
Abstract
Molecular ions are ubiquitous and play pivotal roles1,2,3 in many reactions, particularly in the context of atmospheric and interstellar chemistry4,5,6. However, their structures and conformational transitions7,8, particularly in the gas phase, are less explored than those of neutral molecules owing to experimental difficulties. A case in point is the halonium ions9,10,11, whose highly reactive nature and ring strain make them short-lived intermediates that are readily attacked even by weak nucleophiles and thus challenging to isolate or capture before they undergo further reaction. Here we show that mega-electronvolt ultrafast electron diffraction (MeV-UED)12,13,14, used in conjunction with resonance-enhanced multiphoton ionization, can monitor the formation of 1,3-dibromopropane (DBP) cations and their subsequent structural dynamics forming a halonium ion. We find that the DBP+ cation remains for a substantial duration of 3.6 ps in aptly named ‘dark states’ that are structurally indistinguishable from the DBP electronic ground state. The structural data, supported by surface-hopping simulations15 and ab initio calculations16, reveal that the cation subsequently decays to iso-DBP+, an unusual intermediate with a four-membered ring containing a loosely bound17,18 bromine atom, and eventually loses the bromine atom and forms a bromonium ion with a three-membered-ring structure19. We anticipate that the approach used here can also be applied to examine the structural dynamics of other molecular ions and thereby deepen our understanding of ion chemistry.
Similar content being viewed by others


Main
We use in this study MeV-UED, which can directly map molecular structural changes by means of the information contained in time-resolved electron scattering patterns. UED was used to examine ionic species produced by non-resonant strong-field ionization of toluene molecules (see discussion in the Supplementary Information (ref. 20)), but extracting structural dynamics proved challenging owing to the intricate nature of simultaneously addressing the following two aspects: (1) achieving soft ionization of molecules to generate a specific, desired ionic species while minimizing undesired fragmentations and (2) producing a considerable quantity of ions. To tackle this challenge, we implemented resonance-enhanced multiphoton ionization (REMPI), a well-established technique known for its soft ionization capabilities and high ionization efficiencies, which can reach up to 10% (ref. 21). Specifically, we used [2 + 1] REMPI on 1,3-DBP to investigate the formation of cations and their subsequent structural changes. Quantitative analysis of the experimental scattering data indicated the successful generation of a notable quantity of ionic species, leading to noticeable signals. Detailed information about the structural changes of the ionized DBP molecule was uncovered through the analysis of the UED data collected from the experiment described in Fig. 1.

Photoexcitation by an intense femtosecond ultraviolet laser pulse ionizes 1,3-DBP molecules by means of [2 + 1] REMPI. The molecular structures of 1,3-DBP before and after ionization are captured by using the near-relativistic electron pulse as the probe and measuring the time-resolved diffraction patterns. The sub-picosecond and sub-angstrom temporal and structural resolution of MeV-UED enables direct visualization of the ultrafast structural changes in 1,3-DBP on photoinduced ionization. At a negative time delay, the molecule remains un-ionized and neutral (lower panel) and most of the electrons in the electron beam (grey arrows) travel along a straight path, whereas the remaining electrons undergo diffraction through interaction with the molecules, generating the usual well-centred symmetric diffraction pattern. As the unpumped signal coincides with the signal at the reference time delay (another negative time delay), the difference scattering pattern shown in the lower panel lacks any distinctive features. By contrast, at a positive time delay following ionization and acquisition of a charge (upper panel), the electron beam experiences deflection (red arrow) owing to a slight mismatch between the paths of the electron and laser beams. This deflection causes the electron beam to deviate from its original trajectory (grey arrows), resulting in an off-centred asymmetric diffraction pattern. The difference scattering pattern shown in the upper panel provides information about the structure of the generated ions, as well as its shift owing to the charge of the generated ions. The grey and red arrows in the lower and upper panels depict the paths of the electron beams, highlighting only the trajectory of the direct beam, while omitting the paths of the diffracted beams.
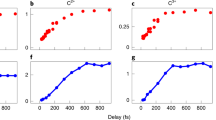
In the MeV-UED experiment, the ionization of DBP was initiated using a pump pulse with a wavelength of 267 nm. Figure 2a shows selected difference scattering patterns at various time delays, whereas the patterns for all time delays are shown in Extended Data Fig. 1. The signals are proportional to the laser fluence with the order of 2.7 ± 0.1, confirming that the UED signal reflects the reaction intermediates formed by means of a three-photon process22, namely, [2 + 1] REMPI (see the ‘Data collection’ section in Methods for details).

a, 2D difference scattering images at selected time delays. The deflection of the electron beam by generated ions results in asymmetry in the scattering images over the azimuthal angle. Each asymmetric image was decomposed into two components: the isotropic component (ΔI0) and the asymmetric component (ΔI1). b,c, False-colour plots of sΔI0 (b) and sΔI1 (c) extracted from the experimental difference scattering images. To emphasize the signal at high momentum-transfer values, ΔI0 and ΔI1 were multiplied by the magnitude of the momentum-transfer vector, s, to give sΔI0 and sΔI1. d, Comparison of the temporal behaviours of sΔI0 and sΔI1. The low-s region (s = 1.3–2.3 Å−1) of sΔI0 and sΔI1 was integrated for each time delay, and the profiles of these integration values are shown. The vertical bar at each point indicates one standard deviation. The instrument response function (IRF) is also indicated. The plots show that the ionization, contributing to sΔI1, occurs instantly on photoexcitation, whereas notable structural changes of the ionized species, contributing to sΔI0, occur gradually after an induction period of approximately 4 ps. a.u., arbitrary units.
The difference scattering pattern was decomposed into two components using Legendre decomposition, one for the scattering signal from the molecular structural changes (ΔI0(s,t)) and the other for the signal owing to the ionic-species-induced beam deflection (ΔI1(s,t)) (see the Supplementary Information for details). Figure 2b depicts the resulting sΔI1(s,t). The time trace of its amplitude, presented in Fig. 2d and Extended Data Fig. 2, demonstrates a rapid increase immediately after time zero, indicating the formation of ions, consistent with rapid photoinduced ionization (<40 fs)23. By contrast, sΔI0(s,t), shown in Fig. 2c, starts showing noticeable signals only after an unusually long induction period of approximately 4 ps. Because the absence of a difference signal in sΔI0(s,t) implies that there is no change in molecular structure, the data indicate that there is no immediate structural response to ionization and that structural changes occur only after 4 ps. The initially generated ions thus have a molecular structure identical to that of the reactant neutral molecule, within the current signal-to-noise ratio. The long induction period we see is uncommon and has not been observed in a previous time-resolved study of DBP that used mass spectrometry24 (see further discussion in the ‘Comparison of the observed induction period with previous studies’ section in the Supplementary Information).
To obtain detailed kinetic information, we conducted kinetic analysis on sΔI0(s,t). First, we performed singular value decomposition (SVD), which decomposes the original data into time-invariant features (left singular vectors, LSVs), their relative contributions (singular values) and their time profiles (right singular vectors, RSVs)25 (see the ‘Singular value decomposition’ section in the Supplementary Information for details). The SVD of sΔI0(s,t) shows that the experimental data comprise two primary components. For the quantitative analysis, we fit the two RSVs globally using a sum of an induction time and two exponential functions. The time constants for two RSVs were shared, resulting in a satisfactory fit with two exponential time constants of 15 ± 2 ps and 77 ± 15 ps and an induction period of 3.6 ± 0.3 ps. On this basis, we performed kinetics-constrained analysis (KCA)25,26 and the results are shown in Fig. 3. To explain an induction period, the kinetic model contains (E+)*, which represents the transient excited state of the ion generated on photoexcitation, and, after the induction period, (E+)* relaxes to the D+ ion. Both (E+)* and D+ are structurally dark states, whose molecular structure is indistinguishable from that of the ground state of the neutral DBP, within the signal-to-noise ratio of the current MeV-UED experiment (see the ‘Details of the kinetic analysis using SVD’ section in the Supplementary Information for details). Among two kinetic models that satisfy the conditions of having three kinetic components (two decay constants and one induction time constant) and two kinetic species, a sequential model in which the first species (A+) is formed from D+, followed by its conversion to the second species (B+), explains sΔI0(s,t) better than the other, parallel model, in which two intermediates are generated from the D+ simultaneously (Extended Data Fig. 4). Figure 3a shows the diagram of the final determined sequential model and Fig. 3b shows the population dynamics of all species involved in the reaction. Using the optimized kinetic model, which is shown in Fig. 3a, obtained from the KCA, we fit ΔI0(s,t) to extract species-associated difference scattering curves (SADS(s)) for all species using linear combination fitting. Figure 3b illustrates the population changes of intermediates. A SADS(s) in s-space can be converted into a difference radial distribution function (ΔRDF(r)) in the real space through a sine Fourier transformation (see Fig. 3c). By using the time-dependent concentrations and two ΔRDF(r)s of A+ and B+ (Fig. 3b,c), we can reconstruct ΔRDF(r,t) for all time delays. These reconstructed ΔRDF(r,t)s satisfactorily reproduce the experimental ΔRDF(r,t) for all time delays, as shown in Fig. 3d and Supplementary Fig. 6, demonstrating that our kinetic model accurately describes the experimental data.

a, The kinetic model used for the analysis, based on the two components (species) and two decay constants obtained from SVD analysis on the UED data (Extended Data Fig. 3). The sequential model provides the best fit for our experimental results (details shown in Extended Data Figs. 3 and 4). b, Time-resolved populations of species A+ and B+ and the intensity of sΔI1(s,t). The error bars for populations are multiplied by 10 for clear visualization. Solid lines represent the kinetics of sΔI0(s,t) based on the kinetic model in a and dots with one-standard-deviation error bars are obtained by independently fitting the experimental data for each time delay as a linear combination of species-associated difference scattering curves. c, RDF for the ground state and the difference RDF (ΔRDF) for species A+ and B+. The experimentally obtained RDF(r) for the ground state (black) is compared with the theoretically calculated one (red). d, Comparison of experimentally obtained ΔRDF(r,t) with the simulated ΔRDF(r,t) based on the best-fit kinetic model determined from the kinetic analysis. a.u., arbitrary units.
Next we qualitatively analysed the structural features of the two species by examining their ΔRDF(r). As detailed in the Supplementary Information, this qualitative analysis already shows that (1) ΔRDF(r) of A+ with a peak at an unusually long distance of about 6 Å suggests that the most probable form of A+ is DBP+ with a loosely bound Br and (2) ΔRDF(r) of B+, lacking positive peaks, indicates that it corresponds to MBP+, for which the loosely bound Br is eventually dissociated. To quantitatively analyse the changes in ΔRDF(r,t) that represent structural changes before and after a reaction, the static RDF(r) of the ground state was analysed to first determine the fractions of conformers (GG, AG and AA, with their geometric parameters listed in Supplementary Table 3). According to the analysis (Supplementary Fig. 7), the ground-state DBP exists in the ratio of 66 ± 2%:20 ± 2%:14 ± 3% for GG:AG:AA, which is similar to the ratio from previous studies (67%:30%:3%)27. On the basis of the static analysis of the ground-state DBP, we extracted structural information on A+ and B+ by quantitatively analysing their SADS(s) (Extended Data Fig. 6). To do so, we investigated several candidate models for the structure of the transient ionic species using density functional theory (DFT) calculations (Fig. 4). For A+, we tested two cationic DBP structures, iso-DBP+ and 1,3-DBP+, and one cationic MBP structure, 4-mem MBP+, that is, MBP+ with a four-membered ring. For B+, we tested three cationic MBP structures, bromonium MBP+, 4-mem MBP+ and 1-MBP+. Starting from the optimized candidate structures obtained from the DFT calculations, we refined the structures through a global fitting approach to simultaneously fit the experimentally measured SADS(s) for species A+ and B+. Figure 4 shows the results of the structural refinement for various candidate structures in the real space, ΔRDF(r). For A+, iso-DBP+ gives the best agreement with the experimental ΔRDF(r) (Fig. 4a, top). Specifically, iso-DBP+ depicts a dissociated Br atom bound to the MBP+ molecule, which possesses a four-membered ring structure, with the dissociated Br atom maintained at a long distance (r ≈ 5.9 Å) from the Br atom of MBP+ (Fig. 4d). The other two candidate structural models (4-mem MBP+ and 1,3-DBP+ in Fig. 4a) lack the long atomic pair distance of approximately 5.9 Å and are therefore unable to accurately fit the ΔRDF(r) of A+. A discussion about the loosely bound nature of iso-DBP+ is provided in the ‘Loosely bound nature of iso-DBP+ supported by calculated vibrational frequencies’ section in the Supplementary Information. The CA1–BrA1 bond distance was optimized to be 1.76 ± 0.01 Å, which is contracted compared with the typical C–Br distance observed in neutral DBP. The contraction can be attributed to the strong interaction between the negatively charged carbon and positively charged Br (Supplementary Table 4). A notable feature is also observed in rCA1CA2, which has a substantially shorter value (1.28 ± 0.03 Å) than the known bond length of 1.5 to 1.6 Å for a C–C single bond. These indicate that iso-DBP+ has stronger C–Br and C–C bonds than a typical neutral molecule. For B+, bromonium MBP+ best describes the experimental ΔRDF(r) (Fig. 4b, top). It has a Br atom forming a triangle with two C atoms, corresponding to a well-known halonium ion structure. The other candidates (4-mem MBP+ and 1-MBP+) were unable to satisfactorily explain the features of ΔRDF(r) at the low r (r < 3.0 Å) region. Although bromonium MBP+ has a rCB3BrB1 of 1.96 ± 0.01 Å, which is similar to the C–Br distance (2.0 Å) of the ground-state DBP, rCB1CB2 (1.74 ± 0.05 Å) and rCB2CB3 (1.68 ± 0.05 Å) were found to be longer than the typical C–C distance (Fig. 4e). Such C–C bond elongation can occur in cationic molecules, as it leads to a decrease in bond order owing to the positive charge28. The fitted parameters for the optimized structures obtained from the simultaneous fitting of the SADSs of the two species (iso-DBP+ and bromonium MBP+) are listed in Supplementary Table 1. The conformer fractions of the neutral DBP were also used as fitting parameters and the optimized fractions (63 ± 3%:20 ± 4%:17 ± 5% for GG:AG:AA) are highly similar to those obtained from the fitting of the static curve of DBP (66 ± 2%:20 ± 2%:14 ± 3%).

a,b, Structural refinements for various candidate structures in the real space, ΔRDF(r). The experimental ΔRDF(r) (black) and theoretical ΔRDF(r) (red) calculated for three candidate structural models of species A+ (a) and species B+ (b) are shown. The DFT-optimized structures were further refined to minimize the discrepancy between the experimental and theoretical data during the refinement process (fitted results in s-space are in Extended Data Fig. 6). For species A+, the iso-DBP+ model (top), which considers a Br radical loosely bound to a four-membered ring MBP+ moiety, fits the UED data much better than the other models, the model considering a complete dissociation of a Br radical (middle, 4-mem MBP+) and the model considering a minimum structural deviation from that of the ground-state 1,3-DBP (bottom, 1,3-DBP+). For species B+, the bromonium MBP+ model (top) featuring Br with bridging character as a bromonium ion provides a much better fit to our UED data than the other two models, 4-mem MBP+ (middle) and MBP+ with Br in a non-bridging configuration (bottom, 1-MBP+). c–e, The refined structures of the ground-state DBP (c), species A+ (d) and species B+ (e). Key atomic pairs contributing markedly to the ΔRDF(r) are highlighted with coloured arrows and their distances are indicated by bans of the same colour in a and b.
A bromonium ion is a well-known intermediate formed during the addition reaction of bromine to an alkene species with a C–C double bond, but its structure has not been directly determined. Instead, the bromonium ions were stabilized in the salt form in crystals and their structures were determined through crystallography19. The gas-phase structure of bromonium MBP+ determined through UED provides the reference for comparison with those in crystals. The comparison reveals that the structure of the bromonium cation as an intermediate in the chemical reaction differs substantially from the structure of the bromonium salt in the crystal in its stable form: in the presence of the counterion in the crystal, rCB1CB2 is 1.50 Å and rCB1BrB1 is 2.1–2.2 Å, whereas their gas-phase counterparts are 1.74 ± 0.05 Å and 1.96 ± 0.01 Å, respectively.
The presence of an induction period implies that the ion species during this state maintains its molecular structure and conformer ratios (see the ‘Existence of the induction period’ section in the Supplementary Information for details). Furthermore, the induction period provides valuable insight into the molecular structure of the excited DBP population in the Rydberg state generated by two-photon absorption, as discussed in the ‘Structure of the Rydberg state’ section in the Supplementary Information.
To corroborate the observed photoreaction pathways that involve a long induction period on photoexcitation29,30 and subsequent Br dissociation, we performed ab initio calculations at various levels of theory (details in the Supplementary Information). First, we explored the potential energy surfaces (PESs) of the electronic ground state (S0) of DBP and the first four doublet states (D0, D1, D2 and D3) of DBP+ by using the extended multistate complete active space second-order perturbation theory (XMS-CASPT2) method. The resulting PESs, drawn as functions of the Br1–C1–C2–C3 and Br2–C3–C2–C1 dihedral angles (Fig. 5a) and as functions of the C–Br distance (Supplementary Fig. 13), provide clues for assigning which cationic excited state is responsible for the initial induction period. PESs of DBP and DBP+ show remarkable similarities, indicating that DBP+ generated at the Franck–Condon region is at a local or global minimum in all doublet states and thus likely to retain the structure identical to that of S0 before transitioning to iso-DBP+. Furthermore, the norms of the Dyson orbitals (Extended Data Table 1), representing the ionization strength from the Rydberg state, highlight that, among D0, D1 and D2 states, which exhibit relatively large norms for at least one conformer, only D2 shows similar norms across all three conformers. On the basis of these considerations, we conclude that D2, characterized by PESs similar to those of S0 and substantial transition rates from the Rydberg state for all three conformers, is the most probable candidate for the initially populated state, (E+)*. To investigate the dynamics from D2 state to iso-DBP+, we calculated the conversion yields of radiative and nonradiative pathways from D2 state to D0. As a result, neither of the pathways were probable, as the lifetime of the former was too long (approximately 1 μs), as shown in Supplementary Table 6, and the thermal energy of the latter was too high, which is not compatible with the observed data. Detailed information can be found in the ‘Reaction pathways from D2 state to iso-DBP+’ section in the Supplementary Information. Therefore, we suggest that the most probable route for the formation of iso-DBP+ starts from D2 to D1, followed by a conical intersection connecting D1 to iso-DBP+ (Fig. 5b). The interpretation of the Dyson orbitals not only facilitates the estimation of transition probabilities but also offers a chemically insightful explanation for the absence of notable structural changes on ionization. As illustrated in Supplementary Fig. 11, these orbitals reveal that, during the cation-formation process, an electron is ejected from an orbital predominantly localized on the bromine atom. Notably, this specific orbital demonstrates nonbonding character, with minimal involvement in the bonding interactions between bromine and carbon atoms. Consequently, the removal of an electron from this orbital exerts only a negligible influence on the molecular structure.

a, PESs of S0, D0, D1, D2 and D3 using XMS-CASPT2 corrections of the SA-CASSCF(8,8) energies. All the PESs in a are plotted on the basis of the energy difference (ΔE) in comparison with the global minima of each state. The functions of the Br1–C1–C2–C3 and Br2–C3–C2–C1 dihedral angles depicted in the PESs offer insights into identifying the cationic excited state that initiates the induction period. b, Overall structural dynamics of DBP followed by the [2 + 1] REMPI process. Initially, no substantial structural change is observed for a period of about 3.6 ps. Afterwards, a transient species, iso-DBP+, is formed with a time constant of 15 ps. Finally, the loosely bound Br in iso-DBP+ escapes, yielding the bromonium MBP+ with a time constant of 77 ps.
To obtain further support for the observed induction period, surface-hopping simulations were carried out up to 1 ps, considering D2 as the initial active state. The trajectories show that DBP+ does not exhibit noticeable structural changes for several hundred femtoseconds after excitation, as evidenced by the computed averaged difference scattering curve (Extended Data Fig. 7). Comprehensive discussions are provided in the ‘Results of surface hopping simulations’ section of the Supplementary Information. Furthermore, we conducted intrinsic reaction coordinate (IRC) calculations to enhance our understanding of the reaction dynamics (Extended Data Fig. 8 and Supplementary Fig. 18). These calculations involved the determination of transition states and an intermediate species, and the results of vibrational frequency calculations for the transition states and intermediate species are provided in Supplementary Tables 7 and 8. On the basis of the results of IRC calculations, we conducted calculations to locate a conical intersection (CI1) that connects the D1 state to iso-DBP+. A detailed description of the computational methods is provided in the ‘Computational details’ and ‘Details of surface hopping simulation’ sections in the Supplementary Information. The plausible reaction coordinates and pathways for the formation of iso-DBP+ and MBP+ can be proposed as detailed in the ‘Reaction pathways to form iso-DBP+ and MBP+’ section in the Supplementary Information.
By introducing a protocol to generate ions suitable for time-resolved scattering and to analyse scattering patterns from ionized species, this study represents a notable step forward in understanding the ultrafast structural dynamics of ionic species in the gas phase. This research addresses a previously unexplored area of study owing to experimental limitations and lays the foundation for identifying the structural dynamics of ions.
Methods
Data collection
The data were collected using the MeV-UED facility at the SLAC National Accelerator Laboratory12,31,32,33. The kinetic energy of the electron pulses is 3.7 MeV, and each pulse contains approximately 104 electrons, focused to a diameter of 200 µm full width at half maximum (FWHM). The sample, 1,3-DBP (99%), was purchased from Sigma-Aldrich and used without further purification. The gas was introduced into the vacuum chamber with a flow cell 100-µm nozzle and the nozzle was heated to 40 °C to prevent clogging of the molecules. The electron and laser beams were aligned to co-propagate with a 5° angle, intersecting the gas jet at roughly 250 µm underneath the nozzle exit. The overall instrumental response was estimated to be around 104 fs FWHM (Supplementary Fig. 2). Diffraction patterns at each time delay were accumulated for 20 min. For photoexcitation, a 565-µJ pump laser pulse with a centre wavelength of 267 nm and a 1.3 nm FWHM bandwidth was linearly polarized and elliptically focused to a spot of dimensions 280 × 200 µm2, giving a fluence of 1,200 mJ cm−2. The electron detector consisted of a P43 phosphor screen with a centre hole, a 45° mirror with a centre hole, an imaging lens and an electron-multiplying charge-coupled device (see the Supplementary Information for details).
For the MeV-UED experiment, the ionization of DBP was initiated using a pump pulse with a wavelength of 267 nm. Despite its low single-photon absorption cross-section (4.73 × 10−20 cm2) at 267 nm (Supplementary Fig. 1), strong pump laser intensity can cause substantial photoinduced ionization through [2 + 1] REMPI (ref. 22). After a time delay (Δt), a 3.7-MeV electron pulse (probe) was sent to the sample and the structural changes of ionic species over time were observed by measuring the diffracted patterns of scattered electrons on a 2D area detector (details in the Supplementary Information). Figure 2a shows selected difference scattering patterns at various time delays, whereas the patterns for all time delays are shown in Extended Data Fig. 1. The time-resolved difference patterns show negligible signals at negative time delays, gradually showing a clear signal as time passes after time zero. At negative time delays, the electron-beam profile and difference patterns are isotropic, whereas at positive time delays, they exhibit asymmetric features with respect to the beam centre (Extended Data Fig. 1), indicating the generation of ionic species capable of affecting the beam path of electrons34. The signals are proportional to the laser fluence with the order of 2.7 ± 0.1 (Supplementary Fig. 3), confirming that the UED signal reflects the reaction intermediates formed by means of a three-photon process, namely, [2 + 1] REMPI (ref. 22).
Data processing and analysis
The scattering patterns (Extended Data Fig. 1) are highly asymmetric and exhibit notable positional shifts of the undiffracted electron beam, unlike typical ones observed in UED data from neutral intermediates. The asymmetry arises from the e-beam deflection caused by the Coulomb interaction with the ion product. As this artefact generated by the beam deflection can distort the structural information of the molecule34, it is a hindering factor in extracting the structural information of transient species from the diffraction-pattern analysis. To remove the artefact corresponding to cosθ in the low-s region, we introduced a method that removes the existing anisotropic scattering signal based on symmetry35,36.
To remove the beam-shift effect caused by the formation of cations and eliminate the high-s side artefact, we used Legendre decomposition, a decomposition based on the Legendre series. The difference scattering image was decomposed using Legendre decomposition into a linear combination of two components with scattering intensity distributions of the zeroth-order and first-order Legendre polynomials along the φ angle, the zeroth-order term (ΔI0) and the first-order term (ΔI1), respectively. The relationship between the original difference scattering image and the three terms obtained from the decomposition is expressed by the following equation:
In this equation, s is the momentum transfer vector, P1 is the first-order Legendre polynomial and φ is the azimuthal angle on the charge-coupled device plane. The momentum transfer vector s is defined as follows:
in which λ is the de Broglie wavelength of the incident electrons and θ is the angle between the incident and scattered electrons.
To ensure that the shift of the electron beam has a minimal influence on both the shape of the ΔI0 curves and the structural parameters derived from the analysis of ΔI0, we tested the effect of beam shifts on our correction method (Supplementary Figs. 4 and 5 and Supplementary Table 2). Detailed validation and discussion on the decomposition method are presented in the Supplementary Information.
General scheme for the kinetic analysis using SVD
To extract kinetics information of intermediates and their structures from ΔI(s,t), we followed the well-established procedure, which had been applied to time-resolved X-ray liquidography studies on small molecules, consisting of kinetic analysis using SVD (refs. 25,26).
We obtained clues about the number of intermediates associated with temporal dynamics by performing the SVD analysis on the whole data. The LSVs from the SVD analysis are shown in Extended Data Fig. 3a,b. We fitted the two notable RSVs with exponential functions and one induction delay constant. The exponential fitting results are shown in Extended Data Fig. 3d. Two notable components are fitted with the one induction delay (td = 3.6 ± 0.3 ps) and two exponential functions (t1 = 15 ± 2 ps, t2 = 77 ± 15 ps). Then, we conducted KCA, which is a method for generating theoretical difference scattering curves using time-dependent population changes of the intermediates expressed with a set of variable kinetic parameters, with an assumed candidate kinetic model from the results of SVD on the data matrix25,26 (Extended Data Fig. 4). Further details on the kinetic analysis using SVD can be found in the ‘Details of the kinetic analysis using SVD’ section in the Supplementary Information.
Generation of theoretical static diffraction curve
Theoretical scattering curves used for the structural analysis were calculated under the independent atom model. In this model, the static diffraction curve, I(s), can be expressed as the sum of the interference terms for all atomic pairs, the molecular scattering, Imol(s), and the sum of the atomic scattering, Iat(s), for each atom in the molecule. A detailed explanation of the process for calculating the theoretical static difference curve, along with the relevant equations, can be found in the ‘Detailed procedure for generating theoretical static diffraction curve’ section in the Supplementary Information.
Generation of difference signals, sΔI(s,t), ΔsM(s,t) and ΔRDF(r,t)
We use the difference-diffraction method37 to calculate the difference signal, sometimes referred to as sΔI(s), and difference sM(s,t) and RDF(r,t), ΔsM(s,t) and ΔRDF(r,t), respectively. A detailed method for calculating the difference signals, accompanied by the relevant equations, is described in the ‘Detailed procedure for generating difference signals’ section of the Supplementary Information.
Structure refinements by structural fitting analysis
Through structural fitting analysis, we determined the structures of the intermediates (species A+ and B+) that best describe the experimentally measured difference scattering curves (see Extended Data Fig. 5). The detailed procedure is described in the ‘Details of structural refinement’ section of the Supplementary Information, along with related figures in Supplementary Figs. 8 and 9.
Structural analysis
We extracted structural information on species A+ and B+ by quantitatively analysing their SADS(s). Further details on the structural analysis process can be found in the ‘Details of structural analysis’ section of the Supplementary Information.
Potential energy profiles
A series of relaxed PES scans were calculated for the ground state as well as the first four ionic doublet states (D0, D1, D2 and D3) using XMS-CASPT2 corrections applied to the SA-CASSCF(8,8) (state-averaged complete active space self-consistent field) energies. The resulting PESs are shown in Fig. 5a and Supplementary Fig. 10a, whereas the corresponding complete active space orbitals can be found in Supplementary Fig. 12. The PESs obtained with CAM-B3LYP functionals are shown in Supplementary Fig. 10b for comparison. The PESs for 1,3-DBP are compared with those of other molecules with similar structures, including 1,3-dichloropropane and propane, in Extended Data Fig. 9. Further details on the PES scans can be found in the ‘Details of potential energy scans’ section of the Supplementary Information.
Dyson orbitals
Dyson orbitals were used to determine which DBP+ doublet state is more readily populated after the ionization of the DBP molecule. We calculated the Dyson orbitals between the first Rydberg state and the first eight doublet states38,39. Further details on the calculation and interpretation of Dyson orbitals can be found in the ‘Details of Dyson orbitals’ section of the Supplementary Information.
Surface-hopping simulation
To investigate the reasons behind the induction time, we performed surface-hopping simulations and examined the reaction pathway and transition rate of DBP+ (refs. 40,41). For the most stable conformer of DBP, the simulations were carried out up to 1 ps, considering D2 as the initial active state (Supplementary Figs. 14–17). The details are presented in the ‘Details of surface hopping simulation’ section in the Supplementary Information.
Data availability
All data supporting the study, excluding raw 2D diffraction data, are available as source data. Raw 2D diffraction data, the sizes of which are too large to be provided in the form of source data, can be obtained from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
The necessary scripts to run IRC and DFT calculations using Gaussian 16, CASSCF, CASPT2, and XMS-CASPT2 calculations using Bagel code, and the ab initio molecular dynamics simulations conducted using the Newton-X package, interfaced with Gaussian 16, as well as other non-commercial analysis codes, are available from the corresponding author on reasonable request.
References
Hubbard, C. D., Illner, P. & van Eldik, R. Understanding chemical reaction mechanisms in ionic liquids: successes and challenges. Chem. Soc. Rev. 40, 272–290 (2011).
Hayyan, M., Hashim, M. A. & AlNashef, I. M. Superoxide ion: generation and chemical implications. Chem. Rev. 116, 3029–3085 (2016).
Snow, T. P. & Bierbaum, V. M. Ion chemistry in the interstellar medium. Annu. Rev. Anal. Chem. 1, 229–259 (2008).
Lu, Q. B. & Sanche, L. Effects of cosmic rays on atmospheric chlorofluorocarbon dissociation and ozone depletion. Phys. Rev. Lett. 87, 078501 (2001).
Belic, D. S., Urbain, X. & Defrance, P. Electron-impact dissociation of ozone cations to O+ fragments. Phys. Rev. A 91, 012703 (2015).
Smith, D. The ion chemistry of interstellar clouds. Chem. Rev. 92, 1473–1485 (1992).
Heo, J. et al. Determining the charge distribution and the direction of bond cleavage with femtosecond anisotropic x-ray liquidography. Nat. Commun. 13, 522 (2022).
Kim, J. G. et al. Mapping the emergence of molecular vibrations mediating bond formation. Nature 582, 520–524 (2020).
Roberts, I. & Kimball, G. E. The halogenation of ethylenes. J. Am. Chem. Soc. 59, 947–948 (1937).
McManus, S. P. & Peterson, P. E. Solvent effects on halonium ion ∡ carbonium ion equilibria. Tetrahedron Lett. 16, 2753–2756 (1975).
Ruasse, M. F. Bromonium ions or .beta.-bromocarbocations in olefin bromination. A kinetic approach to product selectivities. Acc. Chem. Res. 23, 87–93 (1990).
Yang, J. et al. Imaging CF3I conical intersection and photodissociation dynamics with ultrafast electron diffraction. Science 361, 64–67 (2018).
Yang, J. et al. Direct observation of ultrafast hydrogen bond strengthening in liquid water. Nature 596, 531–535 (2021).
Wolf, T. J. A. et al. The photochemical ring-opening of 1,3-cyclohexadiene imaged by ultrafast electron diffraction. Nat. Chem. 11, 504–509 (2019).
Barbatti, M. et al. Non-adiabatic dynamics of pyrrole: dependence of deactivation mechanisms on the excitation energy. Chem. Phys. 375, 26–34 (2010).
Shiozaki, T., Győrffy, W., Celani, P. & Werner, H.-J. Communication: Extended multi-state complete active space second-order perturbation theory: energy and nuclear gradients. J. Chem. Phys. 135, 081106 (2011).
Choi, E. H. et al. Filming ultrafast roaming-mediated isomerization of bismuth triiodide in solution. Nat. Commun. 12, 4732 (2021).
Mereshchenko, A. S., Butaeva, E. V., Borin, V. A., Eyzips, A. & Tarnovsky, A. N. Roaming-mediated ultrafast isomerization of geminal tri-bromides in the gas and liquid phases. Nat. Chem. 7, 562–568 (2015).
Brown, R. S. et al. Stable bromonium and iodonium ions of the hindered olefins adamantylideneadamantane and bicyclo[3.3.1]nonylidenebicyclo[3.3.1]nonane. X-ray structure, transfer of positive halogens to acceptor olefins, and ab initio studies. J. Am. Chem. Soc. 116, 2448–2456 (1994).
Xiong, Y. et al. Strong-field induced fragmentation and isomerization of toluene probed by ultrafast femtosecond electron diffraction and mass spectrometry. Faraday Discuss. 228, 39–59 (2021).
Boesl, U. & Zimmermann, R. in Photoionization and Photo‐Induced Processes in Mass Spectrometry (eds Zimmermann, R. & Hanley, L.) 23–88 (Wiley, 2021).
Fell, C. P., Steinfeld, J. J. & Miller, S. M. Detection of N(4S) by resonantly enhanced multiphoton ionizanon spectroscopy. Spectrochim. Acta A Mol. Spectrosc. 46, 431–434 (1990).
Loh, Z. H. & Leone, S. R. Ultrafast strong-field dissociative ionization dynamics of CH2Br2 probed by femtosecond soft x-ray transient absorption spectroscopy. J. Chem. Phys. 128, 204302 (2008).
Brogaard, R. Y., Møller, K. B. & Sølling, T. I. Real-time probing of structural dynamics by interaction between chromophores. J. Phys. Chem. A 115, 12120–12125 (2011).
Choi, M. et al. Effect of the abolition of intersubunit salt bridges on allosteric protein structural dynamics. Chem. Sci. 12, 8207–8217 (2021).
Ki, H., Gu, J., Cha, Y., Lee, K. W. & Ihee, H. Projection to extract the perpendicular component (PEPC) method for extracting kinetics from time-resolved data. Struct. Dyn. 10, 034103 (2023).
Farup, P. & Stolevik, R. Conformational analysis. V. The molecular structure, torsional oscillations, and conformational equilibria of gaseous 1,3-dibromopropane, (CH2Br)2CH2, as determined by electron diffraction and compared with semi-empirical (molecular mechanics) calculations. Acta Chem. Scand. A 28, 680–692 (1974).
Tamulienė, J. et al. On the influence of low-energy ionizing radiation on the amino acid molecule: Valine case. Lith. J. Phys. 58, 135–148 (2018).
Abedi, M., Pápai, M., Mikkelsen, K. V., Henriksen, N. E. & Møller, K. B. Mechanism of photoinduced dihydroazulene ring-opening reaction. J. Phys. Chem. Lett. 10, 3944–3949 (2019).
Banerjee, A. et al. Photoinduced bond oscillations in ironpentacarbonyl give delayed synchronous bursts of carbonmonoxide release. Nat. Commun. 13, 1337 (2022).
Weathersby, S. P. et al. Mega-electron-volt ultrafast electron diffraction at SLAC National Accelerator Laboratory. Rev. Sci. Instrum. 86, 073702 (2015).
Wang, X. J., Xiang, D., Kim, T. K. & Ihee, H. Potential of femtosecond electron diffraction using near-relativistic electrons from a photocathode RF electron gun. J. Korean Phys. Soc. 48, 390–396 (2006).
Zhu, P. et al. Femtosecond time-resolved MeV electron diffraction. New J. Phys. 17, 063004 (2015).
Chen, L. et al. Mapping transient electric fields with picosecond electron bunches. Proc. Natl Acad. Sci. USA 112, 14479–14483 (2015).
Baskin, J. S. & Zewail, A. H. Oriented ensembles in ultrafast electron diffraction. ChemPhysChem 7, 1562–1574 (2006).
Biasin, E. et al. Anisotropy enhanced X-ray scattering from solvated transition metal complexes. J. Synchrotron Radiat. 25, 306–315 (2018).
Ihee, H., Cao, J. & Zewail, A. H. Ultrafast electron diffraction: structures in dissociation dynamics of Fe(CO)5. Chem. Phys. Lett. 281, 10–19 (1997).
Humeniuk, A., Wohlgemuth, M., Suzuki, T. & Mitrić, R. Time-resolved photoelectron imaging spectra from non-adiabatic molecular dynamics simulations. J. Chem. Phys. 139, 134104 (2013).
Melania Oana, C. & Krylov, A. I. Dyson orbitals for ionization from the ground and electronically excited states within equation-of-motion coupled-cluster formalism: theory, implementation, and examples. J. Chem. Phys. 127, 234106 (2007).
Barbatti, M. et al. Newton-X: a surface-hopping program for nonadiabatic molecular dynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 26–33 (2014).
Barbatti, M. et al. The on-the-fly surface-hopping program system Newton-X: application to ab initio simulation of the nonadiabatic photodynamics of benchmark systems. J. Photochem. Photobiol. A Chem. 190, 228–240 (2007).
Acknowledgements
We thank M.-F. Lin for his help in the initial stages of preparing the experiment. We thank A. Raid, M. C. Hoffmann, A. R. Attar, D. Luo, S. P. Weathersby and X. Shen for their help in conducting the experiment. MeV-UED is operated as part of the Linac Coherent Light Source at the SLAC National Accelerator Laboratory, supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. This work was supported by the Institute for Basic Science (IBS-R033).
Author information
Authors and Affiliations
Contributions
H.I. directed the project. J.H., D.-S.A. and H.I. conceptualized and designed the experiment. J.H., D.K., S.L., J.K., Y.C., K.W.L. and H.I. conducted the UED experiment. J.Y., J.P.F.N. and X.W. supported conducting UED experiments. J.H. developed a data-analysis strategy. J.H. and D.K. analysed data. D.K. and A.S. conducted theoretical simulations. J.H., D.K., A.S., H.K. and H.I. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks German Sciaini and Jasper van Thor for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 2D difference scattering patterns at all time delays.
The 2D difference scattering patterns show the changes in the scattering intensities over time. The patterns, which contain structural information and beam shifts induced by the ion formation, were analysed using the Legendre decomposition to separate the isotropic structural information (ΔI0) and anisotropic ion formation signal (ΔI1).
Extended Data Fig. 2 Comparison of the beam shift and the sΔI1 curves extracted using Legendre decomposition.
The electron beam shifts as it interacts with the ions generated by the pump pulse. The electron-beam shift is an artefact that needs to be addressed in extracting structural information from the scattering signal. The effect of the beam shift can be separated as the sΔI1 curves through Legendre decomposition, which was used to decompose the original difference pattern into one for the scattering signal from the molecular structural changes (ΔI0(s,t)) and the other for the signal owing to the ionic-species-induced beam deflection (ΔI1(s,t)). The beam shifts as a function of time overlap well with the time-dependent intensities of sΔI1, which are integrated values of sΔI1 over the 1.3 < s < 2.0 Å−1 region. This confirms that the Legendre decomposition effectively removes the effects of the beam shift.
Extended Data Fig. 3 SVD results of MeV-UED data for DBP.
a,b, The first four left (a) and right (b) singular vectors (LSVs and RSVs) of ΔI0(s,t). The RSVs are weighted by the corresponding singular values. c, The singular values and the autocorrelation values for RSVs and LSVs. The singular values and autocorrelation values indicate that there are two signal components contributing notably to ΔI0(s,t). d, Temporal profiles of two notable components fitted with two exponential functions (t1 = 15 ± 2 ps, t2 = 77 ± 15 ps) with a constant for the induction period (td = 3.6 ± 0.3 ps).
Extended Data Fig. 4 Results of kinetic analysis.
a,b, Kinetic models for the two scenarios with the two species and two time constants, a sequential model (a) and a branching model (b). c,d, Time-resolved difference scattering curves, sΔI0(s,t), (black curves) and theoretical curves (red curves) generated by linear combinations of LSVs based on the sequential model (a) and the branching model (b). The calculated curves from the sequential model show better agreement with the experimental data.
Extended Data Fig. 5 Fit results for the isotropic difference scattering curves (sΔI0(s)).
Experimental curves with the corresponding standard errors (vertical bars) and the theoretical curves are shown in black and red lines, respectively.
Extended Data Fig. 6 Structural analysis of species A+ and B+.
a,b, Structural refinements for various candidate structures in s-space. Experimentally measured SADSs (sΔI0(s)), which were extracted from the kinetic analysis, and theoretically calculated difference scattering curves are presented in black and red lines, respectively. The comparison between the experimental and theoretical curves calculated for three candidate structural models of species A+ (a) and species B+ (b) are shown. The DFT-optimized structures were further refined to minimize the discrepancy between the experimental and theoretical data during the refinement process. For species A+, the iso-DBP+ model (top), which considers a Br radical loosely bound to a four-membered ring MBP+ moiety, fits the UED data much better than the other models, the model considering a complete dissociation of a Br radical (middle, 4-mem MBP+) and the model considering a minimum structural deviation from that of the ground state 1,3-DBP (bottom, 1,3-DBP+). For species B+, the bromonium MBP+ model (top) featuring Br with bridging character as a bromonium ion provides a much better fit to our UED data than the other two models, 4-mem MBP+ (middle) and MBP+ with Br in a non-bridging configuration (bottom, 1-MBP+).
Extended Data Fig. 7 Simulated difference scattering curves (sΔI0) using trajectories from surface-hopping simulation.
a, Simulated sΔI0(s,t) using the trajectories starting from the GG conformer of DBP. The simulated curves are based on surface-hopping simulation and represent the averaged dynamics of DBP+ within 1 ps after photoexcitation. The instrumental response function of the MeV-UED instrument, which is approximately 100 fs, was used to describe the signal around time zero for the simulated curves. Colour scales in the figures represent the amplitude of the signal in arbitrary units and the maximum intensity of the iso-DBP+ was set as the limit of the colour bar. b, Comparison between the simulated scattering curves from the trajectories and the two species observed in our experiment. The averaged difference scattering curve for the simulated trajectories is shown in blue, whereas the difference scattering curves for the two species identified in the UED experiment are presented in black and red. The absence of any discernible feature in a and in the blue curve, calculated from the ab initio molecular dynamics trajectories and averaged over time, indicates that the photoionized DBP undergoes no immediate structural change after ionization.
Extended Data Fig. 8 IRC calculation.
The IRC calculation for the transition states TS1, TS2 and TS3 along the reaction pathway. The structures of the transition states used for the IRC calculation are shown in Supplementary Table 5. By analysing the results of the IRC calculation, it is possible to confirm that the transition state identified is indeed relevant to the reaction of interest and plays a critical role in the reaction mechanism.
Extended Data Fig. 9 PESs of the S0 (neutral ground state), D0 (first doublet state), D1, D2 and D3 states of 1,3-DBP, 1,3-dichloropropane and propane.
The energies here are calculated using (XMS-)CASPT2 corrections of the (SA-)CASSCF(8,8) energies.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Supplementary Discussion and Supplementary References; it includes 18 Supplementary Figures and 8 Supplementary Tables.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heo, J., Kim, D., Segalina, A. et al. Capturing the generation and structural transformations of molecular ions. Nature 625, 710–714 (2024). https://doi.org/10.1038/s41586-023-06909-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06909-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
