
Abstract
Clinical outcomes of catheter ablation for atrial fibrillation (AF) are suboptimal due, in part, to challenges in achieving durable lesions. Although focal point-by-point ablation allows for the creation of any required lesion set, this strategy necessitates the generation of contiguous lesions without gaps. A large-tip catheter, capable of creating wide-footprint ablation lesions, may increase ablation effectiveness and efficiency. In a randomized, single-blind, non-inferiority trial, 420 patients with persistent AF underwent ablation using a large-tip catheter with dual pulsed field and radiofrequency energies versus ablation using a conventional radiofrequency ablation system. The primary composite effectiveness endpoint was evaluated through 1 year and included freedom from acute procedural failure and repeat ablation at any time, plus arrhythmia recurrence, drug initiation or escalation or cardioversion after a 3-month blanking period. The primary safety endpoint was freedom from a composite of serious procedure-related or device-related adverse events. The primary effectiveness endpoint was observed for 73.8% and 65.8% of patients in the investigational and control arms, respectively (P < 0.0001 for non-inferiority). Major procedural or device-related complications occurred in three patients in the investigational arm and in two patients in the control arm (P < 0.0001 for non-inferiority). In a secondary analysis, procedural times were shorter in the investigational arm as compared to the control arm (P < 0.0001). These results demonstrate non-inferior safety and effectiveness of the dual-energy catheter for the treatment of persistent AF. Future large-scale studies are needed to gather real-world evidence on the impact of the focal dual-energy lattice catheter on the broader population of patients with AF. ClinicalTrials.gov identifier: NCT05120193.
Similar content being viewed by others
Main
Atrial fibrillation (AF) is the most common type of cardiac arrhythmia and is the leading cardiac cause of stroke1. Catheter-based ablation is an effective and safe treatment for patients with AF1,2. The cornerstone of this procedure is the electrical isolation of the pulmonary veins (PVs)3. In patients with persistent AF, catheter ablation often results in less favorable clinical outcomes compared to those in patients with paroxysmal AF4. This disparity has been commonly ascribed to a broader and more complex arrhythmogenic substrate in persistent AF5,6,7.
Conventional catheter ablation technologies are optimized for treating paroxysmal AF but have major shortcomings in treating persistent AF8. Specifically, focal radiofrequency catheter-based ablation procedures typically involve the use of a specialized mapping catheter to create a three-dimensional electro-anatomical map of the left atrium. Subsequently, a separate focal ablation catheter with a solid metal tip applies radiofrequency energy in a sequential point-by-point fashion to form a contiguous set of ablation lesions. Major shortcomings of these AF ablation procedures include (1) limited effectiveness due to the technical challenges of placing contiguous lesions, leading to conduction gaps and subsequent arrhythmia recurrence9,10; (2) risk of atrio-esophageal fistula, phrenic nerve paralysis and PV narrowing11; and (3) the need to use two separate catheters for mapping and ablation, which increases procedural complexity and cost2. ‘Single-shot’ ablation catheters are designed to create a circular lesion pattern for pulmonary vein isolation (PVI) using one or a few applications, but they fall short in generating the additional linear ablations often required for persistent AF.
The novel technological platform investigated in this study integrates high-density electro-anatomical mapping of the heart with dual-energy ablation, employing either radiofrequency or pulsed field energies, within a single lattice-tip catheter system. Pulsed field ablation (PFA), a non-thermal energy source, ablates tissue by using microsecond pulses of electric fields to destabilize cell membranes12,13. Notably, PFA has a preferential effect on myocardial tissue while minimizing impact on adjacent non-cardiac tissues, such as the esophagus and the phrenic nerve14,15,16. Preclinical and first-in-human studies have demonstrated that this lattice-tip catheter with its large footprint generates full-thickness atrial myocardial lesions and achieves PVI faster, with fewer applications and with greater durability compared to conventional catheters, and does so without causing thermal injury to surrounding structures17,18,19,20,21,22,23. Accordingly, SPHERE Per-AF was a randomized, single-blind, non-inferiority clinical trial that compared the lattice-tip dual-energy ablation platform with a conventional radiofrequency ablation platform in the treatment of drug-refractory persistent AF.
Results
Participants
From December 2021 to December 2022, patients were screened for the trial, and 469 were enrolled. After a roll-in phase that included 37 patients (up to two patients per site to familiarize the operators with the technology), 432 patients were randomly assigned to undergo ablation using either the investigational (219 patients) or the control (213 patients) systems, as shown in the participant flow chart (Fig. 1). After accounting for 12 dropouts (seven investigational and five control), a total of 420 patients (212 investigational and 208 control) received the intended treatment across 20 centers by 40 operators, with each operator treating an average of 6 ± 7 patients using the investigational system. The gap between randomization and procedure was 4.9 ± 5.7 d for the investigational arm and 6.0 ± 10.7 d for the control arm. Premature exit from the study occurred in 12 participants (three investigational and nine control). Details on participant dropouts and premature study exits are provided in Fig. 1 and Extended Data Table 2.

Of the 432 patients randomized to either treatment, 219 were assigned to undergo treatment with the investigational system and 213 were assigned to undergo treatment with the control system. Before the ablation procedure, seven patients in the investigational arm and five patients in the control arm were withdrawn. The primary analysis consisted of 212 patients in the investigational arm and 208 patients in the control arm. Of these 420 patients, 408 (97.1%) completed the trial with 12 months of follow-up.
Baseline characteristics are provided in Table 1 and were largely balanced between the two groups, including age, comorbidities, time since AF diagnosis, left atrial dimension, history of cardioversions and usage of class I/III anti-arrhythmic drugs.
For both arms, overall adherence to trial follow-up visits was 97% (2,303/2,385). Compliance to Holter and electrocardiogram (ECG) monitoring was 84% (696/828) and 85% (1,062/1,247), respectively. Compliance to trans-telephonic transmissions was 92% (3,417/3,706) and resulted in a rate of close to two transmissions per patient per month for both arms (Extended Data Table 3). Adherence was similar between groups.
Primary effectiveness and safety endpoints
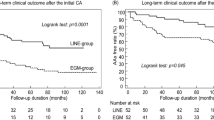
The primary effectiveness analysis included 210 investigational patients and 202 control patients in the primary analysis cohort with primary effectiveness outcome data available. The primary effectiveness endpoint success rate was 73.8% for the investigational arm and 65.8% for the control arm. The observed difference in primary effectiveness success was 8.0% in favor of the investigational arm (95% confidence interval (CI): −0.9% to 16.8%), meeting the criteria for non-inferiority (P < 0.0001; Table 2 and Fig. 2b). The 1-year Kaplan–Meier estimates were 73.5% for the investigational arm and 65.2% for the control arm (Fig. 2a). A post hoc analysis showed that freedom from atrial arrhythmias for the investigational arm (76.7%, 161/210) was non-inferior to the control arm (72.8%, 147/202) (P < 0.0001; Extended Data Table 4). All patients in both study groups underwent PVI. In the investigational arm, acute PVI was successfully achieved using only the assigned study device. In the control arm, one PV could not be isolated using the assigned device and was ultimately treated with adjunctive cryo-balloon ablation. In the investigational arm, acute block was achieved across all ablation lines using only the assigned study device. In the control arm, one mitral line could not be completed using the assigned device and required alcohol ablation of the vein of Marshall.

a, Kaplan–Meier analysis of the primary effectiveness endpoint. Shown are the Kaplan–Meier estimates of freedom from the primary effectiveness endpoint, which is a composite of the freedom from initial procedural failure, repeat ablation at any time and arrhythmia recurrence, anti-arrhythmic drug initiation or escalation or cardioversion after a 3-month blanking period. Comparison of the investigational arm versus control was performed using the two-sided log-rank test. b, Farrington–Manning analysis of the primary effectiveness endpoint. Trial success with respect to effectiveness was defined as non-inferiority of the primary effectiveness endpoint based on binomial proportions using the one-sided Farrington–Manning test with a non-inferiority margin of 15% and a one-sided alpha of 0.025. The observed difference in primary effectiveness success was 8.0% in favor of the investigational arm (95% two-sided CI: −0.9% to 16.8%), based on primary effectiveness for 210 investigational and 202 control patients. Visualized here is the pre-specified 15% non-inferiority margin, the point estimate of the difference between treatment and control and the two-sided 95% CI of the difference. c, Procedural characteristics. Left, energy application time includes both radiofrequency and PFA for the investigational device and radiofrequency time for the control device. Visualized here is the mean and 95% CI for the investigational arm (7.1 (6.8, 7.4), n = 212) and the control arm (36.4 (33.9, 38.8), n = 206) (P < 0.0001). Middle, transpired ablation time is the time between the first and last application, which includes the elapsed time for both PVI and any additional linear ablation. Visualized here is the mean and 95% CI for the investigational arm (46.7 (44.0, 49.4), n = 212) and the control arm (73.5 (68.8, 78.2), n = 208) (P < 0.0001). Right, skin-to-skin procedure time is the time elapsed from first venous access to last sheath removal. Visualized here is the mean and 95% CI for the investigational arm (100.9 (96.8, 105.1), n = 212) and the control arm (126.1 (119.4, 132.8), n = 208) (P < 0.0001). Contingent upon trial success, sequential testing with an overall one-sided alpha of 0.025 was performed on a pre-specified set of endpoints to further examine superiority of the investigational arm versus control. Adjustments were made for multiple comparisons with the sequential testing method.
The primary safety analysis included 212 investigational patients and 208 control patients in the primary analysis cohort. Primary safety events occurred in three (1.4%) patients in the investigational arm and in two (1.0%) patients in the control arm (difference: 0.4%; 90% CI: −2.8% to 3.7%; P < 0.0001 for non-inferiority; Table 3). The events in the investigational arm included hospitalizations for pulmonary edema due to hypertensive urgency, exacerbation of chronic obstructive pulmonary disease and hemoptysis (with no signs of active bleeding by endoscopy and no recurrence). In the control arm, two hospitalizations occurred due to pulmonary edema. In both groups, there were no reports of atrial-esophageal fistula, PV stenosis, tamponade or permanent phrenic nerve paralysis. Based on the pre-defined non-inferiority safety endpoint, treatment with the investigational device was non-inferior to the control device.
A full list of adverse events that were related or possibly related to the study procedure or device is provided in Extended Data Table 5. After ablation, five deaths occurred in the trial that were not related to the procedure or device. In the investigational arm, one patient died due to monoclonal gammopathy and systemic amyloidosis, and another patient died due to choking while eating followed by cardiopulmonary arrest. In the control arm, one patient died due to ampullary adenocarcinoma, the second due to heart failure and coronary artery disease and the third due to heart failure.
Secondary superiority analyses
Pre-specified superiority testing showed shorter procedural durations for the investigational device compared to the control device. This included shorter total energy application time (7.1 ± 2.0 min versus 36.4 ± 17.7 min; difference: −29.2 min, 95% CI −31.7 to −26.8, P < 0.0001) (Table 4 and Fig. 2c); shorter transpired ablation time, defined as the time elapsing between the first and last ablation application (46.7 ± 20.0 min versus 73.5 ± 34.4 min; difference: −26.8 min, 95% CI: −32.2 to −21.4, P < 0.0001) (Table 4 and Fig. 2c); and shorter skin-to-skin procedural time (100.9 ± 30.8 min versus 126.1 ± 49.2 min; difference: −25.1 min, 95% CI −33.0 to −17.3, P < 0.0001) (Table 4 and Fig. 2c). Pre-specified superiority testing did not demonstrate superiority of the primary effectiveness endpoint in the investigational arm compared to the control arm (two-sided P = 0.078, which was greater than the two-sided alpha of 0.05; Fig. 2b).
Treatment characteristics
Procedural characteristics are presented in Table 4. Lower fluoroscopy usage was observed in the investigational arm (4.9 ± 6.6 min) compared to the control arm (6.3 ± 9.1 min). Lower fluid delivery from the ablation catheter was observed with the investigational device compared to the control device (482.0 ± 142.6 ml versus 727.1 ± 378.7 ml, respectively). Furthermore, the use of an esophageal temperature probe was less frequently observed in the investigational arm than in the control arm (29.7%, 63/212 versus 76.0%, 158/208, respectively). Similarly, use of an esophageal deviation device was less frequently observed in the investigational arm than in the control arm (1.4%, 3/212 versus 16.3%, 34/208, respectively). In the investigational group, a single transseptal access approach was used in 95.3% (202/212) of cases compared to 62% (129/208) in the control group. Improved quality of life was observed after ablation in both the investigational and control groups, as indicated by the mental component of the SF-12v2 Health Survey (3.2 ± 8.1 and 4.3 ± 8.8 increase from baseline to 12 months in the investigational and control arms, respectively); the physical component of the SF-12v2 Health Survey (4.7 ± 7.6 and 4.7 ± 8.5 increase from baseline to 12 months in the investigational and control arms, respectively); and the Atrial Fibrillation Effect on Quality-of-Life (AFEQT) survey (22.3 ± 19.5 and 22.2 ± 19.3 increase from baseline to 12 months in the investigational and control arms, respectively) (Extended Data Table 6). At the 1-year follow-up or study exit, 37 out of 212 patients (17.5%) in the investigational arm and 33 out of 208 patients (15.9%) in the control arm were taking class I or class III anti-arrhythmic drugs.
Additional ablation lesion sets
Most patients in both arms received additional linear ablation beyond PVI (95.8% and 85.6% for the investigational and control groups, respectively). Left atrial posterior wall isolation including roof lines was performed in 93.4% and 65.9% in the investigational and control groups, respectively. A cavo-tricuspid isthmus line was created in 54.2% and 47.1% in the investigational and control groups, respectively. A mitral line was created in 34.0% in the investigational group and in 10.6% in the control group, respectively (Table 4). In post hoc analyses performed to assess the heterogeneity with respect to additional ablation lines on clinical outcomes, a regression approach was employed to compare subgroups with and without mitral or posterior/roof lines. The analysis did not reveal any heterogeneity of treatment effects based on the presence or absence of these linear ablations, as indicated by P > 0.1 for both, as shown in Extended Data Table 7.
PVI durability
During the study period, a total of 26 patients underwent a redo catheter ablation procedure, with 10 in the investigational arm and 16 in the control arm (one additional control patient had redo surgical ablation). At this repeat procedure, PVI durability was 50% per patient and 66.7% per vein in the investigational arm compared to 18.8% per patient and 48.4% per vein in the control arm.
Neurological substudy analysis
In brain magnetic resonance imaging (MRI) that examined the presence of silent ischemic lesions after ablation, three out of 37 patients in the investigational group and two out of 35 patients in the control group were found to have fluid-attenuated inversion recovery (FLAIR)-hyperintense acute lesions (Extended Data Table 8). Follow-up MRI scans performed 90 d later for these patients with silent ischemic lesions showed that two out of the three patients in the investigational group and one out of the two patients in the control group demonstrated full resolution.
Discussion
The SPHERE Per-AF trial was a randomized, single-blind, non-inferiority trial of patients with persistent AF comparing an all-in-one mapping and dual-energy (radiofrequency and pulsed field) large-footprint ablation catheter to conventional radiofrequency ablation (Extended Data Fig. 1). The investigational system was non-inferior to the conventional system in both safety and effectiveness. The investigational system was superior to the conventional system in measures of procedural efficiency, with shorter procedural duration, time from the first-to-last application and total energy application time.
Historical outcomes of catheter ablation in patients with persistent AF have been suboptimal, with 1-year success rates ranging between 45% and 62% in different multi-center trials, including the PRECEPT trial, which evaluated the same radiofrequency ablation system used in this trial as the control device4,5,24,25,26.
The present trial confirmed non-inferiority of effectiveness when compared to the standard-of-care mapping and ablation system and represented single-procedure success rate. The observed difference in effectiveness between the two groups was 8.0% (CI: −0.9% to 16.8%). The Kaplan–Meier curves show a visual separation between the two arms that emerged immediately after the 90-d blanking period and remained consistent throughout the follow-up period. However, a pre-defined secondary analysis of effectiveness did not demonstrate superiority, as indicated by a two-sided P value of 0.078, which is higher than the two-sided alpha threshold of 0.05.
The observed difference between the investigational and control arms was not driven by lower performance of the control arm. That is, in PRECEPT, which enrolled a similar cohort of patients with persistent AF who underwent catheter ablation using the same radiofrequency control catheter, the primary effectiveness success rate was 59.3% at 15 months, after a 9-month effectiveness evaluation period27. Although comparisons cannot be made across different trials given differences in clinical study design and patient baseline characteristics, the effectiveness observed in the control arm is similar to historical studies.
Both treatment arms demonstrated a low rate of primary safety events (1.4% for the investigational arm and 1.0% for the control arm) with no evidence of major complications, such as stroke, tamponade, atrio-esophageal fistula or permanent phrenic nerve paralysis. In comparison, the PRECEPT study reported a 4.7% primary safety event rate, including cardiac tamponade, stroke, phrenic nerve injury, pulmonary edema, pericarditis and major vascular access complications4. The lower frequency of major complications in both arms may reflect the extensive experience of the operators with focal ablation and may also attest to the rapid learning curve associated with the investigational device. The rate of FLAIR-hyperintense acute lesions was also relatively low (8%) compared to other technologies, ranging from 0% to 19% (refs. 28,29,30,31,32,33,34).
The design of the investigational system may have potential benefits that may contribute to safety and effectiveness17,18,20,35,36. In terms of safety, the integration of high-density mapping and dual-energy ablation into a single catheter, unlike the current standard that requires at least two separate catheters, reduces the number of transseptal punctures and/or catheter exchanges. This simplifies the procedural workflow and reduces the time involved. Indeed, 95.3% of procedures performed with the investigational system used a single transseptal puncture. In comparison, 62% of control procedures used a single transseptal puncture, with the remaining 38% using a double transseptal access. The wide and compressible lattice tip results in lower tissue pressure compared to a small, solid metal tip, thereby potentially reducing the risk of perforation. The ability to toggle between pulsed field and radiofrequency ablation allows the flexibility of using pulsed field on the posterior wall, an energy source shown to avoid the risk of thermal injury to adjacent organs, such as the esophagus26.
In terms of effectiveness, the wide footprint facilitates the creation of contiguous ablation lesions, decreasing the likelihood of gaps in the ablation line. In a previous clinical study, which included a second re-mapping procedure approximately 3 months after the index AF ablation procedure with this investigational device, the durability of PVI was 97% on a per-vein basis, with all four veins remaining isolated in 90% of patients23.
Procedural times with the investigational device were favorable also when compared to single-shot PFA technologies, typically ranging between 106 min and 145 min26,37. Additionally, the investigational device offers the flexibility to map and treat focal and reentrant atrial tachycardias, which are commonly encountered in this patient population38. Furthermore, this efficiency was noted despite the limited experience with the investigational system; before the study, only five out of 40 operators had clinical experience in the first-in-human study23,35. On average, these 40 operators treated 6 ± 7 patients each with the investigational system, whereas all operators had extensive experience with the conventional system.
Our trial has several limitations. There is a potential for under-detection of asymptomatic atrial tachyarrhythmias due to the absence of continuous invasive monitoring. Nevertheless, the randomized nature of the study suggests that any missed asymptomatic events would likely have impacted both groups equally. Furthermore, adherence to follow-up visits, as well as to Holter and trans-telephonic monitoring, was consistently high across both study groups. Although the ablation protocol, uniformly applied to both groups, mandated PVI and permitted the treatment of documented macro-reentrant tachycardias, it generally discouraged empiric ablation. Nevertheless, there was a relatively high rate of additional ablation lines in both groups, particularly in the interventional arm. Although this study was not designed or powered to assess the value of additional empiric ablation lines, a post hoc analysis indicated that this heterogeneity in treatment did not affect the primary clinical outcome. This finding aligns with the cumulative evidence from randomized clinical studies, suggesting limited clinical value in additional ablation beyond PVI5,39,40. One potential explanation for the higher rate of ablation lines in the investigational arm could be the ease of use of the investigational catheter, which may facilitate physicians’ ablation strategies by making it easier to deliver the ablation. However, this hypothesis warrants further investigation in dedicated clinical studies specifically designed and powered to compare different ablation strategies for treating persistent AF using lattice-tip technology.
The SPHERE Per-AF trial demonstrated that, for patients with persistent AF resistant to anti-arrhythmic drugs, using an all-in-one high-density mapping and ablation catheter with a dual-energy ablation system is non-inferior to the conventional standard of care.
Methods
Trial design
SPHERE Per-AF (NCT05120193) was a pivotal, multicenter, randomized, single-blind, non-inferiority trial. The trial protocol is available in the Supplementary Information. The trial was funded by the manufacturer of the investigational mapping and ablation device, Affera, Inc. (later acquired by Medtronic). The trial received approval from the US Food and Drug Administration (FDA) and from the institutional review board at each participating center and was conducted in accordance with the principles of the Declaration of Helsinki.
The study’s design was developed by the sponsor, incorporating suggestions from several of the authors as well as the FDA. An autonomous board responsible for data and safety monitoring supervised participant safety and the execution of the trial, and an independent clinical events committee that was blinded to the randomization evaluated all outcomes of clinical significance. For evaluations of rhythm monitoring and brain MRI, independent core laboratories, blinded to the mapping and ablation platform, were used.
Data collection and monitoring for the trial were carried out by the sponsor, which also conducted the outcome analyses in line with the statistical methods. All study measurements were taken from distinct samples, with each trial participant as an independent sample. The Statistical Analysis Plan is available in the Supplementary Information. The authors were granted complete access to all data and analyses. The first draft of this manuscript was written by the first author, with subsequent reviews and edits by the other authors. Although the sponsor contributed suggestions, the final decision on the content of the manuscript rested with the first author. The authors collectively vouch for the data’s accuracy and completeness as well as the trial’s adherence to the established protocol.
Study participants
All study participants provided written informed consent. Adults aged 18–80 years experiencing symptomatic persistent AF and who were refractory or intolerant to at least one class I or class III anti-arrhythmic drug were eligible for enrollment. Major inclusion and exclusion criteria are available in Extended Data Table 1; for a full list of inclusion and exclusion criteria, refer to the protocol. To ensure operator familiarity with the investigational mapping and ablation technology, each participating center was permitted to treat up to two roll-in patients. Subsequent patients were randomly assigned in a 1:1 ratio to undergo catheter-based ablation using either the lattice-tip or conventional radiofrequency technology. Randomization was completed via an electronic data capture system, where randomization was blocked and stratified by site and by enrollment in a neurological substudy. Randomized patients were blinded to their procedural assignment. An exploratory neurological substudy examined the effects of each treatment group on silent neurological events, as assessed by brain MRI (including diffusion-weighted imaging (DWI) and FLAIR sequences) and cognitive tests. Imaging targeted 24–48 h after the ablation procedure. Follow-up imaging was performed on day 90 in patients with post-procedure acute ischemia. A total of 23 centers across three countries (United States, Czech Republic and Israel) participated in the trial. The list of participating centers and investigators is provided in Supplementary Table 1 of the Supplementary Information.
Interventions
The investigational technology includes a lattice-tip catheter (Sphere-9 catheter, Medtronic) with a compatible proprietary electro-anatomical mapping system (Affera Mapping and Ablation System, Medtronic) as previously described22,23,21. After creating a high-density electro-anatomical map of the left atrium, the same lattice-tip catheter was used for ablation, using either radiofrequency or pulsed field energies. Radiofrequency ablation applications were delivered in a temperature-controlled mode with an application duration of 5 s, with a target surface temperature of 73 °C and a current limit varying between 80% and 90%. The PFA applications consisted of a train of microsecond-scale pulses delivered for 4 s22,23,21. Operators were instructed to use pulsed field energy on the posterior wall, around the left inferior pulmonary vein and near the phrenic nerve but had discretion to use either type of energy in other areas.
In the control arm, operators employed a commercially available technology comprising an electro-anatomical mapping system (Carto 3, Biosense Webster), a multi-electrode mapping catheter and a contact force-sensing ablation catheter (THERMOCOOL SMARTTOUCH, Biosense Webster)41. Proprietary mapping catheters, including the Lasso, Pentaray and Octaray mapping catheters, were used for high-density mapping.
All procedures required high-density mapping performed with either the investigational or control mapping system. The ablation protocol was similar for both arms, requiring a wide-area circumferential PVI, with a procedural endpoint being a documented acute entrance block in each vein after a minimum 20-min observation period or infusion of adenosine or isoproterenol. Cavo-tricuspid isthmus linear ablation was required in cases with documented typical right atrial flutter either before or during the procedure. Additional linear ablation was permitted for treating documented macro-reentrant tachycardias. Although empiric linear lesion sets were generally discouraged, operators retained the freedom to adhere to their standard of care for treating persistent AF. Assessing block across an ablation line was conducted using differential pacing maneuvers and activation mapping, in line with the operator’s standard approach. This method was consistently applied across both study groups. Operators pursued standard of care per their medical discretion for procedural strategies, such as transseptal puncture, esophageal management, use of fluoroscopy and intra-cardiac echocardiography.
Follow-up
Patients were discharged on oral anti-coagulation according to standard guidelines. The use of class I or class III anti-arrhythmic drugs was allowed but recommended to be discontinued before the end of a 90-d blanking period. Patients were followed for 1 year with office visits at 1 month, 3 months, 6 months and 12 months. After a 90-d blanking period, trans-telephonic ECG monitoring was required at least monthly with additional transmissions triggered by symptoms. A 24-h Holter monitor was performed at 6 months and 12 months, and 12-lead ECGs were performed at 3 months, 6 months and 12 months. Quality of life was evaluated at baseline and 12 months using the SF-12v2 Health Survey and the AFEQT survey42,43,44. In a neurological assessment substudy to assess for silent cerebral lesions, brain MRI (including DWI and FLAIR sequences) was performed within 72 h after the ablation procedure.
Endpoints
The pre-specified primary effectiveness endpoint was freedom from a composite of multiple failure modes, including failure to acutely isolate all targeted PVs and complete all left atrial ablation with the assigned study device during the index procedure; repeat ablation at any time after the index procedure; and, after a 3-month blanking period, documented occurrence of atrial tachyarrhythmia, escalation or initiation of class I or class III anti-arrhythmic drugs or cardioversion. Documented recurrence of AF, atrial tachycardia or atrial flutter was based on either (1) an episode ≥30 s in duration documented by ECG, trans-telephonic monitor or Holter monitor or (2) an episode covering an entire 12-lead ECG recording lasting at least 10 s. The study protocol included additional superiority testing contingent upon non-inferiority of the primary endpoints. Energy application time, elapsed treatment time, total procedure time and primary effectiveness were sequentially tested for superiority of the investigational device compared to the control device.
The pre-specified primary safety endpoint was a composite of pre-specified device-related or procedure-related serious adverse events, including death, atrio-esophageal fistula, stroke, myocardial infarction, cardiac tamponade/perforation, PV stenosis, phrenic nerve paralysis, transient ischemic attack, thromboembolism, major vascular access complications/bleeding, heart block, gastroparesis, severe pericarditis or new or extended hospitalization for a cardiovascular or pulmonary adverse event. Adverse events were determined as serious if they (1) led to death; (2) led to serious deterioration in the health of the patient (including life-threatening illness or injury, permanent impairment of a body structure or function, >24-h hospitalization, chronic disease or medical or surgical intervention to prevent injury or permanent impairment of a body structure or function); and (3) led to fetal distress/death or a congenital abnormality or birth defect. Hospitalizations for pre-existing conditions or procedures without serious deterioration in health were not defined as serious. All primary adverse events were pre-specified, and their severity and association with the device or procedure were adjudicated by an independent clinical events committee. Pre-specified secondary effectiveness and performance endpoints included assessment of changes in quality of life, use of anti-arrhythmic drugs during the effectiveness evaluation period, procedure times, fluoroscopy time and ablation lesion sets delivered. All other endpoints were based on post hoc analyses.
Statistical analysis
This trial aimed to assess for non-inferiority of the investigational device safety and effectiveness compared to the control device. To achieve power greater than 80% for testing each primary endpoint using the Farrington–Manning method, a sample size of 350 evaluable patients (175 per arm) was required for the primary analysis cohort (that is, randomized and treated patients), with assumed underlying rate of 8%, non-inferiority margin of 8% and one-sided alpha of 0.05 for the primary safety endpoint and assumed underlying rate of 60%, non-inferiority margin of 15% and one-sided alpha of 0.025 for the primary effectiveness endpoint. A total of 410 randomized patients was planned based on a conservative 15% attrition estimate. Trial success was defined by demonstrating both non-inferiority of the primary safety endpoint and non-inferiority of the primary effectiveness endpoint based on binomial proportions using the Farrington–Manning method. Contingent upon trial success, secondary sequential testing with an overall one-sided alpha of 0.025 was performed on a pre-specified set of endpoints to further examine superiority of the investigational device compared to the control device.
Quantitative variables were summarized using standard descriptive statistics, including number of non-missing observations, mean and s.d. Categorical variables were summarized using classical frequency statistics: number of non-missing observations, frequency and percentage by category. Kaplan–Meier analysis of the freedom from primary effectiveness failure events (including atrial tachyarrhythmia recurrence) was performed along with the log-rank test. Statistical analyses were performed using the SAS version 9.4 software package (SAS Institute).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All supporting data are available within the article and the Supplementary Information. Source data will not be shared due to patient privacy and informed consent, including the potential for release of protected health information.
Code availability
No custom code was used.
References
Hindricks, G. et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): the Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 42, 373–498 (2021).
Joglar, J. A. et al. 2023 ACC/AHA/ACCP/HRS guideline for the diagnosis and management of atrial fibrillation: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 149, e1–e156 (2024).
Haissaguerre, M. et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659–666 (1998).
Mansour, M. et al. Persistent atrial fibrillation ablation with contact force-sensing catheter: the prospective multicenter PRECEPT trial. JACC Clin. Electrophysiol. 6, 958–969 (2020).
Verma, A. et al. Approaches to catheter ablation for persistent atrial fibrillation. N. Engl. J. Med. 372, 1812–1822 (2015).
Verma, A. & Macle, L. Persistent atrial fibrillation ablation: where do we go from here? Can. J. Cardiol. 34, 1471–1481 (2018).
de Groot, N. M. et al. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: epicardial breakthrough. Circulation 122, 1674–1682 (2010).
Terricabras, M., Piccini, J. P. & Verma, A. Ablation of persistent atrial fibrillation: challenges and solutions. J. Cardiovasc. Electrophysiol. 31, 1809–1821 (2020).
Muller, J. et al. Recurrent atrial fibrillation ablation after initial successful pulmonary vein isolation. J. Clin. Med. 12, 7177 (2023).
Das, M. et al. Pulmonary vein re-isolation as a routine strategy regardless of symptoms: the PRESSURE randomized controlled trial. JACC Clin. Electrophysiol. 3, 602–611 (2017).
Calkins, H. et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation. Heart Rhythm 14, e275–e444 (2017).
Kotnik, T., Rems, L., Tarek, M. & Miklavcic, D. Membrane electroporation and electropermeabilization: mechanisms and models. Annu. Rev. Biophys. 48, 63–91 (2019).
Batista Napotnik, T., Polajžer, T. & Miklavčič, D. Cell death due to electroporation—a review. Bioelectrochemistry 141, 107871 (2021).
Neven, K. et al. Acute and long-term effects of full-power electroporation ablation directly on the porcine esophagus. Circ. Arrhythm. Electrophysiol. 10, e004672 (2017).
Koruth, J. S. et al. Pulsed field ablation vs radiofrequency ablation: esophageal injury in a novel porcine model. Circ. Arrhythm. Electrophysiol. 13, e008303 (2020).
Cochet, H. et al. Pulsed field ablation selectively spares the oesophagus during pulmonary vein isolation for atrial fibrillation. Europace 23, 1391–1399 (2021).
Yavin, H. et al. Pulsed field ablation using a lattice electrode for focal energy delivery: biophysical characterization, lesion durability, and safety evaluation. Circ. Arrhythm. Electrophysiol. 13, e008580 (2020).
Barkagan, M. et al. Expandable lattice electrode ablation catheter: a novel radiofrequency platform allowing high current at low density for rapid, titratable, and durable lesions. Circ. Arrhythm. Electrophysiol. 12, e007090 (2019).
Shapira-Daniels, A. et al. Novel irrigated temperature-controlled lattice ablation catheter for ventricular ablation: a preclinical multimodality biophysical characterization. Circ. Arrhythm. Electrophysiol. 12, e007661 (2019).
Anter, E. et al. A lattice-tip temperature-controlled radiofrequency ablation catheter for wide thermal lesions: first-in-human experience with atrial fibrillation. JACC Clin. Electrophysiol. 6, 507–519 (2020).
Anter, E. N. P. et al. An expandable lattice electrode catheter for rapid and titratable temperature-controlled radiofrequency ablation: a first-in-human multicenter trial. JACC Clin. Electrophysiol. (in the press).
Reddy, V. Y. et al. A lattice-tip temperature-controlled radiofrequency ablation catheter: durability of pulmonary vein isolation and linear lesion block. JACC Clin. Electrophysiol. 6, 623–635 (2020).
Reddy, V. Y. et al. A focal ablation catheter toggling between radiofrequency and pulsed field energy to treat atrial fibrillation. JACC Clin. Electrophysiol. 9, 1786–1801 (2023).
Boveda, S. et al. Single-procedure outcomes and quality-of-life improvement 12 months post-cryoballoon ablation in persistent atrial fibrillation: results from the multicenter CRYO4PERSISTENT AF trial. J. Am. Coll. Cardiol. 4, 1440–1447 (2018).
Su, W. W. et al. Cryoballoon ablation of pulmonary veins for persistent atrial fibrillation: results from the multicenter STOP Persistent AF trial. Heart Rhythm 17, 1841–1847 (2020).
Verma, A. et al. Pulsed field ablation for the treatment of atrial fibrillation: PULSED AF pivotal trial. Circulation 147, 1422–1432 (2023).
Summary of Safety and Effectiveness Data (SSED). THERMOCOOL SMARTTOUCH SF Bi-Directional Navigation Catheter. https://www.accessdata.fda.gov/cdrh_docs/pdf3/P030031S100B.pdf
Gaita, F. et al. Radiofrequency catheter ablation of atrial fibrillation: a cause of silent thromboembolism? Magnetic resonance imaging assessment of cerebral thromboembolism in patients undergoing ablation of atrial fibrillation. Circulation 122, 1667–1673 (2010).
Kimura, T. et al. Asymptomatic cerebral infarction during catheter ablation for atrial fibrillation: comparing uninterrupted rivaroxaban and warfarin (ASCERTAIN). JACC Clin. Electrophysiol. 4, 1598–1609 (2018).
Herrera Siklódy, C. et al. Incidence of asymptomatic intracranial embolic events after pulmonary vein isolation: comparison of different atrial fibrillation ablation technologies in a multicenter study. J. Am. Coll. Cardiol. 58, 681–688 (2011).
Neumann, T. et al. MEDAFI-Trial (micro-embolization during ablation of atrial fibrillation): comparison of pulmonary vein isolation using cryoballoon technique vs. radiofrequency energy. Europace 13, 37–44 (2011).
Halbfass, P. et al. Safety of very high-power short-duration radiofrequency ablation for pulmonary vein isolation: a two-centre report with emphasis on silent oesophageal injury. Europace 24, 400–405 (2022).
Gaita, F. et al. Incidence of silent cerebral thromboembolic lesions after atrial fibrillation ablation may change according to technology used: comparison of irrigated radiofrequency, multipolar nonirrigated catheter and cryoballoon. J. Cardiovasc. Electrophysiol. 22, 961–968 (2011).
Miyazaki, S. et al. Silent cerebral events/lesions after second-generation cryoballoon ablation: how can we reduce the risk of silent strokes? Heart Rhythm 16, 41–48 (2019).
Reddy, V. Y. et al. Lattice-tip focal ablation catheter that toggles between radiofrequency and pulsed field energy to treat atrial fibrillation: a first-in-human trial. Circ. Arrhythm. Electrophysiol. 13, e008718 (2020).
Haines, D. E. Can an expanding lattice electrode catheter expand our success in catheter ablation? Circ. Arrhythm. Electrophysiol. 12, e007306 (2019).
Reddy, V. Y. et al. Pulsed field or conventional thermal ablation for paroxysmal atrial fibrillation. N. Engl. J. Med. 389, 1660–1671 (2023).
Ohlrogge, A. H., Brederecke, J. & Schnabel, R. B. Global burden of atrial fibrillation and flutter by national income: results from the Global Burden of Disease 2019 database. J. Am. Heart Assoc. 12, e030438 (2023).
Dixit, S. et al. Randomized ablation strategies for the treatment of persistent atrial fibrillation: RASTA study. Circ. Arrhythm. Electrophysiol. 5, 287–294 (2012).
Kistler, P. M. et al. Effect of catheter ablation using pulmonary vein isolation with vs without posterior left atrial wall isolation on atrial arrhythmia recurrence in patients with persistent atrial fibrillation: the CAPLA randomized clinical trial. JAMA 329, 127–135 (2023).
Chinitz, L. A. et al. Safety and efficiency of porous-tip contact-force catheter for drug-refractory symptomatic paroxysmal atrial fibrillation ablation: results from the SMART SF trial. Europace 20, f392–f400 (2018).
Dorian, P. et al. Interpreting changes in quality of life in atrial fibrillation: how much change is meaningful? Am. Heart J. 166, 381–387.e8 (2013).
Holmes, D. N. et al. Defining clinically important difference in the atrial fibrillation effect on quality-of-life score. Circ. Cardiovasc Qual. Outcomes 12, e005358 (2019).
Pezawas, T., Ristl, R., Schukro, C. & Schmidinger, H. Health-related quality of life changes in patients undergoing repeated catheter ablation for atrial fibrillation. Clin. Res. Cardiol. 105, 1–9 (2016).
McHorney, C. A., Ware, J. E. Jr., Lu, J. F. & Sherbourne, C. D. The MOS 36-item Short-Form Health Survey (SF-36): III. Tests of data quality, scaling assumptions, and reliability across diverse patient groups. Med. Care 32, 40–66 (1994).
Hurst, N. P., Ruta, D. A. & Kind, P. Comparison of the MOS short form-12 (SF12) health status questionnaire with the SF36 in patients with rheumatoid arthritis. Br. J. Rheumatol. 37, 862–869 (1998).
Der-Martirosian, C., Cordasco, K. M. & Washington, D. L. Health-related quality of life and comorbidity among older women veterans in the United States. Qual. Life Res. 22, 2749–2756 (2013).
Acknowledgements
The authors would like to thank the SPHERE Per-AF sites and their dedicated staff as well as V. Low, C. Shea, V. Nejedlo, J. David, K. Newman, B. Dittrich, S. Hryciuk and all members of the SPHERE Per-AF team who supported product development and study execution. This study was sponsored by Affera, Inc., which was acquired by Medtronic in August 2022.
Author information
Authors and Affiliations
Consortia
Contributions
E.A. wrote the manuscript, with input from all authors (M.M., D.G.N., D.S., T.L.T., P.N., E.L.K., J.K., J.O., S.M., A.N., J.D.H., A.K.A., U.R.S., D.H., P.H., S.L., B.O., K.G.T. and V.Y.R.). E.A. wrote both the first and final drafts of the manuscript. P.H. and D.H. developed the investigational system and conceived of the study design and statistical protocol. S.L. performed all statistical analyses. E.A., M.M., D.G.N., D.S., T.L.T., P.T., E.L.K., J.K., J.O., S.M., A.N., J.D.H., A.A., U.S. and V.Y.R. performed the clinical research at their respective institutions. All authors (E.A., M.M., D.G.N., D.S., T.L.T., P.N., E.L.K., J.K., J.O., S.M., A.N., J.D.H., A.K.A., U.R.S., D.H., P.H., S.L., B.O., K.G.T. and V.Y.R.) contributed to the interpretation of the results and to manuscript development.
Corresponding author
Ethics declarations
Competing interests
E.A. is a consultant to and has received equity from Affera-Medtronic. Unrelated to this manuscript, he serves in consulting and advisory capacities for Biosense Webster, Boston Scientific and Abbott Medical. He has received research grants from Biosense Webster and Medtronic. V.Y.R. is a consultant to and has received equity from Affera-Medtronic. Unrelated to this manuscript, V.Y.R. has served as a consultant for and has equity in Ablacon, Acutus Medical, Anumana, Apama Medical-Boston Scientific, APN Health, Aquaheart, Atacor, Autonomix, Axon Therapies, Backbeat, BioSig, CardiaCare, Cardiofocus, CardioNXT/AFTx, Circa Scientific, CoRISMA, Corvia Medical, Dinova-Hangzhou DiNovA EP Technology, East End Medical, EPD-Philips, EP Frontiers, Epix Therapeutics-Medtronic, EpiEP, Eximo, Farapulse-Boston Scientific, Field Medical, Focused Therapeutics, HRT, Intershunt, Javelin, Kardium, Keystone Heart, Laminar Medical, LuxMed, Medlumics, Middlepeak, Neutrace, Nuvera-Biosense Webster, Oracle Health, Restore Medical, Sirona Medical, SoundCath and Valcare. Unrelated to this work, V.Y.R. has served as a consultant for Abbott, Adagio Medical, Append Medical, AtriAN, Biosense Webster, BioTel Heart, Biotronik, Boston Scientific, Cairdac, Cardionomic, CoreMap, Fire1, Gore & Associates, Impulse Dynamics, Medtronic, Novartis, Novo Nordisk, Philips and Pulse Biosciences. Unrelated to this work, V.Y.R. has equity in Atraverse, DRS Vascular, Manual Surgical Sciences, Newpace, Nyra Medical, Surecor and Vizaramed. A.N. has served as a consultant for iRhythm, Boston Scientific, Biosense Webster, Abbott and Biotronik. M.M. has served as a consultant for Boston Scientific, Biosense Webster, Abbott, Medtronic, Siemens and Sentre Heart/Atricure and has equity in EPD-Philips (divested) and NewPace, Ltd. A.A. has served in consulting and advisory capacities for Medtronic, Boston Scientific and Biosense Webster and in medical education for Siemens. A.A. has equity in Biostar Ventures and has served in consulting and medical education for and been supported by research grants from Philips. S.M. has received Medtronic research grants and honoraria. T.T. has served as a consultant for Biosense Webster and Medtronic. D.N. reports the following disclosures: Abbott Medical: consultant, advisory board and research grants; Boston Scientific: consultant, advisory board and research grants; Medtronic: consultant, advisory board and research grants; Biosense Webster: consultant, advisory board and research grants; Adagio: consultant and research grants; Laminar: research grants; and TerraRecon: consultant. E.K. has served in consulting and advisory capacities for Biosense Webster, Medtronic and Philips, unrelated to this manuscript or technology. J.K. reports personal fees from Biosense Webster, Boston Scientific, GE Healthcare, Medtronic and St. Jude Medical (Abbott) for participation in scientific advisory boards and has received speaker honoraria from Biosense Webster, Biotronik, Boston Scientific, Medtronic and St. Jude Medical (Abbott). D.S. reports grant/research support from Medtronic and consultant/advisory board participation for Biosense Webster, Medtronic, EBR Inc., AltaThera Pharmaceutical and Attune Medical. J.O. reports the following disclosures: Medtronic: consulting; Biosense Webster: research grants, consulting and advisory board; Boston Scientific: consulting, research grants and advisory board; and Abbott: consulting and research grants. J.H. has served as a consultant for Medtronic, Abbott and Volta. D.H., P.H., S.L., B.O. and K.G.T. are employees of Medtronic. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Andreas Goette, Thomas Rostock and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Michael Basson, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Comparison of catheter ablation systems in the SPHERE Per-AF clinical trial.
The investigational Sphere-9TM lattice-tip dual energy catheter with AfferaTM Mapping and Ablation System is shown in the left panel. This system utilizes the Sphere-9TM catheter for both electroanatomical mapping and ablation, employing radiofrequency (red circles) or pulsed field (green circles) energies to create a line of electrical isolation around the pulmonary veins. The control system, depicted in the right panel, consists of a multielectrode mapping catheter for electroanatomical mapping and a separate catheter for radiofrequency ablation (THERMOCOOL® SMARTTOUCH® Surround Flow). As shown in the inserts, the investigational Sphere-9TM catheter has a wider footprint capable for creating wider lesions, resulting in a more contiguous ablation line. LIPV=left inferior pulmonary vein. LSPV=left superior pulmonary vein.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 2, Clinical Investigation Plan and Statistical Analysis Plan.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Anter, E., Mansour, M., Nair, D.G. et al. Dual-energy lattice-tip ablation system for persistent atrial fibrillation: a randomized trial. Nat Med 30, 2303–2310 (2024). https://doi.org/10.1038/s41591-024-03022-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-024-03022-6
This article is cited by
-
Pulsed-field ablation: a revolution in atrial fibrillation therapy
Nature Reviews Cardiology (2024)
-
Pulsed-Field-Ablation – zukünftig die einzige Energiequelle für Ablationen?
Herzschrittmachertherapie + Elektrophysiologie (2024)
-
Erstablation von Vorhofflimmern − reicht nur eine Pulmonalvenenisolation?
Herzschrittmachertherapie + Elektrophysiologie (2024)
