
Abstract
Patient-reported outcomes (PROs) are increasingly used in healthcare research to provide evidence of the benefits and risks of interventions from the patient perspective and to inform regulatory decisions and health policy. The use of PROs in clinical practice can facilitate symptom monitoring, tailor care to individual needs, aid clinical decision-making and inform value-based healthcare initiatives. Despite their benefits, there are concerns that the potential burden on respondents may reduce their willingness to complete PROs, with potential impact on the completeness and quality of the data for decision-making. We therefore conducted an initial literature review to generate a list of candidate recommendations aimed at reducing respondent burden. This was followed by a two-stage Delphi survey by an international multi-stakeholder group. A consensus meeting was held to finalize the recommendations. The final consensus statement includes 19 recommendations to address PRO respondent burden in healthcare research and clinical practice. If implemented, these recommendations may reduce PRO respondent burden.
Similar content being viewed by others

Main
Millions of individuals provide PRO data regularly in a variety of settings1. The substantial benefits of utilizing PRO data for various purposes, including healthcare research, clinical practice, regulatory purposes and value-based healthcare decisions have been demonstrated and extensively documented2,3,4,5,6,7,8; however, the completion of PROs places a potential burden on respondents (patients), especially if responses are requested on a regular basis9,10,11,12,13,14. Respondent burden is the degree to which a respondent perceives their participation in a task as difficult, time consuming or emotionally stressful15.
With regard to the completion of PRO measures, there are several factors that may influence respondent burden, including patient characteristics (such as literacy levels and cognitive impairment) and features of the chosen measure (including length, wording, content, sensitivity of items and formatting)1,11,12. Response burden could also be linked to the mode of administration of PROs (whether electronic, paper or any other format) and frequency of collection in both healthcare research and clinical practice settings16,17; however, these factors are likely to be inter-related and their associations with respondent burden may be nuanced and context-specific. For instance, recent studies have shown that the administration of longer PRO measures may not necessarily be associated with a perception of increased respondent burden, especially if respondents have a clear understanding of the purpose of collection and how their data would be utilized1,17,18.
Failure to address respondent burden may lead to poor PRO completion rates, missing PRO data or trial participant withdrawal17. The US Food and Drug Administration (FDA) advises that sponsors should consider missing data and poor PRO completion rates as possible indicators of inappropriate respondent burden, item content or response options11. The implication of such missing data is that poor quality or nonrepresentative PRO information could be deemed as not sufficiently robust to evaluate treatment benefit, inform clinical care or regulatory decision-making. Therefore, to optimize PRO assessments, it is important that the potential benefits of PRO collection are weighed against the potential burden on respondents19,20,21,22.
Given that there are no international guidelines that address this critical issue, the aim of this international effort was to develop consensus-based recommendations to facilitate the minimization of respondent burden for individuals completing PROs in both healthcare research and clinical practice.
Methods
The recommendations were developed through an international Delphi and consensus process as described in the COMET Handbook v.1.0 (ref. 23). The steering group (O.L.A., S.C.R., J.R., P.K. and M.J.C.) oversaw the design and conduct of the study.
Ethical approval
Ethical approval was granted by the University of Birmingham Ethical Review Board (ERN_ 22-0276). Study information was electronically provided to participants before survey completion and before the consensus meeting. Delphi participants provided electronic informed consent and written consent was obtained from consensus meeting delegates.
Generation of candidate recommendations
Twenty-six candidate recommendations were initially generated based on the findings of a comprehensive literature review conducted and published in 2022 by members of the steering group1. In brief, PubMed was searched on the 22 November 2021 to identify eligible studies (further details are provided in Supplementary Information). There were no restrictions on study design or language of publication. Title, abstract and full-text screening was conducted independently by two reviewers. Analysis of the qualitative data was performed using the framework method24.
International Delphi process
The stakeholders for this project consisted of trialists, PRO-focused clinical researchers and statisticians, patient partners and advocates, healthcare professionals, journal editors, policymakers, industry experts and other professionals who are involved in the implementation of PROs for healthcare research, drug approvals and clinical practice. These individuals were identified through personal networks and suggestions from known experts.
In 2023, the steering group sent invitations to 168 international stakeholders to participate in an online Delphi process to vote on the candidate recommendations and propose additional recommendations. Stakeholder grouping and characteristics are described in the Supplementary Information.
The two online Delphi surveys were delivered using the DelphiManager software (v.5.0) developed and maintained by the COMET initiative. The invitation emails provided information about the study, who to contact for further information and a link to the consent form and survey for those who wished to take part. Voting on the importance of the 26 candidate recommendations was anonymous and scored using a nine-point scale (1–3, not important; 4–6, important but not critical; and 7–9, important and critical). A total of 127 responses were received for Round 1 of the Delphi survey and 106 responses (83% of participants from Round 1) were received for Round 2. The Delphi participants had the option in Round 1 to provide qualitative feedback on the suitability of each recommendation, suggest modifications and propose additional recommendations. The feedback was reviewed and seven additional recommendations were proposed and taken forward to Round 2. Participants were subsequently sent a document detailing how the feedback from Round 1 was addressed by the steering group. Anonymized item-level Round 1 ratings by stakeholder group were also shared with the Delphi participants for their consideration before voting in Round 2. The participants were informed that their Round 1 responses would be retained if they did not complete Round 2. Delphi participants who agreed to be named are listed in Supplementary Information.
Prespecified threshold for inclusion of recommendations
For inclusion, a recommendation was required to meet the prespecified threshold of ≥70% of the Round 2 Delphi participants rating it as ‘important and critical’ (7–9) and ≤15% rating it as ‘not important’ (1–3). Recommendations that achieved consensus were reviewed and ratified at the consensus meeting. Recommendations that did not achieve overall consensus but were rated by ≥70% of any stakeholder group as ‘important and critical’ (7–9) were discussed at the consensus meeting. A summary of qualitative feedback from the Delphi participants on these recommendations was presented at the meeting. Recommendations that did not meet any of the above criteria were proposed for exclusion.
International consensus meeting
The aim of the meeting was to reach consensus on the content of the recommendations. Following Round 2, the steering group collated and reviewed the ratings and the qualitative feedback from the Delphi participants. They proposed the inclusion or exclusion of recommendations based on the Delphi data and sent these to the consensus meeting delegates ahead of the meeting (Supplementary Information).
A consensus meeting was hosted online via Zoom by the University of Birmingham, UK, in September 2023. The meeting was attended by 36 international delegates who had participated in the Delphi study. The delegates were selected in a manner that ensured good representation across stakeholder groups. There were 28 voters and 8 nonvoters. The nonvoters were members of the steering group and experts from institutions already represented. The delegates consisted of 12 trialists/academic researchers/statisticians, 7 industry experts, 6 regulators/policymakers, 5 healthcare professionals, 5 patients/patient advocates and members of the public and 1 journal editor (Supplementary Information).
Delegates discussed the importance of the recommendations that met the prespecified threshold for consensus (overall) as well as the recommendations that only reached consensus in one or more stakeholder groups. The wording and explanatory text of recommendations were also discussed as required. Following group discussion, delegates were invited to vote anonymously on the candidate recommendations using the Zoom Poll tool. The voting options were to include or exclude with response options of ‘yes’, ‘no’ or ‘further discussion required’. Supplementary Information provides further details of how the voting was conducted specifically for each recommendation.
Final consultation
Following the consensus meeting, delegates were sent the draft recommendations for their comments and suggestions on the wording and approval of the final version. Supplementary Information provides further information on the methods.
Results
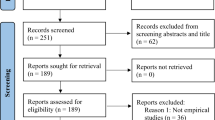
Following the Delphi surveys, 19 recommendations achieved overall consensus and were proposed by the steering group for inclusion; 8 recommendations only achieved consensus in one or more stakeholder groups and required discussion at the consensus meeting; and 6 recommendations did not reach consensus in any stakeholder group and were proposed for exclusion. Further details of the voting and decisions at the consensus meeting can be found in Fig. 1 and Supplementary Information. The 19 recommendations were ratified for inclusion during the consensus meeting (there was consensus to merge two of these recommendations). Of the eight recommendations that were individually discussed, two were voted in for inclusion as standalone recommendations; and there was consensus to merge two other recommendations with recommendations that had been ratified. Two recommendations were merged based on suggestions received on the initial draft of the manuscript. Further details are provided in Supplementary Information.

The flow chart illustrates the process, which culminated in 19 recommendations on how to address respondent burden associated with PRO assessment.
The final consensus statement provides 19 recommendations for consideration by anyone involved in designing and implementing PRO assessment strategies for healthcare research and clinical practice (Table 1). An elaboration describing each recommendation with supporting evidence is presented below. The recommendations are presented in accordance with the categories from the published review1, namely (1) rationale and schedule for PRO assessment; (2) measure selection; and (3) measure delivery.
Rationale and schedule for PRO assessment
Recommendation 1: involve patients, clinicians and other relevant stakeholders in the formulation of the PRO research question(s) or clinical objectives to ensure that they are important and relevant
Effective involvement of patients, clinicians and other relevant stakeholders in the formulation of PRO research question(s) and clinical objectives can help ensure the assessment of outcomes that are relevant and valued by all stakeholders in healthcare research and clinical practice25,26,27. Stakeholder involvement will vary depending on the context of PRO use. For example, patients, caregivers and clinicians can provide valuable perspectives both in healthcare research and in clinical practice settings, while early input from regulatory agencies may be particularly useful in clinical trials of investigational medicinal products. PRO assessments may be perceived as less burdensome if the research questions or clinical objectives are considered relevant and important by patients and other relevant stakeholders28,29.
Recommendation 2: consider the degree of burden that any data collection may impose on respondents and carefully balance this with the quantity and quality of data required
The rationale for collecting PRO data for healthcare research and clinical practice should be evidence-based and should demonstrate that the data collection justifies the burden and potential risks of data collection, such as the time required, emotional angst, distress or fatigue30.
Recommendation 3: ensure that patients, clinicians and other relevant stakeholders are involved in decisions about the PRO assessment schedule and the frequency of assessment
Consultation with patients, clinicians and other relevant stakeholders will help ensure that the PRO assessment schedule captures clinically relevant periods during treatment or clinical management25. The schedule of PRO assessments, including overall duration of assessment, will depend on the research or clinical objective; however, the potential respondent burden should be considered, while maximizing the collection of clinically relevant data31. The assessment schedule may not necessarily be tied to clinic visits; considerations for the mode of administration are described in Recommendation 16 (ref. 25).
The frequency of PRO administration should consider disease trajectory and balance this with respondent burden. Data should only be collected if they are essential to addressing the research objective or informing patient care. In a clinical practice setting, patients with a stable disease/condition may require less frequent PRO administration. Long-term monitoring may be burdensome and may lead to reduced PRO completion rates, but may be warranted in some instances (for example, for chronic disease monitoring or real-world evidence generation)32.
Consider the time points for assessing PRO measures within an allowable window and their relationship to clinical events (for example, treatment, clinical assessments and other assessments). Depending on the research or clinical objective, it may not be necessary to deliver all PRO measures at every time point28. A modular approach could be taken, in which different assessment frequencies are selected to reduce patient burden9. For example, more general quality-of-life aspects (for example, social or emotional well-being) may be assessed less frequently than the presence and severity of symptoms.
Measure selection
Recommendation 4: review the literature to identify relevant concept(s) of interest
To minimize burden, the concept(s) measured by the PRO measure should be relevant to the target population, disease setting and context of use (healthcare research or clinical practice). A literature review and/or surveys or qualitative work (Recommendation 5) can be conducted to identify concept(s) of interest.
Recommendation 5: qualitative and quantitative methods may be used to obtain input from patients and clinicians on selecting or developing PRO measures so that they are fit for purpose
Patient, clinician and other stakeholder input may be obtained using qualitative and/or quantitative methods, including interviews, focus groups and surveys28,33. Patient engagement and involvement is helpful to inform selection of PRO measures that capture meaningful outcomes, while reducing burden26,34. This may help avoid overly paternalistic approaches that are clinician- or researcher-driven35.
Recommendation 6: consider the complexity of the format of PRO measures and their instructions
PRO measures with greater complexity that require more cognitive effort to understand, such as those with complicated instructions, phrasing and reverse response options, may be more burdensome for respondents11,17,33. Discussions with patients from the target population may be used to explore these issues and ascertain the level of burden that may be associated with the PRO measures being considered33. The use of multiple measures with different formats may further increase complexity and should be avoided if possible.
Recommendation 7: consider the literacy level of respondents
Where possible, promote inclusion of individuals with all levels of reading, writing and problem-solving abilities11,35. Ensure that PRO content and training is easy to understand for respondents with different literacy levels and educational experience by conducting relevant readability assessments (for example, Flesch–Kincaid grade level or SMOG (simple measure of gobbledygook) index score)35. It is recommended that PRO items be at the reading level of 11–12 years of age or lower; however, this criterion should be contextualized to the intended target population and justified16.
Recommendation 8: ensure that the selected PRO measures are culturally and linguistically relevant for the target population
If PRO measures are translated into other languages, ensure that they have undergone linguistic validation with cognitive debriefing27,36. Linguistic validation is the testing of translated PRO measures with patients or lay individuals who are representative of the cultural group intending to use the measure to check understandability, interpretation and cultural relevance of the translation37.
Recommendation 9: consider the length of PRO measures and decide whether the use of a relatively longer measure is justified
Measure length is often considered as a contributing factor to PRO respondent burden; however, measure length should be balanced with patient and clinician input on what outcomes are most relevant to the population and context30. Relatively shorter measures may reduce respondent burden and increase patients’ willingness to complete forms38; however, brevity should not outweigh the utilization of PRO measures with appropriate measurement properties (reliability and validity) to assess outcomes that are relevant to key stakeholders, the research question(s)/PRO objectives and purpose of collection30,39,40.
There is evidence that the length of a PRO measure may not necessarily be associated with respondent burden16,18,30 and high response rates could be achieved with administration of relatively longer PRO measures if they are meaningful to respondents41. Furthermore, patients may prefer longer forms to shorter versions if they capture concepts that matter to them and can meaningfully inform care1,17,18. Ultimately, evaluations of PRO measure length should consider the context of use of the data, the views of those living with the condition and those responsible for using the data. Early patient involvement in selection of the measures is crucial (Recommendation 5).
Linked to the issue of PRO measure length is estimated completion time. The needs of the target population (for example, age, disease severity and comorbid conditions) and aspects of design (for example, mode and place of PRO measure administration), may impact overall completion times. Relevant stakeholder input should be sought on the anticipated completion time and its appropriateness in terms of the research or clinical context and the patient population. For instance, a PRO measure that generally requires more time to complete might not be suitable for use in a busy outpatient clinic. The same PRO measure might be appropriate for use if completed remotely, before clinical appointments. In terms of patient population, a PRO measure that requires less time to complete may be preferable for patients with osteoarthritis of the hand. In a research context it has been suggested that completion time of baseline PRO assessments should ideally be limited to 20 min and 10–15 min for subsequent assessments10,22.
Recommendation 10: if selecting more than one PRO measure, avoid overlapping constructs
The use of more than one PRO measure requires careful consideration to avoid duplication, overlap or redundancy of constructs9,42. The administration of several PRO measures may lead to respondent burden and a higher likelihood of missing data in those measures administered later, particularly if the constructs overlap. For research purposes, it is advisable that measures to support the primary and/or secondary outcomes are prioritized over those supporting exploratory outcomes.
Recommendation 11: consider the recall periods for measures, as longer timeframes may be burdensome for some respondents
When selecting PRO measures, it is important to consider the recall period (for example, ‘In the last 7 days…’) and whether characteristics of the disease/condition will affect the respondents’ ability to recall the information easily and correctly11,43. The majority of PRO measures will often have a specified validated recall period, which should not be changed without consultation and approval from the instrument developer. If multiple recall periods have been validated by developers for a particular measure, then input from relevant stakeholders, including clinicians and patients, is recommended to decide which is most appropriate for respondents and the disease/condition of interest43.
Measure delivery
Recommendation 12: ensure that respondents understand why the data are being collected, who will have access, how it will be used and why it is important for them to complete the PRO measures
It is important to inform patients about why PRO data are being collected, making it clear how the data they report could help improve their own care in clinical practice and the future treatment of patients in healthcare research10,26,44. Perceptions of the intrusiveness of items and their usefulness may influence respondents’ perception of burden14. Explanations of the importance of PRO collection and the challenge of missing PRO data, may encourage respondents to complete PROs on a regular basis26. This recommendation applies not only in healthcare research settings, where informed consent is formally obtained, but also in clinical practice where PROs are being used as part of standard care (and patients typically do not sign consent forms).
Recommendation 13: provide clear instructions, training and support for respondents on the completion of PROs as needed
It is important that patients are provided with clear instructions on how to provide their PRO responses and be given ongoing support as needed. This may enhance the quality and completeness of the data collected.
Recommendation 14: provide training and guidance for research staff and clinicians in clinical practice so that they understand the value of PROs and respondent burden
PROs may be perceived to be burdensome by research personnel, clinical teams and research ethics committees, particularly if there are numerous measures or participants are very ill12,26. Qualitative interviews suggest that trialists may be reluctant to collect PROs due to the perceived respondent burden, even when participants may be willing to complete them26. Appropriate training for staff might help alleviate their concerns and avoid an overly paternalistic approach, and may help them address any questions raised by participants regarding PRO collection. It may also help them provide information on the importance and value of data collection, which may motivate participants to complete PRO measures.
For clinical trials, site manuals or protocols should provide specific guidance on PRO administration and management and highlight the importance of facilitating adherence and completeness of data31.
Recommendation 15: specify the level or type of support that can be provided to respondents to facilitate the completion of PRO measures
For respondents unable to complete PRO measures on their own, consider and specify what help can be provided to support completion by the respondent (for example, holding a pen, assistance with a telephone or computer keyboard, scrolling/turning pages or reading out text)11. Responses to the PRO questions should be decided by the patient and not an assisting person.
Recommendation 16: offer flexible modes of administration to meet the needs of target populations and underserved groups
Modes of PRO administration may include paper, mobile device applications (apps), web-based completion, telephone interviews, interactive voice response, audio-computer-assisted interviews and other modes10,28,36. The needs of the target population and their individual preferences should be considered, such as paper or electronic delivery and whether multiple modes are needed to reach all respondent groups36. For example, in older people or respondents with low literacy or visual impairment, interactive voice response, provision of grip-pens or interviewer-administered PRO measures could be considered to reduce the burden10. Patients can also provide feedback on the acceptability of a bring-your-own device versus provisioned devices and additional options such as tablets or paper versions in the waiting room for those who cannot complete PROs electronically at home. Practical ways to reduce the burden at clinic or study visits should be considered28.
Consider the implications of using different modalities when preparing data for analyses. If multiple modes are used for data collection to minimize burden and facilitate diversity and adherence, consider how data from different sources will be integrated. For more information on measurement comparability, see the updated recommendations from The Professional Society for Health Economics and Outcomes Research (ISPOR) Task Force on measurement comparability between modes of data collection for PRO measures45.
Recommendation 17: where possible, consider the use of ePROs, which may help reduce respondent burden, but must be balanced with the needs and preferences of the target population
With patient populations who have access to and are comfortable with electronic devices, the use of electronic PROs (ePROs) may offer additional functionality, which could help reduce burden and improve adherence in healthcare research and clinical practice46. This could include allowing completion on their own devices, with real-time reminders, notifications and responses from the research or clinical team, either to thank them for completion or to respond to issues identified on the PRO, depending on the context of use. Furthermore, ePROs make the use of innovations such as computerized adaptive testing possible. ePROs may also facilitate symptom monitoring between visits12; however, patients may face barriers to using digital services, including a lack of digital skills/low computer literacy or lack of access to reliable information technology infrastructure. Estimates suggest that 37% of the world’s 7.8 billion population are digitally excluded, with older people, people on low incomes and other marginalized groups most likely to be affected35. It is important that these potential barriers and the preferences of the patient population in terms of mode of collection (as described Recommendation 16) are carefully considered with patient input when developing PRO strategies, to ensure that PRO assessments are as inclusive and equitable as possible35.
Recommendation 18: if developing new ePRO systems or modifying an existing one for a new context of use, involve patients and clinicians in the co-design of the ePRO system
Ensure that the patients providing input to the development or modification of the ePRO system include representatives from the target population and are diverse in terms of computer literacy and internet access, considering attributes as appropriate to the research question or clinical context47. Examples may include but are not limited to: sex; gender; socioeconomic background; race/ethnicity; age; health literacy; computer literacy and internet access; and disease characteristics. There may be country-specific regulatory expectations and requirements that may also need to be considered when developing ePRO systems48. The Electronic Clinical Outcome Assessment Consortium has published best practices for the electronic implementation and migration of paper PRO measures to ePROs49.
Recommendation 19: explore the functionality of ePROs with diverse representatives from the target population where possible
Several ePRO features may facilitate completion and help to minimize burden46. Patient involvement in the study co-design and usability testing with the target population can be used to identify appropriate formats50,51. Depending on the context and where permissible, consider providing the following elements in the platform: estimated completion time, progress tracker, graphical results that are easy to interpret, positive messaging/reminders, completion rate and a thank you message after completion42,46. In terms of the format consider using underlining and capitalization where appropriate, easy-to-read fonts and font sizes, one question per screen, back/next buttons and location, branching logic and adaptive web design (where multiple versions of a web page are created to fit different devices)11,42,49. Seek patient preferences on how they receive requests and reminders to complete ePRO measures (for example, emails and/or text messages).
Discussion
Discussions about respondent burden frequently arise when health researchers, trialists and clinical teams are considering the use of PROs for healthcare research or clinical practice. This consensus statement provides accessible information in the form of consensus-based recommendations for addressing PRO-related respondent burden. The 19 recommendations are organized into three categories: rationale and schedule of assessments, measure selection and measure delivery.
The use of these recommendations by stakeholders such as trialists, researchers, clinicians and healthcare providers may facilitate the identification of factors that could influence PRO-related respondent burden and support the formulation of mitigating efforts appropriate for the context of use. Research and clinical teams are encouraged to seek input from PRO experts and utilize these recommendations, as well as the resources we have highlighted in Table 2, when considering the implementation of PROs for healthcare research or clinical practice.
While the recommendations are nonmandatory and not all may be relevant to every context, stakeholders are urged to use and reference this consensus statement to demonstrate explicitly how they have considered and addressed the issue of respondent burden. The consensus statement may also be a useful reference for those involved in scientific and ethical review of protocols and supporting materials such as peer reviewers, funding panels and ethics committees.
Although the recommendations focus on PROs in adult patients within healthcare research and clinical practice, they could be considered for use in other settings or populations such as pediatric populations and measures such as patient-reported experience measures; however, further considerations may be relevant in these contexts beyond the scope of the present work.
These recommendations have some limitations. First, while the initial review that informed the generation of the candidate recommendations was comprehensive (with 89 articles included1), only one database (PubMed) was searched. There is a possibility that some relevant articles might have been missed and that some potential recommendations were not identified; however, the international Delphi participants had the opportunity to provide qualitative feedback and propose additional recommendations not identified by the review during Round 1 of the Delphi process.
Second, these recommendations do not consider burden from the perspective of research or clinical staff; separate recommendations are needed to address burden concerns for these stakeholders.
Addressing PRO respondent burden could help to ensure the collection of more representative and high-quality PRO data to inform regulatory decisions and patient care. This work is complementary to existing resources to support the collection of high-quality PROs in healthcare research and clinical care. These include the resources available on the PROTEUS trials and practice website52,53,54,55, equity, diversity and inclusion in the collection and utilization of PROs35, PRO ethics guidelines22, the US FDA guidance11 and European Medicines Agency guidance10 (Table 2).
The use of the recommendations in this consensus statement and related guidance could lead to high-quality PRO data collection that carefully considers the needs of respondents, promoting inclusive data collection. The impact of these recommendations, when implemented in different clinical contexts, should be evaluated in future research.
References
Aiyegbusi, O. L. et al. Key considerations to reduce or address respondent burden in patient-reported outcome (PRO) data collection. Nat. Commun. 13, 6026 (2022).
Aiyegbusi, O. L., Nair, D., Peipert, J. D., Schick-Makaroff, K. & Mucsi, I. A narrative review of current evidence supporting the implementation of electronic patient-reported outcome measures in the management of chronic diseases. Ther. Adv. Chronic Dis. 12, 20406223211015958 (2021).
Cruz Rivera, S. et al. The impact of patient-reported outcome (PRO) data from clinical trials: a systematic review and critical analysis. Health Qual. Life Outcomes 17, 156 (2019).
Cruz Rivera, S. et al. Patient-reported outcomes in the regulatory approval of medical devices. Nat. Med. 27, 2067–2068 (2021).
Calvert, M., Kyte, D., Price, G., Valderas, J. M. & Hjollund, N. H. Maximising the impact of patient-reported outcome assessment for patients and society. BMJ 364, k5267 (2019).
Calvert, M. J., O’Connor, D. J. & Basch, E. M. Harnessing the patient voice in real-world evidence: the essential role of patient-reported outcomes. Nat. Rev. Drug Discov. 18, 731–732 (2019).
Denis, F. et al. Two-year survival comparing web-based symptom monitoring vs routine surveillance following treatment for lung cancer. JAMA 321, 306–307 (2019).
Basch, E. et al. Overall survival results of a trial assessing patient-reported outcomes for symptom monitoring during routine cancer treatment. JAMA 318, 197–198 (2017).
US Food and Drug Administration. Core Patient-Reported Outcomes in Cancer Clinical Trials. Guidance for Industry https://www.fda.gov/media/149994/download (2021).
European Medicines Agency. The Use of Patient-reported Outcome (PRO) Measures in Oncology Studies https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-regulatory-guidance-use-healthrelated-quality-life-hrql-measures-evaluation_en.pdf (2016).
US Food and Drug Administration. Guidance for Industry. Patient-Reported Outcome Measures: Use in Medicinal Product Development to Support Labeling Claims https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf (2009).
Aiyegbusi, O. L. et al. Patient and clinician perspectives on electronic patient-reported outcome measures in the management of advanced CKD: a qualitative study. Am. J. Kidney Dis. 74, 167–178 (2019).
Snyder, C. F., Jensen, R. E., Geller, G., Carducci, M. A. & Wu, A. W. Relevant content for a patient-reported outcomes questionnaire for use in oncology clinical practice: putting doctors and patients on the same page. Qual. Life Res. 19, 1045–1055 (2010).
Bingham, C. O. 3rd et al. Montreal accord on patient-reported outcomes (PROs) use series. Paper 4: patient-reported outcomes can inform clinical decision making in chronic care. J. Clin. Epidemiol. 89, 136–141 (2017).
Lavrakas, P. J. (ed.) Encyclopedia of Survey Research Methods (Sage Publications, 2008).
Francis, D. O., McPheeters, M. L., Noud, M., Penson, D. F. & Feurer, I. D. Checklist to operationalize measurement characteristics of patient-reported outcome measures. Syst. Rev. 5, 129–129 (2016).
Atkinson, T. M. et al. Perceptions of response burden associated with completion of patient-reported outcome assessments in oncology. Value Health 22, 225–230 (2019).
Rolstad, S., Adler, J. & Rydén, A. Response burden and questionnaire length: is shorter better? A review and meta-analysis. Value Health 14, 1101–1108 (2011).
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 310, 2191–2194 (2013).
National Health and Medical Research Council. National Statement on Ethical Conduct in Human Research (National Health and Medical Research Council, 2018).
NIH. Guiding Principles for Ethical Research https://www.nih.gov/health-information/nih-clinical-research-trials-you/guiding-principles-ethical-research (2016).
Cruz Rivera, S. et al. Ethical considerations for the inclusion of patient-reported outcomes in clinical research: the PRO ethics guidelines. JAMA 327, 1910–1919 (2022).
Williamson, P. R. et al. The COMET handbook: version 1.0. Trials 18, 280 (2017).
Gale, N. K., Heath, G., Cameron, E., Rashid, S. & Redwood, S. Using the framework method for the analysis of qualitative data in multi-disciplinary health research. BMC Med. Res. Method. 13, 117 (2013).
King-Kallimanis, B. L. et al. Patient-reported outcomes after treatment discontinuation: commercial clinical trial data from four cancer types. Value Health 24, 1302–1307 (2021).
Retzer, A. et al. International perspectives on suboptimal patient-reported outcome trial design and reporting in cancer clinical trials: a qualitative study. Cancer Med. 10, 5475–5487 (2021).
Skevington, S. M. & McCrate, F. M. Expecting a good quality of life in health: assessing people with diverse diseases and conditions using the WHOQOL-BREF. Health Expect. 15, 49–62 (2012).
Turner, R. R., Quittner, A. L., Parasuraman, B. M., Kallich, J. D. & Cleeland, C. S. Patient-reported outcomes: instrument development and selection issues. Value Health 10, S86–S93 (2007).
Lawrance, R., Skaltsa, K., Regnault, A. & Floden, L. Reflections on estimands for patient-reported outcomes in cancer clinical trials. J. Biopharm. Stat. https://doi.org/10.1080/10543406.2023.2280628 (2023).
Ettridge, K. et al. A randomised online experimental study to compare responses to brief and extended surveys of health-related quality of life and psychosocial outcomes among women with breast cancer. Qual. Life Res. 30, 407–423 (2021).
Biber, J. et al. Patient-reported outcomes: experiences with implementation in a university health care setting. J. Patient-Rep. Outcomes 2, 34–34 (2018).
Low, C. A. et al. Estimation of symptom severity during chemotherapy from passively sensed data: exploratory study. J. Med. Internet Res. 19, e420 (2017).
Ploughman, M., Austin, M., Stefanelli, M. & Godwin, M. Applying cognitive debriefing to pre-test patient-reported outcomes in older people with multiple sclerosis. Qual. Life Res. 19, 483–487 (2010).
US Food and Drug Administration. Patient-Focused Drug Development: Selecting, Developing, or Modifying Fit-for-Purpose Clinical Outcome Assessments: Guidance for Industry, Food and Drug Administration Staff, and Other Stakeholders https://www.fda.gov/media/159500/download (2022).
Calvert, M. J. et al. Patient-reported outcome assessment must be inclusive and equitable. Nat. Med. 28, 1120–1124 (2022).
Trick, W. E., Deamant, C., Smith, J., Garcia, D. & Angulo, F. Implementation of an audio computer-assisted self-interview (ACASI) system in a general medicine clinic: patient response burden. Appl Clin. Inf. 6, 148–162 (2015).
Wild, D. et al. Principles of good practice for the translation and cultural adaptation process for patient-reported outcomes (PRO) measures: report of the ISPOR Task Force for Translation and Cultural Adaptation. Value Health 8, 94–104 (2005).
Shetty, P. N., Hawken, J., Sanghavi, K. K. & Giladi, A. M. Correlation of patient-reported outcomes measurement information system questionnaires with the brief michigan hand questionnaire in patients with 5 common hand conditions. J. Hand Surg. Am. 46, 709 (2021).
Kroenke, K., Monahan, P. O. & Kean, J. Pragmatic characteristics of patient-reported outcome measures are important for use in clinical practice. J. Clin. Epidemiol. 68, 1085–1092 (2015).
Allen, M. J. & Yen, W. M. Introduction to Measurement Theory (Waveland Press, 2001).
Shepshelovich, D. et al. Feasibility assessment of using the complete patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) item library. Oncologist 24, e146–e148 (2019).
Mercieca-Bebber, R. et al. Design, implementation and reporting strategies to reduce the instance and impact of missing patient-reported outcome (PRO) data: a systematic review. BMJ Open 6, e010938 (2016).
Norquist, J. M., Girman, C., Fehnel, S., DeMuro-Mercon, C. & Santanello, N. Choice of recall period for patient-reported outcome (PRO) measures: criteria for consideration. Qual. Life Res. 21, 1013–1020 (2012).
Kyte, D., Ives, J., Draper, H. & Calvert, M. Current practices in patient-reported outcome (PRO) data collection in clinical trials: a cross-sectional survey of UK trial staff and management. BMJ Open 6, e012281 (2016).
O’Donohoe, P. et al. Updated recommendations on evidence needed to support measurement comparability among modes of data collection for patient-reported outcome measures: a good practices report of an ISPOR Task Force. Value Health 26, 623–633 (2023).
Dumais, K. M. et al. Preferences for use and design of electronic patient-reported outcomes in patients with chronic obstructive pulmonary disease. Patient 12, 621–629 (2019).
Kyte, D. et al. Development of an electronic patient-reported outcome measure (ePROM) system to aid the management of patients with advanced chronic kidney disease. J. Patient-Rep. Outcomes 4, 55 (2020).
Walker, P. Medicines and Healthcare products Regulatory Agency (MHRA) Inspectorate: ePRO – An Inspector’s Perspective https://mhrainspectorate.blog.gov.uk/2016/07/07/epro-an-inspectors-perspective/ (2016).
Mowlem, F. D. et al. Best practices for the electronic implementation and migration of patient-reported outcome measures. Value Health https://doi.org/10.1016/j.jval.2023.10.007 (2024).
Aiyegbusi, O. L. Key methodological considerations for usability testing of electronic patient-reported outcome (ePRO) systems. Qual. Life Res. 29, 325–333 (2020).
McMullan, C., Hughes, S. E., Aiyegbusi, O. L. & Calvert, M. Usability testing of an electronic patient-reported outcome system linked to an electronic chemotherapy prescribing and patient management system for patients with cancer. Heliyon 9, e16453 (2023).
Reeve, B. B. et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual. Life Res. 22, 1889–1905 (2013).
Calvert, M. et al. Guidelines for inclusion of patient-reported outcomes in clinical trial protocols: the SPIRIT-PRO extension. JAMA 319, 483–494 (2018).
Proteus Consortium. SPIRIT-PRO PROtocol Reporting Template: A Template Based on Recommendations for Writing Clinical Trial Protocols with Patient-reported Outcomes https://theproteusconsortium.org/wp-content/uploads/2023/02/230220-SPIRIT-PRO-PROtocol-template.pdf (2021).
Crossnohere N, B. M., Snyder C. & the Advisory Group. The PROTEUS Guide to Implementing Patient-Reported Outcomes in Clinical Practice: A Synthesis of Resources (PROTEUS Consortium, 2023).
Calvert, M. et al. SPIRIT-PRO extension explanation and elaboration: guidelines for inclusion of patient-reported outcomes in protocols of clinical trials. BMJ Open 11, e045105 (2021).
Cruz Rivera, S. et al. ‘Give us the tools!’: development of knowledge transfer tools to support the involvement of patient partners in the development of clinical trial protocols with patient-reported outcomes (PROs), in accordance with SPIRIT-PRO extension. BMJ Open 11, e046450 (2021).
Snyder, C. et al. Recommendations for including or reviewing patient-reported outcome endpoints in grant applications. BMJ 373, n1367 (2021).
US Food and Drug Administration. Guidance for Industry, Food and Drug Administration Staff, and Other Stakeholders: Patient-Focused Drug Development: Methods to Identify What Is Important to Patients. https://www.fda.gov/media/131230/download (2019).
US Food and Drug Administration. Guidance for Industry, Food and Drug Administration Staff, and Other Stakeholders: Patient-Focused Drug Development: Collecting Comprehensive and Representative Input https://www.fda.gov/media/139088/download (2020).
US Food and Drug Administration. Patient-Focused Drug Development: Incorporating Clinical Outcome Assessments into Endpoints For Regulatory Decision-Making. Guidance for Industry, Food and Drug Administration Staff, and Other Stakeholders https://www.fda.gov/media/166830/download (2023).
Acknowledgements
We thank the participants who were involved in the Delphi study (Supplementary Information). The views expressed in this publication are those of the authors and Delphi participants and may not represent the views of the broader stakeholder group or host institutions.
Author information
Authors and Affiliations
Contributions
O.L.A., M.J.C., S.C.R., J.R. and P.K. were responsible for concept and design, acquisition and data analysis. O.L.A. and M.J.C. had full access to the data in the study and are responsible for the integrity of the data and the accuracy of the data analysis. All authors were responsible for interpretation of data. O.L.A. and M.J.C. were responsible for drafting the manuscript. All authors were responsible for critical revision of the manuscript for intellectual content.
Corresponding author
Ethics declarations
Competing interests
O.L.A. receives funding from the National Institute for Health and Care Research (NIHR) Birmingham Biomedical Research Centre (BRC), NIHR Blood and Transplant Research Unit (BTRU) in Precision Transplant and Cellular Therapeutics, NIHR Applied Research Collaboration (ARC) West Midlands, UK Research and Innovation (UKRI), Health Foundation, Merck, Gilead, Anthony Nolan, Sarcoma UK and GSK. He declares personal fees from Gilead Sciences Ltd, Merck and GSK outside the submitted work. S.C.R. receives funding from UK SPINE, Merck and declares personal fees from Merck. M.J.C. is Director of the Birmingham Health Partners Centre for Regulatory Science and Innovation, Director of the Centre for the Centre for Patient-Reported Outcomes Research and is an NIHR Senior Investigator. M.J.C. receives funding from the NIHR, UKRI, NIHR Birmingham BRC, the NIHR Surgical Reconstruction and Microbiology Research Centre, NIHR ARC West Midlands, UK SPINE, European Regional Development Fund – Demand Hub and Health Data Research UK at the University of Birmingham and University Hospitals Birmingham NHS Foundation Trust, Innovate UK (part of UKRI), Macmillan Cancer Support, UCB Pharma, Janssen, GSK and Gilead. M.C. has received personal fees from Astellas, Aparito, CIS Oncology, Takeda, Merck, Daiichi Sankyo, Glaukos, GSK, Pfizer, the Patient-Centered Outcomes Research Institute (PCORI) and Vertex outside the submitted work. In addition, a family member owns shares in GSK. J.R., through her institution, has received consultancy fees from the University of Birmingham Enterprise. Through her institution, she is supported by an unrelated Select Foundation Fellowship and has received unrelated research funding from Pfizer, the EuroQol Foundation, PCORI, the Royal Hobart Hospital Research Foundation and the US FDA. In addition, she is Chair of the ISPOR Patient-Centered Special Interest Group and the ISOQOL Australia and New Zealand Special Interest Group and Associate Editor for the Journal of Patient-Reported Outcomes. N.A. receives funding from NIHR ARC West Midlands. S.E.H. receives funding from the NIHR, NIHR BTRU in Precision Transplant and Cellular Therapeutics, NIHR Birmingham BRC, NIHR (ARC) West Midlands, UKRI and UK SPINE. She declares personal fees from Cochlear, Pfizer, Rinri Therapeutics, AstraZeneca, Aparito and CIS Oncology outside the submitted work. J.D.P. has received unrelated research funding from the National Cancer Institute, the National Institutes of Health, the Food and Drug Administration, the ECOG-ACRIN Medical Research Foundation, the Peter G. Peterson Foundation, Veloxis Pharmaceuticals, Pfizer and the Northwestern University George M. O’Brien Kidney Core Center. He has received unrelated personal fees from AstraZeneca, IMPAQ International and FACIT.org. In addition, he is part of the ISOQOL, Psychometric Special Interest Group Chair. F.E. received personal fees from AbbVie, Incyte, Syros and Novartis, outside the submitted work. C.M. receives funding from NIHR Surgical Reconstruction and Microbiology Research Centre, UKRI, NIHR, NIHR BTRU in Precision Transplant and Cellular Therapeutics and declares personal fees from Aparito outside the submitted work. E.H.D. owns an ePRO software platform called Atom5 through Aparito. A.B. is the founder of Bottomley Consulting Group. He has received fees, payments from Sevier, Pfizer, Ferring, Bayer, GSK, Merck, BMS, EMD Serano, Mirati, Boehringer Ingelheim, Vertex, FWA, European Commission and MD Anderson and is a Board member of the PROTEUS consortium and receives bursary from John Hopkins University. He is a member of ISOQOL and ISPOR. B.K.K. received unrelated research funding from grants from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Jazz Pharma, Genentech, Eli Lilly, Janssen, Takeda, Dachii Sankyo, Blueprint Medicines, Janssen, Amgen and Seagen. She also served as a paid consultant to Eli Lilly, AbbVie, Bristol Myers Squibb, Gilead outside of the submitted work. A.M. is partner of QC Medica, a health economics and outcomes research consultancy company. C.S. receives research funding from Pfizer (current) and Genentech (previous) through her institution and personal fees from Janssen (previous) and Shionogi (current). A.M.S. receives unrelated research funding through her institution from the National Institutes of Health, PCORI, AHRQ, Pfizer, UroGen Pharma and the American Society of Clinical Oncology. She has received past funding from Sivan Innovation, Bladder Cancer Advocacy Network, Hematology/Oncology Pharmacy Association and the Cancer and Aging Research Group. She also served as a paid consultant to Navigating Cancer (personal fees). M.B. receives research funding from Pfizer through another institution. A.D. is Deputy Director of the Birmingham Health Partners Centre for Regulatory Science and Innovation and is an NIHR Senior Investigator. He receives funding from the NIHR, UKRI and NIHR Birmingham BRC. No other disclosures are reported. J.M. is chief editor at Nature Medicine and has not taken part in the peer review and editorial process of this consensus statement. This work was funded by Merck Healthcare and along with the study sponsor, the University of Birmingham, had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript; or decision to submit the manuscript for publication. Several authors are employees of this organization; however, beyond the declared author contributions, the sponsor had no additional role. The views expressed in this article are those of the authors and not necessarily those of the NIHR, US FDA, Medicines and Healthcare products Regulatory Agency, European Medicines Agency and Health Canada.
Peer review
Peer review information
Nature Medicine thanks Aaron Hansen, Thomas Atkinson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Karen O’Leary, in collaboration with the Nature Medicine team.
Supplementary information
Supplementary Information
Supplementary Information.
Supplementary Data
Pre-meeting information: consensus meeting.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aiyegbusi, O.L., Cruz Rivera, S., Roydhouse, J. et al. Recommendations to address respondent burden associated with patient-reported outcome assessment. Nat Med 30, 650–659 (2024). https://doi.org/10.1038/s41591-024-02827-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-024-02827-9
