03 haplotype is associated with the risk for late-onset Alzheimer’s disease in APOE \({{\varepsilon }}\)4–negative Japanese adults
npj Aging (2024)
