
Abstract
The coronavirus SARS-CoV-2 is the cause of the ongoing pandemic of COVID-191. Coronaviruses have developed a variety of mechanisms to repress host mRNA translation to allow the translation of viral mRNA, and concomitantly block the cellular innate immune response2,3. Although several different proteins of SARS-CoV-2 have previously been implicated in shutting off host expression4,5,6,7, a comprehensive picture of the effects of SARS-CoV-2 infection on cellular gene expression is lacking. Here we combine RNA sequencing, ribosome profiling and metabolic labelling of newly synthesized RNA to comprehensively define the mechanisms that are used by SARS-CoV-2 to shut off cellular protein synthesis. We show that infection leads to a global reduction in translation, but that viral transcripts are not preferentially translated. Instead, we find that infection leads to the accelerated degradation of cytosolic cellular mRNAs, which facilitates viral takeover of the mRNA pool in infected cells. We reveal that the translation of transcripts that are induced in response to infection (including innate immune genes) is impaired. We demonstrate this impairment is probably mediated by inhibition of nuclear mRNA export, which prevents newly transcribed cellular mRNA from accessing ribosomes. Overall, our results uncover a multipronged strategy that is used by SARS-CoV-2 to take over the translation machinery and to suppress host defences.
Similar content being viewed by others


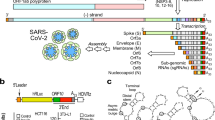
Main
Upon SARS-CoV-2 infection, two overlapping open reading frames (ORFs) are translated from the positive-sense genomic RNA to generate continuous polypeptides that are cleaved into 16 nonstructural proteins (NSP1 to NSP16), which then facilitate the transcription of genomic and subgenomic RNA8. Subgenomic RNAs are translated into structural and accessory proteins8,9. The translation of viral proteins relies on the cellular translation machinery, and coronaviruses have evolved diverse mechanisms—including the degradation of host mRNA and inhibition of host translation2,3—to hijack the translation machinery and to inhibit antiviral defence mechanisms (including the interferon (IFN) response). The extent to which SARS-CoV-2 suppresses the IFN response is a key characteristic that distinguishes it from other respiratory viruses10, and the IFN response seems to have a critical role in the pathogenesis of SARS-CoV-211,12. Although several proteins of SARS-CoV-2 have previously been implicated in shutting off host expression4,5,6,7, a comprehensive depiction of the effect of SARS-CoV-2 infection on cellular gene expression and the underlying molecular mechanism is lacking.
To gain a detailed view of the changes that occur in translation over the course of SARS-CoV-2 infection, we infected a human lung epithelial cell line (Calu3 cells) with SARS-CoV-2 at multiplicity of infection of 3, which resulted in infection of the majority of the cells (Extended Data Fig. 1a) and thus a synchronous cell population. At 3, 5 and 8 h after infection, we collected infected cells as well as uninfected cells for RNA sequencing (RNA-seq) and ribosome profiling (Fig. 1a). To assess the reproducibility of our experiments, we prepared two independent biological replicates for the uninfected cells and cells collected at 8 h after infection: both the mRNA and footprint measurements were reproducible (Extended Data Fig. 1b, c). Footprint read-length distribution peaked at around 29 nt, consistent with previous analyses9,13 (Extended Data Fig. 1d). Metagene analysis revealed the expected profiles of footprints and mRNAs (Extended Data Fig. 1e-h). Using this data, we quantitatively assessed the expression pattern of 8,627 cellular transcripts and 12 viral ORFs that are expressed from the genomic and subgenomic RNAs along the course of SARS-CoV-2 infection. Our analysis of the mRNAs and footprints that originate from cellular and viral transcripts illustrates the dominance of SARS-CoV-2 over the mRNA pool. At 8 h after infection, viral mRNAs comprise almost 80% of the mRNAs in infected cells (Fig. 1b). At the same time point, viral mRNAs account for only about 34% of the RNA fragments engaged with ribosomes in the cells (Fig. 1c). To quantitatively evaluate the ability of SARS-CoV-2 to co-opt host ribosomes, we calculated the translation efficiency (the ratio of footprints to mRNAs for a given gene) of viral and cellular RNA over the course of infection. We then compared the translation efficiency of human genes to that of viral genes at each of the time points along infection. At 3 h after infection, the translation efficiencies of viral genes fall within the general range of cellular gene translation (Fig. 1d). This indicates that, when infection initiates, viral transcripts are translated with efficiencies similar to those of host transcripts. As infection progresses, the translation efficiency of viral genes is substantially reduced relative to that of cellular genes. Because double-membrane replication compartments are formed to accommodate viral genome replication and transcription14, one possibility is that, as infection proceeds, these compartments encompass a sizable fraction of the viral RNA molecules and thus prevent them from being translated.

a, Cells were left uninfected or infected with SARS-CoV-2 for 3, 5 or 8 h, and then collected for RNA-seq and ribosome profiling. b, c, Percentage of reads that aligned to human or viral coding regions for mRNAs (b) and footprints (c) in uninfected cells (un) and at 3, 5 and 8 h after infection (hpi, hours post-infection). Uninfected and 8 h after infection, n = 2; 3 and 5 h after infection, n = 1. Duplicate values for 8 h after infection are presented. d, Cumulative frequency of human (black) and viral (colours) genes, according to their relative translation efficiency (TE) at 3 h (left), 5 h (middle) and 8 h (right) after infection. e, Protein synthesis measurement by flow cytometry of uninfected cells or cells infected with SARS-CoV-2 (multiplicity of infection (MOI) = 3) for 3, 5 and 8 h after infection, following incorporation of O-propargyl puromycin. OPP fl., O-propargyl puromycin fluorescence. f, Percentage of reads that aligned to the human or viral transcripts as in b, but from total aligned RNA reads. g, Heat map of relative mRNA and footprint levels during SARS-CoV-2 infection of human transcripts that showed the strongest changes in their mRNA levels. Expression levels scaled by gene after partitioning clustering are shown.
Deep sequencing measurements inherently provide relative values, but not absolute quantification of RNA and translation levels. Because the SARS-CoV-2-encoded protein NSP1 has recently been shown to interfere with translation by blocking the mRNA entry channel of ribosomes6,7, and because the extent to which SARS-CoV-2 interferes with the overall levels of cellular mRNA has not been assessed, we next examined whether SARS-CoV-2 infection affects global translation and RNA levels. To quantify absolute translation levels, we measured nascent protein synthesis levels by quantifying the incorporation of O-propargyl puromycin into elongating polypeptides15 (Extended Data Fig. 2a, b). We infected Calu3 cells with SARS-CoV-2 and measured protein synthesis in uninfected cells, and at 3, 5 and 8 h after infection. We observed a major reduction in global translation levels at 3 h after infection, which was augmented over time: at 8 h after infection, translation activity was reduced by 70% (Fig. 1e). In parallel, we measured the levels of rRNA and of total RNA extracted from uninfected cells and along the course of infection, which showed there are no major changes in total RNA or in rRNA levels during SARS-CoV-2 infection (Extended Data Fig. 3a, b). Because mRNA comprises only a small fraction of the cellular RNA, we assessed the relative abundance of cellular and viral mRNAs in uninfected and infected cells by sequencing total RNA (without rRNA depletion). Our analysis reveals an increase in the pool of mRNAs during infection, owing to the massive production of viral transcripts; however, the relative fraction of cellular mRNA is reduced by approximately twofold (Fig. 1f). This suggests that during infection there is a massive production of viral transcripts and a concomitant substantial reduction in the levels of cellular transcripts. We next assessed the expression pattern of cellular genes along the course of SARS-CoV-2 infection. We clustered the mRNA levels of genes that showed the strongest changes over the course of infection, which enabled us to group cellular transcripts into four main classes on the basis of temporal expression profiles from RNA-seq. Expression of the majority of transcripts was reduced during infection (with different kinetics), accompanied by a concurrent reduction in footprints, but there were also numerous transcripts that showed increased expression (Fig. 1g). The clustering of genes that showed elevated expression along the course of infection revealed mRNAs that were upregulated early or late, or which showed transient induction (Extended Data Fig. 4a). The cluster of genes that was induced in late kinetics was significantly enriched with genes related to the immune response, including Toll receptor signalling, and chemokine and cytokine signalling (Extended Data Fig. 4b, Supplementary Table 1). These genes include IL6 and IL8 (also known as CXCL8), which have an important role in the pathogenesis of SARS-CoV-216, as well as several IFN-stimulated genes (such as IFIT1, IFIT2, IFIT3 and ISG15) and genes encoding TNF-induced proteins.
Our results indicate that levels of the majority of cellular RNAs are reduced during SARS-CoV-2 infection and that this reduction probably contributes to the shutting off of cellular protein synthesis. The reduction in cellular RNA levels could be due to interference with RNA production and/or accelerated RNA degradation. To explore the molecular mechanism, we examined whether the reduction of cellular transcript levels is associated with the subcellular localization of these transcripts. We found that the levels of transcripts that mostly localize to the cytoplasm are reduced more in infected cells, as compared to transcripts that are mostly nuclear (Fig. 2a, Extended Data Fig. 4c), and there was a clear correlation between subcellular localization and the extent of reduction in transcript levels (Extended Data Fig. 4d). Furthermore, mitochondrially encoded transcripts are much less affected by infection as compared to nuclear-encoded transcripts (Fig. 2b). The specific sensitivity of cytosolic transcripts indicates these transcripts may be targeted for degradation during SARS-CoV-2 infection. To directly evaluate mRNA decay, we used thiol (SH)-linked alkylation for the metabolic sequencing of RNA (SLAM-seq)17 on uninfected and SARS-CoV-2-infected cells (Extended Data Fig. 5a). This approach allowed us to measure the half-lives of endogenous mRNAs on the basis of the incorporation of 4-thiouridine (4sU) into newly synthesized RNA, the conversion of 4sU into a cytosine analogue and the quantification of these U-to-C conversions by RNA-seq17,18. We obtained all of the characteristics of a high-quality SLAM-seq library: over 3,000 quantified genes, rising U-to-C mutation rates and an increase in the portion of labelled RNA over time, which was stronger in infected cells (indicating a faster turnover of RNA in infected cells) (Extended Data Fig. 5b–e). There was a strong correlation between the half-lives estimated from our measurements and estimates that were previously conducted19 in a different cell type (Extended Data Fig. 5f). Importantly, we observed a substantial reduction in the half-lives of cellular mRNAs upon SARS-CoV-2 infection (Fig. 2c), which indicates increased degradation of cellular mRNA in infected cells. Furthermore, the reduction in half-lives correlated with the reduction that we measured in RNA expression, which indicates that mRNA decay dominates changes in mRNA during infection (Extended Data Fig. 5g). Consistent with the changes in mRNA expression, the half-lives of cytoplasmic transcripts were more reduced than those of nuclear transcripts (Fig. 2d, Extended Data Fig. 5h). In coronaviruses, the most prominent and well-characterized protein linked to cellular shutoff is NSP120. Previous studies on NSP1 of SARS-CoV-2 have demonstrated that it restricts translation by directly binding to the ribosome 40S subunit6,7, thereby globally inhibiting the initiation of translation. For SARS-CoV, in addition to this translation effect, the interaction of NSP1 with 40S has been shown to induce cleavage of translated cellular mRNAs, thereby accelerating their turnover21,22,23. We therefore examined whether the degradation of cellular transcripts in SARS-CoV-2-infected cells is related to their translation. We observed a weak, but significant, correlation between the translation efficiency of cellular genes and the reduction in their mRNA half-life after infection (Extended Data Fig. 5i), which indicates that accelerated turnover of cellular transcripts in infected cells may be related to their translation. To assess the role of NSP1 in RNA degradation, we analysed RNA-seq data from cells transfected with NSP124, which revealed that ectopic NSP1 expression leads to weaker but similar signatures to those that we identified in infected cells: a stronger reduction of cytosolic transcripts compared to nuclear transcripts, and a stronger sensitivity of nuclear-encoded transcripts (Extended Data Fig. 6a, b).

a, log2-transformed fold change in RNA level of cellular RNAs at 5 h after infection, relative to uninfected cells. RNAs were grouped into ten bins on the basis of their cytosol-to-nucleus localization ratio. P values calculated using two-sided t-test comparing the first and last bins. n = 5,650 genes. b, log2-transformed fold change in RNA levels of nuclear-encoded (n = 6,757) or mitochondrially encoded transcripts (n = 7) over the course of infection, relative to uninfected cells. p-values calculated using two-sided Wilcoxon tests. c, Scatter plot of mRNA half-life in infected cells relative to uninfected cells, as calculated from SLAM-seq. d, Scatter plot depicting log2-transformed fold changes in transcript half-life between infected and uninfected cells, relative to the log2-transformed cytosol/nuclear ratio of cellular transcripts in uninfected cells. Pearson’s R and two-sided P value are shown. e, Aligned RNA reads at the end of IL32 from uninfected cells and at 8 h after infection. Rep., replicate. f, Box plots of ratio of intronic to exonic reads for genes (n = 9,123) over the course of infection, relative to uninfected cells. P values calculated using two-sided t-test. g, Percentage of reads that align to exonic or intronic regions relative to rRNA abundance, over the course of infection. h, Scatter plot of log2-transformed fold change in the ratio of intronic to exonic reads of cellular genes between 7 h after infection and uninfected cells, relative to log2-transformed fold changes in transcript half-life in infected cells relative to uninfected cells. Pearson’s R and two-sided P value are shown. Box plots show median, first to third quartile, 1.5× interquartile range and outliers.
We noticed that SARS-CoV-2 infection also leads to increased levels of intronic reads in many cellular transcripts (Figs. 2e, f), which indicates that SARS-CoV-2 may interfere with cellular mRNA splicing (as has recently been suggested5). However, considerable degradation of mature cytosolic mRNAs may also generate a relative increase in intronic reads. We therefore analysed the ratio of intronic and exonic reads to rRNA, which we found to be unperturbed in infection (Extended Data Fig. 3b). Whereas exonic reads showed a marked reduction along the course of SARS-CoV-2 infection (relative to rRNA levels), the intronic read levels showed only a subtle change (Fig. 2g). Additionally, in our SLAM-seq measurements, we did not detect major changes in the turnover of intronic RNA in infected cells (Extended Data Fig. 7a). Furthermore, we observed a correlation between the reduction in transcript half-lives and the relative increase in intronic reads (Fig. 2h). Likewise, the increase in the ratio of intronic to exonic reads was greater in genes with expression that was reduced along infection, as compared to genes with expression that was induced (Extended Data Fig. 7b). These results imply that the increase in intronic reads compared to exonic reads during SARS-CoV-2 infection is driven mostly by the accelerated degradation of mature cellular transcripts, which leads to a relative reduction in exonic reads. Overall, these findings demonstrate that SARS-CoV-2 infection leads to accelerated degradation of mature cytosolic cellular mRNAs.
An important aspect of host shutoff is the ability of the virus to hamper the translation of cellular transcripts, while recruiting the ribosome to its own transcripts. It has previously been suggested that SARS-CoV-2 mRNAs are refractory to the translation inhibition induced by NSP15,25. However, our measurements indicate that RNA degradation—which is probably mediated by NSP1—has a prominent role in remodelling the mRNA pool in infected cells, and that SARS-CoV-2 dominates the mRNA pool. All of the subgenomic RNAs that are encoded by SARS-CoV-2 contain a common 5′ leader that is added during negative-strand synthesis. We therefore explored whether the genomic 5′ untranslated region (UTR) or the common 5′ leader protect viral mRNAs from NSP1-induced degradation. We fused the viral 5′ leader, the genomic 5′ UTR or a control host 5′ UTR to a GFP reporter (Extended Data Fig. 8a) and transfected these constructs together with vectors encoding NSP1 or one of two controls (NSP2 or mCherry) into 293T cells (Extended Data Fig. 8b). We found that NSP1 expression suppresses the production of the control GFP but not of the GFP containing the viral 5′ UTR or 5′ leader (Fig. 3a, b, Extended Data Fig. 8c–e). We extracted RNA from these cells and observed that the NSP1-induced reduction in the level of control GFP was associated with an approximately 12-fold reduction in the GFP mRNA levels, whereas the levels of GFP mRNA fused to the viral 5′ UTR or to the 5′ leader were only mildly reduced by NSP1 expression (Fig. 3c, Extended Data Fig. 8f). The GFP reporter plasmid we used also contains an mCherry reporter that is expressed from an independent promoter. NSP1 also induces a reduction in mCherry protein and RNA levels when compared to NSP2 (Extended Data Fig. 8g–i). These results indicate that the 5′ leader of viral RNAs provides protection from NSP1-induced degradation and that this protection contributes to the ability of the virus to dominate the mRNA pool in infected cells.

a, b, Microscopy images (a) and flow cytometry analysis (b) of 293T cells co-transfected with mCherry or NSP1 together with a GFP reporter that includes control 5′ UTR, SARS-CoV-2 5′ UTR or SARS-CoV-2 5′ leader. Representative of four independent replicates are shown. Scale bars, 100 μm. c, Relative GFP RNA levels for the various GFP reporters in cells expressing NSP1 or mCherry, as measured by quantitative real-time PCR. Points show measurement of biological replicates.
The results discussed so far exemplify how SARS-CoV-2 remodels the transcript pool in infected cells. To quantitatively evaluate the role of translational control during SARS-CoV-2 infection, we calculated the translation efficiency of cellular genes along the course of infection. We then clustered the genes that showed the strongest reduction or increase in their relative translation efficiency over the course of infection, which generated four clusters that largely reflect either increased or decreased relative translation efficiency during infection. The mRNA and footprint temporal profiles of these genes revealed a clear signature; the genes with a relative translation efficiency that decreased over the course of infection were genes with mRNA levels that increased during infection without a corresponding increase in footprints (Fig. 4a, Extended Data Fig. 9a). These clusters were enriched in immune-response genes (false-discovery rate < 10−4) such as IRF1, IL6 and CXCL3. Comparing changes in mRNA and translation efficiency levels of cellular genes along the course of infection demonstrates that, generally, transcripts that are transcriptionally induced after infection show a reduction in their relative translation efficiency, and vice versa (Fig. 4a, b). These data indicate that newly generated transcripts are less likely to engage with ribosomes. One molecular mechanism that could explain these measurements is inhibition of nuclear mRNA export. To test whether SARS-CoV-2 interferes with nuclear mRNA export, we left Calu3 cells uninfected or infected them with SARS-CoV-2 and assessed the subcellular localization of polyadenylated transcripts by cytoplasmic/nuclear fractionation followed by RNA-seq. We obtained a strong correlation between our cytoplasmic/nuclear fractionation measurements and measurements that were previously conducted19 (Extended Data Fig. 9b). Infection led to a relative nuclear enrichment of most cellular transcripts (Fig. 4c). Furthermore, genes with relative translation efficiency that was reduced in infection showed a stronger nuclear enrichment, which suggests that the inability of induced transcripts to reach the ribosome may be explained by nuclear retention. Because there is considerable degradation of cytosolic mRNA in infected cells, the relative nuclear enrichment may be expected even with no interference in the export of nuclear mRNA. To more accurately assess whether this nuclear enrichment is also related to the inhibition of nuclear export, we used whole-cell extract samples to normalize the cytoplasmic/nuclear ratios, which enabled us to obtain absolute RNA localization values for each compartment26. We observed that transcripts that are transcriptionally induced show a significant increase specifically in the nuclear fraction in infected cells (Fig. 4d), which indicates that SARS-CoV-2 infection disrupts their nucleocytoplasmic export. Because cytokine and IFN-induced genes are induced upon infection, this inability of nascent transcripts to exit the nucleus and reach the ribosomes may explain why infected cells do not launch a robust antiviral response11.

a, Heat map of relative translation efficiency, mRNA and footprints of the human genes that showed the strongest changes in their relative translation efficiency over the course of infection. Relative expression ratios, after partitioning clustering on the basis of changes in relative translation efficiency, are shown. Several immune-related genes are labelled. CXCL1/3, CXCL1 and CXCL3; NFKBIA/E, NFKBIA and NFKBIE; TNFAIP2/3, TNFAIP2 and TNFAIP3. b, Scatter plot of cellular transcript levels in uninfected cells and at 8 h after infection. Genes are coloured on the basis of the relative change in their translation efficiency between cells 8 h after infection and uninfected cells. Central cytokines and IFN-stimulated genes are labelled. RPKM, reads per kilobase of transcript per million mapped reads. c, Scatter plot of the cytosolic/nuclear ratio in infected (7 h after infection, MOI = 3) and uninfected cells. Genes that showed a reduced translation efficiency in a are labelled in purple. d, Effects of infection on the normalized abundance of cytosolic and nuclear RNA. Transcripts are divided according to their induction kinetics (Extended Data Fig. 4a): induced late (n = 91), induced early (n = 28), induced mix (n = 56) and all the rest (n = 7,283). Coloured triangles represent the direction of the change between infected and uninfected samples. P values were calculated using a linear regression model. Box plots show median, first to third quartile, 1.5× interquartile range and outliers. e, Scheme of SARS-CoV-2 RNAs and ORFs. f, Relative translation efficiency of viral ORFs along infection. Genes are divided into three groups (‘beginning’, ‘middle’ and ‘end’) on the basis of their physical location along the genome. Mean at 8 h after infection (n = 2), and values at 3 and 5 h after infection (n = 1), are shown. g, Model of SARS-CoV-2 suppression of host gene expression: (1) global translation reduction; (2) degradation of cytosolic cellular mRNAs; and (3) translation inhibition of induced cellular mRNAs (probably through inhibition of the export of nuclear mRNA).
Finally, we used our measurements to examine viral translation dynamics along the course of infection. Viral ORFs are translated from the genomic RNA or from a nested series of subgenomic RNAs that contain a common 5′ leader (Fig. 4e). We examined how the translation of viral genes is distributed between viral transcripts at different times after infection. This analysis revealed that the relative translation efficiency of ORFs that are located at the 5′ end of the genome increases along infection. By contrast, the relative translation efficiency of ORFs that are encoded towards the 3′ end of the genome decreases, and ORFs located in the middle of the genome show no major changes in their relative translation efficiency (P = 0.002 for differences in the slope of the translation efficiency changes using a linear mixed model) (Fig. 4f). We used published RNA-seq and ribosome profiling data13 to perform an analysis of the expression and translation of ORFs of mouse hepatitis virus strain A59 along the course of infection, which revealed a trend that was weaker than—but paralleled—that of our SARS-CoV-2 results (Extended Data Fig. 10a, b). Because subgenomic RNAs share the same 5′ UTR, one possible explanation for these potential differences in translation is an unappreciated role for the 3′ UTR or for viral transcript length, which varies greatly between viral transcripts (Fig. 4e).
Most SARS-CoV-2 ORFs are 3′ proximal and translated from dedicated subgenomic mRNAs (Fig. 4e). However, several subgenomic RNAs encode additional out-of-frame ORFs (including ORF2b, ORF3c, ORF7b and ORF9b27), probably via a leaky scanning mechanism. The translation of these out-of-frame ORFs correlated with the expression of the 3′-proximal main ORF, which indicates that there are no major changes in the efficiency of ribosome scanning of viral transcripts over the course of infection (Extended Data Fig. 10c).
Using unbiased measurements of translation and RNA expression along the course of SARS-CoV-2 infection, we identified three major routes by which SARS-CoV-2 interferes with cellular gene expression in infected cells: (1) global inhibition of protein translation, (2) degradation of cytosolic cellular transcripts and (3) specific translation inhibition of newly transcribed mRNAs, which is probably explained by inhibition of nuclear mRNA export. The disruption of cellular protein production using these three components represents a multipronged mechanism that synergistically acts to suppress the host antiviral response (Fig. 4g). These mechanisms may explain the molecular basis of the potent suppression of IFN response that has been observed in animal models and in patients with severe COVID-1910,12.
We reveal that, similar to what has previously been described for NSP1 of SARS-CoV, NSP1 of SARS-CoV-2 shuts down host protein translation by two mechanisms: first, it stalls mRNA translation (as has previously been reported5,6,7), which leads to a general reduction in the translation capacity of infected cells. Second, NSP1 leads to accelerated degradation of cellular mRNA. NSP1 of SARS-CoV induces endonucleolytic cleavage and the subsequent degradation of host mRNAs, and this activity depends on the binding of NSP1 to the 40S ribosome subunit2. Our results are consistent with a similar mechanism operating in SARS-CoV-2-infected cells, as we show that cytosolic RNAs are more susceptible to SARS-CoV-2-mediated degradation. Several previous studies have shown that mRNAs with the viral 5′ leader are translated more efficiently in the presence of NSP15,25, but a further study has previously demonstrated that NSP1 inhibits the translation of reporter mRNA containing both cellular and viral 5′ UTRs6, which implies that viral mRNAs may not simply evade translation inhibition in the context of the 5′ UTR sequence. Our results support a model in which NSP1 acts as a strong inhibitor of translation and at the same time leads to the accelerated degradation of cellular, but not of viral, mRNAs—thus providing the means for viral mRNA to quickly dominate the mRNA pool. This accumulation of SARS-CoV-2 mRNAs may explain how infected cells divert their translation towards viral mRNAs. Overall, our study provides an in-depth depiction of how SARS-CoV-2 efficiently interferes with cellular gene expression, leading to the shutdown of host protein production through a multipronged strategy.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Cells and viruses
Calu3 cells (ATCC HTB-55) were cultured in 6-well or 10-cm plates with RPMI supplemented with 10% fetal bovine serum (FBS), MEM non-essential amino acids, 2 mM l-glutamine, 100 units per ml penicillin and 1% Na-pyruvate. Monolayers were washed once with RPMI without FBS and infected with SARS-CoV-2 virus, at an MOI of 3, in the presence of 20 μg per ml TPCK trypsin (Thermo scientific). Plates were incubated for 1 h at 37 °C to allow viral adsorption. Then, RPMI medium supplemented with 2% FBS, was added to each well. SARS-CoV-2 BavPat1/2020 Ref-SKU: 026V-03883 was provided by C. Drosten. It was propagated (5 passages) and titred on Vero E6 cells and then sequenced9 before it was used. Handling and working with SARS-CoV-2 virus was conducted in a BSL3 facility in accordance with the biosafety guidelines of the Israel Institute for Biological, Chemical and Environmental sciences. The Institutional Biosafety Committee of Weizmann Institute approved the protocol used in these studies. Calu3 and 293T cells were authenticated by ATCC using STR profiling. All cell lines tested negative for mycoplasma.
Preparation of ribosome profiling and RNA-seq samples
SARS-CoV-2-infected cells were collected at 3, 5 and 8 h post infection (hpi): two independent biological replicates were done for the 8-hpi time point and for uninfected cells that were collected in parallel at 3 and 5 hpi. For RNA-seq, cells were washed with PBS and then collected with Tri-Reagent (Sigma-Aldrich), total RNA was extracted and poly-A selection was performed using Dynabeads mRNA DIRECT Purification Kit (Invitrogen). The mRNA sample was subjected to DNaseI treatment and 3′ dephosphorylation using FastAP Thermosensitive Alkaline Phosphatase (Thermo Scientific) and T4 PNK (NEB) followed by 3′ adaptor ligation using T4 ligase (NEB). The ligated products were used for reverse transcription with SSIII (Invitrogen) for first-strand cDNA synthesis. The cDNA products were 3′-ligated with a second adaptor using T4 ligase and amplified for 8 cycles in a PCR for final library products of 200–300 bp. For ribosome profiling libraries, cells were treated with 100 μg ml−1 cycloheximide for 1 min. Cells were then placed on ice, washed twice with PBS containing 100 μg ml−1 cycloheximide, scraped from 10-cm plates, pelleted and lysed with lysis buffer (1% triton in 20 mM Tris 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol supplemented with 10 U ml−1 Turbo DNase and 100 μg ml−1 cycloheximide). After lysis, samples stood on ice for 2 h and subsequent ribosome profiling library generation was performed, as previously described28. In brief, cell lysate was treated with RNaseI for 45 min at room temperature followed by SUPERase-In quenching. Sample was loaded on sucrose solution (34% sucrose, 20 mM Tris 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol and 100 μg ml−1 cycloheximide) and spun for 1 h at 100,000 rpm using TLA-110 rotor (Beckman) at 4 °C. The pellet was collected using TRI reagent and the RNA was collected using chloroform phase separation. For size selection, 15 μg of total RNA was loaded into 15% TBE-UREA gel for 65 min, and 28–34-nt footprints were excised using 28-nt and 34-nt flanking RNA oligonuclotides, followed by RNA extraction and ribosome profiling library construction as previously described28.
Sequence alignment and metagene analysis
Sequencing reads were aligned as previously described9. In brief, linker (CTGTAGGCACCATCAAT) and poly-A sequences were removed and the remaining reads were aligned to the hg19 and to the SARS-CoV-2 genome (Genebank NC_045512.2) with 3 changes to match the used strain (BetaCoV/Germany/BavPat1/2020 EPI_ISL_406862), (241:C>T, 3037:C>T and 23403:A>G). Alignment was performed using Bowtie v.1.1.229 with maximum two mismatches per read. Reads that were not aligned to the genome were aligned to the transcriptome (using the known canonical isoform UCSC gene annotations) and to SARS-CoV-2 junctions that were recently annotated30. The aligned position on the genome was determined as the 5′ position of RNA-seq reads, and for ribosome profiling reads the p-site of the ribosome was calculated according to read length using the offset from the 5′ end of the reads that was calculated from canonical cellular ORFs. The offsets used were +12 for reads that were 28 or 29 bp and +13 for reads that were 30–33 bp. Footprint reads that were of other lengths were discarded. In all figures presenting ribosome density data, only footprint lengths of 28–33 nt are presented.
For the metagene analysis, only genes with CDS length of at least 300 nucleotides, UTRs of at least 50 nucleotides and more than 50 reads in the analysed window around the start or the stop codon were used. For each gene, reads were normalized to the sum of reads in the analysed window and then averaged.
Gene filtering, quantification and RPKM normalization
For cellular gene quantification, genes were filtered according to the number of reads as follows. The number of reads aligned to the CDS of each gene in each replicate, from at least one of the extreme conditions (uninfected or 8 hpi) had to be greater than 50 reads for the mRNA libraries and greater than 25 for the footprint libraries. In addition, genes with zero reads in any of the samples (mRNA or footprint of any of the time points) were excluded. Histone genes (which are not polyadenylated) were excluded from the analysis. RNA-seq read coverage on CDS was normalized to units of RPKM to normalize for CDS length and for sequencing depth. For analysis in which host and viral gene expression were compared (Fig. 1b–d, f), RPKM was calculated on the basis of the total number of uniquely aligned reads to the coding regions of both the host and the virus. For analysis that was focused on cellular gene expression, for RNA expression, the RPKM values were calculated on the basis of the total number of uniquely aligned reads to the cellular coding regions. RPKM values were further scaled according to the ratio of the aligned host mRNA reads to the total aligned reads, including viral, rRNA and tRNA reads in the total RNA sequencing (without polyA selection), to keep the total reads equal across samples. For footprint libraries, read coverage of cellular and viral genes was normalized to units of RPKM normalizing to the total CDS aligned ribosome profiling reads, including both viral and host reads.
Because the viral RNAs are widely overlapping, RNA-seq RPKM levels of viral genes were computed with deconvolution as was previously described for mouse hepatitis virus (MHV)13. First, values for each gene were calculated by subtracting the RPKM of an ORF from the RPKM of the ORF located just upstream of it in the genome. Then, for subgenomic RNAs, leader–body junctions were quantified on the basis of the number of uniquely mapped reads that span each canonical junction using STAR 2.5.3a aligner31. Finally, on the basis of the correlation between the deconvoluted RPKM and junction abundance of the subgenomic RNAs, the RPKM levels of all viral RNAs (including the genomic RNA) were estimated. Viral and host gene translation efficiency was calculated as the ratio between footprint RPKM and RNA RPKM. To compare viral translation efficiency along infection, the translation efficiency of viral genes was further normalized by dividing the translation efficiency of each viral gene by the sum of the viral gene translation efficiency in each sample and multiplying by 100. For comparing the relative translation levels of canonical ORFs and overlapping viral ORFs decoded from the same subgenomic RNA, we used ORF-RATER32. To estimate the error in our expression measurements of out-of-frame ORFs, for each of these ORFs, we defined 500 random partial ORF regions, which range in length between 50% and 100% of the original ORF, and used ORF-RATER to quantify the expression from these regions while keeping all other ORFs unmodified. On the basis of these values, we have added a standard deviation for the ORF expression measurement. Previous data13 were analysed for calculating changes in relative viral gene translation efficiency. Owing to differences in the per cent of aligned viral reads between the replicates, we analysed only replicate 1, which showed the expected gradual increase in viral mRNA and footprint reads along infection.
Clustering and heat maps
We performed clustering on 2,000 cellular genes that showed the strongest change on the basis of the fold change in RNA-seq expression levels between 8 hpi and uninfected samples (both averaged across duplicates). For clustering of upregulated genes, cellular genes that showed at least 1.5-fold increase in expression levels between 8 hpi and uninfected samples were used. RNA-seq and footprint measurements for each of these genes were scaled so that the minimum level across samples is 0 and the maximum is 100. Hierarchical clustering of these normalized values was performed using ward.D2 method on Pearson correlations between scaled RNA-seq measurements using the means of the uninfected and 8-hpi samples.
For presenting changes in relative translation efficiency, we performed clustering on the 35 most-increased and 35 most-decreased genes on the basis of the fold change in relative translation efficiency between 8 hpi and uninfected samples (averaged across duplicates, fold change > 1.7 or fold change < −2.4). Translation efficiency, RNA-seq and footprint measurements for these genes were scaled so that the average level for each gene across samples is one, using mean expression from uninfected and 8-hpi duplicates. Hierarchical clustering of genes was performed using ward.D2 method on Pearson correlations between scaled relative translation efficiency measurements. With this clustering, we obtained four homogenous groups of genes, each one of which showed a clear different pattern of behaviour with time.
Quantification of intronic reads
Read density for introns was calculated as described for exons, with intron annotations based on the known canonical isoform UCSC gene annotations. To avoid biases from intron read count, genes without introns or genes for which one of the introns overlaps with an exon of another gene were excluded. In addition, genes with low number of reads (<20 on the exons and <2 on the introns) were omitted. The number of reads on exons and introns was normalized by the total length of the exons and introns, respectively, to get a quantification proportional to the number of molecules. Finally, the normalized number of reads on introns was calculated as percentage of the normalized number of reads on exons. Statistical significance (Fig. 2f) was tested using a paired t-test on the log values of the percentage (with offset of 0.1 to overcome zero values).
Protein synthesis measurement using OPP
The OPP assay (OPP, Thermo Fisher Scientific) was carried out following the manufacturer’s instructions. In brief, cells were collected following treatment with 10 μM OPP for 30 min at 37 °C. The cells were then fixed for 15 min in 3.7% formaldehyde, and permeabilized in 0.1% Triton X-100 for 15 min. OPP was then fluorescently labelled by a 30-min incubation in Click-iT Plus OPP reaction cocktail with Alexa Fluor594 picolyl azide (Thermo Fisher Scientific). Cells were analysed using BD LSRII flow cytometer. The decrease in translation levels was calculated according to the median Alexa 594 fluorescence intensity between the 8 hpi and uninfected samples.
Pathway enrichment analysis
Enrichment analysis of cellular pathways in specific gene clusters (Fig. 4a, Extended Data Fig. 4a) was done with PANTHER version 15.0, with default settings and the PANTHER pathways dataset33,34.
Fractionation assay
Uninfected or SARS-CoV-2-infected (MOI = 3) Calu3 cells at 7 hpi were washed in PBS, trypsinized and resuspended in cold PBS. A fraction of 10% of the cells was then transferred to a new tube and RNA was extracted in Tri-reagent to obtain whole cellular extract. Remaining cells were pelleted for 5 min at 300g. Cells were resuspended in 150 μl fractionation buffer A (15 mM Tris-Cl pH 8, 15 mM NaCl, 60 mM KCl, 1 mM EDTA pH 8, 0.5 mM EGTA pH 8, 0.5 mM spermidine, and 10 U ml−1 RNase inhibitor), and 150 μl 2× lysis buffer (15 mM Tris-Cl pH 8, 15 mM NaCl, 60 mM KCl, 1 mM EDTA pH 8, 0.5 mM EGTA pH 8, 0.5 mM spermidine, 10 U ml−1 RNase inhibitor and 0.5% NP-40) was added followed by 10 min incubation on ice. The extract was pelleted for 5 min at 400g and the supernatant containing the cytoplasmic fraction was removed to a new tube. This was centrifuged again at 500g for 1 min, the supernatant was transferred to a new tube and RNA was extracted with Tri-reagent. The nuclear pellet was resuspended in 1 ml RLN buffer (50 mM Tris-Cl pH 8, 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 10 mM EDTA and 10 U ml−1 RNase inhibitor) and incubated on ice for 5 min. The nuclear fraction was then pelleted for 5 min at 500g, the supernatant was removed and RNA was extracted from the pellet with Tri-reagent. RNA-seq libraries were then prepared from all three fractions as described in ‘Preparation of ribosome profiling and RNA-seq samples’.
Fractionation assay analysis
RNA-seq reads from total, nuclear and cytosolic fractions were aligned to the human and viral reference as described in ‘Sequence alignment and metagene analysis’. Human gene read counts were adjusted to RPKM as described in ‘Gene filtering, quantification and RPKM normalization’, and then converted to transcripts per million (TPM) by normalizing to the sum of RPKM in each sample, so that the expression levels in each sample sum up to the same value.
A list of 3,884 average-expressed genes was defined. These genes were genes with 25 or more reads across all samples and with a sum of TPM values in the total RNA samples across replicates within quantiles 0.4 and 0.9. On the basis of this list, for each replicate, a linear regression model was calculated of the total fraction as a linear combination of the cytosolic and the nuclear fractions. The regression coefficients were used to normalize the cytosolic and nuclear TPM values to obtain absolute localization values26. To correct for changes in total mRNA levels, the absolute values were further scaled by a factor calculated from total RNA-seq as described in ‘Gene filtering, quantification and RPKM normalization’. To statistically compare the effect of infection on nuclear–cytosolic distribution of mRNAs from different clusters, P values were calculated from the interaction term in a linear model.
RNA labelling for SLAM-seq
For metabolic RNA labelling, growth medium of infected Calu3 cells (MOI = 3) at 3 hpi or uninfected cells was replaced with medium containing 4sU (T4509, Sigma) at a final concentration of 200 μM (a concentration that did not induce substantial cell cytotoxicity at 4 h labelling). Cells were collected with Tri-reagent at 1, 2, 3 and 4 h after medium replacement (corresponding to 4, 5, 6 and 7 hpi for infected cells). RNA was extracted under reducing conditions and treated with iodoacetamide (A3221, Sigma) as previously described17. RNA-seq libraries were prepared and sequenced as described in ‘Preparation of ribosome profiling and RNA-seq samples’and paired-end reads were sequenced with 51 cycles for each end.
SLAM-seq data analysis and half-life calculation
Alignment of SLAM-seq reads was performed using STAR31, with parameters that were previously described9. First, reads were aligned to a reference containing human rRNA and tRNA, and all reads that were successfully aligned were filtered out. The remaining reads were aligned to a reference of the human and the virus as described in ‘Sequence alignment and metagene analysis’. Reads mapped to the virus were discarded and reads mapped to the human were used in the next steps. Output .bam files from STAR were used as input for the GRAND-SLAM analysis18 with default parameters and with trimming of 5 nucleotides in the 5′ and 3′ends of each read. Infected and uninfected samples were analysed in separate runs. Each one of the runs also included an unlabelled sample (no 4sU) that was used for estimating the linear model of the background mutations. The output of GRAND-SLAM is the estimated ratio of newly synthesized out of total molecules for each gene (new to total ratio (NTR)). The old transcript fraction for each gene in each sample is 1 – NTR; this number reflects the pre-existing mRNA molecules (not labelled) and these values were used for half-life estimation of cellular genes. In the case of uninfected samples, we compared two approaches for calculating mRNA half-life: in the first, we assumed steady state and in the second, we analysed gene composition in each sample and the old fraction was normalized to the gene composition. These two approaches yielded highly similar values and the half-life values in uninfected cells that are presented in the figures are based on the calculation that assume steady state. In the case of the infected samples, the gene composition from the total RNA levels were used normalize the expression of the old mRNA fraction as follows. The total number of reads in each sample was scaled according to the ratio of cellular mRNA to rRNA and tRNA, as calculated on the basis of sequencing of total RNA-seq without poly-A selection. These normalized ratios were used to normalize the old transcript fractions in each sample.
The half-life of each gene in uninfected and infected cells was calculated by linear regression of the log values of the calculated old transcript fraction. Estimated variance of the values as calculated by GRAND-SLAM were used as weights in the linear regression. The regression coefficient lambda was converted to half-life as −log(2)/lambda. For further analysis, only genes for which the P value in the regression was < 0.01 and the adjusted r2 > 0.8 were used.
For analysis of intronic RNA turnover, reads that were aligned to any transcript annotation in Ensembl hg19 annotations were filtered out (may represent exonic reads). The rest of the reads were aligned to hg19 genome and were used as input for GRAND-SLAM using intron annotations on the basis of the known canonical UCSC genes.
Immunofluorescence
Cells were plated on ibidi slides, infected as described in ‘Cells and viruses’ or left uninfected and at the indicated time point washed once with PBS, fixed in 3% paraformaldehyde for 20 min, washed in PBS, permeabilized with 0.5% Triton X-100 in PBS for 2 min, and then blocked with 2% FBS in PBS for 30 min. Immunostaining was performed with rabbit anti-SARS-CoV-2 serum35 at a 1:200 dilution. Cells were washed and labelled with anti-rabbit FITC conjugated antibody at a 1:200 dilution and with DAPI (4′,6-diamidino-2-phenylindole) at a 1:200 dilution. Imaging was performed on a Zeiss AxioObserver Z1 wide-field microscope using a ×40 objective and Axiocam 506 mono camera.
Plasmids and cloning
pLVX-EF1alpha-SARS-CoV-2-nsp1-2XStrep-IRES-Puro and pLVX-EF1alpha-SARS-CoV-2-nsp2-2XStrep-IRES-Puro were provided by N. Krogan. mCherry–Flag was cloned in to the lentiCRISPR v.2 plasmid (Addgene no. 52961) instead of the Cas9 cassette. The viral genomic 5′ UTR was constructed on the basis of nucleotides 4–265 of the reported sequence of SARS-CoV-2 isolate Wuhan-Hu-1 (NC_045512.2) by sequential annealing of DNA oligonucleotides (IDT, 5′UTR oligo_1-5 listed in Supplementary Table 2). The coding sequence for the first 12 amino acids of ORF1a as well as the GFP homology region were added to the 5′ UTR by two PCR amplifications. The viral 5′ UTR with the 12 amino acid region was cloned into pAcGFP1-C1 (Takara Biotech) using restriction-free cloning. The entire expression cassette from the promoter to the poly-A site was amplified and cloned into pDecko-mCherry (Addgene plasmid no. 78534) using restriction-free cloning. Primers for PCR amplification of fragments were ordered from Sigma-Aldrich. The viral subgenomic 5′ leader was amplified from the viral 5′ UTR plasmid using primers containing homologous regions to clone the 5′ leader back into the plasmid. This was subsequently done using restriction-free cloning. For the human beta-globin (HBB) 5′ UTR control plasmid, the HBB 5′ UTR with GFP homology was ordered from Sigma-Aldrich as two oligonucleotides. These were used in a self-priming PCR reaction and inserted upstream of GFP in place of the viral 5′ UTR using restriction-free cloning. All primers and oligonucleotides used for cloning are listed in Supplementary Table 2.
Reporter assay
293T cells were transfected using JetPEI (Polyplus-transfection) following the manufacturer’s instructions. Twenty-four h after transfection cells were imaged on a Zeiss AxioObserver Z1 wide-field microscope using a ×20 objective and Axiocam 506 mono camera and assayed for reporter expression by flow cytometry on a BD Accuri C6 flow cytometer. In parallel, cells were assayed for expression of NSP1, NSP2 and mCherry–Flag and reporter mRNA levels as detailed in ‘Flow cytometry analysis of Strep and Flag tags’ and ‘Quantitative real-time PCR analysis’.
Flow cytometry analysis of Strep and Flag tags
The expression of NSP1, NSP2 and mCherry was verified by staining of the fused tags, Strep tag for NSP1 and NSP2 and Flag tag for mCherry, followed by flow cytometry. Cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X100, and stained using either Strep-TactinXT DY-649 (IBA-lifesciences) or Alexa Fluor647 anti-DYKDDDDK Tag Antibody (BioLegend). Flow cytometry analysis was performed on BD Accuri C6 and analysed on FlowJo. Normalization to the mode is presented in the histograms.
Quantitative real-time PCR analysis
Total RNA was extracted using Direct-zol RNA Miniprep Kit (Zymo Research) following the manufacturer’s instructions. cDNA was prepared using qScript FLEX cDNA Synthesis Kit with random primers (Quanta Biosciences) following the manufacturer’s instructions. Real-time PCR was performed using the SYBR Green PCR master-mix (ABI) on the QuantStudio 12K Flex (ABI) with the following primers (forward, reverse): GFP (TGACCCTGAAGTTCATCTGC, GAAGTCGTGCTGCTTCATGT); mCherry (ACCGCCAAGCTGAAGGTGAC, GACCTCAGCGTCGTAGTGGC); and 18S (CTCAACACGGGAAACCTCAC, CGCTCCACCAACTAAGAACG)
GFP and mCherry mRNA levels were calculated relative to 18S rRNA.
Graphics
Figure 4g was created with BioRender. Fluorescence-activated cell sorting figures were created with FlowJo and the rest of the figures were drawn with ggplot2 in R.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All next-generation sequencing data files have been deposited in Gene Expression Omnibus under accession number GSE162323. NSP1 expression data analysed in this study are available from Gene Expression Omnibus with the accession number GSE158374. MHV infection data are available from ArrayExpress database under the accession number E-MTAB-4111.
References
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Kamitani, W., Huang, C., Narayanan, K., Lokugamage, K. G. & Makino, S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 16, 1134–1140 (2009).
Lokugamage, K. G. et al. Middle East respiratory syndrome coronavirus Nsp1 inhibits host gene expression by selectively targeting mRNAs transcribed in the nucleus while sparing mRNAs of cytoplasmic origin. J. Virol. 89, 10970–10981 (2015).
Addetia, A. et al. SARS-CoV-2 ORF6 disrupts bidirectional nucleocytoplasmic transport through interactions with Rae1 and Nup98. mBio 12, e00065-21 (2021).
Banerjee, A. K. et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339.e21 (2020).
Schubert, K. et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 27, 959–966 (2020).
Thoms, M. et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 369, 1249–1255 (2020).
V’kovski, P., Kratzel, A., Steiner, S., Stalder, H. & Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 19, 155–170 (2021).
Finkel, Y. et al. The coding capacity of SARS-CoV-2. Nature 589, 125–130 (2021).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045.e9 (2020).
Sa Ribero, M., Jouvenet, N., Dreux, M. & Nisole, S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 16, e1008737 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Irigoyen, N. et al. High-resolution analysis of coronavirus gene expression by RNA sequencing and ribosome profiling. PLoS Pathog. 12, e1005473 (2016).
Wolff, G. et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 369, 1395–1398 (2020).
Liu, J., Xu, Y., Stoleru, D. & Salic, A. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc. Natl Acad. Sci. USA 109, 413–418 (2012).
Del Valle, D. M. et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 26, 1636–1643 (2020).
Herzog, V. A. et al. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 14, 1198–1204 (2017).
Jürges, C., Dölken, L. & Erhard, F. Dissecting newly transcribed and old RNA using GRAND-SLAM. Bioinformatics 34, i218–i226 (2018).
Zuckerman, B., Ron, M., Mikl, M., Segal, E. & Ulitsky, I. Gene architecture and sequence composition underpin selective dependency of nuclear export of long RNAs on NXF1 and the TREX complex. Mol. Cell 79, 251–267.e6 (2020).
Nakagawa, K., Lokugamage, K. G. & Makino, S. in Advances in Virus Research, vol. 96 (ed. Ziebuhr, J.) 165–192 (Academic, 2016).
Kamitani, W. et al. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl Acad. Sci. USA 103, 12885–12890 (2006).
Narayanan, K. et al. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 82, 4471–4479 (2008).
Huang, C. et al. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. 7, e1002433 (2011).
Rao, S. et al. Genes with 5′ terminal oligopyrimidine tracts preferentially escape global suppression of translation by the SARS-CoV-2 NSP1 protein. Preprint at https://doi.org/10.1101/2020.09.13.295493 (2020).
Tidu, A. et al. The viral protein NSP1 acts as a ribosome gatekeeper for shutting down host translation and fostering SARS-CoV-2 translation. RNA, https://doi.org/10.1261/rna.078121.120 (2020).
Carlevaro-Fita, J. & Johnson, R. Global positioning system: understanding long noncoding RNAs through subcellular localization. Mol. Cell 73, 869–883 (2019).
Jungreis, I. et al. Conflicting and ambiguous names of overlapping ORFs in the SARS-CoV-2 genome: a homology-based resolution. Virology 558, 145–151 (2021).
Finkel, Y. et al. Comprehensive annotations of human herpesvirus 6A and 6B genomes reveal novel and conserved genomic features. eLife 9, e50960 (2020).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921 (2020).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Fields, A. P. et al. A regression-based analysis of ribosome-profiling data reveals a conserved complexity to mammalian translation. Mol. Cell 60, 816–827 (2015).
Mi, H. & Thomas, P. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 563, 123–140 (2009).
Mi, H., Muruganujan, A., Ebert, D., Huang, X. & Thomas, P. D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019).
Yahalom-Ronen, Y. et al. A single dose of recombinant VSV-ΔG-spike vaccine provides protection against SARS-CoV-2 challenge. Nat. Commun. 11, 6402 (2020).
Acknowledgements
We thank members of the laboratory of N.S.-G. for providing valuable feedback; N. Krogan for the SARS-CoV-2 ORF expression plasmids; G. Jona and Weizmann Bacteriology and Genomic Repository Units for technical assistance; members of the virology research group at the IIBR for their contribution and support; and S. Weiss for biosafety guidance. This study was supported by a research grant from Weizmann Corona Response Fund. Work in the laboratory of N.S.-G. is supported by a European Research Council consolidator grant (CoG-2019-864012). N.S.-G. is an incumbent of the Skirball Career Development Chair in New Scientists and is a member of the European Molecular Biology Organization (EMBO) Young Investigator Program.
Author information
Authors and Affiliations
Contributions
Y.F., R.W., A.N., A.G., N.P., M.S. and N.S.-G. conceptualized the study; A.G., R.W., T.F., B.R., O.M. and B.Z. performed experiments; Y.L. and B.S. provided reagents. Y.F., A.N. and R.W. undertook data analysis; Y.Y-R., H.T., T.I. and N.P. performed experiments with SARS-CoV-2; Y.F., A.G., T.F., A.N., I.U., N.P., M.S. and N.S.-G. interpreted data; and Y.F., M.S. and N.S.-G. wrote the manuscript with contributions from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Lars Dölken and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Ribosome profiling and RNA-seq along SARS-CoV-2 infection.
a, Calu3 cells were infected with SARS-CoV-2 at an MOI = 3 and at 8 hpi the cells were fixed and stained with antisera against SARS-CoV-2 (green) and DAPI (blue). Representative images of two independent replicates are shown. Scale bars, 100 μm. b, c, Scatter plots depicting gene read counts derived from our two independent biological replicates for mRNA (b) and footprints (FP) (c), demonstrating reproducibility between our replicates. Pearson’s R and two-sided P values from log values are presented. Coloured dots represent the different viral ORFs. d, Length distribution of ribosome footprint fragments from SARS-CoV-2 genes (blue) or cellular genes (pink). e–h, Metagene analysis of read densities around the start and stop codon of cellular protein-coding genes (e, f) and viral genes (g, h) in ribosome profiling (e, g) or mRNA (f, h) libraries at different time points after infection. The x axis shows the nucleotide position relative to the start codon or the stop codon. The average normalized read densities of well-expressed genes (with more than 50 reads mapped to the CDS) are shown, with different colours indicating the three relative phases. The phase aligned with the translated frame is labelled in red (red, phase 0; black, phase +1; grey, phase +2).
Extended Data Fig. 2 Measurement of protein synthesis using the OPP assay.
a, Scheme of labelling and detection of nascent protein synthesis by OPP incorporation followed by fluorescent labelling. OPP is efficiently incorporated into newly translating proteins, releases the polypeptides and terminates translation. Following fixation, OPP is fluorescently labelled using Click reaction and can then be measured by flow cytometry. b, Protein synthesis measurement by flow cytometry of untreated 293T cells and 293T cells treated with cycloheximide leading to efficient translation inhibition. Unlabelled cells are shown as control.
Extended Data Fig. 3 Total RNA and ribosomal RNA levels in infected and uninfected Calu3 cells.
a, Total RNA was measured in uninfected Calu3 cells and in cells at 4, 5, 6 and 7 hpi by Qubit Fluorometer. b, rRNA concentration was measured in uninfected and infected Calu3 cells at 5 and 7 hpi by measuring the concentration in the 18S and 28S ribosomal RNA peaks using a TapeStation system.
Extended Data Fig. 4 Upregulated genes in SARS-CoV-2 infected cells.
a, Heat map presenting mRNA and footprints levels of transcripts with mRNA levels at 8 hpi relative to uninfected that were increased by at least 1.5-fold. Expression levels scaled by gene after partitioning clustering are presented. b, Summary of pathway enrichment analysis of genes in cluster A, induced late. Dot size reflects the number of genes from each pathway, and dot colour reflects the false-discovery rate (FDR) of the pathway enrichment. c, RNA-level fold change (FC) of cellular RNAs at 3 and 8 hpi relative to uninfected cells. RNAs were grouped to ten bins on the basis of their cytosol-to-nucleus localization ratio in uninfected Calu3 cells. P values calculated using two-sided t-test comparing the first and last bins in each time point, n = 5,650 genes. d, Scatter plots depicting log2-transformed cytosol-to-nuclear ratios of cellular transcripts relative to log2-transformed fold changes in transcript levels between infected cells at the different time points during SARS-CoV-2 infection and uninfected cells. Pearson’s R and two-sided P value on log values are presented.
Extended Data Fig. 5 SLAM-seq measurements in uninfected and SARS-CoV-2-infected cells.
a, Experimental design of the SLAM-seq measurements and the half-life calculation. b, c, Rates of nucleotide substitutions demonstrate efficient conversion rates in 4sU-treated samples compared to non-treated cells (no 4sU) for reads originating from both uninfected (b) and infected (c) cells. d, e, Proportion of new-to-total RNA (NTR) in uninfected (d) and infected (e) samples as calculated by GRAND-SLAM, a Bayesian method that computes the ratio in a quantitative manner18. f, Scatter plot of mRNA half-life in hours calculated from our SLAM-seq measurements in uninfected Calu3 cells relative to SLAM-seq based measurements of mRNA half-life in MCF7 cells19. Pearson’s R and two-sided P value on log values are presented. g, Scatter plot of the changes due to infection in mRNA half-lives relative to changes in mRNA expression. Pearson’s R and two-sided P value on log values are presented. h, Change in cellular transcripts half-lives in infected cells relative to uninfected cells. RNAs were grouped to ten bins on the basis of their cytosol-to-nucleus localization ratio. P value was calculated using t-test between the most-nuclear and the most-cytosolic bins, n = 2,750 with 275 genes in each bin. Box plots show median, first to third quartile, 1.5× interquartile range and outliers. i, Scatter plot depicting the changes in transcript half-life between uninfected and infected cells relative to translation efficiency of cellular transcripts in uninfected cells. Pearson’s R and two-sided P value on log values are presented.
Extended Data Fig. 6 Analysis of RNA expression in NSP1-expressing cells.
a, mRNAs were binned on the basis of their cytosol-to-nucleus localization ratio. Fold change of each bin in RNA expression in cells transfected with NSP1 relative to cells transfected with NSP2 is presented. P value was calculated using two-sided t-test between the most-nuclear and the most-cytosolic bins, n = 470 with 47 genes in each bin. b, The change in mRNA expression of nuclear-encoded or mitochondrially encoded mRNAs in cells transfected with NSP1 relative to cells transfected with NSP2. P value was calculated using Wilcoxon’s test, n = 10 mitochondrial genes, n = 503 nuclear genes. RNA-seq data of NSP1 and NSP2 transfected cells is a previous publication24. Box plots show median, first to third quartile, 1.5× interquartile range and outliers.
Extended Data Fig. 7 Analysis of intronic reads.
a, Proportion of newly synthesized intronic RNA (NTR) in uninfected and infected samples as calculated by GRAND-SLAM. Values shown are for one replicate of each time point and mean value for the 5-hpi sample. The 6-hpi time point was omitted from the analysis as it did not contain enough intronic reads. b, Heat map presenting relative mRNA levels of well-expressed human transcripts (more than 50 reads) that showed the strongest changes in their mRNA levels at 8 hpi relative to uninfected, across time points during SARS-CoV-2 infection (as presented in Fig. 1g). Left, fold change in the ratio of intron to exon reads for each transcript in 8 hpi compared to uninfected.
Extended Data Fig. 8 SARS-CoV-2 5′ leader protects mRNA from NSP1-mediated degradation.
a, 293T cells were co-transfected with an expression vector containing either NSP1–2×Strep, NSP2–2×Strep or mCherry–Flag (the two latter as controls) and with a GFP reporter that includes either the HBB 5′ UTR as control (control-5′UTR), the viral genomic 5′ UTR (CoV2-5′UTR) or the viral 5′ leader (CoV2-leader). The reporter plasmids also contained an independent mCherry reporter. b, Flow cytometry analysis of the Flag tag in cells co-transfected with mCherry–Flag and for the Strep tag in cells co-transfected with NSP1 or NSP2. c, Microscopy images of GFP in cells co-transfected with NSP2 (top) or NSP1 (bottom), together with control 5′ UTR reporter, SARS-CoV-2 5′ UTR reporter or SARS-CoV-2 5′ leader reporter. Representative images of four independent replicates are shown. Scale bars, 100 μm. d, Gating strategy used to determine the single-cell population for all flow cytometry analyses. e, Flow cytometry analysis of GFP in cells co-transfected with NSP1 or NSP2 together with control 5′ UTR reporter, SARS-CoV-2 5′ UTR reporter or SARS-CoV-2 5′ leader reporter. f, Relative GFP RNA levels in cells expressing NSP1 or NSP2 together with control 5′ UTR reporter, SARS-CoV-2 5′ UTR reporter or SARS-CoV-2 5′ leader reporter, as measured by quantitative real-time PCR. Data points show the measurement of biological replicates. One representative experiment out of two performed is shown. g, Microscopy images of mCherry in cells co-transfected with NSP2 (top) or NSP1 (bottom) together with the SARS-CoV-2 5′ UTR reporter or SARS-CoV-2 5′ leader reporter. Representative images of four independent replicates are shown. Scale bars, 100 μm. h, Flow cytometry analysis of mCherry in cells co-transfected with NSP1 or NSP2 together with SARS-CoV-2 5′ UTR reporter or SARS-CoV-2 5′ leader reporter. i, Relative mCherry RNA levels in cells expressing NSP1 or NSP2 together with SARS-CoV-2 5′ UTR reporter or SARS-CoV-2 5′ leader reporter, as measured by quantitative real-time PCR. Data points show the measurement of biological replicates. One representative experiment out of two performed is shown.
Extended Data Fig. 9 The translation of induced transcripts is impaired during infection.
a, mRNA and footprint levels of the indicated genes in uninfected cells and at 3, 5 and 8 hpi. Thirty-five genes that showed the strongest reduction in their relative translation efficiency are presented. b, Scatter plot of cytosolic-to-nuclear RNA ratio calculated from our fractionation experiment in uninfected Calu3 cells relative to fractionation-based measurements of cytosolic to nuclear RNA ratio in MCF7 cells19. Pearson’s R and two-sided P value on log values are shown.
Extended Data Fig. 10 Translation of viral genes along the course of infection.
a, Schematic of the MHV genomic and subgenomic mRNAs, and the main ORFs. ORFs that are not expressed in the strain that was used for ribosome profiling (MHV-A59) are marked by empty rectangles. b, Relative translation efficiency of each canonical MHV ORF along infection. Genes are divided to two groups on the basis of their location on the genome. Expression was calculated from a previous publication13. c, Relative translation levels of the main ORFs (ORF7a, S and ORF3a, labelled by circle) and the out-of-frame ORFs (ORF7b, ORF2b and ORF3c, labelled by diamond) of the ORF7a, S, and ORF3a subgenomic transcripts, respectively, along the course of SARS-CoV-2 infection. ORF9b expression was low in our measurements and it was therefore excluded from this analysis. Translation levels were calculated from ribosome densities using ORF-RATER32. Duplicate values of 8 hpi are shown as lines. Standard deviation for our ORF expression measurement shown as rectangles calculated using 500 random partial ORF regions.
Supplementary information
Supplementary Table 1
Expression values (RPKM) of genes induced more than 1.5-fold during infection, and their division into clusters. This table lists genes induced by more than 1.5-fold at 8 hours post infection compared to uninfected cells, with their respective expression values at the different times points normalized to reads per kilobase per million (RPKM) and to total mRNA levels (see methods for normalization methods). Genes are divided into clusters (A-C) as shown in extended data figure 4a.
Supplementary Table 2
Primers used for cloning. This table lists primers used for cloning of constructs described in the manuscript and their sequence.
Rights and permissions
About this article

Cite this article
Finkel, Y., Gluck, A., Nachshon, A. et al. SARS-CoV-2 uses a multipronged strategy to impede host protein synthesis. Nature 594, 240–245 (2021). https://doi.org/10.1038/s41586-021-03610-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03610-3
This article is cited by
-
Decoding HiPSC-CM’s Response to SARS-CoV-2: mapping the molecular landscape of cardiac injury
BMC Genomics (2024)
-
A guide to complement biology, pathology and therapeutic opportunity
Nature Reviews Immunology (2024)
-
Coronavirus takeover of host cell translation and intracellular antiviral response: a molecular perspective
The EMBO Journal (2024)
-
SARS-CoV-2 biology and host interactions
Nature Reviews Microbiology (2024)
-
COVID-19 immune signatures in Uganda persist in HIV co-infection and diverge by pandemic phase
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
