
Abstract
Antiviral CD8+ T cell immunity depends on the integration of various contextual cues, but how antigen-presenting cells (APCs) consolidate these signals for decoding by T cells remains unclear. Here, we describe gradual interferon-α/interferon-β (IFNα/β)-induced transcriptional adaptations that endow APCs with the capacity to rapidly activate the transcriptional regulators p65, IRF1 and FOS after CD4+ T cell-mediated CD40 stimulation. While these responses operate through broadly used signaling components, they induce a unique set of co-stimulatory molecules and soluble mediators that cannot be elicited by IFNα/β or CD40 alone. These responses are critical for the acquisition of antiviral CD8+ T cell effector function, and their activity in APCs from individuals infected with severe acute respiratory syndrome coronavirus 2 correlates with milder disease. These observations uncover a sequential integration process whereby APCs rely on CD4+ T cells to select the innate circuits that guide antiviral CD8+ T cell responses.
Similar content being viewed by others


Main
Antigen-presenting cells (APCs) depend on capturing and presenting viral antigens through major histocompatibility complex (MHC) molecules to prime naive T cells and restimulate antigen-experienced T cells during virus infections1,2,3. Effective T cell responses also hinge on a variety of non-antigenic signals that are relayed from APCs to T cells by co-stimulatory molecules and soluble mediators. It is well established that such contextual cues broadly reflect the exposure of APCs to inflammatory cytokines, such as interferon-α/interferon-β (IFNα/β) and danger signals that stimulate the NF-κB pathway3,4. Yet, the number of co-stimulatory molecules and soluble mediators that APCs use to convey these cues to T cells is discrete, and the expression of many of these factors continues to change as the APCs interact with T cells. For example, CD4+ T cells responding to antigen rapidly increase the expression of CD40L and provide stimulation back to the APC via CD40 and the NF-κB pathway5. Such ‘T cell help’ involves cooperation with innate stimuli6,7, but how APCs integrate these different signals at the cellular level and whether such cooperation requires prolonged interactions with CD4+ T cells or follows more dynamic patterns is currently unclear. Resolving how APCs integrate and relay these different signals to CD8+ T cells is important for our general understanding of how the innate–adaptive cross-talk regulates T cell responses and will provide key insights required to improve CD8+ T cell responses during infection and vaccination.
Here, we systematically dissected how APCs integrate stimulation through IFNα/β and CD40 from CD4+ T cells. We identified an iterative process whereby APCs require IFNα/β-dependent rewiring of the signaling cascade downstream of CD40 that enables the subsequent partition of NF-κB-, IRF1- and FOS-dependent genes into distinct patterns of co-stimulatory molecule expression and mediator provision. This carefully sequenced integration process is critical for antiviral CD8+ T cell responses in a mouse virus infection model, and its activity in APCs from individuals infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) correlates with CD8+ T cell responses and milder forms of coronavirus disease 2019 (COVID-19).
Results
IFNα/β and CD40 induce distinct responses by dendritic cells
To dissect how APCs integrate signals from IFNα/β and CD40 stimulation, we initially focused on type 1 conventional dendritic cells (cDC1s), known platforms for T cell help8,9. We exposed bone marrow-derived CD24hiCD11blo cDC1s (hereafter, BMDC1s) to IFNαA and an antibody that mimics T cell help by cross-linking CD406. RNA sequencing (RNA-seq) revealed that CD40 induced some changes in BMDC1s, but this response was limited compared to >1,000 differentially expressed (false discovery rate (FDR) > 0.05, 1.5-fold change) genes induced by IFNαA (Fig. 1a and Supplementary Table 1). Most IFN-stimulated genes (ISGs)10 remained unaffected by additional CD40 stimulation (Fig. 1a, ‘CD40-unresponsive genes’). However, a subgroup of genes, which included Ccl4 and Il15, was further increased when IFNαA and CD40 antibody were applied together (Fig. 1a, ‘amplified genes’). We also observed genes that could not be induced by either stimulus alone but were strongly increased in BMDC1s exposed to both IFNαA and CD40 antibody (Fig. 1a, ‘combinatorial genes’). This response included Ccl5 and Tnf and other genes with known roles in the interplay between APCs and T cells, such as Cd83 and Cxcl16 (Fig. 1a). We validated these distinct response patterns in separate experiments, focusing on interleukin-15 (IL-15) and CCL4 as examples for the amplified response and tumor necrosis factor-alpha (TNF-α) and CCL5 for the combinatorial synergy between IFNαA and CD40 stimulation (Fig. 1b,c and Extended Data Fig. 1a). Comparable responses could also be elicited when CD40 synergized with IFNβ (Extended Data Fig. 1b) or other innate stimuli, such as polyinosinic–polycytidylic acid (poly(I:C)), lipopolysaccharide (LPS) or cytosine–phosphate–guanine (CpG), which triggered Toll-like receptor 3 (TLR3), TLR4 and TLR9, respectively (Extended Data Fig. 1c). These findings indicate that CD40 synergizes with various innate stimuli in inducing ‘amplified’ and ‘combinatorial’ responses in BMDC1s.

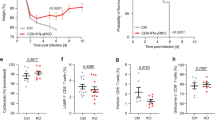
a, Changes in gene expression of BMDC1s stimulated for 4 h with IFNαA (n = 6 independent experiments), CD40 antibody (Ab) (n = 3 independent experiments) or IFNαA + CD40 antibody (n = 3 independent experiments) compared to unstimulated BMDC1s (n = 6 independent experiments). Differentially expressed genes are displayed as heat maps after z-score transformation of counts per million (CPM). Statistical differences were assessed by one-way analysis of variance (ANOVA); FDR-adjusted P ≤ 0.05 and fold change greater than 1.5. b,c, Relative expression of Il15 and Ccl4 (b) and Tnf and Ccl5 (c) over time in BMDC1s stimulated with IFNαA and/or CD40 antibody. Data are mean ± s.e.m. from two to three independent experiments. Statistically significant differences between conditions were assessed by one-way ANOVA; adjusted P values are indicated. d, IFNγ production after ex vivo antigen restimulation for 5 h by splenic wild-type (WT) and Cxcr6–/– HSV-1-specific CD8+ T cells 10 days after HSV-1 skin infection of wild-type mice that were irradiated and reconstituted with an equal mix of Cxcr6–/– and wild-type bone marrow cells 6–8 weeks earlier. Data are mean ± s.e.m. from three independent experiments (n ≥ 5 per experiment). e, IFNγ production after ex vivo antigen restimulation for 5 h by transgenic HSV-1-specific CD8+ T cells (gBT-I cells) transferred as naive gBT-I cells into wild-type or Ccl5–/– mice 1 day before HSV-1 infection and isolated from the spleen 7 days after infection. Data are mean ± s.e.m.from three independent experiments (n ≥ 1 per experiment). Statistical significance between conditions in d and e was assessed by two-tailed Wilcoxon rank-sum test, and respective P values are indicated.
cDC1s require in vivo stimulation from both IFNα/β and CD4+ T cells through CD40 to ‘amplify’ their capacity to provide IL-15 to herpes simplex virus (HSV)-specific CD8+ T cells6. To investigate whether priming of HSV-specific CD8+ T cells requires mediators that can only be induced by the synergy between IFNα/β and CD40 (such as CXCL16 and CCL5), we transferred Cxcr6+/+ and Cxcr6–/– bone marrow cells into irradiated hosts and infected them 6–8 weeks later with HSV-1 on the skin. Seven days later, splenic HSV-specific Cxcr6–/– CD8+ T cells produced less IFNγ in response to ex vivo antigen restimulation than their Cxcr6+/+ counterparts (Fig. 1d). CCL5-competent transgenic HSV-specific CD8+ T cells transferred into Ccl5–/– mice also had a significant, albeit more subtle, defect in IFNγ production in response to ex vivo antigen restimulation compared to wild-type recipients of HSV-specific transgenic CD8+ T cells (Fig. 1e), indicating that multiple genes required stimulation through both IFNα/β and CD40 for optimal helper-dependent DC–CD8+ T cell interactions in vivo.
IFNα/β change transcription downstream of CD40
Next, we tested whether IFNα/β and CD40 antibody acted concurrently or in sequence. To first investigate whether CD40 stimulation conditioned a more efficient response of BMDC1s to IFNα/β, we stimulated BMDC1s with CD40 antibody for 4 h and added IFNαA for the last 15, 30, 60, 120 or 180 min of the stimulation. Il15 expression increased after 1–2 h of IFNαA stimulation, and this expression increased more than twofold in the presence of CD40 antibody (Fig. 2a). Tnf, Cxcl16 and Cd83 were also induced in BMDC1s after 2–3 h of IFNαA stimulation (Fig. 2a), indicating that BMDC1s required ~2 h of IFNα/β exposure before they responded to CD40 triggering. We then determined whether IFNα/β conditioned the BMDC1s for CD40 responses by exposing BMDC1s to IFNα/β over 4 h and adding CD40 antibody for the last 15, 30, 60, 120 or 180 min. Tnf, Cxcl16 and Cd83 increased after 30–60 min (Fig. 2a), which showed that BMDC1s responded rapidly to CD40 stimulation if exposed to IFNα/β for ~2 h and suggested that BMDC1 need to be exposed to IFNα/β prior to CD40 stimulation IFNα/β conditions the dendritic cells (DCs) to become receptive to T cell help. We also tested this requirement in vivo using HSV-1 skin infection6. CD8+ cDC1s residing in the brachial lymph nodes of wild-type mice increased MHC class II expression 2 days after infection, but this increase was absent in Ifnar2–/– mice (Extended Data Fig. 1d). Because lack of CD4+ T cells does not impact MHC class II expression by CD8+ cDC1s in the brachial lymph nodes of wild-type mice infected 2 days earlier with HSV-1 on the skin6, these findings indicate that IFNα/β signals also need to precede CD40-mediated T cell help in vivo.

a, Changes in the expression of Il15, Tnf, Cd83 and Cxcl16 in BMDC1s stimulated with IFNαA for 4 h and stimulated additionally with CD40 antibody for the last 15, 30, 60, 120, 180 and 240 min (‘IFNαA conditioning’) or with CD40 antibody for 4 h and stimulated additionally with IFNαA for the last 15, 30, 60, 120, 180 and 240 min (‘CD40 conditioning’) compared to BMDC1s exposed to IFNαA alone (at least three independent experiments for each time point). Error bars represent mean ± s.e.m. Statistically significant differences between the corresponding conditions and IFNαA at 4 h alone were assessed by one-way ANOVA. b,c, Heat map (b) and SOM (c) of differentially expressed genes between BMDC1s left unstimulated or stimulated with IFNαA alone (IFNαA; n = 6 independent experiments) or with IFNαA for 4 h + CD40 for 15 min (IFNαA + CD40-15min; n = 3 independent experiments), 30 min (IFNαA + CD40-30min; n = 3 independent experiments) or 4 h (IFNαA + CD40-4h; n = 3 independent experiments). The 341 genes were clustered into 25 SOM clusters that follow similar patterns of expression with the identification of select genes in their respective clusters. d, Gene Ontology (GO) terms enriched in IFNαA + CD40-30min SOM clusters (clusters 1–4 and 6–8, as in panel c) and IFNαA + CD40-4h SOM clusters (clusters 5, 9, 10, 13–15, 18–20 and 23–25, as in panel c). Nodes represent the GO term, edges represent a score inversely proportional to the genes shared between the two GO terms, and the size of the nodes is proportional to the enrichment score. GO terms with a P value of ≤0.005 that relate to three or more GO terms are displayed; TH1, type 1 helper T cell. e, Correlation coefficients of weighted gene correlation network analysis (WGCNA) eigengenes combining transcriptional results from RNA-seq of BMDC1s that were unstimulated (n = 6 independent experiments) or stimulated with CD40 for 15 min (CD40-15min; n = 3 independent experiments), CD40 for 30 min (CD40-30min; n = 3 independent experiments), CD40 for 4 h (CD40-4h; n = 3 independent experiments), IFNαA alone for 4 h (IFNαA; n = 6 independent experiments), IFNαA for 4h + CD40 for 15 min (IFNαA + CD40-15min), IFNαA for 4 h + CD40 for 30 min (IFNαA + CD40-30min) or IFNαA for 4 h + CD40 for 4 h (IFNαA + CD40-4h; n = 3 independent experiments).
Next, we tested whether IFNα/β prepared DCs for T cell help by increasing CD40 expression7. IFNαA-stimulated and unstimulated BMDC1s increased the expression of CD40 over time similarly (Extended Data Fig. 1e), indicating that surface CD40 expression was not rate limiting in these responses. To investigate whether the ‘amplified’ and ‘combinatorial’ responses resulted from the effect of IFNα/β on the pathways downstream of CD40, we performed RNA-seq of BMDC1s stimulated with IFNαA for 4 h (BMDC1-IFN-αA) and compared gene expression to BMDC1s additionally stimulated with CD40 antibody for the last 15 min (BMDC1-IFNαA + CD40-15min) or 30 min (BMDC1-IFNαA + CD40-30min) or over the entire 4-h period (BMDC1-IFNαA + CD40-4h). Overall, BMDC1s changed expression of 341 genes over the 15-min, 30-min and 4-h time points compared to BMDC1-IFNαA, BMDC1-CD40 or unstimulated BMDC1s (Fig. 2b). Unsupervised self-organizing maps (SOMs) of these 341 genes identified smaller groups of genes appearing transiently at 15 min and 30 min (that is, Ifi44, Ifit3 and Fos), while different and larger sets of genes clustered at 4 h (that is, Cd83, Il15, Cxcl16, Il27 and Cd80) (Fig. 2c and Supplementary Table 2). General cellular processes, such as ‘enhanced survival’ and ‘increased mRNA stability’, were enriched in BMDC1-IFNαA + CD40-30min, while more specific responses, including ‘regulation of cytokine production’, characterized BMDC1-IFNαA + CD40-4h (Fig. 2d). We also performed coexpression analysis11 to identify similarly expressed groups of genes (‘modules’) independently of fold change cutoffs used to define differentially expressed genes across all time points (Fig. 2e and Supplementary Table 3). Genes in modules 1 and 3, such as Cxcl16 and Tnf, responded to the combination of IFNαA and CD40 antibody at 30 min and 4 h (Extended Data Fig. 2a). Modules 2 and 4 grouped genes that were induced by IFNαA (that is, Oasl1, Isg20 and Il2rg) or CD40 antibody (that is, Cxcr4, Apol7c and Il12b), respectively, while modules 5 and 6 contained genes with little responsiveness to either stimulation (that is, Itga3, Sox4 and Irak1) (Fig. 2e). These modules also differed in GO term enrichments (Fig. 2d). Together, these analyses indicate that IFNα/β changed how BMDC1s responded at the transcriptional level to CD40 stimulation.
IFNαA enable CD40 to activate p65, FOS and IRF1
Next, transcription factor binding motif prediction analysis of the‘amplified’ genes suggested the involvement of overlapping transcriptional regulators, including members of the IRF and STAT families (Fig. 3a). More specifically, BMDC1-IFNαA + CD40-30min and BMDC1-IFNαA + CD40-4h were enriched for binding sites for NF-κB, including NFKB1, REL, RELA (p65 subunit) and RELB (Fig. 3a). To test these predictions, we examined canonical and non-canonical NF-κB signaling cascades in the interplay between IFNαA and CD40 antibody. The induction of amplified genes (Il15 and Ccl4) and combinatorial genes (Tnf and Cxcl16) in BMDC1-IFNαA + CD40-4h was similar between Nfkb2−/− and wild-type BMDC1s (data not shown), indicating that the non-canonical NF-κB pathway was not required. BMDC1-IFNαA + CD40-15min resulted in IκBα degradation and p65 phosphorylation (Fig. 3b and Extended Data Fig. 2c), and the NF-κB inhibitor ammonium pyrrolidinedithiocarbamate (PDTC)12 impaired the increased expression of Tnf and Ccl4 in BMDC1-IFNαA + CD40-4h (Fig. 3c). These findings highlight that IFNα/β conditioning enabled CD40 to trigger the canonical NF-κB pathway in BMDC1s.

a, Transcription factor (TF) motifs enriched for ‘CD40-unresponsive genes’ and ‘amplified genes’ at 30 min and 4 h (as in Fig. 2b) and displayed as Venn diagrams. b, IκBα degradation (n = 3 independent experiments) and p65 phosphorylation (n = 2 independent experiments) in BMDC1s, as in Fig. 2b. c, Tnf and Ccl4 expression in BMDC1s stimulated as in Fig. 2b in the absence or presence of inhibitors for ERK (nimbolide) and the NF-κB pathway (PDTC). Data are displayed relative to treatment alone and are pooled from three independent experiments. Error bars represent mean ± s.e.m. Adjusted P values of the statistically significant differences as assessed by two-way ANOVA are indicated; Nimb, nimbolide; NS, not significant. d, Fos expression based on RNA-seq of BMDC1s, stimulated as Fig. 2b, expressed as CPM. e, Relative expression of select genes (Il15, Il27, Il15ra, Tnf, Nfkb2, Ccl4, Ccl5, Traf6, Cd83, Rela, Relb, Cxcl16 and Irf1) in BMDC1s transduced with Fos single guide RNA (sgRNA) or NTC and stimulated as in Fig. 2b (n = 3 independent experiments). f, Phosphorylation of ERK1/ERK2 (p-ERK1/ERK2) and p38 MAPK (p-p38 MAPK) (n = 3–4 independent experiments) in BMDC1s as in Fig. 2b. Pooled data and representative histograms are displayed. g, Expression of ‘combinatorial genes’ in BMDC1s as in Fig. 2b. h, Expression of Irf1 in RNA-seq data from BMDC1s stimulated as in Fig. 2b, expressed as CPM. i, Enrichment of IRF1 binding signals in combinatorial genes in BMDC1-IFN-αA + CD40-4h (P < 0.0001; n = 2 replicates). j, IRF1 binding signals analyzed using CUT&Tag in BMDC1s stimulated as in Fig. 2b (n = 2 replicates). k, Expression of Cxcl16 in wild-type or Irf1−/− BMDC1s stimulated as in Fig. 2b. Data are mean ± s.e.m. from three independent experiments. Adjusted P values were assessed by two-way ANOVA. l, p65 binding motif enrichment in DNA bound to IRF1 in BMDC1s as in Fig. 2b, expressed as target relative to the entire genomic background data. Statistical testing was performed using the cumulative binomial distribution to compare target binding to background signal. Data are mean ± s.e.m. from two replicates. Error bars in b, d, f and h represent mean ± s.e.m. Adjusted P values of the statistically significant differences between the corresponding conditions as assessed by one-way ANOVA are indicated.
The transcriptional regulator FOS was induced in BMDC1-IFNαA + CD40-15min compared to in BMDC1-IFNαA, BMDC1-CD40 and BMDC1-IFNαA + CD40-4h (Fig. 3d and Supplementary Table 2). We therefore deleted FOS from FLT3L-propagated BMDCs using CRISPR–Cas9 and stimulated these cells for 4 h with IFNαA and CD40 antibody. Compared to non-targeting guide control (NTC) BMDCs, Il15ra and Il27, but not Cxcl16 or Nfkb2, were reduced in the absence of FOS (Fig. 3e). ERK13 and CD40 signaling14 can activate FOS, and we found phosphorylated p38 and ERK in BMDC1-IFNαA + CD40-15min (Fig. 3f). Inhibition of ERK by nimbolide prevented the increase in Ccl4 expression and partially reduced Tnf expression in BMDC1-IFNαA + CD40-4h compared to in BMDC1-IFNαA (Fig. 3c). Together, these findings indicate that IFNα/β conditioning enables CD40 to activate FOS, likely through activation of ERK and p38.
The ‘combinatorial’ genes induced by IFNα/β and CD40 antibody were enriched in IRF1 binding sites (Fig. 3g), and expression of Irf1 was increased in BMDC1-IFNαA + CD40-30min and BMDC1-IFNαA + CD40-4h compared to in BMDC1-IFNαA (Fig. 3h). IRF1 binding signals were enriched in combinatorial genes in BMDC1-IFNαA + CD40-4h compared to in BMDC1-NS, BMDC1-IFNαA and BMDC1-CD40, as revealed by cleavage under targets and tagmentation (CUT&TAG) analysis (Fig. 3i). Endogenous IRF1 was bound to the promoter region of Cxcl16 in BMDC1-IFNαA + CD40-4h but not in BMDC1-IFNαA, BMDC1-CD40 or BMDC1-NS (Fig. 3j). Moreover, Irf1–/– BMDC1s did not induce the expression of Cxcl16 in response to 4 h of combined IFNα/β and CD40 antibody stimulation (Fig. 3k), and transcription factor binding motifs in the vicinity of IRF1 binding sites were enriched for motifs recognized by p65 (Fig. 3l). Together, these findings show that IFNα/β conditioning enhances the capacity of cDC1s to degrade IκBα and phosphorylate p65, p38 and ERK downstream of CD40, thus enabling CD4+ T cells to induce p65-, IRF1- and FOS-dependent transcriptional programs.
Mild COVID-19 is associated with IFNα/β and CD40 synergy
Imbalances in IFNα/β provision15 and low-avidity CD4+ T cell responses16 are associated with severe COVID-19 (refs. 17,18), while milder outcomes correlate with virus-specific CD8+ T cells18 and the ability of individuals to respond to CCL5 (ref. 19) and CXCL16 (ref. 20). To investigate the synergy between IFNα/β and CD40 during SARS-CoV-2 infection, we isolated CD14−HLA-DR+ DCs from the blood of individuals with COVID-19 4 to 35 days after symptom onset21. This included mild to moderate disease (WHO (World Health Organization) score of 2–5) and severe disease (WHO score of 6–8) (Supplementary Table 7). CD14−HLA-DR+ DCs from individuals with severe disease had significantly reduced expression of MHC class II (HLA-DR) compared to CD14−HLA-DR+ DCs from individuals with mild disease (Fig. 4a). A similar pattern was observed in CD14+CD11c+ monocytes, with a significant reduction in MHC class II expression compared to that observed in mild COVID-19 cases (Fig. 4a). To test whether IFNα/β signals contribute to MHC class II expression, we collected blood samples 4 to 35 days after symptom onset from individuals with COVID-19 who had developed neutralizing antibodies against type I IFN (IFN-AAB)22. CD14−HLA-DR+ DCs and CD14+CD11c+ monocytes from IFN-AAB+ individuals had an even stronger reduction of MHC class II than observed in those from individuals with mild disease (Fig. 4a). The expression of CD40 on CD14−HLA-DR+ DCs in individuals with severe disease increased irrespective of IFN-AAB but was reduced in CD14+CD11c+ monocytes in individuals with IFN-AAB (Fig. 4a), suggesting that IFNα/β regulate the ability of DCs and monocytes to receive T cell help through the expression of MHC class II5,23,24.

a, HLA-DR expression of CD14+CD11c+ monocytes and CD14−HLA-DR+ DCs from individuals with COVID-19 with mild or moderate symptoms (WHO score of 2–5; n = 11) or severe disease with (WHO score of 6–8; n = 3–6) and without (n = 10) IFN-AAB. Data are displayed as box and whisker plots showing the median and the 25th and 75th percentiles and two whiskers at 1.5× the interquartile range (IQR) of the mean fluorescence intensity (MFI) and percentage of CD40+ cells. b, Single-sample gene set variation analysis (GSVA) of the ‘CD40-unresponsive’, ‘amplified’ and ‘combinatorial’ gene signatures in scRNA-seq data from CD14+ monocytes and CD1C+ DCs from PBMCs of individuals with mild (WHO score of 3; n = 16), moderate (WHO score of 4–5; n = 11) or severe (WHO score of 7; n = 23) COVID-19 and healthy control (HC) individuals (n = 13; reanalyzed from ref. 25). Box plots as in panel a. Data points are colored and shaped according to disease severity and stage based on days after onset of symptoms, respectively. Wilcoxon rank-sum test P values are shown. c, Gene set enrichment analysis plots (top) showing enrichment curves of the ‘amplified’ and ‘combinatorial’ signatures in the differentially expressed genes (two-sided Wilcoxon rank-sum test, minimum percentage = 0.1, log2(fold change) > 0.2) in CD14+ monocytes from mild compared to severe COVID-19 cases as in b. The log10(FDR P values) and the log2(fold change) values of the differentially expressed genes are shown as a volcano plot (bottom). Genes are colored according to the ‘CD40-unresponsive’, ‘amplified’ or ‘combinatorial’ signature; Padj, adjusted P value. d, Differential expression of ‘CD40-unresponsive’, ‘amplified’ and ‘combinatorial’ signature genes in CD1C+ DCs from healthy control individuals and individuals with mild, moderate and severe cases of COVID-19, as in a, determined using a two-sided Wilcoxon rank-sum test. e, Heat map showing GeneScores for disease-specific, significantly differentially accessible genes in scATAC-seq data of CD14+ monocytes from PBMC samples derived from individuals with mild (WHO score of 1–3; n = 7 samples), moderate (WHO score of 4–5; n = 4) or severe (WHO score of 6–7; n = 6) COVID-19 and healthy control individuals (n = 6; reanalyzed from ref. 29) determined using a two-sided Wilcoxon rank-sum test (FDR ≤ 0.01 and log2 (fold change) ≥ 0.58). f, Imputed GeneScores of IL15, CD83, CXCL16 and TNF in CD14+ monocytes grouped according to COVID-19 severity as in e. Data are displayed as violin plots with overlaying box and whisker plots showing the median and 25th and 75th percentiles and two whiskers at 1.5× IQR. g, Differentially accessible genes (FDR ≤ 0.01 and log2(fold change) ≥ 0.58) in CD14+ monocytes from individuals with mild COVID-19 compared to healthy control individuals, as in e, visualized as a volcano plot showing –log10(FDR) and log2(fold change) values (left) and the corresponding enriched Hallmark terms for the 789 genes with increased accessibility in mild COVID-19 CD14+ monocytes compared to healthy control monocytes displayed as dot plots showing gene counts and adjusted P values per term (right). h, Secretion of TNF in hDC1s stimulated with IFNα and/or CD40 antibody for 18 h. Data are mean ± s.e.m. from six donors. Statistical significance for differences between conditions was assessed by one-way ANOVA, and adjusted P values are indicated.
Next, we used published single-cell RNA-seq (scRNA-seq) data from individuals with COVID-19 (ref. 25) to examine the expression of the ‘CD40-unresponsive’, ‘amplified’ and ‘combinatorial’ gene signatures identified above. This included peripheral blood mononuclear cells (PBMCs) from individuals with COVID-19 (mild, WHO score of 3, n = 16; moderate, WHO score of 4–5, n = 11; severe, WHO score of 7, n = 23) collected within the first 25 days after symptom onset before availability of vaccination. These were compared to samples from healthy or otherwise hospitalized individuals who tested negative for SARS-CoV-2, were serologically negative or had no indication of acute COVID-19 disease based on clinical or laboratory parameters (HC; n = 13)25. We analyzed 31,736 classical monocytes and 722 myeloid DCs using reference-based cell-type annotation and clustering (Methods), referred to here as CD14+ monocytes and CD1C+ DCs, respectively. CD14+ monocytes from individuals with mild disease25 were significantly enriched for the ‘amplified’ and ‘combinatorial’ responses (that is, CD83, CXCL16, NFKB2 and JUND) compared to CD14+ monocytes from individuals with moderate or severe disease or from healthy control individuals (Fig. 4b,c and Supplementary Table 5). Also, CD1C+ DCs from individuals with mild COVID-19 had increased transcription of genes of the ‘amplified’ and ‘combinatorial’ responses, such as CD83, EGR1 and REL, compared to CD1C+ DCs from individuals with severe COVID-19, which in turn had increased expression of CD40-unresponsive genes, such as IFIT3, MX1 and IRF7 (Fig. 4d and Supplementary Table 6). Similar patterns were observed in scRNA-seq data of a second cohort26, which included three individuals with moderate disease (respiratory symptoms and pneumonia), four individuals with severe disease (supplemental oxygen requirement) collected 2–16 days after symptom onset and five asymptomatic healthy control individuals from whom samples were collected before the widespread circulation of SARS-CoV-2 (Extended Data Fig. 3a).
We also performed scRNA-seq on PBMC samples from the COVID-19 cohort above, which included IFN-AAB+ individuals22 (Supplementary Table 7). CD14+ monocytes from IFN-AAB+ individuals had lower induction of prototypical ISGs, such as ISG15 and IFIT2, than CD14+ monocytes from healthy individuals and individuals with disease without IFN-AAB (Extended Data Fig. 3b). Furthermore, the expression of HLA-DRA, HLA-DRB1, TNF, CD83 and CCL4 was reduced in CD14+ monocytes from individuals with severe COVID-19 and in IFN-AAB+ individuals compared to in healthy individuals and in individuals with COVID-19 without IFN-AAB (Extended Data Fig. 3b). To gain more robust insights into data distribution, we integrated our data with comparable published scRNA-seq data sets21,27,28, including a study examining four IFN-AAB+ individuals28. This yielded 179,012 single-cell CD14+ monocyte transcriptomes across 263 samples (HC, n = 39; WHO score of 1–3, mild, n = 79; WHO score of 4–5, moderate, n = 82; WHO score of 6–8, severe, n = 52; WHO score of 7–8, severe + IFN-AAB, n = 11). HLA-DRB1, CD83 and TNF were significantly reduced in individuals with COVID-19 with increasing disease severity, reaching a minimum in individuals with IFN-AAB (Extended Data Fig. 3b). Furthermore, the ‘amplified’ and ‘combinatorial’ signatures were reduced in CD14+ monocytes from individuals with severe COVID-19, with and without IFN-AAB, compared to in CD14+ monocytes from individuals with mild disease (Extended Data Fig. 3c). Together, these findings indicate that IFNα/β signals are critical drivers of ‘amplified’ and ‘combinatorial’ responses during SARS-CoV-2 infection.
Reanalysis of published single-cell assay for transposase-accessible chromatin with sequencing (scATAC-seq) data sets29 from PBMCs of individuals with COVID-19 indicated that CD14+ monocytes from individuals with mild disease had significantly increased accessibility of more than 300 genes, including IL15, CD83, TNF and CXCL16, compared to CD14+ monocytes from individuals with moderate and severe COVID-19 (Fig. 4e,f). Furthermore, Hallmark enrichment analysis of more accessible genes in CD14+ monocytes from individuals with mild COVID-19 compared to CD14+ monocytes from healthy control individuals identified ‘IFNγ response’ and ‘TNF signaling via NF-κB’ as major pathways differentially regulated in mild COVID-19 (Fig. 4g). To investigate whether these responses can be elicited in vitro in human cDC1s, we differentiated human CD141+CADM1+CLEC9A+ cDC1s (hDC1s) from blood-derived CD34+ stem cells using FLT3L, stem cell factor and IL-4 (ref. 30) and stimulated them with human recombinant IFNα and human CD40 Ab separately or in combination for 18 h. hDC1s secreted TNF in response to IFNα when aided by CD40 triggering, but not after treatment with IFNα alone (Fig. 4h). These observations indicated that APCs from individuals with mild, but not severe, COVID-19 had increased chromatin accessibility and transcription of genes requiring the synergy between IFNα/β and CD40 described in the mouse experiments.
CD40 triggers NF-κB and FOS-dependent transcription in mild COVID-19
To explore whether IFNα/β also affected the signaling cascade downstream of CD40 in human APCs, we subjected the differentially expressed genes that were significantly higher in CD14+ monocytes from individuals with mild COVID-19 than in CD14+ monocytes from individuals with severe COVID-19 to enrichment analyses using the Hallmark database31 and transcription factor binding motifs32. There was a significant enrichment of genes associated with the NF-κB pathway, including CD83, CD86, TNFAIP3, IL1B, DUSP2, NFKB2 and REL (Fig. 5a and Supplementary Table 5). We also observed preferential involvement of the NF-κB family (NFKB1, RELA and RELB) and the FOS and JUN families (AP-1; Fig. 5b). Visualization of the links between predicted transcription factors and their target genes within the differentially expressed genes between mild and severe COVID-19 indicated a dense regulatory network controlled by NF-κB, FOS and JUN transcription factors (Fig. 5b).

a, Differentially expressed genes between disease severities and stages in CD14+ monocytes representing the significantly enriched Hallmark terms ‘IFNγ response’, ‘IFNα response’ and ‘TNF signaling via NF-κB’, displayed as dot plots. b, RcisTarget transcription factor binding motif enrichment based on differentially expressed genes (two-tailed Wilcoxon rank-sum test, minimum percentage = 0.1, log2(fold change) > 0.2) in CD14+ monocytes from individuals with mild compared to severe COVID-19, as in Fig. 4a. Data are visualized as a dot plot (left) showing the number of enriched genes and the normalized enrichment score per motif. The inner circle (right) shows the enriched transcription factors for all differentially expressed (DE) genes, and the outer circle shows the respective target genes responsible for their enrichments. Transcription factors enriched for the genes overlapping with the ‘amplified’ and ‘combinatorial’ gene signatures and the target genes are colored in red. NES, normalized enrichment score. c, Transcription factor binding motif enrichment based on significantly differentially accessible peaks in CD14+ monocytes from individuals with mild or severe COVID-19 compared to CD14+ monocytes from healthy control individuals. Data are based on scATAC-seq data29 and are displayed as dot plots showing FDR-adjusted P values of the enrichments and the number of target regions per transcription factor binding motif. d, Target regions of the top 10 highest enriched transcription factor binding motifs and motifs corresponding to IRF1 and p65 (RELA), shown as UpSet plots comparing the number of target regions.
Similarly, transcription factor binding motif enrichment analyses in differentially accessible chromatin regions of CD14+ monocytes from individuals with mild or severe COVID-19 compared to those from healthy control individuals29 predicted members of the FOS family as key regulators (Fig. 5c). The enrichment score and number of accessible target regions of the predicted transcription factors, including FRA1/FRA2, FOSL2 and JUN, were higher in CD14+ monocytes from individuals with mild COVID-19 than in CD14+ monocytes from individuals with severe COVID-19 (Fig. 5c,d). Furthermore, the top 10 predicted transcription factor binding motifs and motifs corresponding to IRF1 and p65 (RELA) revealed large and distinct sets of target regions for the identified key regulator families among more accessible chromatin regions in CD14+ monocytes from mild COVID-19 cases than those from healthy control individuals. There were also substantially lower numbers of target regions with increased accessibility in CD14+ monocytes from individuals with severe COVID-19 than in CD14+ monocytes from healthy control individuals (Fig. 5d). These findings suggest that the amplified and combinatorial responses enriched in CD14+ monocytes in individuals with mild COVID-19 are regulated by signal integration through transcription factors of the NF-κB, FOS and JUN families.
Mild COVID-19 is associated with ‘helped’ CD8+ T cells
To test whether IFNα/β-dependent provision of T cell help to DCs and monocytes affects the CD8+ T cell response, we used the PBMC scRNA-seq data set from the cohort of individuals with COVID-19 and healthy control individuals defined above (Fig. 4b). We analyzed 11,734 CD8+ T cells using reference-based cell-type annotation and clustering (Methods) and compared their transcriptional profiles to published gene signatures that reflected CD8+ T cell priming in the presence (‘helped’) or absence (‘unhelped’) of CD4+ T cell help for DCs33. CD8+ T cells from individuals with moderate and severe COVID-19 were enriched for ‘unhelped’ profiles (including CD200, CD200R1, BTLA, ID3 and PDCD1) compared to CD8+ T cells from individuals with mild COVID-19 (Fig. 6a,b). Clustering analysis further indicated that individuals with mild and moderate COVID-19 were enriched in CD8+ T cell subsets with transcriptional profiles (IL7R, TCF7, JUNB and JUND) indicative of early effector or activated memory T cells34,35 (Fig. 6c,d and Supplementary Table 8, cluster 3). CD8+ T cells with characteristics of terminal differentiation (CX3CR1 and ISG15, cluster 5) also dominated in individuals with severe COVID-19 (Fig. 6c,d) and had a reduction in ‘helped’ signatures (that is, CD69, IL2RA and TNF) and a corresponding gain in the ‘unhelped’ signature (that is, IL6R, CD9, ISG15 and PDCD1) (Fig. 6e). We found comparable patterns in published scRNA-seq data from two other cohorts of individuals with COVID-19 (Extended Data Fig. 4a–c)36,37. We used cytometry by time of flight (CyTOF) to examine protein expression in CD8+ T cells from blood samples of 9 healthy control individuals and individuals with COVID-19 with mild (WHO score of 2–3; n = 25) or severe (WHO score of 7–8; n = 18) disease and IFN-AAB+ individuals with severe disease (WHO score of 7–8; n = 9)21 collected 4 to 30 days after symptom onset (Supplementary Table 7). Dimensionality reduction using uniform manifold approximation and projection (UMAP) and clustering with the FlowSOM algorithm indicated differences in the composition of CD8+ T cells between individuals with COVID-19 with different disease severity (Fig. 6f). Individuals with mild COVID-19 had increased proportions of CD27+CD8+ T cells with memory potential6 (Fig. 6f and Extended Data Fig. 4d,e, clusters 17, 19 and 21), while individuals with severe COVID-19 had greater proportions of CD27−KLRG1+CD8+ T cells (clusters 3 and 8) than healthy individuals or individuals with mild disease (Fig. 6f and Extended Data Fig. 4d,e). Principal component analysis (PCA) of CD8+ T cells from individuals with mild and severe COVID-19 identified the expression of CD27 and KLRG1 as distinct features of CD8+ T cells from individuals with mild and severe disease, respectively (Fig. 6g,i), and CD8+ T cells from individuals with severe disease were enriched for LAG3 (Fig. 6h), a molecule induced by priming with unhelped DCs33. These findings indicate that severe outcomes of COVID-19 are associated with unhelped phenotypes of CD8+ T cells.

a, UMAP visualization of scRNA-seq profiles of 11,734 CD8+ T cells from individuals with mild (WHO score of 3; n = 16), moderate (WHO score of 4–5; n = 11) or severe (WHO score of 7; n = 23) COVID-19 and healthy control individuals (n = 13; reanalyzed from ref. 25). Cells are split and colored according to disease severity. b, GSVA of ‘helped’ and ‘unhelped’ T cell signatures derived from published gene expression33 profiles of mouse CD8+ T cells primed in the presence or absence of CD4+ T cells. Data are displayed as box and whisker plots showing the median and 25th and 75th percentiles and two whiskers at 1.5× IQR. Two-sided Wilcoxon rank-sum test P values are shown. c, UMAP of CD8+ T cells segregated into clusters 0–5 (left) and heat map of the respective proportionate cluster occupancy per disease severity (right) as in a. d, Expression of key genes associated with clusters 0–5 as in c. e, AUCell enrichment of genes derived from published gene expression33 profiles of mouse CD8+ T cells primed in the presence or absence of CD4+ T cells as in b, grouped according to the clustering as in c and displayed as violin plots of area under the curve (AUC) scores. FDR-corrected Dunn’s multiple comparison test P values are indicated. f, UMAP visualization of CD8+ T cells from whole-blood samples from healthy control individuals (n = 10) and individuals with COVID-19 (mild, n = 22; severe, n = 21; severe with IFN-AAB, n = 9) analyzed by CyTOF. The rightmost plot shows the UMAP colored according to FlowSOM clustering, while the four plots on the left show the distribution of events across the groups. g, PCA analysis plot showing average PC1 and PC2 values for all the events per individual as in f, colored according to the sample group. Ellipses show an estimated region of group accumulation, arrows represent correlation of the respective marker with either of the PC axes, and arrow length represents correlation strength. h, Mean scaled signal intensities for KLRG1, CXCR3 and CD69 (left) and LAG3 (right) displayed as box and whisker plots showing the median and 25th and 75th percentiles and two whiskers at 1.5× IQR. i, Relative abundance of CD27−KLRG1+ cells in the total CD8+ T cell fraction displayed as box and whisker plots showing the median and the 25th and 75th percentiles and two whiskers at 1.5× IQR. Statistics in h and i show two-sided Benjamini–Hochberg-corrected pairwise Wilcoxon P values.
Discussion
Our findings uncovered an iterative consolidation process, in which innate stimuli, such as IFNα/β or TLR agonists, determined broad response options in APCs, and CD4+ T cells subsequently partitioned these into distinct sets of co-stimulatory molecules, cytokines and chemokines through CD40L. Together, these consecutive signals endowed APCs with optimal capacities to orchestrate effective antiviral CD8+ T cell responses in mouse HSV-1 infections and during community-acquired SARS-CoV-2 infections, where effective consolidation of IFNα/β and CD40 signals in APCs correlated with milder outcomes of COVID-19.
The conditioning of APCs by IFNα/β to become receptive to T cell help involved increased expression of MHC class II and distinct changes in how the APCs responded to CD40 stimulation. The changes in CD40 responsiveness were not just a function of increased expression of CD40 alone8, as spontaneously matured CD40hi DCs in mice and CD40hi APCs in individuals with severe COVID-19 were unable to engage ‘helper’-dependent programs. Instead, the capacity to receive help depended on additional changes in the signaling cascade downstream of CD40. These endowed APCs with the capacity to rapidly engage a network of transcription factors, including p65, IRF1 and FOS, and likely others, such as JUN, to select a distinct group of genes that provide the DCs with optimal capacities to prime CD8+ T cells responding to antigen. Some of the transcription factors were directly regulated by IFNα/β and CD40 stimulation, suggesting that conditioning also enhanced the availability of relevant transcription factors. These responses were not exclusive to the cooperation between IFNα/β and CD40, as similar patterns of CD40-dependent calibration also occurred in DCs stimulated through different TLRs. Together with increased chromatin accessibility at binding sites for the above-mentioned transcription factors in promoter regions of key genes regulated through IFNα/β and CD40, our study revealed a multitude of transcriptional and post-translational changes as a functional basis for how innate cues condition APCs to become receptive to T cell help, thus enabling CD4+ T cells to calibrate APCs for optimal stimulation of CD8+ T cell responses.
We have investigated the relevance of these findings for antiviral CD8+ T cell immunity in a mouse model of HSV-1 skin infection and showed that optimal HSV-specific CD8+ T cell responses depended on contextual cues that require IFNα/β and NF-κB signal integration by DCs. Notably, we translated these experimental insights to individuals with SARS-CoV-2 infection and demonstrated that the consecutive activation of APCs by IFNα/β and CD4+ T cells played an important role in regulating how APCs orchestrate CD8+ T cell responses during COVID-19. This interpretation not only helps align a number of currently unlinked findings in COVID-19, such as an association of milder disease with effective provision of CXCL16 (ref. 20) and CCL5 (ref. 19), high-avidity CD4+ T cells16 and effective CD8+ T cell responses18, but also raises the prospect of ‘unhelped’ APCs launching too many terminally differentiated CD8+ T cells that contribute to immunopathology in individuals with severe COVID-19. It is important to acknowledge limitations around our findings in individuals with COVID-19. Our study cannot discern if the observed failures in signal integration by APCs and preponderance of terminally differentiated CD8+ T cells are a ‘cause’ or ‘effect’ of severe COVID-19 or are more likely a complex combination of both. Moreover, it is possible that interindividual differences in T cell antigen receptor epitopes, precursor frequencies of antigen-specific CD8+ T cells and a great number of many other covariates (that is, age, gender and comorbidities) influence the interaction between APCs and CD8+ T cells in individuals with COVID-19. However, having validated our findings across multiple unrelated clinical data sets, it is unlikely that our findings simply represent the confounding effects of any one of these covariates. We likely also missed some of the more nuanced aspects of the interaction between APCs and naive CD8+ T cells that take place in lymph nodes before symptom onset, which are difficult to capture as the precise time point of infection is unknown in community-acquired infections, and lymph nodes are not as amenable as blood for routine sampling, especially in individuals with mild disease.
Collectively, our findings demonstrate the reliance of antiviral immunity on a step-wise, carefully orchestrated consolidation process, whereby APCs combine and integrate innate signals and, after selection by CD4+ T cells, produce a discrete set of co-stimulatory molecules and soluble mediators that adapt responding CD8+ T cells to the specific challenge. In showing how innate and adaptive signals cooperate to partition tailored responses from multiple broad and overlapping innate pathways and demonstrating functional relevance of these processes in mouse and human virus infections, our study provides critical new insights into how the host mounts effective antiviral immunity.
Methods
Mice
C57BL/6, Ccl5–/–, Cxcr6–/–, Ifnar2–/–, Irf1–/– and CD45.1+ gBT-I mice were bred and maintained at the animal facility of the Department of Microbiology and Immunology, The University of Melbourne. All animal experiments were approved by The University of Melbourne Animal Ethics Committee.
Human samples
This study includes a subset of individuals enrolled between March 2020 and April 2021 in the Pa-COVID-19 study, a prospective observational cohort study assessing pathophysiology and clinical characteristics of individuals with COVID-19 at Charité Universitätsmedizin, Berlin38. The study was approved by the Institutional Review board of Charité (EA2/066/20). Written informed consent was provided by all individuals or legal representatives for participation in the study. Specifics about the participants per application (flow cytometry, CyTOF and scRNA-seq), including COVID-19 status, time point of sampling after onset of symptoms, sex, age and outcome, are listed in Supplementary Table 7 and are described elsewhere21,22. Human umbilical cord blood was obtained with written informed consent from the Queensland Cord Blood Bank and approval from the Mater Human Research Ethics Committee (HREC13/MHS/86).
In vitro generation of BMDC1s
Single-cell suspensions from mouse bone marrow were cultured with FLT3L to generate BMDCs6. Red blood cells were removed using 1 ml of red blood cell lysis buffer (Sigma-Aldrich) per mouse for 90 s. Cells were cultured at 1.5 × 106 cells per ml in complete medium supplemented with 1.32 mM l-glutamine, 10% fetal calf serum, 90 µM β-mercaptoethanol, 100 U ml–1 penicillin, 0.2 g liter–1 streptomycin and 150 ng ml–1 FLT3L (BioXCell). Following 8 days of culture at 37 °C, cells were stained for 30 min on ice with CD45R/B220 (RA3-6B2), SIRPα (P84), CD11c (N418), CD11b (M1/70), I-A/E (M5114) and CD24 (M1/69) antibodies. cDC1 or CD8+ DC equivalents were identified by CD24highSIRPαlowCD11blowCD45R/B220− and were sorted using a FACS Aria III (BD Biosciences). Following sorting, BMDC1s were washed and resuspended before stimulation. Stimulation was performed on bulk BMDCs or sorted BMDC1s with IFNαA (PBL; 1,000 U ml–1), IFNβ (R&D Systems; 1 µg ml–1), LPS (Sigma-Aldrich; 10 µg ml–1), CpG (1668, GeneWorks; 1.6 nmol ml–1) or poly(I:C) (InvivoGen; 10 µg ml–1) in the presence or absence of monoclonal antibody to CD40 (CD40 Ab; FGK45.5, Miltenyi Biotec; 10 µg ml–1). Cells and supernatants were collected at different time points thereafter. Pharmacological inhibition of NF-κB and ERK pathways was achieved with 1 h pretreatment using PDTC (ab141406, 10 µM) and nimbolide (ab142138, 10 µM), respectively.
Cytokine and chemokine determination
Supernatants were subjected to BD Cytometric Bead Array measurement of CCL4 (limit of detection of 4.88 pg ml–1), CCL5 (limit of detection of 1.22 or 4.88 pg ml–1) and TNF-α (limit of detection of 39.07 or 9.7 pg ml–1), according to the manufacturer’s instructions. Samples were assessed using an LSRFortessa and FACS Diva software 6.1.3, and all concentrations were determined relative to a standard curve.
Real-time PCR
Cells were resuspended in TRIzol (Life Technologies), and mRNA was extracted using a Direct-zol RNA MicroPrep kit (Zymo Research) following the manufacturer’s instructions. cDNA was synthesized with an Omniscript RT kit for reverse transcription (Qiagen) using oligo(dT) primers (Promega) and RNaseOUT recombinant ribonuclease inhibitor (Thermo Fisher Scientific). Real-time PCR was performed with Taqman Universal PCR master mix (Life Technologies) with primers/probes for 18S (Mm03928990_g1), B2m (Mm00437762_m1), Ccl4 (Mm00443111_m1), Ccl5 (Mm01302427_m1), Cd83 (Mm00486868_m1), Cxcl16 (Mm00469712_m1), Gapdh (Mm99999915_g1), Hprt (Mm00446968_m1), Il15 (Mm00434210_m1), Il15ra (Mm04336046_m1), Il27 (Mm00461162_m1), Irf1 (Mm01288580_m1), Nfkb2 (Mm00479807_m1), Rela (Mm00501346_m1), Relb (Mm00485664_m1), Tnf (Mm00443258_m1) and Traf6 (Mm00493836_m1).
RNA-seq and data analysis
Gene expression changes were investigated using RNA-seq. Up to 100 ng of total RNA was used for library preparation, according to the manufacturer’s protocol, and was either sequenced in a 125-base pair (bp) paired-end run on a HiSeq HT sequencer (Illumina) or in a 50-bp single-read QuantSeq 3′-mRNA (Lexogen) run. Reads were aligned against the mouse genome mm10 by STAR v2.5.3a. Gene quantification was performed via the E/M algorithm in PartekFlow (v8.0.19.0707) and normalized as CPM. Genes with a mean expression of ≤1 CPM under all conditions were excluded from further analysis, resulting in 10,222 present genes for ANOVA in the Partek Genomics Suite (PGS, v7.18.0402). Genes with a fold change of 1.5 and an FDR-adjusted P value of ≤0.05 were defined as differentially expressed between two tested conditions. GO enrichment for the modules was performed using DAVID39 with the GOTERM_BP_DIRECT annotation. GO terms were filtered by unadjusted P ≤ 0.05 and visualized using ggplot2. Biological interpretation of differentially expressed genes was performed with the following tools. Gene set enrichment analysis was performed using the GSEA application (v4.0.3) and the Hallmark gene set published by the Broad Institute. Enrichments were plotted using ggplot2 (v3.3.3)40. Cytoscape was used to visualize enriched GO terms as a network with the two plugins BiNGO (v3.0.3) and EnrichmentMap (v3.2.1). WordCloud plugin (v3.1.3) was used to visualize the most frequent annotation associated within a cluster of GO terms. All present genes were used as input for a WGCNA, performed using the WGCNA R package (v1.70-3), to identify correlations of gene expression within the data set in an unbiased approach. The β-value was set at 23. For the module dissimilarity, a threshold of 0.42 was chosen, and the minimal cluster size was set to 30 genes. The prediction of transcription factor binding motifs was performed using the Cytoscape plugin iRegulon (v1.3) with a minimum normalized enrichment score of 3 and a maximum FDR on motif similarity of 0.001. All potential transcription factors annotated to the enriched binding motifs were used in the Venn diagram to illustrate their overlaps.
CUT&Tag and analysis
The CUT&Tag experiments were performed as previously described41 with a hyperactive in situ ChIP library prep kit purchased from Epicypher (CUTANA CUT&Tag Assays) following the manufacturer’s recommendations. A minimum of 1 × 105 stimulated BMDC1s were bound to activated concanavalin A-coated magnetic beads and were subjected to immunoprecipitation with 0.5 µg of primary antibody (anti-IRF1, D5E4, Cell Signaling Technology; rabbit anti-mouse IgG control). Immunoprecipitated DNA was amplified with high-fidelity 2× PCR mix (Epicypher) using universal barcodes i5 and uniquely barcoded i7 primers and 21 cycles. PCR products were purified with AMPure XP beads and eluted in water. Libraries were sequenced on an Illumina NextSeq platform, and 150-bp paired-end reads were generated. Fastq reads for each sample were aligned to the mm39 reference genome using bwa (v0.7.17). PCR duplicates were removed using picard tools’ MarkDuplicates (v2.25.0), and peaks were called using macs2 (v2.2.7.1) with the ‘—nomodel’ parameter. To establish consensus peaks between all conditions, peak sets were merged using homer’s mergePeaks (v4.11.1), and reads in consensus peaks were counted for each replicate using subread’s featureCounts (v2.0.0). PCA plots were generated using R (v4.1) and the prcomp function. Differentially occupied peaks were established using the limma package (v3.46.0) and its voom, lmFit and eBayes functions. Motif occupancy at peaks was established with homer and the findMotifsGenome function (v4.11.1).
Immunoblotting
BMDC1s were lysed in resuspension with RIPA buffer containing 50 mM Tris-HCl (pH 8), 150 mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS (Sigma-Aldrich) supplemented with PhosSTOP phosphatase inhibitor cocktail tablets (Roche) and cOmplete protease inhibitor cocktail tablets (Roche). Cell lysates were rotated at 4 °C for 30 min and clarified at 4 °C at 13,000g for 10 min. Proteins were denatured for 5 min at 90 °C with sample buffer containing 350 mM Tris-HCl (pH 6.8–5), 5% β-mercaptoethanol, 10% SDS, 36% glycerol and 0.0012% bromophenol blue. Proteins were then separated using NuPAGE 4–12% Bis-Tris gels (Thermo Fisher Scientific). Proteins were transferred onto nitrocellulose membranes (Bio-Rad) and blocked for 30 min with either 5% milk or 5% bovine serum albumin (BSA; for phosphorylated proteins) in PBS or TBS (for phosphorylated proteins) with 0.1% Tween 20. The following primary antibodies were used: rabbit anti-NF-κB p65 (D14E12), mouse anti-phospho S536 NF-κB p65 (7F1), rabbit anti-IκBα (44D4) and rabbit anti-β-actin (13E5), all purchased from Cell Signaling Technology. Membranes were incubated with horseradish peroxidase-conjugated secondary antibodies goat anti-rabbit IgG and horse anti-mouse IgG (Cell Signaling Technology) and subsequently with a Novex ECL chemiluminescent substrate reagent kit before imaging. Quantitative analysis of the signal intensity was performed using ImageJ software.
PhosFlow cytometry
Following in vitro stimulation of BMDCs, 100 µl of warm PhosFlow Lyse/Fix Buffer (BD Biosciences) was directly added to the samples and incubated for 10 min at 37 °C. Samples were then resuspended in PhosFlow Perm Buffer III (BD Biosciences) and incubated for 30 min on ice. After being washed twice, samples were stained for 1 h at room temperature with the antibodies described above supplemented with antibody to phospho-P44/42 MAPK (ERK1/ERK2; Thr 202/Tyr 204; 197G2; Cell Signaling Technology) and phospho-p38 MAPK (Thr 180/Tyr 182; 4NIT4KK; Thermo Fisher Scientific). A Biosciences Cytek Aurora was used for the measurement of samples, and FlowJo software (TreeStar) was used for analysis.
CRISPR–Cas9 gene editing
Freshly isolated bone marrow precursors were edited via electroporation before culture with FLT3L, as described previously42. In brief, per 10 × 106 mouse bone marrow precursors to be electroporated, 61 pmol of Cas9 nuclease (IDT) and 300 pmol of sgRNA (Synthego) were combined and incubated for 10 min at room temperature, generating Cas9–sgRNA ribonucleoprotein complex. Bone marrow precursors (10 × 106) were then washed with 1× PBS twice and resuspended in 20 µl of P3 buffer (Lonza) combined with the Cas9–sgRNA complex and electroporated using 4D-Nucleofector (Lonza) using the pulse code CM-137. Prewarmed medium was immediately added in electroporation wells to allow cells to recover for 10 min at 37 °C. Cells were subsequently cultured for 8 days in complete medium supplemented with FLT3L, as described above. sgRNA sequences used were Fos (UAGUGCCAACUUUAUCCCCA) and NTC (GCACUACCAGAGCUAACUCA).
Virus infection and viral titers
HSV-1 KOS was grown using Vero cells (CSL). Mice were epicutaneously infected with 106 plaque-forming units of HSV-1, as previously described6.
Flow cytometry analysis of in vivo HSV-1 responses
Endogenous HSV-specific CD8+ T cells were analyzed using H-2Kb-restricted gB498–505-specific tetramers, as previously described6. In some experiments, Ccl5−/− and wild-type mice were transferred with 50,000 naive HSV-specific CD8+ T cells (gBT-I cells) before infection, and their expansion was measured 10 days later in the spleen, as described previously6. IFNγ production in gB498–505-specific CD8+ T cells was measured after restimulation for 5 h ex vivo in the presence of brefeldin A. Single-cell suspensions were stained with antibodies to CD16/CD32 (2.4G2, Fc block), CD8 (53-6.7), CD44 (IM7), CD45.2 (104) and CD3 (145-2C11) and, when necessary, with either CD45.1 monoclonal antibody (A20) or tetramer staining. After fixation and permeabilization with BD Cytofix/Cytoperm (BD Biosciences), cells were stained for 20 min at room temperature with antibodies to IFNγ (XMG1.2) in BD Perm/Wash buffer (BD Biosciences) before analysis on a flow cytometer. Dead cells were excluded by using a LIVE/DEAD fixable dead cell stain kit (Thermo Fisher Scientific). A BD LSRFortessa and a FACS Diva or Biosciences Cytek Aurora and SpectroFlo were used for measurement of samples, and FlowJo software (TreeStar) was used for analysis. In some experiments, CD8+ DCs from wild-type versus Ifnar2−/− mice were analyzed in the brachial lymph node 2 days after HSV-1 skin infection, as previously described6. Cells were stained with antibodies to CD11c (N418), CD8 (53-6.7), CD3 (145-2C11, BD Biosciences), CD19 (1D3), NK1.1 (PK136) and IA/E (2G9), and CD8+ DCs were then processed on an analytic flow cytometer (LSRFortessa BD Biosciences).
BM chimeras
Mixed chimeras were generated as previously described6. C57BL/6 mice were lethally irradiated with 2 × 550 cGy and were reconstituted with a total of 5 × 106 bone marrow cells, previously depleted for T cells, from Cxcr6−/− and wild-type mice in a 1:1 ratio.
scRNA-seq data generation and analysis
scRNA-seq data of PBMCs from individuals with COVID-19 and healthy control individuals collected from April to July 2020 in Bonn, Germany, were used, as previously reported26. Samples were classified by disease severity according to the WHO ordinal scale (WHO score of 3, mild; WHO score of 4–5, moderate; WHO score of 7, severe) and by the time after onset of first symptoms (early: days 0–10, late: >day 11) at the date of sampling. Details about sample procurement and processing, sequencing and data analysis have been previously described25, and an extensive description of the protocol has also been published43. Processed and annotated scRNA-seq data25 were used as published previously and are available at https://beta.fastgenomics.org/p/schulte-schrepping_covid19. The data were imported into R version 4.0.3 and were mainly analyzed using Seurat v3.9.9.
Subset analysis of DCs and monocytes
PBMCs were subjected to Seurat v4 reference mapping following the developer vignette (satijalab.org/seurat/articles/multimodal_reference_mapping.html) using the multimodal PBMC reference data set44. Only those cells classified as DC or monocyte subsets were selected to remove any possible cellular contaminations in the data set. Subsequently, the remaining 37,100 cells were reclustered after scaling and regressing for unique molecular identifier (UMI) count per cell, identification of variable genes and PCA in this cellular subspace using the Louvain algorithm with a resolution of 0.2 based on the first 10 PCs. Clusters representing DCs or classical CD14+ monocytes were then subsetted, respectively, and the resulting 31,736 monocytes and 722 DCs were analyzed in detail, including rescaling, identification of variable genes, PCA and subsequent UMAP based on the first 10 PCs. Disease severity-specific marker gene analysis was performed using the Wilcoxon rank-sum test with the following cutoffs: genes had to be expressed in more than 10% of the cells of the respective condition and exceed a logarithmic fold change cutoff of at least 0.2. Before dot plot visualization and functional enrichment analyses, sets of differentially expressed genes were filtered for ribosomal protein-coding genes (RPL/RPS), mitochondrial genes (MT-) and hemoglobin genes (HBA1, HBA2 and HBB). Hallmark enrichment analysis of differentially expressed gene sets was performed using the Hallmark v7.3 database and the enricher function implemented in the R package clusterProfiler v3.18.0 (ref. 45). Gene set enrichment analyses of ‘CD40-unresponsive’, ‘amplified’ and ‘combinatorial’ gene signatures in the differentially expressed genes in monocytes from individuals with mild COVID-19 compared to those from individuals with severe disease were performed using the fgsea package v1.16.0. Single-sample GSVA using the ‘CD40-unresponsive’, ‘amplified’ and ‘combinatorial’ signatures derived from the mouse bulk RNA-seq analysis of this study was performed using GSVA v1.38.2 (ref. 46). For this, aggregated expression values of all cells of each sample were calculated using the AggregateExpression function in Seurat and were used as input for the sample-specific analysis. Of note, the IFNαA response signature was intersected with the top 100 IFN-response genes derived from an integrated analysis of eight microarray data sets on IFN response of myeloid cells listed in the Interferome database (http://www.interferome.org/)10 ranked by their combined log2(fold change) values to reduce the signature to a length comparable to the amplified and combinatorial signatures. Transcription factor binding motif enrichment analysis based on the significantly differentially expressed genes in monocytes derived from individuals with mild COVID-19 compared to cells from individuals with severe disease and those differentially expressed genes that intersected with the ‘amplified’ and ‘combinatorial’ gene signatures was performed using RcisTarget32, the hg38__refseq-r80__10kb_up_and_down_tss.mc9nr.feather database and a normalized enrichment score threshold of 4. Enriched transcription factor binding motifs were filtered for the transfac_pro, cisbp and swissregulon databases and those motifs with high-confidence transcription factor annotation (TF_highConf). A network linking enriched target genes and predicted transcriptional regulators based on the Rcistarget transcription factor binding motif enrichment results was constructed and visualized in a circular layout using Cytoscape v3.7.1.
scRNA-seq analysis of CD14+ monocytes from individuals with IFN-AAB and corresponding healthy individuals
PBMC scRNA-seq data were produced from five control samples, five samples from individuals with moderate COVID-19, five samples from individuals with severe COVID-19 and seven samples from individuals with severe COVID-19 with IFN-AAB, which were tested for each individual in virus neutralization assays described in Akbil et al.22. On the day of the experiment, frozen live PBMCs were thawed in prewarmed medium (RPMI 1640 (Gibco), 2% fetal calf serum (Sigma) and 0.01% Pierce Universal Nuclease (Thermo Fisher)). The PBMCs were then labeled with 0.5 µg of TotalSeq-C hashtag antibodies (Biolegend) in DPBS supplemented with 0.5% BSA and 2 mM EDTA for 30 min at 4 °C and washed at least three times with DPBS + 1% BSA. Subsequently, the PBMCs were counted, and up to seven different samples were pooled in equal proportions. The resulting cell pool was filtered through a 40-µm mesh (Flowmi Cell Strainer, Merck) and super loaded with 50,000 cells per lane in the Chromium Controller for partitioning single cells into nanoliter-scale Gel Bead-In-Emulsions (GEMs). For reverse transcription, cDNA amplification and library construction of the gene expression libraries, the Chromium Next GEM Single Cell kit 5′ v2 (10x Genomics) was used. The Chromium Single Cell 5′ Feature Barcode Library kit (10x Genomics) was used for preparing additional hashtag libraries. All libraries were prepared according to the protocols provided by 10x Genomics, quantified by Qubit Flex fluorometer (Thermo Fisher) and quality checked using the 4150 TapeStation system. Sequencing was performed in paired-end mode (R1 26 cycles, R2 90 cycles) on a NovaSeq 6000 (Illumina) with a NovaSeq 6000 S2 reagent kit (100 cycles). After demultiplexing, raw sequencing data were processed with CellRanger v5 and aligned against the GRCh38 reference, including TotalSeq-C hashtag barcodes. scRNA-seq UMI count matrices were imported into R 4.0.3, and gene expression data analysis was performed using the R/Seurat package 3.9.9. Cells from pooled samples were demultiplexed using a combination of HTODemux implemented in Seurat and vireo (v0.5.6)47 after scoring common variants from the 1000Genomes project with cellsnp-lite (v1.2.0)48. Events classified as ‘negative’ and ‘doublet’ by the HTODemux algorithm were assigned an ID via vireo classification. Subsequently, cells were filtered by number of features (over 200 and less than 5,000), percentage of mitochondrial genes (<10% mitochondrial UMIs) and number of counts per cell (<20,000) to exclude debris and doublets. Gene expression values were normalized by total UMI counts per cell, multiplied by 10,000 (TP10K) and log transformed by log10 (TP10k + 1). For cell-type annotation, cells were subjected to Seurat v4 reference mapping following the developer vignette using the multimodal PBMC reference data set44. Cells classified as CD14+ classical monocytes were selected and reclustered after scaling and identification of variable genes using vst and PCA using the Louvain algorithm with a resolution of 0.2 based on the first 10 PCs. A cluster characterized by the expression of T cell marker genes was removed to exclude potential T cell contamination in the CD14+ monocyte subset. Averaged gene expression values per sample of selected key genes were visualized as box plots across disease severity groups.
To increase the number of samples per severity group, scRNA-seq data of PBMCs from other COVID-19 cohorts produced using the same scRNA-seq protocol (10x Genomics, 5′) by us21 and others27,28 were included in the analysis and processed as described above. The total number of samples combined in this analysis was 263. All samples were grouped according to their WHO ordinal scale classification into mild (WHO score of 1–3), moderate (WHO score of 4–5) and severe (WHO score of 6–8) COVID-19 disease. In addition, samples known to be derived from individuals with IFN-AAB were subgrouped accordingly. PBMC scRNA-seq data from Van der Wijst et al.28 were downloaded and filtered for the earliest sample available per donor, resulting in 11 control samples and 35 samples from individuals with moderate COVID-19, 26 samples from individuals with severe COVID-19 and 4 samples from individuals with severe COVID-19 with IFN-AAB. PBMC scRNA-seq data from Su et al.27 were downloaded and filtered for the earliest sample available per donor, resulting in 17 control samples and 69 samples from individuals with mild COVID-19, 45 samples from individuals with moderate COVID-19 and 15 samples from individuals with severe COVID-19. PBMC scRNA-seq data from Georg et al.21 included six control samples, five samples from individuals with mild COVID-19, two samples from individuals with moderate COVID-19 and six samples from individuals with severe COVID-19. Single-sample GSVA using the ‘CD40-unresponsive’, ‘amplified’ and ‘combinatorial’ signatures derived from the mouse bulk RNA-seq analysis of this study was performed using GSVA v1.38.2.
For validation, we additionally analyzed scRNA-seq data from DCs from PBMC data enriched for DCs as previously published26. After downloading the respective data from the public domain, we selected those cells originally classified as monocytes and DCs and followed the same procedure of filtering the cells using the Seurat v4 reference mapping approach, as outlined above. Differential gene expression analyses and signature enrichment analyses of the ‘CD40-unresponsive’, ‘amplified’ and ‘combinatorial’ signatures were performed as described above.
Subset analysis of CD8+ T cells
For detailed analysis of the CD8+ T cell compartment, cells classified as T cells according to the original annotation provided were selected from the PBMC data set. These cells were subjected to Seurat v4 reference mapping following the developer vignette (satijalab.org/seurat/articles/multimodal_reference_mapping.html) using the multimodal PBMC reference data set44. Only those cells classified as T cells were selected to remove any possible cellular contaminations in the T cell data set. Subsequently, the remaining 45,516 cells were reclustered after scaling, regressing for UMI count per cell, identification of variable genes and PCA in this cellular subspace using the Louvain algorithm with a resolution of 0.2 based on the first 10 PCs. Cluster 1, representing CD8+ T cells, was then subsetted, and the resulting 12,386 cells were analyzed in detail, including rescaling, identification of variable genes, PCA and subsequent UMAP49 based on the first 10 PCs. Ribosomal protein-coding genes (RPL/RPS), mitochondrial genes (MT-) and hemoglobin genes (HBA1, HBA2 and HBB) were excluded from the set of variable features to remove potential sources of technical differences. Single-sample GSVA using the ‘helped’ and ‘unhelped’ T cell signatures derived from RNA-seq analysis of CD8+ T cells primed in the presence or absence of CD4+ T cell responses was performed using GSVA v1.38.2. For this, aggregated expression values of all CD8+ T cells of each sample were calculated using the AggregateExpression function in Seurat and were used as input for the sample-specific analysis. Clustering of the CD8+ T cells was performed using the Louvain algorithm with a resolution of 0.4 based on the first 10 PCs, and cells identified as γδT cells were removed. To investigate proportional cluster occupancy per disease severity, cell counts per condition were normalized before calculation of per-cluster percentages. Single-cell gene set enrichment analysis across cells of each CD8+ T cell subcluster using the ‘helped’ and ‘unhelped’ T cell signatures derived from RNA-seq analysis of CD8+ T cells primed in the presence or absence of CD4+ T cell responses was performed using AUCell v1.12.0 (ref. 32). For validation, we analyzed CD8+ T cells from two other data sets36,37. After downloading the respective data from the public domain, we followed the same procedure of filtering the cells using the Seurat v4 reference mapping approach and performed signature enrichment analysis using the above-mentioned T cell signatures.
Analysis of scATAC-seq data
scATAC-seq data of PBMCs from individuals with COVID-19 and healthy individuals produced using a Chromium Next GEM Single Cell ATAC reagent kit version 1.1 (10x Genomics, PN-1000175) was used, as previously published29. Processed and annotated scATAC-seq data from Wilk et al.29 were downloaded from Gene Expression Omnibus (GEO) under accession number GSE174072 and https://github.com/ajwilk/COVID_scMultiome and were imported to R version 4.1.0. After creation of Arrow files and a respective ArchRproject using the R package ArchR version 1.0.1 (ref. 50), the resulting single-cell data were filtered based on the published cell annotation and subsetted to CD14+ monocytes. Imputation weights on GeneScores were calculated using MAGIC51 implemented in ArchR’s addImputeWeights function. Severity-specific accessible genes were identified using the Wilcoxon rank-sum test comparing gene scores of monocytes from individuals with mild COVID-19 to cells from control donors with the following cutoffs: FDR ≤ 0.05 and log2(fold change) ≥ 0.58. Hallmark enrichment analyses were performed using clusterProfiler version 4.0.5 and the Hallmark gene set v6.2. After generation of pseudo-bulk replicates across cells of each COVID-19 severity group, peaks were called using MACS3 (ref. 52) and annotated using ChIPseeker version 1.28.3 (ref. 53). Subsequently, transcription factor binding motifs were identified in the peak regions using the homer motif set. After calculation of severity-specific differentially accessible peak regions (FDR ≤ 0.01 and log2(fold change) ≥ 2) comparing chromatin profiles of monocytes from individuals with mild and severe COVID-19 to cells from control donors, motif enrichment analysis was performed using ArchR’s peakAnnoEnrichment function.
CyTOF data and analysis
For mass cytometry data from a publicly available publication21, please refer to the Methods part of the work for detailed descriptions of the cohort, data collection and analysis workflows. Here, CD8+ T cells were separately reanalyzed and pregated using OMIQ cloud-based cytometry analysis software, also in relation to the presence of IFN-AAB22. In addition to the steps described21, we performed a PCA using R (4.0.2), where principal components were first calculated for all the events, and averages of principal component values per individual were used in plotting. Figures were rendered with the help of the R package ggfortify and function autoplot, which allows plotting of eigenvectors of input variables when used on precalculated principal components. An ellipse was calculated with ggplot2 to visually estimate the localization of different groups. Marker intensity box plots show average z-score-normalized intensity signals for all the CD8+ T cells per individual. Z-score normalization was performed beforehand over all the immune cells acquired in CyTOF.
Flow cytometry analysis of monocytes and DCs in individuals with COVID-19
Fixed whole-blood samples from individuals with COVID-19 were collected, processed and stored, as previously described25. The samples were subsequently thawed to room temperature, and erythrocytes were lysed with Thaw-Lysis buffer (Smart Buffer). After 5 min of treatment with 50 U ml–1 Pierce Universal Nuclease for Cell Lysis (Thermo Scientific) and 20 min of blocking with 1 mg ml–1 beriglobin (CSL Behring), the samples were stained for 30 min at 4 °C with antibodies to CD45 (HI30), CD11c (Bu15), CD14 (MφP9), CD3 (UCHT1), CD19 (SJ25C1), CD40 (5C3), CD83 (HB15e), CD86 (IT2.2), HLA-DR (G46-6), CD16 (3G8), CD141 (1A4) and CD163 (GHI/61). A BD LSRFortessa was used for the measurement of samples, and FlowJo software (TreeStar) was used for analysis. Expression of CD45, CD3, CD19 and HLA-DR was used for granulocyte, T cell, B cell and natural killer cell exclusion, respectively. Monocytes were gated as CD14+CD11c+ and DCs as CD14−HLA-DR+ events.
In vitro generation of human CD34+ stem cell-derived cDC1s
Human umbilical cord blood was obtained with written informed consent from the Queensland Cord Blood Bank and approval from the Mater Human Research Ethics Committee (HREC13/MHS/86). cDC1s were differentiated in a 9- to 10-d culture of in vitro expanded cord blood CD34+ progenitors in 100 ng ml–1 FLT3L (Peprotech), 100 ng ml–1 stem cell factor (Peprotech), 2.5 ng ml–1 IL-4 (Invitrogen) and 2.5 ng ml–1 granulocyte–macrophage colony-stimulating factor (Invitrogen), as previously described30, but with the addition of an irradiated OP9-DL1 stromal cell feeder layer to maximize cDC1 yields54. CD141+CADM1+CLEC9A+ cDC1s were enriched to >80% purity by labeling with biotinylated antibodies to human CADM1 (CM004-6) or CD141 (M80) and anti-biotin microbeads, followed by positive selection on an LS column according to manufacturer’s instructions (Miltenyi). Purified cDC1s were cultured at a density of 1 × 106 per ml in the presence of 1,000 U ml–1 human IFNα2a (PBL), 5 µg ml–1 CD40 agonistic antibody11 (34G12-h2, a gift from M. Cragg at University of Southhampton) or a combination. TNF-α was detected in the supernatant after 18 h using a LegendPlex kit (Biolegend) on a CytoFLEX-S (Beckman Coulter) flow cytometer.
Quantification and statistical analysis
Prism v8.4.3 (GraphPad Software) was used to assess statistical significance of non-RNA-seq data; z score = (x – mean)/s.d. The sample size (n), statistical significance and statistical tests are indicated in the legends. Data distribution was assumed to be normal, but this was not formally tested. Data collection and analysis were not performed blind to the conditions of the experiments and no formal randomization was used. No data points were excluded.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data set generated in this study can be accessed via the GEO accession number GSE171690.
Code availability
Code used for the analysis of scRNA-seq and scATAC-seq data is available at https://github.com/schultzelab/Gressier_2022. We also provide the scRNA-seq data sets used in this study and the code to analyze the respective data sets via FASTGenomics (https://beta.fastgenomics.org/p/gressier_2022).
References
Chow, A., Brown, B. D. & Merad, M. Studying the mononuclear phagocyte system in the molecular age. Nat. Rev. Immunol. 11, 788–798 (2011).
Joffre, O. P., Segura, E., Savina, A. & Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 12, 557–569 (2012).
Cabeza-Cabrerizo, M., Cardoso, A., Minutti, C. M., Pereira da Costa, M. & Reis e Sousa, C. Dendritic cells revisited. Annu. Rev. Immunol. 39, 131–166 (2021).
Ardouin, L. et al. Broad and largely concordant molecular changes characterize tolerogenic and immunogenic dendritic cell maturation in thymus and periphery. Immunity 45, 305–318 (2016).
Borst, J., Ahrends, T., Babala, N., Melief, C. J. M. & Kastenmuller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 18, 635–647 (2018).
Greyer, M. et al. T cell help amplifies innate signals in CD8+ DCs for optimal CD8+ T cell priming. Cell Rep. 14, 586–597 (2016).
Schulz, O. et al. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity 13, 453–462 (2000).
Eickhoff, S. et al. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell 162, 1322–1337 (2015).
Hor, J. L. et al. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity 43, 554–565 (2015).
Rusinova, I. et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–D1046 (2013).
Yu, X. et al. Isotype switching converts anti-CD40 antagonism to agonism to elicit potent antitumor activity. Cancer Cell 37, 850–866 (2020).
Schreck, R., Meier, B., Mannel, D. N., Droge, W. & Baeuerle, P. A. Dithiocarbamates as potent inhibitors of nuclear factor κB activation in intact cells. J. Exp. Med. 175, 1181–1194 (1992).
Lavoie, H., Gagnon, J. & Therrien, M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 21, 607–632 (2020).
Kashiwada, M. et al. Tumor necrosis factor receptor-associated factor 6 (TRAF6) stimulates extracellular signal-regulated kinase (ERK) activity in CD40 signaling along a Ras-independent pathway. J. Exp. Med. 187, 237–244 (1998).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Bacher, P. et al. Low-avidity CD4+ T cell responses to SARS-CoV-2 in unexposed individuals and humans with severe COVID-19. Immunity 53, 1258–1271 (2020).
Schultze, J. L. & Aschenbrenner, A. C. COVID-19 and the human innate immune system. Cell 184, 1671–1692 (2021).
Sette, A. & Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 184, 861–880 (2021).
Galani, I. E. et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 22, 32–40 (2021).
Pairo-Castineira, E. et al. Genetic mechanisms of critical illness in COVID-19. Nature 591, 92–98 (2021).
Georg, P. et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell 185, 493–512 (2022).
Akbil, B. et al. Early and rapid identification of COVID-19 patients with neutralizing type I interferon auto-antibodies. J. Clin. Immunol. 42, 1111–1129 (2022).
Bedoui, S., Heath, W. R. & Mueller, S. N. CD4+ T-cell help amplifies innate signals for primary CD8+ T-cell immunity. Immunol. Rev. 272, 52–64 (2016).
Wu, R. & Murphy, K. M. DCs at the center of help: origins and evolution of the three-cell-type hypothesis. J. Exp. Med. 219, e20211519 (2022).
Schulte-Schrepping, J. et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182, 1419–1440 (2020).
Arunachalam, P. S. et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 369, 1210–1220 (2020).
Su, Y. et al. Multi-omics resolves a sharp disease-state shift between mild and moderate COVID-19. Cell 183, 1479–1495 (2020).
van der Wijst, M. G. P. et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci. Transl. Med. 13, eabh2624 (2021).
Wilk, A. J. et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J. Exp. Med. 218, e20210582 (2021).
Pearson, F. E. et al. Human CLEC9A antibodies deliver Wilms’ tumor 1 (WT1) antigen to CD141+ dendritic cells to activate naive and memory WT1-specific CD8+ T cells. Clin. Transl. Immunol. 9, e1141 (2020).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) Hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Ahrends, T. et al. CD4+ T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity 47, 848–861 (2017).
Kaech, S. M. & Wherry, E. J. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 27, 393–405 (2007).
Pipkin, M. E. Runx proteins and transcriptional mechanisms that govern memory CD8 T cell development. Immunol. Rev. 300, 100–124 (2021).
Bernardes, J. P. et al. Longitudinal multi-omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity 53, 1296–1314 (2020).
Chua, R. L. et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020).
Kurth, F. et al. Studying the pathophysiology of coronavirus disease 2019: a protocol for the Berlin prospective COVID-19 patient cohort (Pa-COVID-19). Infection 48, 619–626 (2020).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Freund, E. C. et al. Efficient gene knockout in primary human and murine myeloid cells by non-viral delivery of CRISPR–Cas9. J. Exp. Med. 217, e20191692 (2020).
De Domenico, E. et al. Optimized workflow for single-cell transcriptomics on infectious diseases including COVID-19. STAR Protoc. 1, 100233 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell https://doi.org/10.1016/j.cell.2021.04.048 (2021).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013).
Huang, Y., McCarthy, D. J. & Stegle, O. Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 20, 273 (2019).
Huang, X. & Huang, Y. Cellsnp-lite: an efficient tool for genotyping single cells. Bioinformatics 37, 4569–4571 (2021).
McInnes, L., Healy, J. & Melville, J. UMAP: Uniform Manifold Approximation and Projection for dimension reduction. Preprint at https://doi.org/10.48550/arXiv.1802.03426 (2018).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
van Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729 (2018).
Zhang, Y. et al. Model-based analysis of ChIP–seq (MACS). Genome Biol. 9, R137 (2008).
Yu, G., Wang, L. G. & He, Q. Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Balan, S. et al. Large-scale human dendritic cell differentiation revealing notch-dependent lineage bifurcation and heterogeneity. Cell Rep. 24, 1902–1915 (2018).
Acknowledgements
We thank L. Loyal, A. Thiel, C. Iwert, C. Meisel, R. Rudraraju and K. Subbarao for discussions, F. Koay and D. Godfrey for Cxcr6–/– mice and M. Cragg for the human CD40 antibody. The technical expertise in breeding, maintaining and manipulating specific pathogen-free mice by the Doherty Bioresources facility is gratefully acknowledged. We also thank D. Kunkel and J. Keye from the BIH Flow and Mass Cytometry Core Facility for sample acquisition. We are grateful to the Genomics platform at the Walter & Eliza Hall Institute for Medical Research in Melbourne. Our research is supported by the National Health and Medical Research Council of Australia (APP1124815, APP1071916, APP1103895 and APP1154540), the Sylvia & Charles Viertel Charitable Foundation, a 350th Anniversary Research Grant from Merck KgGA, The Advanced Genomic Collaboration and the International Research Training Group (IRTG2168) funded by the German Research Council and The University of Melbourne. B.S. received support from the European Union’s Horizon 2020 research and innovation program (INsTRuCT, 860003) and the German Federal Ministry of Education and Research (BMBF) project RECAST (01KI20337). A.H. is supported by the Jürgen Manchot Foundation. E.L. and S.V.S were supported by the German Federal Ministry of Education and Research through the COVIMMUN project (grant 01KI20343). Furthermore, E.L. received support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), grant 397484323, TRR259. We thank the NGS Core Facility of the University Hospital Bonn for library preparation and the generation of the sequencing data. We also would like to thank the German COVID-19 OMICS Initiative (DeCOI) for providing access to scRNA-seq data. J.L.S. was supported by the DFG (IRTG2168, INST 217/1011-1 and INST 217/1017-1, Excellence Cluster ImmunoSensation2 (EXC2151/1) under project number 390873048) and SYSCID, receiving funding from the European Union’s Horizon 2020 research and innovation program under grant agreement number 733100. We are indebted to the participants, their families and the hospital staff for support, without whom this study would not have been possible.
Author information
Authors and Affiliations
Contributions
Conceptualization: S. Bedoui, E.G., J.S.-S. and S.V.S. Methodology: P.G.W., A.B., K.H., M.K., M. Clarke, T.H.O.N., P.S., K.W., C.V.L.O., B.O., C.v.d.S., Y.-C.E.C., K.J.R., T.M., M. Chopin, S. Brumhard, S.S.G., K.K. and S.L.L. Formal analysis: E.G., J.S.-S., A.O., J. Spitzer, L.J.G., P.J.H., L.P., T.K., T.A., F.K., J. Schroeder and B.S. Investigation: E.G., J.S.-S., P.G.W., A.B., M.G. and F.K. Writing, original draft: S. Bedoui and E.G. Writing, review and editing: S. Bedoui, E.G., J.S.-S., S.V.S., J.L.S., W.K., A.K., T.G., E.L., C.K. and L.E.S. Funding acquisition: S. Bedoui, T.G., E.L., J.L.S. and S.V.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks Teunis Geijtenbeek, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ioana Visan, in collaboration with the Nature Immunology team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 CD40 synergizes with varying inflammatory stimuli BMDC1.
a, ‘BMDC1-IFN-αA+CD40’ increase secretion of CCL4, TNF-α and CCL5 (from left to right) over time compared to ‘BMDC1-IFN-αA’, ‘BMDC1-CD40’ and ‘BMDC1-unstimulated’. Data are presented as mean ± s.e.m pooled from 3 independent experiments. Adjusted p-value of statistically significant differences between conditions as assessed by one-way ANOVA indicated. b, Changes in Il15 and Cxcl16 expression in ‘BMDC1-IFN-αA+CD40’ and ‘BMDC1-IFN-β+CD40’ compared to ‘BMDC1-IFN-αA’ or ‘BMDC1-IFN-β’ respectively and to ‘BMDC1-CD40’ and ‘BMDC1-unstimulated’. c. Tnf and Ccl4 in expression in BMDC1s stimulated with LPS, CpG or poly(I:C) for 6 h with or without CD40 Ab for the last 30 min. b-c, Data are presented as mean ± s.e.m pooled from 3 independent experiments. Adjusted p-value of statistically significant differences between conditions as assessed by one-way ANOVA indicated; ns = non-significant. d, Percent of MHC-IIhi CD8+ DCs from IFNαR-deficient (Ifnar2−/−) and WT mice naïve or 2 days after epicutaneous HSV-1 infection. Data are presented as mean ± s.e.m pooled from 7 independent experiments (n≥5 per experiment). Statistically significant differences between conditions as assessed by Mann-Whitney test; two-tailed p-value indicated; ns = non-significant. e. ‘BMDC1-IFN-αA’ and ‘BMDC1-unstimulated’ increase CD40 expression to comparable levels over time. Data are presented as mean ± s.e.m pooled from 3 independent experiments. Two-way ANOVA performed between the corresponding conditions ns = non-significant.
Extended Data Fig. 2 CD40 stimulation induces successive waves of transcriptional regulation in IFN-αA-conditioned BMDC1.
a, Genes included in modules 1, 2 and 3 from the co-expression analysis (Fig. 2e) displayed as heatmap. b, Top GO-terms associated with the genes included in modules 1, 2 and 3 (Fig. 2e). c, Representative immunoblotting of IκBα degradation and P65 phosphorylation in 'BMDC1-IFNαA+CD40-15min', 'BMDC1-IFNαA-30min' and ‘BMDC1-IFN-αA+CD40-4h’ compared to ‘BMDC1-IFN-αA’, ‘BMDC1-CD40’ and ‘BMDC1-unstimulated’. Full gels of the two independent experiments are displayed below. Probing of β-actin and/or total P65 served as loading control.
Extended Data Fig. 3 Enrichment of APC with ‘help’-dependent transcriptional profiles in patients with moderate COVID-19.
a, Differentially expressed genes in DCs comparing disease severity and disease stage that correspond to the ‘CD40 unresponsive’, ‘amplified’ and ‘combinatorial’. Data from published DC-enriched scRNAseq data26. b, Average gene expression in CD14+ monocytes per sample across selected key genes in a cohort of control (n=5), mild (n=5) and severe (n=5) COVID-19 patients and 7 samples derived from patients with IFN-AAB. c. Combined data set across 263 samples including controls (n=39), mild COVID-19 (WHO 1-3, n=79), moderate COVID-19 (WHO 4-5, n=82), severe COVID-19 (WHO 6-8, n=52), severe COVID-19 with IFN-AAB (WHO 7-8, n=11). Samples are stratified by disease severity according to the WHO ordinal scale as indicated and segregated by time point of sample collection relative to the onset of symptoms where available. c, Single-sample GSVA of the ‘CD40 unresponsive’, ‘amplified’ and ‘combinatorial’ gene signatures in monocytes from COVID-19 and control samples of the combined data set in b. stratified by disease severity and plotted as box plots of the enrichment scores. Wilcoxon rank-sum test p-value is shown.
Extended Data Fig. 4 Enrichment of CD8+ T cells with ‘help’-dependent transcriptional profiles in patients with moderate COVID-19.
a, Differential expression of selected key genes in CD8+ T cells derived from PBMCs scRNA-seq data of moderate and severe cases of COVID-19 and healthy HC originally as published36. b, AUCell enrichment of CD8+ T cells for ‘helped’ and ‘unhelped’ T cell gene signatures derived from RNA-seq analysis of CD8+ T cells primed in the presence or absence of CD4+ T cell help. Data are stratified by disease severity and plotted as violin plots of the ‘Area Under the Curve’ (AUC) scores. c. AUCell enrichment of CD8+ T cells for ‘helped’ and ‘unhelped’ T cell gene signatures derived from RNA-seq analysis of CD8+ T cells primed in the presence or absence of CD4+ T cell help. Data are derived from scRNA-seq of nasopharyngeal and bronchial samples stratified by disease severity and plotted as violin plots of the ‘Area Under the Curve’ (AUC) scores37. d, Heatmap showing z-scaled expression values of indicated proteins across the clusters identified in the CyTOF data of individuals with COVID-19 and HCs. e. Box plots showing relative cluster abundances of selected clusters across COVID-19 and control samples stratified according to disease severity and presence of IFN-AAB. Benjamini-Hochberg corrected pairwise Wilcoxon p-values are shown.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gressier, E., Schulte-Schrepping, J., Petrov, L. et al. CD4+ T cell calibration of antigen-presenting cells optimizes antiviral CD8+ T cell immunity. Nat Immunol 24, 979–990 (2023). https://doi.org/10.1038/s41590-023-01517-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-023-01517-x
This article is cited by
-
Epigenetic modulation of myeloid cell functions in HIV and SARS-CoV-2 infection
Molecular Biology Reports (2024)
-
CD4+ T cells produce IFN-I to license cDC1s for induction of cytotoxic T-cell activity in human tumors
Cellular & Molecular Immunology (2024)
