
Abstract
Bacteria in the gut can modulate the availability and efficacy of therapeutic drugs. However, the systematic mapping of the interactions between drugs and bacteria has only started recently1 and the main underlying mechanism proposed is the chemical transformation of drugs by microorganisms (biotransformation). Here we investigated the depletion of 15 structurally diverse drugs by 25 representative strains of gut bacteria. This revealed 70 bacteria–drug interactions, 29 of which had not to our knowledge been reported before. Over half of the new interactions can be ascribed to bioaccumulation; that is, bacteria storing the drug intracellularly without chemically modifying it, and in most cases without the growth of the bacteria being affected. As a case in point, we studied the molecular basis of bioaccumulation of the widely used antidepressant duloxetine by using click chemistry, thermal proteome profiling and metabolomics. We find that duloxetine binds to several metabolic enzymes and changes the metabolite secretion of the respective bacteria. When tested in a defined microbial community of accumulators and non-accumulators, duloxetine markedly altered the composition of the community through metabolic cross-feeding. We further validated our findings in an animal model, showing that bioaccumulating bacteria attenuate the behavioural response of Caenorhabditis elegans to duloxetine. Together, our results show that bioaccumulation by gut bacteria may be a common mechanism that alters drug availability and bacterial metabolism, with implications for microbiota composition, pharmacokinetics, side effects and drug responses, probably in an individual manner.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
All data generated during this study are included in this published Article (and its Supplementary Information files). Supplementary Table 18 provides an overview of the different methods and data associated with all figures. UPLC and mass spectrometry data are deposited at the MetaboLights repository under the accession codes MTBLS1264, MTBLS1757, MTBLS1627, MTBLS1319, MTBLS1791, MTBLS1792, and MTBLS2885. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the dataset identifiers PXD016062 and PXD016064. Source data are provided with this paper.
Code availability
The data analysis codes are available at https://github.com/sandrejev/drugs_bioaccumulation.
References
Zimmermann, M., Zimmermann-Kogadeeva, M., Wegmann, R. & Goodman, A. L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467 (2019).
Forslund, K., Hildebrand, F., Nielsen, T. & Falony, G. A. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266 (2015).
Falony, G. et al. Population-level analysis of gut microbiome variation. Science 352, 560–564 (2016).
Maier, L. & Typas, A. Systematically investigating the impact of medication on the gut microbiome. Curr. Opin. Microbiol. 39, 128–135 (2017).
Jackson, M. A. et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat. Commun. 9, 2655 (2018).
Maier, L. et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623–628 (2018).
Spanogiannopoulos, P., Bess, E. N., Carmody, R. N. & Turnbaugh, P. J. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 14, 273–287 (2016).
Alexander, J. L. et al. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat. Rev. Gastroenterol. Hepatol. 14, 356–365 (2017).
Fuller, A. T. Is p-aminobenzenesulphonamide the active agent in prontosil therapy? Lancet 229, 194–198 (1937).
Goldman, P., Peppercorn, M. A. & Goldin, B. R. Metabolism of drugs by microorganisms in the intestine. Am. J. Clin. Nutr. 27, 1348–1355 (1974).
Chhabra, R. S. Intestinal absorption and metabolism of xenobiotics. Environ. Health Perspect. 33, 61–69 (1979).
Koppel, N., Maini Rekdal, V. & Balskus, E. P. Chemical transformation of xenobiotics by the human gut microbiota. Science 356, eaag2770 (2017).
Sousa, T. et al. The gastrointestinal microbiota as a site for the biotransformation of drugs. Int. J. Pharm. 363, 1–25 (2008).
Klaassen, C. D. & Cui, J. Y. Review: mechanisms of how the intestinal microbiota alters the effects of drugs and bile acids. Drug Metab. Dispos. 43, 1505–1521 (2015).
Haiser, H. J., Seim, K. L., Balskus, E. P. & Turnbaugh, P. J. Mechanistic insight into digoxin inactivation by Eggerthella lenta augments our understanding of its pharmacokinetics. Gut Microbes 5, 233–238 (2014).
Koppel, N., Bisanz, J. E., Pandelia, M.-E., Turnbaugh, P. J. & Balskus, E. P. Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. eLife 7, e33953 (2018).
Wallace, B. D., Wang, H., Lane, K. T. & Scott, J. E. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330, 831–835 (2010).
Tramontano, M. et al. Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat. Microbiol. 3, 514–522 (2018).
Chrystal, E. J. T., Koch, R. L., McLafferty, M. A. & Goldman, P. Relationship between metronidazole metabolism and bactericidal activity. Antimicrob. Agents Chemother. 18, 566–573 (1980).
Mahmood, S., Khalid, A., Arshad, M., Mahmood, T. & Crowley, D. E. Detoxification of azo dyes by bacterial oxidoreductase enzymes. Crit. Rev. Biotechnol. 36, 639–651 (2016).
Khan, A. K. A., Guthrie, G., Johnston, H. H., Truelove, S. C. & Williamson, D. H. Tissue and bacterial splitting of sulphasalazine. Clin. Sci. 64, 349–354 (1983).
Goodman, A. L. et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl Acad. Sci. USA 108, 6252–6257 (2011).
Shu, Y. Z. & Kingston, D. G. A. Metabolism of levamisole, an anti-colon cancer drug, by human intestinal bacteria. Xenobiotica 21, 737–750 (1991).
Schloissnig, S. et al. Genomic variation landscape of the human gut microbiome. Nature 493, 45–50 (2013).
Fenner, K., Canonica, S., Wackett, L. P. & Elsner, M. Evaluating pesticide degradation in the environment: blind spots and emerging opportunities. Science 341, 752–758 (2013).
Gulde, R., Anliker, S., Kohler, H. E. & Fenner, K. Ion trapping of amines in protozoa: a novel removal mechanism for micropollutants in activated sludge. Environ. Sci. Technol. 52, 52–60 (2018).
Congeevaram, S., Dhanarani, S., Park, J., Dexilin, M. & Thamaraiselvi, K. Biosorption of chromium and nickel by heavy metal resistant fungal and bacterial isolates. J. Hazard. Mater. 146, 270–277 (2007).
Bae, W., Chen, W., Mulchandani, A. & Mehra, R. K. Enhanced bioaccumulation of heavy metals by bacterial cells displaying synthetic phytochelatins. Biotechnol. Bioeng. 70, 518–524 (2000).
Becher, I. et al. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 12, 908–910 (2016).
Franken, H. et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 10, 1567–1593 (2015).
Brochado, A. R. et al. Species-specific activity of antibacterial drug combinations. Nature 559, 259–263 (2018).
Rakoff-Nahoum, S., Coyne, M. J. & Comstock, L. E. An ecological network of polysaccharide utilization among human intestinal symbionts. Curr. Biol. 24, 40–49 (2014).
Hooper, L. V., Littman, D. R. & Macpherson, A. J. Interactions between the microbiota and the immune system. Science 336, 1268–1273 (2012).
Zhang, F. et al. Caenorhabditis elegans as a model for microbiome research. Front. Microbiol. 8, 485 (2017).
Vetizou, M. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–107 (2015).
Wu, H. et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 23, 850–858 (2017).
Macedo, D. et al. Antidepressants, antimicrobials or both? Gut microbiota dysbiosis in depression and possible implications of the antimicrobial effects of antidepressant drugs for antidepressant effectiveness. J. Affect. Disord. 208, 22–32 (2017).
Sharon, G., Sampson, T. R., Geschwind, D. H. & Mazmanian, S. K. The central nervous system and the gut microbiome. Cell 167, 915–932 (2016).
Dent, R. et al. Changes in body weight and psychotropic drugs: a systematic synthesis of the literature. PLoS ONE 7, e36889 (2012).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011).
Elias, J. E. & Gygi, S. P. Target–decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Porollo, A. EC2KEGG: a command line tool for comparison of metabolic pathways. Source Code Biol. Med. 9, 19 (2014).
Mateus, A. et al. Thermal proteome profiling in bacteria: probing protein state in vivo. Mol. Syst. Biol. 14, e8242 (2018).
Hughes, C. S. et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757 (2014).
Hughes, C. S. et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 14, 68–85 (2019).
Ortmayr, K., Charwat, V., Kasper, C., Hann, S. & Koellensperger, G. Uncertainty budgeting in fold change determination and implications for non-targeted metabolomics studies in model systems. Analyst, 142, 80–90 (2017).
He, L., Diedrich, J., Chu, Y. Y. & Yates, J. R. 3rd Extracting accurate precursor information for tandem mass spectra by RawConverter. Anal. Chem. 87, 11361–11367 (2015).
Mahieu, N. G., Genenbacher, J. L. & Patti, G. J. A roadmap for the XCMS family of software solutions in metabolomics. Curr. Opin. Chem. Biol. 30, 87–93 (2016).
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R. & Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 (2006).
Vinaixa, M. et al. A guideline to univariate statistical analysis for lc/ms-based untargeted metabolomics-derived data. Metabolites 2, 775–795 (2012).
Smith, C. A. et al. METLIN: a metabolite mass spectral database. Ther. Drug Monit. 27, 747–751 (2005).
Tanabe, M. & Kanehisa, M. Using the KEGG database resource. Curr. Protoc. Bioinfomatics 38, 1.12.1–1.12.43 (2012).
Fuhrer, T., Heer, D., Begemann, B. & Zamboni, N. High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection–time-of-flight mass spectrometry. Anal. Chem. 83, 7074–7080 (2011).
Ponomarova, O. et al. yeast creates a niche for symbiotic lactic acid bacteria through nitrogen overflow. Cell Syst. 5, 345–357 (2017).
Wishart, D. S. et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46, D608–D617 (2018).
Sumner, L. W. et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3, 211–221 (2007).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl Acad. Sci. USA 108, 4516–4522 (2011).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 (2014).
Acknowledgements
This project was supported by the European Union’s Horizon 2020 research and innovation programme under the grant agreement number 686070, and by the UK Medical Research Council (project number MC_UU_00025/11). A.M., L.M., M.T. and V.P. were supported by the EMBL interdisciplinary postdoctoral program. We thank EMBL Genomics, Metabolomics and Proteomics core facilities for their support in respective analyses.
Author information
Authors and Affiliations
Contributions
M. Klünemann, P.B., A.T. and K.R.P. conceived the study. M. Klünemann, S.A., A.T. and K.R.P. planned the overall experiments. S.A. performed the overall data analysis. K.Z. and V.P. performed the drug clustering. M. Klünemann carried out the interaction screen, large-volume validation and UPLC data analysis. A.R.B., L.M., M.T. and M. Banzhaf assisted with the screen set-up. F.H. and C.S. designed and synthesized the clickable drug. M. Klünemann, M.-T.M. and T.B. performed the click chemistry proteomics experiments. M. Beck designed and supervised the click chemistry proteomics analysis. A.M. and M.M.S. planned the TPP experiments. S.B. and A.M. performed the TPP experiments. A.M., M.M.S. and S.A. analysed the TPP data. J.V. and D.C.S. performed the FIA–MS experiments and data analysis. S.B. performed the bacterial culturing experiments and P.P. performed the LC–MS analysis for drug measurements. M. Klünemann and P.P. performed the secreted metabolite LC–MS analysis in buffer. M. Klünemann and S.B. prepared the samples for, and B.S., L.N. and J.H. performed, the NMR analysis. M. Klünemann and D.K. performed growth assays and sample preparation for cross-fed metabolite analysis. S.B. performed growth assays and sample preparation for secreted metabolite analysis in GMM. S.D., E.M., E.K. and M.Z. performed the HILIC–MS/MS analysis. K.Z. analysed the cross-feeding metabolomics data. M. Kumar performed the motif analysis. M. Klünemann performed the community assembly experiments and Y.K. analysed the 16S data. T.A.S. and F.C. performed the C. elegans experiments and data analysis, D.K. measured drug concentrations. M. Klünemann and K.R.P. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
M. Klünemann, S.A., L.M., M.T., Y.K., P.B., A.T. and K.R.P. are inventors in a patent application based on the findings reported in this study (US patent application number 16966322). S.B., A.M., P.P., S.D., J.V., B.S., T.A.S., E.K., D.K., K.Z., E.M., M. Banzhaf, M.-T.M., F.H., L.N., A.R.B., T.B., V.P., M. Kumar, C.S., M. Beck, J.H., M.Z., D.C.S., F.C. and M.M.S. declare no competing interests.
Additional information
Peer review information Nature thanks Kim Lewis, Michael Shapira and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
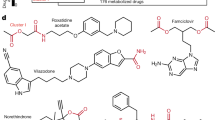
Extended Data Fig. 1 Bacteria and drug selection.
a, Distribution of the selected 25 bacterial strains by their phylogenetic class, and their cumulative metabolic diversity measured as the coverage of annotated enzymes as per the KEGG database64. b, We started with approximately 1,000 annotated drugs from the SIDER side effect database (Kuhn et al. 2016), which were filtered for their gut related side effects. Drug selection was enriched from another database (Saad et al. 2012) for known or suspected interactions with the gut microbiome, before filtered for oral administration and manually curated for overall interest. Final selection was filtered for availability from vendors and establishment of UPLC methods. c, The drugs used in this study span a broad range of structural diversity. Shown is the spread of the selected drugs in a principle coordinate analysis, covering >2,000 drugs from the DrugBank database. Maximum common sub-structure was used to calculate the distances between drug pairs. d, Selected drugs cover several therapeutic classes / indication areas. e, Chemical structures of the 15 drugs used in this study.
Extended Data Fig. 2 Correlation between the screen and validation in higher-volume cultures.
For screen, n≥4 independent replicates (median number of replicates = 17). For validation, n = 3. Error bars = S.E.M. For screening, multiple independent batches were performed as indicated in Supplementary Table 3. Shown R (correlation coefficient) and p-value based on Pearson correlation test.
Extended Data Fig. 3 NMR measurements showing duloxetine depletion by bacterial cells.
a, B. uniformis, b, E. coli ED1A, c, E. coli IAI1, and d, C. saccharolyticum. e, NMR spectrum from C. saccharolyticum cell pellet extract showing that the recovered drug is unmodified duloxetine. Resonances appearing to be out of phase and strong baseline distortions are due to the presence of large solvent signals outside the displayed chemical shift range.
Extended Data Fig. 4 NMR measurements showing unmodified duloxetine recovered from bacterial pellet.
Bacterial cells were incubated with the drug for 4 h in PBS buffer prior to recovery. a, Illustration of the experimental procedure marking the sample collection points. b, NMR spectra of recovered duloxetine from different fractions of E. coli IAI1 and C. saccharolyticum preincubated in PBS. The reference spectrum was scaled to the amount present in the sample to assess the relative amount of free duloxetine present in respective samples. Resonances appearing to be out of phase and strong baseline distortions are due to the presence of large solvent signals outside the displayed chemical shift range.
Extended Data Fig. 5 Duloxetine bioaccumulation by E. coli IAI1 in GMM and recovery from pellet.
a, Procedure and collected samples: S0-S4. b, Recovered duloxetine from different samples (S0-S4) collected as described in a. Different starting duloxetine concentrations, between 0-70 µM, were used. S0 = medium without bacteria (drug only control), S1 = total culture (medium plus bacteria), S2 = supernatant, S3 = wash (pellet was washed with PBS, no drug was found therein supporting intracellular accumulation), S4 = washed pellet. n = 3, error bars = SD, central squares mark the mean. c, MS/MS spectra of duloxetine standard (bottom) and duloxetine detected in a S1 sample (top).
Extended Data Fig. 6 Molecular effects of duloxetine bioaccumulation.
a, Alkynated duloxetine made for the biotin-pull down assay. b, Fold change of proteins detected in the duloxetine pull down assay in C. saccharolyticum lysate using alkynated duloxetine. Four replicates were used in both test and control sets. Significantly enriched (hypergeometric test, FDR corrected p < 0.1, log2(Fc)>2) proteins are shown in red. c, d, Bioaccumulating E. coli strain features larger change in protein abundance in response to drug treatment. Shown are the number of proteins with altered abundance in E. coli ED1A (c, non-bioaccumulating), and E. coli IAI1 (d, bioaccumulating) strains in response to duloxetine exposure at different concentrations. e–h, Comparison of MS/MS spectra of four nucleotide-pathway metabolites from the supernatant of duloxetine-treated C. saccharolyticum with MS/MS spectra of analytical standards. (CE = 10 eV; further details in Methods) Related to Fig. 2d and Supplementary Table 11.
Extended Data Fig. 7 Duloxetine induces a shift in metabolite secretion.
a, Effect of duloxetine treatment on the exo-metabolome of six gut bacterial strains. Shown is the distribution of individual samples over the first two principle components. Principle Component Analysis (PCA) was performed on untargeted FIA–MS data (Methods). The numbers in parentheses of PC1 and PC2 mark the corresponding explained variance for the first and the second principle component, respectively. The dotted block arrow marks the duloxetine induced shift in exo-metabolome of C. saccharolyticum. b, Duloxetine concentration dependent changes in the C. saccharolyticum exo-metabolome. The ion mapping to the deprotonated duloxetine was removed from the PCA analysis shown in a and b. c, The signal for the closest matching ion for deprotonated duloxetine [M-H]- from the exometabolomics data (m/z 296.110079) plotted against initial duloxetine concentration. Data from all six species are pooled together (n = 24 for each initial duloxetine concentration). Overlaid box plots show the interquartile range (IQR), the median value and whiskers extending to include all the values less than 1.5 × IQR away from the 1st or 3rd quartile, respectively. d, Duloxetine signal in the FIA–MS data stratified by species. The signal for the closest matching ion for deprotonated duloxetine [M-H]- from the exometabolomics data (FIA–MS) (m/z 296.110079) plotted against initial duloxetine concentration. Thick transparent line traces medians of replicates (n = 4) at each initial concentration. The dotted lines show linear regression fit.
Extended Data Fig. 8 Duloxetine-induced exo-metabolome changes.
a, Change in C. saccharolyticum exo-metabolome (HILIC-MS data) in response to non-bioaccumulated roflumilast. b, Same as in Fig. 2g, but based on 69 metabolites, whose chemical identity was putatively assigned, and confirmed for 2 metabolites using chemical standards (Supplementary Fig. 3, Supplementary Table 17), using HILIC-MS/MS analysis.
Extended Data Fig. 9 Duloxetine bioaccumulation, community assembly and host response.
a, E. rectale relative abundance in transfer assays based on 16-S amplicon reads. b, E. rectale relative abundance as in a but normalized with respect to equal abundance of each of the five species in the inoculum mixture. Mean values from biological triplicates are shown. c, Duloxetine depletion in community assembly assay. Dashed line indicates mean of control. n = 6 (3 biological replicates, 2 measurements per sample); overlaid box plots show the interquartile range (IQR), the median value and whiskers extending to include all the values less than 1.5 × IQR away from the 1st or 3rd quartile, respectively. d, Metabolic cross-feeding between S. salivarius and E. rectale. Shown are the results of untargeted metabolomics analysis (FIA–MS) of supernatants collected during the growth of S. salivarius in GMM with duloxetine and the subsequent growth of E. rectale in the cell-free conditioned medium. Shown are the profiles of the ions that increased during S. salivarius growth and decreased during E. rectale growth, implying cross-feeding. Ions showing similar pattern in the drug-free solvent (DMSO) control were filtered out. Mean intensities from three biological replicates are shown. e, Dose dependent effects of duloxetine on muscular function in wild type C. elegans animals. Larval stage four (L4) worms were incubated in LB medium in the presence of duloxetine at the indicated concentrations. Each bar represents the mean of six independent experiments, each performed with two technical replicates, ± SD. P values mark difference to the no-drug control, estimated using one-way ANOVA followed by correction for multiple pair-wise comparisons (Tukey’s test). f, Duloxetine concentration in the C. elegans behaviour assay (n = 6; 3 biological replicates, 2 measurements per sample). Overlaid box plots show the interquartile range (IQR), the median value and whiskers extending to include all the values less than 1.5 × IQR away from the 1st or 3rd quartile, respectively
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1–3 and the legends from Supplementary Tables 1–18.
Supplementary Tables 1–7
See main Supplementary Information PDF for table legends.
Supplementary Table 8
See main Supplementary Information PDF for table legend.
Supplementary Tables 9–18
See main Supplementary Information PDF for table legends.
Rights and permissions
About this article

Cite this article
Klünemann, M., Andrejev, S., Blasche, S. et al. Bioaccumulation of therapeutic drugs by human gut bacteria. Nature 597, 533–538 (2021). https://doi.org/10.1038/s41586-021-03891-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03891-8
This article is cited by
-
High-throughput transcriptomics of 409 bacteria–drug pairs reveals drivers of gut microbiota perturbation
Nature Microbiology (2024)
-
The gut ileal mucosal virome is disturbed in patients with Crohn’s disease and exacerbates intestinal inflammation in mice
Nature Communications (2024)
-
High-throughput anaerobic screening for identifying compounds acting against gut bacteria in monocultures or communities
Nature Protocols (2024)
-
Exploring the influence of the microbiome on the pharmacology of anti-asthmatic drugs
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Novel Techniques and Models for Studying the Role of the Gut Microbiota in Drug Metabolism
European Journal of Drug Metabolism and Pharmacokinetics (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
