
Abstract
Artemisinin resistance (delayed P. falciparum clearance following artemisinin-based combination therapy), is widespread across Southeast Asia but to date has not been reported in Africa1,2,3,4. Here we genotyped the P. falciparum K13 (Pfkelch13) propeller domain, mutations in which can mediate artemisinin resistance5,6, in pretreatment samples collected from recent dihydroarteminisin-piperaquine and artemether-lumefantrine efficacy trials in Rwanda7. While cure rates were >95% in both treatment arms, the Pfkelch13 R561H mutation was identified in 19 of 257 (7.4%) patients at Masaka. Phylogenetic analysis revealed the expansion of an indigenous R561H lineage. Gene editing confirmed that this mutation can drive artemisinin resistance in vitro. This study provides evidence for the de novo emergence of Pfkelch13-mediated artemisinin resistance in Rwanda, potentially compromising the continued success of antimalarial chemotherapy in Africa.
Similar content being viewed by others

Main
Malaria represents a major public health issue in the tropics, with an estimated 228 million cases and 405,000 deaths in 2018 (refs. 8,9). Of increasing concern is P. falciparum resistance to artemisinin (ART) derivatives, used worldwide as the core components of ART-based combination therapies (ACTs)10. ART resistance (ART-R), characterized by delayed P. falciparum clearance following treatment with artemisinin monotherapy or an ACT1,11, is now widespread in the Greater Mekong subregion (GMS), which consists of Cambodia, Thailand, Vietnam, Myanmar and Laos12,13. Resistance to the partner drugs piperaquine and mefloquine is also now common in the GMS, causing high rates of ACT treatment failure14,15.
The appearance of ART-R parasites in Africa would pose a major public health threat. Resistance to the former first-line antimalarial chloroquine first arose in the GMS in the 1960s before spreading to Africa. Resistance to pyrimethamine (used in association with sulfadoxine) followed shortly thereafter16. The lost clinical efficacy of these compounds is suspected to have contributed to millions of additional malaria deaths in young African children in the 1980s17. In addition to the risk of imported resistance18, the likelihood of resistance emerging locally in Africa has increased in areas where control measures have reduced the disease transmission intensity. The resulting attenuation in naturally acquired human immunity can increase the frequency of symptomatic infections and the need for treatment, while decreasing parasite genetic diversity and reducing competition between sensitive and resistant parasites19. To date, the efficacy of ACTs has remained high outside Southeast Asia (SEA)2. Early detection of resistance provides the best chance of minimizing its lethal impact.
Mutations in the Pfkelch13 propeller domain (PF3D7_1343700) constitute the primary determinant of ART-R1,5,6. These mutations are suspected to reduce Pfkelch13 function, which is required for parasite-mediated endocytosis of host hemoglobin in the newly invaded intra-erythrocytic ring stages20,21. Pfkelch13 C580Y is the most widespread allele in SEA13,15 and has recently been detected in Guyana22 and Papua New Guinea23. In Africa, slow-clearing infections after ACT treatment have been observed at frequencies of <1%24. Previously we observed nonsynonymous Pfkelch13 mutations in <5% of African isolates, with >50% of the polymorphisms present in only a single P. falciparum infection. The most frequent Pfkelch13 mutation in Africa was A578S, which did not confer ART-R in vivo or in vitro4. Nonsynonymous Pfkelch13 mutations associated with delayed parasite clearance or day 3 positivity (day 3+) in the GMS (F446I, Y493H, R539T, I543T, P553L, R561H, P574L, C580Y, A675V) have only been rarely reported, if at all, in African parasites25,26.
Here we conducted an in-depth genetic analysis of P. falciparum samples collected from 2012 to 2015 at six Rwandan sites and performed gene-editing studies to evaluate the in vitro resistance phenotypes of parasites harboring the Pfkelch13 R561H or P574L mutations identified in these samples.
Results
Clinical drug efficacy trial design and outcomes
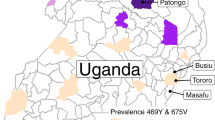
From September 2013 to December 2015, clinical drug efficacy studies to assess the efficacy of artemether-lumefantrine (AL) and dihydroartemisinin-piperaquine (DP) for the treatment of uncomplicated P. falciparum malaria were conducted in patients enrolled at the Masaka and Ruhuha health facilities in Rwanda (Fig. 1; http://www.isrctn.com/ISRCTN63145981). The overall 42-day PCR-corrected efficacies of AL (95.2% (196 of 206); 95% CI, 91.3% to 97.7%) and DP (97.5% (212 of 217); 95% CI, 94.4% to 99.1%) were similar between both sites (P = 0.17, log-rank test)7. The day 3 positivity rate (day 3+), defined as the proportion of patients who were still parasitemic on day 3 after initiation of treatment, was low for both treatments: 1 of 263 (0.4%) for AL and 0 of 264 for DP (Table 1).

Isolates were sourced from the MalariaGEN P. falciparum Community Project (https://www.malariagen.net/apps/pf/4.0). Locations of clinical drug efficacy study sites where Rwandan isolates were collected are indicated. Patients enrolled at Masaka and Ruhuha (black) were treated with AL or DP, whereas patients enrolled at Bugarama, Kibirizi, Nyarurema and Rukara (gray) were treated with AL. Pfkelch13 nonsynonymous mutations identified in these regions and relative proportions of mutant alleles are detailed in Table 1. Each leaf represents one sample and is colored according to the country of collection. Rwandan parasites carrying the Pfkelch13 R561H mutation or the Pfkelch13 WT allele are identified by filled or unfilled red stars at the tip, respectively. Rwandan Pfkelch13 R561H mutants are closely related to other African samples at a genomic level, demonstrating that they are the product of a local emergence event. Scale bar, 0.0001 nucleotide substitutions per character. Only branch confidence supports <95% are indicated.
Pfkelch13 genotyping
Pfkelch13 propeller domain genotyping was performed on 534 pretreatment samples collected at Masaka and Ruhuha. Of the 507 successfully genotyped samples, 35 (6.9%) harbored 1 of 14 different Pfkelch13 nonsynonymous mutations. These included M460I, C469Y, R513L, V555A, R561H, P574L, R575I, A578S, G592E, E605K, A626E, V637I, E651K and P667R. One sample contained two clones, each with a different mutation (G592E or V637I). The Pfkelch13 561H variant, previously associated with delayed parasite clearance following ART monotherapy or ACT treatment in the GMS26, was the most predominant mutant. This R561H mutation was observed exclusively in Masaka, where it was present in 7 of 58 samples in 2013–2014 and 12 of 199 samples in 2015 (19 of 257, 7.4%). We also detected two additional Pfkelch13 mutations (P574L and C469Y) previously associated with delayed parasite clearance26. We did not observe significant changes in the proportion of Pfkelch13 nonsynonymous mutations over time (P = 0.3, chi-squared test, 1.92, d.f., 2) at either study site (Table 1).
Pfkelch13 genotyping was also carried out on 420 additional blood samples collected before AL treatment from patients enrolled in a study following the same clinical protocol that was conducted in 2012–2015 across four sites in Rwanda (ISRCTN63145981; Fig. 1). Among these, ten (2.4%) carried a Pfkelch13 nonsynonymous mutation (C469F, V487I, V555A, R561H, A578S, A578V or P667R). The Pfkelch13 R561H mutation was found in a sample from Rukara from a patient who presented a negative Giemsa-stained blood film on day 3 after treatment (day 3−) but had recrudescent parasitemia on day 21. A blood sample collected on this day was also found to carry R561H parasites (Supplementary Table 1).
Relationship between Pfkelch13 alleles and clinical outcomes
All patients with uncomplicated P. falciparum infections that had Pfkelch13 mutant parasites were day 3−, with the exception of one patient who was day 3+ (80 parasites per µl) and had P. falciparum parasites harboring the Pfkelch13 574L variant (Table 2). Three patients (one in the AL arm and two in the DP arm) presented signs of severe malaria on day 1 and were treated with intravenous artesunate according to national treatment guidelines. These patients were all day 3+ and of these, two had Pfkelch13 mutant infections (either 561H or 626E).
By excluding the intravenous artesunate-treated patients presenting with severe malaria from our final analysis, we did not find any association between Pfkelch13 nonsynonymous mutations and delayed parasite clearance as assessed by day 3+ (P = 0.06, Fisher’s exact test) or by clinical outcome at day 42 (cured versus recrudescent) (P = 1, Fisher’s exact test; Table 2). Furthermore, we did not observe any correlation between mutation status and clinical outcome in the samples from the second study conducted at the four additional sites in Rwanda (P = 1 for day 3+ and P = 0.3 for day 42 clinical outcome, Fisher’s exact test). This analysis included the patient mentioned above (treated with AL) who had a recrudescent parasitemia on day 21 with a Pfkelch13 561H infection (Table 2).
In vitro susceptibility of Pfkelch13 561H and 574L mutants to artemisinin
To test the impact of the Pfkelch13 R561H and P574L mutations on ART-R in vitro, we used CRISPR-Cas9 to introduce these mutations into Dd2 parasites and subjected the recombinant mutant and wild-type (WT) control lines to phenotyping in the ring-stage survival assay (RSA0–3h)6. The Pfkelch13 R561H mutation was found to confer in vitro ART-R (increased RSA survival), with Dd2R561H parasites exhibiting a mean survival rate of 4.3% versus 0.6% for the Dd2WT line expressing WT Pfkelch13 (P < 0.0001, Mann–Whitney U-test). The survival of the 561H line was comparable to that of Dd2C580Y line (mean survival of 4.7%), which harbors the Pfkelch13 C580Y mutation (Fig. 2). These results demonstrate that the Pfkelch13 R561H mutation can yield ring-stage ART-R at a level that is comparable to the C580Y mutation that has swept across SEA13,15.

Mean ± s.e.m. RSA0–3h survival rates (percentage of viable parasites) were as follows: Dd2R561H 4.3 ± 0.1% (n = 7 assays); Dd2P574L 2.1 ± 0.3% (n = 8 assays); Dd2C580Y 4.7 ± 0.4% (n = 9 assays); Dd2WT 0.6 ± 0.1% (n = 13 assays). All assays were performed in duplicate. Mann–Whitney U-tests (two-sided) were used to test for statistically significant differences between Pfkelch13-edited clones and the Dd2WT comparator line. Survival rates of Dd2R561H, Dd2P574L and Dd2C580Y all differed significantly from Dd2WT (**** P < 0.0001). The limit of detection of viable parasites was estimated at 0.1% parasitemia (lower limit of 50 parasitized red blood cells per total number of 50,000 counted for each line in each assay).
Dd2P574L parasites displayed a mean RSA survival rate of 2.1%, which represented a modest but significant increase relative to the Dd2WT line (P < 0.0001, Mann–Whitney U-test; Fig. 2). These results provide evidence that the Pfkelch13 P574L mutation is able to confer a lesser degree of ART-R.
Origins of the Rwandan Pfkelch13 561H haplotype and its relationship to other P. falciparum populations
To study the origin of the Pfkelch13 561H mutants found in Rwanda, we compared whole-genome sequences of 340 samples, comprising 25 Rwandan P. falciparum sequences generated for this study, and an earlier collection of 104 sequences from central, western and southern African locations, 164 from Bangladesh and SEA and 45 from South America, in addition to 2 reference genomes (3D7 from Africa and 7G8 from South America; Supplementary Table 2). Of the 25 Rwandan sequences, 16 were Pfkelch13 561H mutants and 9 were Pfkelch13 WT. The isolates from SEA (Myanmar and Thailand) included 17 561H mutants. All other parasite sequences either had distinct nonsynonymous Pfkelch13 mutations or were WT for Pfkelch13.
A maximum-likelihood phylogenetic tree inferred from the 14 P. falciparum chromosomes showed clear separations between the African, Asian and South American parasites (Fig. 1). Additionally, the Rwandan Pfkelch13 561H mutants clustered unambiguously with the Rwandan Pfkelch13 WT parasites.
We also explored haplotype diversity across a 200-kb region surrounding the R561H mutation. This analysis used sequences from eight Rwandan Pfkelch13 mutant infections that seemed to be predominantly monoclonal (allelic depth of the WT allele <0.05), as well as 17 sequences from Pfkelch13 561H mutants from SEA (Myanmar and Thailand). The presence of a single shared haplotype surrounding the 561H variant in the Rwandan samples was consistent with a single epidemiological origin for this mutation. These results confirmed that the Rwandan 561H mutants share no genetic relatedness to the 561H mutants previously detected in Myanmar and Thailand (Extended Data Fig. 1).
Next, we performed a principal coordinate analysis (PCoA) based on a pairwise genetic distance matrix (computed from a 200-kb window around the Pfkelch13 gene). This analysis confirmed that the African samples (including both the Rwandan Pfkelch13 561H mutants and WT parasites) clustered together and were distinct from Asian samples (Extended Data Fig. 2). In the eight Pfkelch13 561H mutants from Rwanda we identified an extended 494-kb region, encompassing the mutation that was identical across isolates (Extended Data Fig. 3). Although an ancient common ancestry cannot strictly be ruled out, our data provide compelling evidence that Rwandan Pfkelch13 561H is the product of a recent de novo local emergence.
Investigation of the genetic background of Rwandan Pfkelch13 561H mutants
To investigate further the genetic background of the Rwandan Pfkelch13 561H mutants, we searched for molecular signatures associated with resistance to other antimalarials, including the ACT partner drugs piperaquine and lumefantrine. We also screened for mutations that have been identified in founder populations common to SEA ART-R parasites (those that constitute a ‘genetic background’ for ART-R)27.
First, we investigated 233 isolates from Masaka and Ruhuha for amplification of the plasmepsin2 (pfpm2; PF3D7_1408000) and multidrug resistance-1 (pfmdr1; PF3D7_0523000) genes, considered markers of reduced susceptibility to piperaquine and lumefantrine/mefloquine, respectively28,29,30. Of these, we found 4 isolates (1.7%) with two copies of pfpm2 and 12 isolates (5.1%) with two copies of pfmdr1. All isolates carrying two copies of pfpm2 or pfmdr1 were WT for Pfkelch13 (Supplementary Table 3). We also tested 14 of the 20 Pfkelch13 561H mutants for mutations in the chloroquine resistance transporter gene (pfcrt; PF3D7_0709000), whose variants can confer resistance to chloroquine or piperaquine31,32. All 561H mutants carried WT pfcrt (Supplementary Table 4).
Second, we tested whether the proportions of single-nucleotide polymorphisms (SNPs) associated with the emergence of ART-R in the SEA genetic background varied between Rwandan Pfkelch13 561H mutants and WT parasites. For this analysis, we used 14 Rwandan Pfkelch13 561H mutants and 10 randomly selected WT parasites and tested for mutations in the six markers defining the SEA ART-R background. No significant differences were observed between the two groups of isolates. We detected four isolates with the D193Y mutation in the ferredoxin gene (pffd; PF3D7_1318100), two (15.4%) in Pfkelch13 561H mutant samples and two (22.2%) in WT samples (P = 0.69, Fisher’s exact test). No mutations were detected in the P. falciparum apicoplast ribosomal protein S10 precursor (pfarps10, PF3D7_1460900), multidrug resistance protein 2 (pfmdr2, PF3D7_1447900), pfpib7 (PF3D7_0720700), pfpph (PF3D7_1012700) or exonuclease (PF3D7_1362500) genes in either the Pfkelch13 561H or WT isolates (Supplementary Table 4).
Discussion
This study clearly shows early warning signs of ART-R in Rwanda. We provide evidence for the clonal expansion of an indigenous Pfkelch13 561H lineage in two localities 100 km apart in Rwanda (prevalence 7.4% in Masaka and 0.7% in Rukara). This expansion was not linked to delayed parasite clearance in vivo or clinical treatment failure following AL or DP treatments, likely due to the high efficacy of the partner drugs lumefantrine and piperaquine. Genetic analyses indicate that Rwandan Pfkelch13 561H mutants are the product of recent de novo local emergence. These findings contrast with previous scenarios from the 1980s in which the emergence of chloroquine- and pyrimethamine-resistant parasites in Africa resulted from the westward spread of these parasites from SEA16, and confirm that local emergence of ART-R is possible in Africa.
We used gene editing and the RSA0–3h, a clinically validated in vitro phenotypic analysis6,33, to demonstrate that the Pfkelch13 R561H mutation is sufficient to confer ART-R in vitro. These experiments employed Dd2, which has been the most widely used P. falciparum strain for Pfkelch13 gene editing4,6. Our results revealed that in Dd2 parasites, the R561H mutation confers survival at levels comparable to the C580Y mutation that predominates in SEA (with mean survival rates of 4.3% and 4.7%, respectively)1,13,15. Previous studies have shown that Pfkelch13 mutations that afford resistance do so across all strains, with the parasite genetic background modulating resistance levels and with mutations conferring less resistance in Dd2 compared to contemporary SEA strains6. While we did not test the impact of this mutation in Rwandan parasites due to a lack of availability of culture-adapted strains, we are confident that the resistance phenotype observed herein would be maintained across strains.
At a genomic level, Rwandan Pfkelch13 561H mutants were phylogenetically closely related to other African samples and clustered unambiguously with Rwandan Pfkelch13 WT parasites. Haplotype analysis revealed that Rwandan Pfkelch13 561H mutants shared an identical haplotype surrounding the R561H mutation that differed from the haplotypes of SEA 561H mutants, strongly suggesting a single de novo epidemiological origin and recent spread of the mutation. No genetic relatedness was observed between Rwandan Pfkelch13 561H parasites and Pfkelch13 561H mutants previously detected in Myanmar and Thailand by PCoA.
The current rise and expansion of the in vitro ART-R Pfkelch13 R561H mutation in Rwanda is particularly notable in light of the observed absence of clinical outcomes typically associated with ART-R. We suspect that the absence of delayed parasite clearance in Rwandan patients harboring Pfkelch13 561H mutant parasites is due to high levels of naturally acquired immunity to P. falciparum in the study participants. Indeed, it has been shown that P. falciparum antibody titers are strongly associated with faster parasite clearance rates in patients living in high-transmission areas like Rwanda and that antibodies against P. falciparum blood stages enhance antimalarial efficacy34. In our study, the ages of patients enrolled at both sites ranged from 1 to 14 years, with an estimated median age of 8 years (interquartile range (IQR): 5–11 years). Given that immunity is acquired gradually with age, a clinical drug efficacy trial limited to younger populations (≤5 years of age) might reveal a significant association between the presence of Pfkelch13 561H mutants in pretreatment isolates and delayed parasite clearance. We hypothesize that early signs of clinical ART-R can lie undetected in populations with high levels of immunity, calling into question the relevance of the current clinical metrics used to detect ART-R in Africa. This hypothesis is supported by data from population-based mathematical modeling19 that showed that ART-R parasites might be able to circulate up to 10 years longer without detection in high-transmission areas than in low-transmission areas.
To date, the Pfkelch13 R561H mutation has been reported multiple times in SEA (Cambodia until 2006, Myanmar and Thailand)25, once in India35 and a few times in Africa (Democratic Republic of the Congo4, Rwanda36 and Tanzania37), but has only been associated with slow-clearing infections in SEA26. Thus, the degree to which Pfkelch13 561H mutant parasites are able to withstand exposure to ART in vivo and how Pfkelch13 561H is successfully transmitted between patients in the absence of clinical recrudescence (Table 2) requires further elucidation. It is possible that the resistance advantage afforded by the Pfkelch13 561H mutation is slight and undetectable based on day 3+ and recrudescence metrics, and thus would be evident only with ART monotherapy trials. Regarding transmission, we can offer several hypotheses. First, Pfkelch13 561H mutants could be less susceptible to ART due to an ability to enter into a dormant state38 and later produce transmissible gametocytes. Second, Pfkelch13 561H parasites may have a higher capacity to be transmitted due to an unknown genetic feature or Pfkelch13 561H gametocytes may be less susceptible to the gametocytocidal activity of artemisinin. However, it is most likely that the transmission of Pfkelch13 561H mutants in Rwanda is maintained by asymptomatic individuals or mildly symptomatic untreated patients with circulating Pfkelch13 561H mutants that have been selected by low levels of circulating drugs.
We did not detect the combination of background mutations earlier suspected to be linked to the ART-R phenotype in SEA in the Pfkelch13 561H Rwandan isolates27. This suggests that the emergence of mutant Pfkelch13 that drives in vitro resistance is not dependent on the presence of secondary mutations within the parasite genome. So far, no gene-editing and in vitro phenotyping experiments have been performed to test the importance of these secondary mutations for resistance. Data from this study suggest that mutations in fd, mdr2, arps10 and others represent the genetic architecture of regional ART-R in P. falciparum SEA parasite populations rather than secondary determinants of resistance.
The findings of this study have substantial implications for public health in confirming the de novo emergence and clonal expansion of an ART-R Pfkelch13 R561H lineage in Rwanda and in validating this mutation as a mediator of ART-R in vitro. In the absence of effective strategies to contain the spread of resistance across Rwanda and to neighboring countries, we may soon witness a rise of resistance to ACT partner drugs, which will in turn lead to high treatment failure rates, as has occurred in SEA14. Recent studies have predicted that ACT treatment failures in Africa could be responsible for an additional 78 million cases and 116,000 deaths over a 5-year period39.
Molecular surveillance of Pfkelch13-related ART-R currently implemented by the National Malaria Control Programme in Rwanda needs to be sustained and strengthened so that mutations can be identified before clinical phenotypes become apparent. Our findings argue for the need for more rapid collection of data, analysis and dissemination of information using new high-throughput field-based surveillance tools operable at a national level. Likewise, we have to reappraise the performances of the current clinical phenotypic metrics (delayed parasite clearance and day 3+) to detect the warning signs of ART-R in African populations with high immunity early on.
Methods
Clinical drug efficacy trial oversight and blood sample collection
The clinical drug efficacy trial (ISRCTN63145981, http://www.isrctn.com/ISRCTN63145981) was conducted by the Rwanda National Malaria Program between 2013 and 2015 at two health facilities in Rwanda (Masaka and Ruhuha, in the Kicukiro and Bugesera districts, respectively) to assess the efficacy of AL or DP for the treatment of uncomplicated P. falciparum malaria in children 1–14 years of age, presenting with suspected uncomplicated P. falciparum malaria7. Patients at both sites were randomly assigned to receive a full course of AL (Co-artem, 20 mg artemether and 120 mg lumefantrine per tablet) or DP (Duo-cotecxin, 40 mg dihydroartemisinin and 320 mg piperaquine per tablet) according to the manufacturer’s dosing schedule.
The primary outcome of the study was the PCR-adjusted clinical response to the designated treatment on day 42 (ref. 40). The secondary outcome was the day 3+, defined as the proportion of patients who were still parasitemic on day 3 after initiation of treatment as assessed by thick blood smear (Supplementary Methods 1)41.
Pfkelch13 genotyping and whole-genome sequencing
Genomic investigations were carried out on blood samples collected before ACT treatment (AL or DP) from patients enrolled at Masaka and Ruhuha. We also analyzed blood samples collected before AL treatment from patients enrolled in clinical drug efficacy studies conducted at four additional sites (Bugarama, Kibirizi, Nyarurema and Rukara) across Rwanda between 2012 and 2015.
Parasite DNA was extracted from dried blood samples (Fig. 1) using the QIAamp DNA Blood Mini Kit (Qiagen). Mutations in the propeller domain of Pfkelch13 (PF3D7_1343700, codons 440–680, 720 bp) were identified by capillary sequencing of PCR products4. Parasite whole-genome sequences were obtained by Illumina paired-end sequencing after capture-based enrichment of parasite DNA (Supplementary Methods 2)5.
Phylogenetic analysis
For each sequenced sample, read alignments against the chromosome sequences of P. falciparum 3D7 v45 were processed to infer consensus sequences. These consensus sequences were pooled and concatenated, leading to 17,313,072 aligned nucleotide characters that were used to infer a maximum-likelihood phylogenetic tree (Supplementary Methods 3).
Genotyping and haplotype analysis
The Genome Analysis Toolkit Haplotype Caller was used to identify SNPs in each isolate. We assessed the genetic identity of Pfkelch13 561H mutants from Rwanda and Asia by comparing alleles at loci within a 200-kb window around the mutation and recording the number of discrepancies between each sample and the mutant consensus sequence. PCoA was performed by computing pairwise Euclidean genetic distances between samples in an extended 494-kb window (Supplementary Methods 4).
Generation of gene-edited lines and in vitro susceptibility assays
Dd2R561H and Dd2P574L gene-edited parasite lines, as well as Dd2WT and the Dd2C580Y lines used as controls, were generated by CRISPR-Cas9-mediated editing of the Pfkelch13 locus using the pDC2-cam-coSpCas9-U6-hdhfr vector. In vitro ART susceptibilities of these lines were assessed using RSA0–3h (Supplementary Methods 5–7).
Statistical analysis
Sample size calculations and clinical data management methods have been previously described7. PCR-adjusted clinical efficacy rates at day 42 were calculated using Kaplan–Meier survival analysis. Survival curves were compared using the Mantel–Haenszel log-rank test (one-sided). Patients with new infections during the 42-day follow-up period and patients with undetermined or noninterpretable PCR genotyping data were excluded from the final analysis. Data were reported in Microsoft Excel (Office 2016) and analyzed with MedCalc v.12 (MedCalc Software) and Prism 8 (GraphPad Software). Mann–Whitney U-tests (two-sided) were used for nonparametric comparisons. For frequency data (expressed with percentages and 95% CIs), we used chi-squared or Fisher’s exact tests (one-sided). Relative risks were estimated using the Mantel–Haenszel test. All P values <0.05 were deemed significant.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request. Parasite whole-genome sequences have been deposited in the repository https://www.ncbi.nlm.nih.gov/bioproject/PRJEB38946, and the sequence files are accessible under the accession numbers ERS4758427 to ERS4758451.
Change history
27 May 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41591-021-01365-y
References
Ashley, E. A. et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 371, 411–423 (2014).
Conrad, M. D. & Rosenthal, P. J. Antimalarial drug resistance in Africa: the calm before the storm? Lancet Infect. Dis. 19, e338–e351 (2019).
MalariaGEN Plasmodium falciparum Community Project. Genomic epidemiology of artemisinin resistant malaria. eLife 5, e08714 (2016).
Menard, D. et al. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N. Engl. J. Med. 374, 2453–2464 (2016).
Ariey, F. et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505, 50–55 (2014).
Straimer, J. et al. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347, 428–431 (2015).
Uwimana, A. et al. Efficacy of artemether-lumefantrine versus dihydroartemisinin-piperaquine for the treatment of uncomplicated malaria among children in Rwanda: an open-label, randomized controlled trial. Trans. R. Soc. Trop. Med. Hyg. 113, 312–319 (2019).
World Health Organization. World Malaria Report. https://www.who.int/publications-detail/world-malaria-report-2019 (2019).
White, N. J. et al. Malaria. Lancet 383, 723–735 (2014).
Menard, D. & Dondorp, A. Antimalarial drug resistance: a threat to malaria elimination. Cold Spring Harb. Perspect. Med. 7, a025619 (2017).
Dondorp, A. M. et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361, 455–467 (2009).
Amato, R. et al. Origins of the current outbreak of multidrug-resistant malaria in Southeast Asia: a retrospective genetic study. Lancet Infect. Dis. 18, 337–345 (2018).
Imwong, M. et al. The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: a molecular epidemiology observational study. Lancet Infect. Dis. 17, 491–497 (2017).
van der Pluijm, R. W. et al. Determinants of dihydroartemisinin-piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: a prospective clinical, pharmacological, and genetic study. Lancet Infect. Dis. 19, 952–961 (2019).
Hamilton, W. L. et al. Evolution and expansion of multidrug-resistant malaria in Southeast Asia: a genomic epidemiology study. Lancet Infect. Dis. 19, 943–951 (2019).
Blasco, B., Leroy, D. & Fidock, D. A. Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 23, 917–928 (2017).
Murray, C. J. et al. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379, 413–431 (2012).
Huang, Z. & Tatem, A. J. Global malaria connectivity through air travel. Malar. J. 12, 269 (2013).
Scott, N. et al. Implications of population-level immunity for the emergence of artemisinin-resistant malaria: a mathematical model. Malar. J. 17, 279 (2018).
Birnbaum, J. et al. A Kelch13-defined endocytosis pathway mediates artemisinin resistance in malaria parasites. Science 367, 51–59 (2020).
Yang, T. et al. Decreased K13 abundance reduces hemoglobin catabolism and proteotoxic stress, underpinning artemisinin resistance. Cell Rep. 29, e2915 (2019).
Mathieu, L. C. et al. Local emergence in Amazonia of Plasmodium falciparum k13 C580Y mutants associated with in vitro artemisinin resistance. eLife 9, e51015 (2020).
Miotto, O., et al. Emergence of artemisinin-resistant Plasmodium falciparum with kelch13 C580Y mutations on the island of New Guinea. Preprint at bioRxiv https://doi.org/10.1101/621813 (2019).
WWARN Artemisinin-based Combination Therapy Africa Baseline Study Group. et al. Clinical determinants of early parasitological response to ACTs in African patients with uncomplicated falciparum malaria: a literature review and meta-analysis of individual patient data. BMC Med. 13, 212 (2015).
Ocan, M. et al. K13-propeller gene polymorphisms in Plasmodium falciparum parasite population in malaria affected countries: a systematic review of prevalence and risk factors. Malar. J. 18, 60 (2019).
WWARN Genotype-Phenotype Study Group. Association of mutations in the Plasmodium falciparum Kelch13 gene (Pf3D7_1343700) with parasite clearance rates after artemisinin-based treatments-a WWARN individual patient data meta-analysis. BMC Med. 17, 1 (2019).
Miotto, O. et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 47, 226–234 (2015).
Witkowski, B. et al. A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: a phenotype-genotype association study. Lancet Infect. Dis. 17, 174–183 (2017).
Amato, R. et al. Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype-phenotype association study. Lancet Infect. Dis. 17, 164–173 (2017).
Sidhu, A. B. et al. Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J. Infect. Dis. 194, 528–535 (2006).
Dhingra, S. K., Small-Saunders, J. L., Menard, D. & Fidock, D. A. Plasmodium falciparum resistance to piperaquine driven by PfCRT. Lancet Infect. Dis. 19, 1168–1169 (2019).
Ross, L. S. et al. Emerging Southeast Asian PfCRT mutations confer Plasmodium falciparum resistance to the first-line antimalarial piperaquine. Nat. Commun. 9, 3314 (2018).
Witkowski, B. et al. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in vitro and ex vivo drug–response studies. Lancet Infect. Dis. 13, 1043–1049 (2013).
O’Flaherty, K. et al. Contribution of functional antimalarial immunity to measures of parasite clearance in therapeutic efficacy studies of artemisinin derivatives. J. Infect. Dis. 220, 1178–1187 (2019).
Mishra, N. et al. Surveillance of artemisinin resistance in Plasmodium falciparum in India using the kelch13 molecular marker. Antimicrob. Agents Chemother. 59, 2548–2553 (2015).
Wang, X. et al. Molecular surveillance of PfCRT and k13 propeller polymorphisms of imported Plasmodium falciparum cases to Zhejiang Province, China between 2016 and 2018. Malar. J. 19, 59 (2020).
Bwire, G. M., Ngasala, B., Mikomangwa, W. P., Kilonzi, M. & Kamuhabwa, A. A. R. Detection of mutations associated with artemisinin resistance at k13-propeller gene and a near complete return of chloroquine susceptible falciparum malaria in southeast of Tanzania. Sci. Rep. 10, 3500 (2020).
Barrett, M. P., Kyle, D. E., Sibley, L. D., Radke, J. B. & Tarleton, R. L. Protozoan persister-like cells and drug treatment failure. Nat. Rev. Microbiol. 17, 607–620 (2019).
Slater, H. C., Griffin, J. T., Ghani, A. C. & Okell, L. C. Assessing the potential impact of artemisinin and partner drug resistance in sub-Saharan Africa. Malar. J. 15, 10 (2016).
World Health Organization. Methods and techniques for clinical trials on antimalarial drug efficacy: Genotyping to identify parasite populations. https://www.who.int/malaria/publications/atoz/9789241596305/en/ (2008).
World Health Organization. Status report on artemisinin resistance and ACT efficacy (August 2018). https://www.who.int/malaria/publications/atoz/artemisinin-resistance-august2018/en/ (2018).
Acknowledgements
This work was supported by the World Bank through the East African Public Health Laboratory Networking Project (A.U., JL.M.N., A.M., T.M. and JB.M.), the Bill and Melinda Gates Foundation through the World Health Organization (grant OPP1140599, D.M. and P.R.), the US Department of Defense W81XWH-19-1-0086 (D.A.F.) and the National Institutes of Health R01 AI109023 (D.A.F.). This work used the computational and storage services (TARS cluster) provided by the IT department at Institut Pasteur, Paris. We thank all patients who contributed samples and their guardians in the communities of the Kicukiro (Masaka), Bugesera (Ruhuha), Rusizi (Bugarama), Gisagara (Kibirizi), Nyagatare (Nyarurema) and Kayonza (Rukara) and all team members in the health centers. We are also grateful to the members of the Malaria and Other Parasitic Diseases Division at the Rwanda Biomedical Center for their support. We acknowledge L. Duval (National Museum of Natural History, Paris) who furnished the probes for Plasmodium DNA capture and B. Izac who performed Illumina sequencing. We also thank B. Menu, C. Gicquel, N. Jolly, M. Ait Ahmed, V. Pirard and S. Ouchhi (Institut Pasteur, Paris) for their support.
Author information
Authors and Affiliations
Contributions
A.U., JL.M.N., A.M., D.A.F., D.M., E.L., P.R., N.U., D.N., T.M., JB.M., M.W., M.M., E.P. and K.M. contributed to study design. A.U., JL.M.N., A.M., T.M. and JB.M. collected clinical samples and data. D.M. and E.L. prepared the DNA. D.M., E.L., F.A., P.C. and A.C. performed the sequencing and sequence analyses. B.H.S. and D.A.F. performed genome-editing and in vitro assays. A.U., JL.M.N., A.M., D.M., P.R., P.C. and A.C. analyzed data. D.M., B.H.S. and D.A.F. wrote the report. All authors read and approved the final manuscript. P.R. is a staff member of the World Health Organization and is responsible for the views expressed in this publication, noting that they do not necessarily represent the decisions, policy or views of the World Health Organization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Alison Farrell is the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Comparison of mutant pseudo-haplotypes in a 200 kb window around the R561H mutation (100 kb on both sides of the mutation, on chromosome 13).
Each cell represents a single SNP. The blocks of cells (grouped in columns) correspond to SNPs falling into the same 20 kb interval within the 200 kb window. The R561H mutation in Pfkelch13 (PF3D7_1343700) is flagged in the dark red cell at the top. Light blue cells correspond to the reference allele (that is the 3D7 genome), dark blue cells correspond to the alternate allele and grey cells to missing values. Each row corresponds to one isolate, with isolates color-coded according to the country of origin (red for Rwanda, cyan for Thailand and green for Myanmar). Mutant pseudo-haplotypes include eight P. falciparum monoclonal Rwandan samples and 18 Southeast Asian samples (from Myanmar and Thailand, sourced from the Plasmodium falciparum Community Project; https://www.malariagen.net/apps/pf/4.0). The presence of a single shared haplotype surrounding the R561H mutation in Rwandan P. falciparum isolates is consistent with a single epidemiological origin of the genetic background on which the mutation arose. This genetic background demonstrates no genetic relatedness to R561H mutants previously detected in Myanmar and Thailand.
Extended Data Fig. 2 Principal Coordinate Analysis (PCoA) based on pairwise genetic distances in a 494 kb window around the Pfkelch13 gene.
Principal Coordinate Analysis including Pfkelch13 wild type and 561H isolates including those sourced from a public database (small dots, the MalariaGEN Plasmodium falciparum Community Project, https://www.malariagen.net/apps/pf/4.0) and originating from different continents (Asia, Africa or South America). Isolates originating from populations where the Pfkelch13 R561H mutation was found are emphasized (large dots). Empty large dots correspond to Pfkelch13 wild-type isolates and filled large dots correspond to Pfkelch13 561H mutants. While the mutants tend to cluster with individuals of similar origin, axis 1 clearly discriminates African (Rwanda) from Asian (Thailand and Myanmar) Pfkelch13 561H mutants.
Extended Data Fig. 3 Extent of the common core haplotype in the eight Rwandan Pfkelch13 561H isolates (monoclonal isolates).
a, Recombination breakpoints estimated based on the accumulation of discrepancies between the consensus core sequence of mutants and each haplotype on both sides of the Pfkelch13 R561H mutation. The analysis was performed on the eight isolates that appeared monoclonal. Genomic positions are indicated relative to the Pfkelch13 mutation (0 kb). b, Length of the corresponding core mutant haplotypes (obtained based on (A)). Dotted lines delineate a common core region of 494 kb within which all mutant haplotypes appear identical. Genomic positions are indicated relative to the Pfkelch13 mutation (relative position 0 kb). In the larger haplotypes, no clear recombination breakpoint was observed on chromosome 13, indicating a sequence identity along the whole chromosome. Each of the eight isolates are represented by a specific color, consistent between panel (a) and panel (b).
Supplementary information
Supplementary Information
Supplementary Methods 1–7 and Supplementary Tables 1–4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Uwimana, A., Legrand, E., Stokes, B.H. et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat Med 26, 1602–1608 (2020). https://doi.org/10.1038/s41591-020-1005-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-020-1005-2
This article is cited by
-
Therapeutic efficacy and safety of artemether-lumefantrine for uncomplicated Plasmodium falciparum malaria treatment in Metehara, Central-east Ethiopia
Malaria Journal (2024)
-
Expansion of artemisinin partial resistance mutations and lack of histidine rich protein-2 and -3 deletions in Plasmodium falciparum infections from Rukara, Rwanda
Malaria Journal (2024)
-
Molecular survey of pfmdr-1, pfcrt, and pfk13 gene mutations among patients returning from Plasmodium falciparum endemic areas to Turkey
Malaria Journal (2024)
-
The effect of an anti-malarial herbal remedy, Maytenus senegalensis, on electrocardiograms of healthy Tanzanian volunteers
Malaria Journal (2024)
-
Efficacy of artesunate-amodiaquine for treatment of uncomplicated Plasmodium falciparum malaria in mainland Tanzania
Malaria Journal (2024)
