
Abstract
The prevalence of highly repetitive sequences within the human Y chromosome has prevented its complete assembly to date1 and led to its systematic omission from genomic analyses. Here we present de novo assemblies of 43 Y chromosomes spanning 182,900 years of human evolution and report considerable diversity in size and structure. Half of the male-specific euchromatic region is subject to large inversions with a greater than twofold higher recurrence rate compared with all other chromosomes2. Ampliconic sequences associated with these inversions show differing mutation rates that are sequence context dependent, and some ampliconic genes exhibit evidence for concerted evolution with the acquisition and purging of lineage-specific pseudogenes. The largest heterochromatic region in the human genome, Yq12, is composed of alternating repeat arrays that show extensive variation in the number, size and distribution, but retain a 1:1 copy-number ratio. Finally, our data suggest that the boundary between the recombining pseudoautosomal region 1 and the non-recombining portions of the X and Y chromosomes lies 500 kb away from the currently established1 boundary. The availability of fully sequence-resolved Y chromosomes from multiple individuals provides a unique opportunity for identifying new associations of traits with specific Y-chromosomal variants and garnering insights into the evolution and function of complex regions of the human genome.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
All data generated and used in this study were derived from lymphoblast lines available from the Coriell Institute for Medical Research for research purposes (https://www.coriell.org/). Further details are provided in Supplementary Table 1. All data generated by the HGSVC (PacBio HiFi, ONT-UL, Hi-C, RNA-seq, Iso-seq and Bionano Genomics optical genome maps) are available at the International Nucleotide Sequence Database Collaboration (INSDC) under the following NCBI project IDs: PRJEB58376, PRJNA988114, PRJEB41077, PRJEB39684 and PRJEB39750. The HPRC (https://humanpangenome.org/) year 1 PacBio HiFi, ONT long-read sequencing and Bionano Genomics optical mapping data files are available at INSDC (PRJNA701308). Further details are provided in Supplementary Table 1. Existing testis Iso-seq data from seven individuals are available from the European Nucleotide Archive (ENA) under accessions SRX9033926 and SRX9033927. The GEUVADIS expression data are available through ArrayExpress under accession E-GEUV-3. The Genome in a Bottle (GIAB) data can be downloaded from ENA (PRJNA200694). Large supplementary data files such as the assembled genomes are available online (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC3/working/20230412_sigY_assembly_data).
Code availability
Project code implemented to produce the assemblies and the basic quality control/evaluation statistics is available at GitHub (https://github.com/marschall-lab/project-male-assembly). All scripts written and used in the study of the Yq12 subregion are available at GitHub (https://github.com/Markloftus/Yq12).
References
Skaletsky, H. et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825–837 (2003).
Porubsky, D. et al. Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell 185, 1986–2005 (2022).
Charlesworth, B. & Charlesworth, D. The degeneration of Y chromosomes. Philos. Trans. R. Soc. Lond. B 355, 1563–1572 (2000).
Vollger, M. R. et al. Segmental duplications and their variation in a complete human genome. Science 376, eabj6965 (2022).
Altemose, N., Miga, K. H., Maggioni, M. & Willard, H. F. Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput. Biol. 10, e1003628 (2014).
Nakahori, Y., Mitani, K., Yamada, M. & Nakagome, Y. A human Y-chromosome specific repeated DNA family (DYZ1) consists of a tandem array of pentanucleotides. Nucleic Acids Res. 14, 7569–7580 (1986).
Cooke, H. Repeated sequence specific to human males. Nature 262, 182–186 (1976).
Skov, L., The Danish Pan Genome Consortium & Schierup, M. H. Analysis of 62 hybrid assembled human Y chromosomes exposes rapid structural changes and high rates of gene conversion. PLoS Genet. 13, e1006834 (2017).
Kuderna, L. F. K. et al. Selective single molecule sequencing and assembly of a human Y chromosome of African origin. Nat. Commun. 10, 4 (2019).
Rhie, A. et al. The complete sequence of a human Y chromosome. Nature https://doi.org/10.1038/s41586-023-06457-y (2023).
Sahakyan, H. et al. Origin and diffusion of human Y chromosome haplogroup J1-M267. Sci. Rep. 11, 6659 (2021).
Poznik, G. D. et al. Punctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosome sequences. Nat. Genet. 48, 593–599 (2016).
The Y Chromosome Consortium. A nomenclature system for the tree of human Y-chromosomal binary haplogroups. Genome Res. 12, 339–348 (2002).
Karmin, M. et al. A recent bottleneck of Y chromosome diversity coincides with a global change in culture. Genome Res. 25, 459–466 (2015).
Hallast, P., Agdzhoyan, A., Balanovsky, O., Xue, Y. & Tyler-Smith, C. A Southeast Asian origin for present-day non-African human Y chromosomes. Hum. Genet. 140, 299–307 (2021).
Rautiainen, M. et al. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01662-6 (2023).
Wenger, A. M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 37, 1155–1162 (2019).
Lang, D. et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. Gigascience 9, giaa123 (2020).
Mikheenko, A., Bzikadze, A. V., Gurevich, A., Miga, K. H. & Pevzner, P. A. TandemTools: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 36, i75–i83 (2020).
Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, eabf7117 (2021).
Vollger, M. R. et al. Long-read sequence and assembly of segmental duplications. Nat. Methods 16, 88–94 (2019).
Bergman, J. & Schierup, M. H. Evolutionary dynamics of pseudoautosomal region 1 in humans and great apes. Genome Biol. 23, 215 (2022).
Falconer, E. et al. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods 9, 1107–1112 (2012).
Shi, W. et al. Evolutionary and functional analysis of RBMY1 gene copy number variation on the human Y chromosome. Hum. Mol. Genet. 28, 2785–2798 (2019).
Brown, E. J., Nguyen, A. H. & Bachtrog, D. The Drosophila Y chromosome affects heterochromatin integrity genome-wide. Mol. Biol. Evol. 37, 2808–2824 (2020).
Lappalainen, T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013).
Miga, K. H. et al. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 24, 697–707 (2014).
Oakey, R. & Tyler-Smith, C. Y chromosome DNA haplotyping suggests that most European and Asian men are descended from one of two males. Genomics 7, 325–330 (1990).
Miga, K. H. et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020).
Logsdon, G. A. et al. The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107 (2021).
Altemose, N. et al. Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178 (2022).
Gershman, A. et al. Epigenetic patterns in a complete human genome. Science 376, eabj5089 (2022).
Cooke, H. J. & McKay, R. D. Evolution of a human Y chromosome-specific repeated sequence. Cell 13, 453–460 (1978).
Rahman, M. M., Bashamboo, A., Prasad, A., Pathak, D. & Ali, S. Organizational variation of DYZ1 repeat sequences on the human Y chromosome and its diagnostic potentials. DNA Cell Biol. 23, 561–571 (2004).
Pathak, D., Premi, S., Srivastava, J., Chandy, S. P. & Ali, S. Genomic instability of the DYZ1 repeat in patients with Y chromosome anomalies and males exposed to natural background radiation. DNA Res. 13, 103–109 (2006).
Manz, E., Alkan, M., Bühler, E. & Schmidtke, J. Arrangement of DYZ1 and DYZ2 repeats on the human Y-chromosome: a case with presence of DYZ1 and absence of DYZ2. Mol. Cell. Probes 6, 257–259 (1992).
Wilson Sayres, M. A., Lohmueller, K. E. & Nielsen, R. Natural selection reduced diversity on human Y chromosomes. PLoS Genet. 10, e1004064 (2014).
Lange, J. et al. Isodicentric Y chromosomes and sex disorders as byproducts of homologous recombination that maintains palindromes. Cell 138, 855–869 (2009).
Verma, R. S. Heterochromatin: Molecular and Structural Aspects (Cambridge Univ. Press, 1988).
Tyler-Smith, C. & Brown, W. R. Structure of the major block of alphoid satellite DNA on the human Y chromosome. J. Mol. Biol. 195, 457–470 (1987).
Cooper, K. F., Fisher, R. B. & Tyler-Smith, C. Structure of the sequences adjacent to the centromeric alphoid satellite DNA array on the human Y chromosome. J. Mol. Biol. 230, 787–799 (1993).
1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Mendez, F. L. et al. An African American paternal lineage adds an extremely ancient root to the human Y chromosome phylogenetic tree. Am. J. Hum. Genet. 92, 454–459 (2013).
Byrska-Bishop, M. et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440 (2022).
Logsdon, G. HMW gDNA purification and ONT ultra-long-read data generation v3. Protocols.io https://doi.org/10.17504/protocols.io.b55tq86n (2022).
Gong, L., Wong, C.-H., Idol, J., Ngan, C. Y. & Wei, C.-L. Ultra-long read sequencing for whole genomic dna analysis. J. Vis. Exp. https://doi.org/10.3791/58954 (2019).
Sanders, A. D., Falconer, E., Hills, M., Spierings, D. C. J. & Lansdorp, P. M. Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat. Protoc. 12, 1151–1176 (2017).
Sanders, A. D. et al. Single-cell analysis of structural variations and complex rearrangements with tri-channel processing. Nat. Biotechnol. 38, 343–354 (2020).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Poznik, G. D. et al. Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science 341, 562–565 (2013).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Fu, Q. et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514, 445–449 (2014).
Mölder, F. et al. Sustainable data analysis with Snakemake. F1000Res. 10, 33 (2021).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A. & Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121 (2013).
Poplin, R. et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983–987 (2018).
Shafin, K. et al. Haplotype-aware variant calling with PEPPER-margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods 18, 1322–1332 (2021).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Teitz, L. S., Pyntikova, T., Skaletsky, H. & Page, D. C. Selection has countered high mutability to preserve the ancestral copy number of Y chromosome amplicons in diverse human lineages. Am. J. Hum. Genet. 103, 261–275 (2018).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 (2011).
Shepelev, V. A. et al. Annotation of suprachromosomal families reveals uncommon types of alpha satellite organization in pericentromeric regions of hg38 human genome assembly. Genom Data 5, 139–146 (2015).
Altemose, N. A classical revival: human satellite DNAs enter the genomics era. Semin. Cell Dev. Biol. 128, 2–14 (2022).
Pedersen, B. S. & Quinlan, A. R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 34, 867–868 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Waskom, M. seaborn: statistical data visualization. J. Open Source Softw. 6, 3021 (2021).
Hunter, J. D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Guy, L., Kultima, J. R. & Andersson, S. G. E. genoPlotR: comparative gene and genome visualization in R. Bioinformatics 26, 2334–2335 (2010).
Vollger, M. R., Kerpedjiev, P., Phillippy, A. M. & Eichler, E. E. StainedGlass: interactive visualization of massive tandem repeat structures with identity heatmaps. Bioinformatics https://doi.org/10.1093/bioinformatics/btac018 (2022).
Fenner, J. N. Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. Am. J. Phys. Anthropol. 128, 415–423 (2005).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Katoh, K., Misawa, K., Kuma, K.-I. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Helgason, A. et al. The Y-chromosome point mutation rate in humans. Nat. Genet. 47, 453–457 (2015).
Ren, J. & Chaisson, M. J. P. lra: a long read aligner for sequences and contigs. PLoS Comput. Biol. 17, e1009078 (2021).
Heller, D. & Vingron, M. SVIM-asm: structural variant detection from haploid and diploid genome assemblies. Bioinformatics https://doi.org/10.1093/bioinformatics/btaa1034 (2020).
Smolka, M. et al. Comprehensive structural variant detection: from mosaic to population-level. Preprint at bioRxiv https://doi.org/10.1101/2022.04.04.487055 (2022).
Zheng, Z. et al. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. Nat. Comput. Sci. 2, 797–803 (2022).
Jiang, T. et al. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 21, 189 (2020).
Edge, P. & Bansal, V. Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun. 10, 4660 (2019).
Audano, P. A. et al. Characterizing the major structural variant alleles of the human genome. Cell 176, 663–675 (2019).
Xue, Y. & Tyler-Smith, C. An exceptional gene: evolution of the TSPY gene family in humans and other great apes. Genes 2, 36–47 (2011).
Cunningham, F. et al. Ensembl 2022. Nucleic Acids Res. 50, D988–D995 (2022).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 12, 2 (2021).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Trifinopoulos, J., Nguyen, L.-T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Hagberg, A. A., Schult, D. A. & Swart, P. J. Exploring network structure, dynamics, and function using NetworkX. In Proc. 7th Python in Science Conference (SciPy2008) (eds Varoquaux, G. et al.) 11–15 (SciPy, Pasadena, 2008).
Raghavan, U. N., Albert, R. & Kumara, S. Near linear time algorithm to detect community structures in large-scale networks. Phys. Rev. E 76, 036106 (2007).
Zhou, W. et al. Identification and characterization of occult human-specific LINE-1 insertions using long-read sequencing technology. Nucleic Acids Res. 48, 1146–1163 (2020).
Shumate, A. & Salzberg, S. L. Liftoff: accurate mapping of gene annotations. Bioinformatics https://doi.org/10.1093/bioinformatics/btaa1016 (2020).
Larsson, A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014).
Zook, J. M. et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data 3, 160025 (2016).
Snajder, R., Leger, A., Stegle, O. & Bonder, M. J. pycoMeth: a toolbox for differential methylation testing from Nanopore methylation calls. Genome Biol. 24, 83 (2023).
Cuomo, A. S. E. et al. Optimizing expression quantitative trait locus mapping workflows for single-cell studies. Genome Biol. 22, 188 (2021).
Casale, F. P., Rakitsch, B., Lippert, C. & Stegle, O. Efficient set tests for the genetic analysis of correlated traits. Nat. Methods 12, 755–758 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Knight, P. A. & Ruiz, D. A fast algorithm for matrix balancing. IMA J. Numer. Anal. 33, 1029–1047 (2012).
Crane, E. et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244 (2015).
Kruse, K., Hug, C. B. & Vaquerizas, J. M. FAN-C: a feature-rich framework for the analysis and visualisation of chromosome conformation capture data. Genome Biol. 21, 303 (2020).
Dekker, J. et al. The 4D nucleome project. Nature 549, 219–226 (2017).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28, 1102–1104 (2000).
Yadav, S. K., Kumari, A., Javed, S. & Ali, S. DYZ1 arrays show sequence variation between the monozygotic males. BMC Genet. 15, 19 (2014).
Prosser, J., Frommer, M., Paul, C. & Vincent, P. C. Sequence relationships of three human satellite DNAs. J. Mol. Biol. 187, 145–155 (1986).
Babcock, M., Yatsenko, S., Stankiewicz, P., Lupski, J. R. & Morrow, B. E. AT-rich repeats associated with chromosome 22q11.2 rearrangement disorders shape human genome architecture on Yq12. Genome Res. 17, 451–460 (2007).
Nurk, S. et al. The complete sequence of a human genome. Science https://doi.org/10.1101/2021.05.26.445798 (2021).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Minh, B. Q., Nguyen, M. A. T. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195 (2013).
Konkel, M. K., Walker, J. A. & Batzer, M. A. LINEs and SINEs of primate evolution. Evol. Anthropol. 19, 236–249 (2010).
Hoyt, S. J. et al. From telomere to telomere: the transcriptional and epigenetic state of human repeat elements. Science 376, eabk3112 (2022).
Acknowledgements
Funding was provided by National Institutes of Health (NIH) grants U24HG007497 (to C. Lee, E.E.E., J.O.K. and T.M.), U01HG010973 (to T.M., E.E.E. and J.O.K.), R01HG002385 and R01HG010169 (to E.E.E.), and GM123312 (to S.J.H. and R.J.O.); the German Federal Ministry for Research and Education (BMBF 031L0184 to J.O.K. and T.M.); the German Research Foundation (DFG 391137747 to T.M.); the German Human Genome-Phenome Archive (DFG (NFDI 1/1) to J.O.K.); the European Research Council (ERC Consolidator grant 773026 to J.O.K.); the EMBL (to J.O.K. and P. Hasenfeld); the EMBL International PhD Programme (to W.H.); the Jackson Laboratory Postdoctoral Scholar Award (to K.K.); NIH National Institute of General Medical Sciences (NIGMS R35GM133600 to C.R.B.; 1P20GM139769 to M.K.K. and M.L.) and the National Cancer Institute (NCI) (P30CA034196 to C.R.B. and P.A.A.); U24HG007497 (P. Hallast, F.Y., Q.Z., F.T. and J.Y.K.); NIGMS K99GM147352 (to G.A.L.); and Wellcome grant 098051 (to C.T.-S.). This work was also supported, in part, by the P30 CA034196 grant from the NCI. E.E.E. is an investigator of the Howard Hughes Medical Institute. We thank A. Rhie and A. Phillippy for coordination and discussions; Y. Xue for discussions and advice throughout the project; J. Wood and the members of the Genome Reference Informatics Team at the Wellcome Sanger Institute for suggestions and feedback on assembly evaluation; L. Skov for advice and sharing his scripts for gene conversion detection; the members of the HPRC (https://humanpangenome.org) for making their data publicly available; the staff at Clemson University for their allotment of compute time on the Palmetto Cluster; staff at the Center for Information and Media Technology at Heinrich Heine University Düsseldorf and the Scientific Services at the Jackson Laboratory, including the Genome Technologies Service for their assistance with the work described herein and Research IT for providing computational infrastructure and support and the members of the Phillippy laboratory (NIH/NHGRI) for their Verkko support; and the people who contributed samples as part of the 1000 Genomes Project.
Author information
Authors and Affiliations
Consortia
Contributions
PacBio production sequencing: Q.Z., K.M.M., A.P.L. and J.K. ONT production: Q.Z. and K.H. Strand-seq production: P. Hasenfeld. and J.O.K. ONT re-basecalling and methylation calling: P.A.A. and W.T.H. Genome assembly: P.E., F.Y. and T.M. Assembly analysis and evaluation: P.E., P. Hallast, F.Y., W.H. and F.T. Assembly-based variant calling: P.E., P.A.A., P. Hallast and C.R.B. Variant quality control, merging and annotation: P.A.A. and P. Hallast. Short-read calling, phylogeny construction and dating: P. Hallast. Analysis of Bionano Genomics optical maps: F.Y. Strand-seq inversion detection and genotyping: D.P. MEI discovery and integration: W.Z., M.L. and M.K.K. Inversion analysis: P. Hallast, D.P., K.K., M.L. and M.K.K. Gene conversion and evolutionary rate: K.K., P. Hallast and M.K.K. Gene families: M.L., F.Y. and M.K.K. Analyses on Y subregions: P.E., P. Hallast, M.L., F.Y., G.A.L., P.A.A., W.H., K.K., F.T., M.K.K., E.E.E. and C.Lee. RNA-seq analysis: M.J.B. Methylation and meQTL analysis: M.J.B. HiC analysis: C. Li. and X.S. Repeat annotation: S.J.H. and R.J.O. Iso-seq analysis: P.C.D. and E.E.E. Gene annotations F.Y. and P.C.D. Supplementary materials: P. Hallast, P.E., M.L., F.Y., P.A.A., G.A.L., M.J.B., W.Z., W.H., K.K., C. Li, S.J.H., P.C.D., F.T., J.Y.K., Q.Z., K.M.M., P. Hasenfeld, X.S. and M.K.K. Display items: P. Hallast, P.E., M.L., F.Y., G.A.L., W.H., K.K., F.T. and M.K.K. Manuscript writing: P. Hallast, P.E., M.L., P.A.A., G.A.L., M.J.B., W.Z., M.K.K., C.Lee with contributions from all of the other authors. All of the authors contributed to the final interpretation of data. HGSVC co-chairs: C. Lee, J.O.K., E.E.E. and T.M.
Corresponding author
Ethics declarations
Competing interests
E.E.E. is a scientific advisory board member of Variant Bio, Inc. C. Lee is a scientific advisory board member of Nabsys and Genome Insight. The following authors have previously disclosed a patent application (no. EP19169090) relevant to Strand-seq: J.O.K., T.M. and D.P. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Mikkel Heide Schierup, John Lovell and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
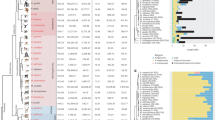
Extended Data Fig. 1 Variation in structure and composition across Y-chromosomal subregions.
a. Overview of the Y chromosome. A three-way comparison of sequence identity between GRCh38 Y, NA19317 (E1b1a1a1a1c1a1a3a1-CTS8030) and the T2T Y (excluding Yq12 and PAR2 subregions), highlighting substantial differences in the size and orientation of some subregions. b. Focus on Yq12. Sequence identity heatmaps of the Yq12 subregion for six contiguously assembled samples (HG01890, HG02666, HG01106, HG02011, HG00358 and HG01952), two samples (NA19705 and HG01928) with a single gap in the Yq12 subregion (gap location marked with asterisk) and the T2T Y using 5kb window size. c. Focus on TSPY repeat array. Sequence identity heatmaps of ~20.3-kbp TSPY repeat units for three males highlighting putative expansion events harbouring both single and multiple repeat units. Red shades from lighter to darker indicate sequence identity from 99–100%, respectively, while white fill indicates sequence identity <99%. The last copy on the right is the single separate repeat unit containing the TSPY2 gene. See Fig. S22 for heatmaps of all samples. d. Dotplots of the TSPY repeat array for HG02666 with 5 kbp of flanking regions showing identical matches of 2, 5, and 10 kbp in size indicating regions with high sequence identity. See Fig. S25 for additional examples.
Extended Data Fig. 2 Distribution of genetic variants across the Y chromosome and repeat elements in PAR1, XDR1 and XTR1 subregions.
a. Distribution of variant sizes for SVs (≥ 50 bp, top), Indels (< 50 bp, middle), and SNVs (bottom) with the Y chromosome coloured by subregion. High peaks in heterochromatin are apparent for SVs, but are absent in SNVs and indels. b. Repeat element distribution across 10 samples with contiguously assembled PAR1 regions and the T2T Y. Repeat elements on sense (+) and antisense (−) strand are shown, coloured according to repeat class. Extensive differences in size can be seen between samples, especially in the satellite arrays located close to the telomere (in dark red), and substantial differences in repeat element composition in PAR1 vs. the male-specific XDR1 and XTR1 regions. The locations of PAR1, XDR1 and XTR1 subregions in each individual are shown in black, red and black, respectively. Please note that the maroon colour of the “Unknown” elements close to the telomere is caused by significant clustering of those elements. DNA: DNA repeat elements, snRNA: small nuclear RNA, tRNA: transfer RNA, rRNA: ribosomal RNA, srpRNA: signal recognition particle RNA, scRNA: small conditional RNA, RC: rolling circle.
Extended Data Fig. 3 Examples of structural variation identified in the assembled Y chromosomes.
a. Inversions identified in the AZFc/ampliconic 7 subregion. Top - comparison between the T2T Y and select de novo assemblies, bottom - GRCh38 Y and the de novo assemblies (see Fig. S34 for details on AZFc/ampliconic 7 subregion composition). Potential NAHR path is shown below the dotplot. b. Inverted duplication affecting roughly two thirds of the 161 kbp unique ‘spacer’ sequence in the P3 palindrome, spawning a second copy of the TTTY5 gene and elongating the LCRs in this region. A detailed sequence view reveals a high sequence similarity between the duplication and its template, and its placement in Y phylogeny supports emergence of this variant in the common ancestor of haplogroup E1a2 carried by NA19239, HG03248 and HG02572 (Fig. 3a).
Extended Data Fig. 4 RBMY1 gene similarity and architecture.
a. A schematic distribution of individual RBMY1 gene copies (filled rectangles) within analysed Y chromosome assemblies (42 + T2T + GRCh38). The RBMY1 gene copies are located in four primary regions (NA19239 carries a partial duplication of gene region 2 and the composition of HG02666 suggests at least one inversion within the RBMY regions). Fill colours refer to the assigned network community (NC) and indicates a similar sequence (Methods). Assembly of this region was not contiguous in HG03065 (brown line) and was not included in the analysis. b. A secondary directed network showing connections between NCs with the most similar consensus sequences. An edge pointing from one node to a second node indicates that the second node was the first’s closest match (i.e., most similar sequence; ties are allowed and shown as multiple edges stemming from a node). The width of the edge represents the sequence similarity between two nodes (i.e., NC consensus sequence similarity; thicker means fewer SNVs). The node size is representative of the total edges pointing to the node. c. RBMY1 phylogenetic analysis of exonic nucleotide sequences. Shown is the unrooted phylogenetic tree of RBMY1 genes constructed using a maximum likelihood approach (Methods). This tree is rooted at the midpoint with the total count of RBMY1 copies shown on the right. The scale bar represents the average number of substitutions per site. RBMY1 copies located in regions 1 and 2 (primarily dark blue, orange, dark/light green, and pink) distinguish themselves from those located downstream in regions 3 and 4 (primarily light blue and purple copies).
Extended Data Fig. 5 TSPY gene similarity and architecture.
a. TSPY array visualization of each sample with contiguous assembly in this region. Individual TSPY gene copies are shown (rectangles), and their colour is based on the assigned network community (NC) (Methods). Sample names with black rectangles (NA19331, HG03732 and HG03492) carry the IR3/IR3 inversion and were re-oriented for visualization. Asterisks within individual gene copies indicate possible gene conversion (GC) or recombination (R) events unique to that gene copy. If a GC/R event is shared by an NC an asterisk is shown in the NC legend rectangle. The TSPY2 gene copy is shown as a red rectangle. b. A secondary directed network showing the sequence similarity between NC consensus sequences. An edge pointing from one node to a second node indicates that the second node was the first’s closest match (i.e., most similar sequence; ties are allowed and shown as multiple edges stemming from a node). The width of the edge represents the sequence similarity between two nodes (i.e., NC consensus sequence similarity; thicker means fewer SNVs). The node size is representative of the total edges pointing to the node. c. TSPY phylogenetic analysis of exonic nucleotide sequences. Shown is the unrooted phylogenetic tree of TSPY genes constructed using a maximum likelihood approach (Methods). This tree is rooted at the midpoint and the total count of TSPY copies is shown on the right. The scale bar represents the average number of substitutions per site. The early split/rise of NC1 within the tree, in conjunction with the secondary directed network and manual comparison of TSPY sequences (as well as their presence across all lineages) suggests that NC1 TSPY copies represent the ancestral TSPY gene sequence.
Extended Data Fig. 6 DNA methylation patterns as determined from the ONT data across the three contiguously assembled Y chromosomes.
Methylation patterns for samples: a. HG1890, b. HG02666 and c. HG00358. The three dot plots (in grey) show the smoothed DNAme levels, in 5 kbp windows for visualization, in beta-scale ranging from 0 (not methylated) to 1 (methylated). The locations of Yq12 repeat arrays (DYZ18, 2.7kb-repeat, 3.1kb-repeat, DYZ1 and DYZ2) and the Y-chromosomal subregions are shown below as bar plots.
Extended Data Fig. 7 Functional analyses on the Y chromosome with DNA-methylation, RNA expression and HiC information as anchored to GRCh38 Y.
a. The top three panels show DNA-methylation levels and variation over the studied chromosomes (n = 41). In black (top dot plot) the average methylation is shown, in green (middle dot plot) the variation in DNAme levels across the studied genomes. The bottom boxplot represents the DNA methylation segmentation using PycoMeth-seg (Methods). In grey shades 2,861 methylation segments, and in red shades the 340 significantly differentially methylated segments (DMS). The CpG sites that fall in a DMS are coloured in a lighter shade in the top two dot plots, the dot plots are in beta-scale, ranging from 0 (not methylated) to 1 (methylated). b. Average insulation scores (top) and variance of insulation scores between any two samples (bottom) across 40 samples with Hi-C data with 10 kbp resolution. Regions with lower insulation scores are more insulated and more likely to be topologically associating domain (TAD) boundaries, while regions with higher scores are more likely to stay inside TADs (the regions between the two adjacent TAD boundaries). The y-axis represents the average insulation scores ranging from −2 (most insulated) to 2 (least insulated) and the variance insulation scores ranging from 0 (no variance) to 8 (more variance). c. The Geuvadis-based gene-expression analysis, shown are the 205 genes on the Y chromosome (grey shades), the 64 genes expressed in the Geuvadis LCLs (blue shades), of which 22 are differentially expressed (red shades, Supplementary Results ‘Functional analysis’ for additional details).
Extended Data Fig. 8 Composition of the Y-chromosomal (peri-)centromeric regions.
a. Organization of the chromosome Y centromeric region from 21 genomes representing all major superpopulations. The structure (top), α-satellite HOR organization (middle), and sequence identity heat map (bottom) for each centromere is shown and reveals the presence of novel HORs in over half of the centromeres. Note - the sizes of the DYZ3 α-satellite array are shown on top as determined using RepeatMasker (Methods). b. Genetic landscape of the Y-chromosomal pericentromeric region for three select samples (see Figs. S47–S48 for all samples). The top panel shows locations and composition of the pericentromeric region with repeat array sizes shown for each Y chromosome (the DYZ3 α-satellite array size as determined using RepeatMasker, Methods). The middle panel shows (UL-)ONT read depth and bottom sequence identity head maps generated using the StainedGlass pipeline74 (using a 5 kbp window size).
Extended Data Fig. 9 Divergence of DYZ18, Yq11/Yq12 transition region and DYZ1 repeat units.
An overview of the Bray-Curtis distance/dissimilarity of k-mer abundance profiles for individual DYZ18 (grey), 3.1-kbp (red), 2.7-kbp (blue) and DYZ1 (black) repeats versus their consensus sequence. The Yq11/transition region/Yq12 are shown for each of the seven samples with a completely assembled Yq12 subregion. Lighter colours indicate less distance/dissimilarity (i.e., more similar) k-mer abundance profiles compared to their consensus sequence. Results indicate that arrays located on the proximal and distal boundaries of the Yq12 subregion contain repeats with k-mer abundance compositions less similar to their consensus sequence (i.e., more diverged). The size of individual lines is a function of the length of the repeat. The repeat unit orientation (above = sense, below = antisense) was determined based on RepeatMasker annotations of satellite sequences within repeats (Methods).
Extended Data Fig. 10 Divergence of Yq12 DYZ2 repeat units.
An overview of the divergence of individual DYZ2 subunits for a. samples with completely assembled Yq12 subregion (HG01890, HG02666, HG01106, HG02011, T2T Y, HG00358, HG01952), and b. the two most closely related genomes (NA19317 and NA19347) with incompletely assembled Yq12 subregions. The size of individual lines is a function of the length of the repeat. The repeat unit orientation (above = sense, below = antisense) was determined based on RepeatMasker annotations of satellite sequences within repeats (Methods). A higher divergence was observed within the subunits located in arrays at the proximal and distal ends of the Yq12 subregion. Additionally, DYZ2 subunits located near the boundaries of individual arrays tend to be more diverged than those located centrally. Between the closely related genomes, the divergence of DYZ2 repeats within the shared DYZ2 arrays are highly similar.
Supplementary information
Supplementary Information
Supplementary Results 1–9 providing more quality control and in-depth analyses of gene content, inversions, the Yq12 region and putatively functional consequences of the Y variation; Supplementary Figs. 1–74, complementing all analyses in the main text and in the Supplementary Results; and Supplementary References 1–46.
Supplementary Tables
Supplementary Tables 1–61, complementing all analyses in the main text and in the Supplementary Results. The file includes a short description of each table in the table of contents (the first table sheet in the document).
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hallast, P., Ebert, P., Loftus, M. et al. Assembly of 43 human Y chromosomes reveals extensive complexity and variation. Nature 621, 355–364 (2023). https://doi.org/10.1038/s41586-023-06425-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06425-6
This article is cited by
-
Unlocking the mystery of the human Y chromosome
Nature Reviews Urology (2024)
-
Complete or partial loss of the Y chromosome in an unselected cohort of 865 non-vasectomized, azoospermic men
Basic and Clinical Andrology (2023)
-
The complete sequence of a human Y chromosome
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
