
Abstract
A newly described coronavirus named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected over 2.3 million people, led to the death of more than 160,000 individuals and caused worldwide social and economic disruption1,2. There are no antiviral drugs with proven clinical efficacy for the treatment of COVID-19, nor are there any vaccines that prevent infection with SARS-CoV-2, and efforts to develop drugs and vaccines are hampered by the limited knowledge of the molecular details of how SARS-CoV-2 infects cells. Here we cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins in human cells and identified the human proteins that physically associated with each of the SARS-CoV-2 proteins using affinity-purification mass spectrometry, identifying 332 high-confidence protein–protein interactions between SARS-CoV-2 and human proteins. Among these, we identify 66 druggable human proteins or host factors targeted by 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are preclinical compounds). We screened a subset of these in multiple viral assays and found two sets of pharmacological agents that displayed antiviral activity: inhibitors of mRNA translation and predicted regulators of the sigma-1 and sigma-2 receptors. Further studies of these host-factor-targeting agents, including their combination with drugs that directly target viral enzymes, could lead to a therapeutic regimen to treat COVID-19.
Similar content being viewed by others
Main
SARS-CoV-2 is an enveloped, positive-sense, single-stranded RNA betacoronavirus of the family Coronaviridae3,4. Coronaviruses that infect humans historically included several common cold viruses, including hCoV-OC43, HKU and 229E5. However, over the past two decades, highly pathogenic human coronaviruses have emerged, including SARS-CoV in 2002, which is associated with 8,000 cases worldwide and a death rate of around 10%, and Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012, which caused 2,500 confirmed cases and had a death rate of 36%. Infection with these highly pathogenic coronaviruses can result in acute respiratory distress syndrome, which may lead to a long-term reduction in lung function, arrhythmia or death. In comparison to MERS-CoV or SARS-CoV, SARS-CoV-2 has a lower case-fatality rate but spreads more efficiently6, making it difficult to contain. To devise therapeutic strategies to counteract SARS-CoV-2 infection and the associated COVID-19 pathology, it is crucial to understand how this coronavirus hijacks the host during infection, and to apply this knowledge to develop new drugs and repurpose existing ones.
Thus far, no clinically available antiviral drugs have been developed for SARS-CoV, SARS-CoV-2 or MERS-CoV. Clinical trials are ongoing for treatment of COVID-19 with the nucleoside-analogue RNA-dependent RNA polymerase (RdRP) inhibitor remdesivir7, and recent data suggest that a new nucleoside analogue may be effective against SARS-CoV-2 infection in laboratory animals8. Clinical trials using several vaccine candidates are also underway9, as are trials of repurposed compounds that inhibit the human protease TMPRSS210. We believe that there is great potential in systematically exploring the host dependencies of the SARS-CoV-2 virus to identify other host proteins that are already targeted by existing drugs. Therapies that target the host–virus interface, where the emergence of mutational resistance is arguably less likely, could potentially present durable, broad-spectrum treatment modalities11. Unfortunately, limited knowledge of the molecular details of SARS-CoV-2 precludes a comprehensive evaluation of small-molecule candidates for host-directed therapies. We sought to address this gap by systematically mapping the interaction landscape between SARS-CoV-2 proteins and human proteins.
Cloning and expression of SARS-CoV-2 proteins
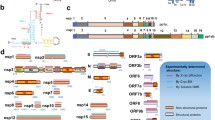
Sequence analysis of SARS-CoV-2 isolates suggests that the 30-kb genome encodes as many as 14 open-reading frames (ORFs). ORF1a and ORF1ab encode polyproteins, which are auto-proteolytically processed into 16 non-structural proteins (NSP1–NSP16) that form the replicase–transcriptase complex (Fig. 1a). The replicase–transcriptase complex consists of multiple enzymes, including the papain-like protease (NSP3), the main protease (NSP5), the NSP7–NSP8 primase complex, the primary RNA-dependent RNA polymerase (NSP12), a helicase–triphosphatase (NSP13), an exoribonuclease (NSP14), an endonuclease (NSP15) and N7- and 2′O-methyltransferases (NSP10 and NSP16)1,12,13. At the 3′ end of the viral genome, as many as 13 ORFs are expressed from 9 predicted sub-genomic RNAs. These include four structural proteins: spike (S), envelope (E), membrane (M) and nucleocapsid (N)13, and nine putative accessory factors1,12 (Fig. 1a). The SARS-CoV-2 genome is very similar to SARS-CoV. Although both viruses have an ORF1ab that encodes the 16 predicted NSPs as well as the four typical structural proteins of coronaviruses, they differ in their complement of 3′ ORFs: SARS-CoV-2 possesses an ORF3b and ORF10, which have limited detectable homology to SARS-CoV proteins1,12 (Extended Data Fig. 1a).

a, SARS-CoV-2 genome annotation. The colour intensity is proportional to the protein sequence similarity with SARS-CoV homologues (when homologues exist). n = 4 structural proteins; n = 16 NSPs; n = 9 accessory factors. b, Experimental workflow for AP-MS studies. MS, mass spectrometry; PPI, protein–protein interaction.
Mature NSPs, with the exception of NSP3 and NSP16, and all predicted proteins expressed from other SARS-CoV-2 ORFs (27 proteins and one mutant) were codon-optimized and cloned into a mammalian expression vector that contained a 2×Strep-tag II affinity tag that can be used for affinity-purification–mass spectrometry (AP-MS)-based proteomics when expressed in HEK-293T/17 cells. High-confidence interacting proteins were identified using SAINTexpress and MiST scoring algorithms14,15.
To verify the expression of viral proteins, we performed western blot using an anti-Strep antibody on the input cell lysate, and with the exception of NSP4, NSP6, NSP11, and ORF3b, we observed bands consistent with the predicted protein sizes (24 out of 28 constructs) (Extended Data Fig. 1b). Despite the lack of detection by western blot, we detected expression of viral peptides NSP4, NSP6 and ORF3b in the proteomic analysis. The fourth construct not confirmed by western blot, the small peptide NSP11, had a predicted molecular mass of 4.8 kDa (including tag) but an apparent mass of approximately 30 kDa (Extended Data Fig. 1b).
Alignment of 2,784 SARS-CoV-2 sequences revealed a premature stop codon at position 14 of ORF3b in 17.6% of isolates (Extended Data Fig. 1c), and two mutations were also observed that resulted in premature stop codons in ORF9c (Extended Data Fig. 1d). These data suggest that ORF3b and ORF9c might not be bona fide SARS-CoV-2 reading frames, or are dispensable for replication. Pending a comprehensive evaluation of viral protein expression, we nevertheless proceeded with the analysis for all possible viral proteins. Out of the 27 bait proteins (Fig. 1b), the affinity purification of ORF7b showed an unusually high number of background proteins and was therefore excluded from protein interaction analysis. We have thus far sent these plasmids to almost 300 laboratories in 35 countries.
Analysis of SARS-CoV-2–host protein interactions
Our AP-MS analysis identified 332 high-confidence protein interactions between SARS-CoV-2 proteins and human proteins, observing correlations between replicate experiments of each viral bait (Pearson’s R = 0.46–0.72) (Extended Data Fig. 2 and Supplementary Tables 1, 2). We studied the interacting human proteins with regards to their biological functions, anatomical expression patterns, expression changes during SARS-CoV-2 infection16 and in relation to other maps of host–pathogen interacting proteins15,17 (Fig. 2a). We analysed each viral protein for Gene Ontology enrichment (Fig. 2b and Extended Data Fig. 3) and identified the major cell processes of the interacting proteins, including lipoprotein metabolism (S), nuclear transport (NSP7) and ribonucleoprotein complex biogenesis (NSP8). To discover potential binding interfaces, we enriched for domain families within the interacting proteins of each viral bait (Extended Data Fig. 4). For instance, DNA polymerase domains are enriched among proteins that interact with NSP1, and bromodomains and extra-terminal domain (BET) family domains are enriched among proteins that interact with E (Supplementary Discussion and Supplementary Methods).

a, Overview of global analyses performed. b, Gene Ontology (GO) enrichment analysis was performed on the human interacting proteins of each viral protein. P values were calculated by hypergeometric test and a false-discovery rate was used to account for multiple hypothesis testing (Methods). The top GO term of each viral protein was selected for visualization. c, Degree of differential protein expression for the human interacting proteins (n = 332) across human tissues. We obtained protein abundance values for the proteome in 29 human tissues and calculated the median level of abundance for the human interacting proteins (top 16 tissues shown). This was then compared with the abundance values for the full proteome in each tissue and summarized as a Z-score from which a P value was calculated. A false-discovery rate was used to account for multiple hypothesis testing. d, The distribution of the correlation of protein level changes during SARS-CoV-2 infection for pairs of viral–human proteins (median, white circles; interquartile range, black bars) is higher than non-interacting pairs of viral–human proteins (P = 4.8 × 10−5; Kolmogorov–Smirnov test). The violin plots show each viral–human protein correlation for preys (n = 210, minimum = −0.986, maximum = 0.999, quartile (Q)1 = −0.468, Q2 = 0.396, Q3 = 0.850) and non-preys (n = 54765, minimum = −0.999, maximum = 0.999, Q1 = −0.599, Q2 = 0.006, Q3 = 0.700). e, Significance of the overlap of human interacting proteins between SARS-CoV-2 and other pathogens using a hypergeometric test (unadjusted for multiple testing). The background gene set for the test consisted of all unique proteins detected by mass spectrometry across all pathogens (n = 10,181 proteins). HCV, hepatitis C virus; HIV, human immunodeficiency virus; HPV, human papillomavirus; KSHV, Kaposi’s sarcoma-associated herpesvirus; Mtb, M. tuberculosis; WNV, West Nile virus.
Although the cell line used for these AP-MS experiments, HEK-293T/17, can be infected with the SARS-CoV-2 virus18, it does not represent the primary physiological site of infection—lung tissue. From 29 human tissues19, we identified the lung as the tissue with the highest expression of the prey proteins relative to the average proteome (Fig. 2c). Consistent with this, the interacting proteins were enriched in the lung relative to other tissues (Extended Data Fig. 5a), and compared to overall RefSeq gene expression in the lung (median transcripts per million (TPM) = 3.198), proteins that interacted with SARS-CoV-2 proteins were expressed at a higher level (median TPM = 25.52, P = 0.0007; Student’s t-test) (Extended Data Fig. 5b), supporting the hypothesis that SARS-CoV-2 preferentially hijacks proteins that are expressed in lung tissue.
We also studied the evolutionary properties of the host proteins bound by SARS-CoV-2 (Supplementary Table 3, Supplementary Methods and Supplementary Discussion). In addition, we analysed changes in protein abundance during SARS-CoV-2 infection16. We calculated, when possible, the correlation between changes in the abundance of viral proteins and their human interaction partners across four time points. Interacting pairs typically had stronger correlated changes than other pairs of viral–human proteins (Fig. 2d) (Kolmogorov–Smirnov test P = 4.8 × 10−5), indicating that the AP-MS-derived interactions are relevant for the target tissue and the infection context. We compared our SARS-CoV-2 interaction map with those of ten other pathogens (Fig. 2e) and found that West Nile virus20 and Mycobacterium tuberculosis21 had the most similar host-protein interaction partners. The association with M. tuberculosis is of particular interest as it also infects lung tissue.
The interactome reveals SARS-CoV-2 biology
Our study highlights interactions between SARS-CoV-2 proteins and human proteins that are involved in several complexes and biological processes (Fig. 3). These included DNA replication (NSP1), epigenetic and gene-expression regulators (NSP5, NSP8, NSP13 and E), vesicle trafficking (NSP2, NSP6, NSP7, NSP10, NSP13, NSP15, ORF3a, E, M and ORF8), lipid modification (S), RNA processing and regulation (NSP8 and N), ubiquitin ligases (ORF10), signalling (NSP7, NSP8, NSP13, N and ORF9b), nuclear transport machinery (NSP9, NSP15 and ORF6), cytoskeleton (NSP1 and NSP13), mitochondria (NSP4, NSP8 and ORF9c) and the extracellular matrix (NSP9).

332 high-confidence interactions between 26 SARS-CoV-2 proteins (red diamonds) and human proteins (circles; drug targets: orange; protein complexes: yellow; proteins in the same biological process: blue). Edge colour proportional to MiST score; edge thickness proportional to spectral counts. Physical interactions among host proteins (thin black lines) were curated from CORUM, IntAct, and Reactome. An interactive protein–protein interaction map can be found at kroganlab.ucsf.edu/network-maps. ECM, extracellular matrix; ER, endoplasmic reticulum; snRNP, small nuclear ribonucleoprotein. n = 3 biologically independent samples.
Approximately 40% of SARS-CoV-2-interacting proteins were associated with endomembrane compartments or vesicle trafficking pathways. Host interactions with NSP8 (signal recognition particle (SRP)), ORF8 (protein quality control in the endoplasmic reticulum), M (morphology of the endoplasmic reticulum) and NSP13 (organization of the centrosome and Golgi) may facilitate the marked reconfiguration of endoplasmic reticulum and Golgi trafficking during coronavirus infection, and interactions in peripheral compartments with NSP2 (WASH), NSP6 and M (vacuolar ATPase), NSP7 (Rab proteins), NSP10 (AP2), E (AP3) and ORF3a (HOPS) may also modify endomembrane compartments to favour coronavirus replication. NSP6 and ORF9c interact with Sigma receptors that have been implicated in lipid remodelling and the stress response of the endoplasmic reticulum; these proteins interact with many human drugs (see ‘Antiviral activity of host-directed compounds’).
Trafficking into the endoplasmic reticulum and mitochondria may also be affected by the main protease of SARS-CoV-2, NSP5. We identified one high-confidence interaction between wild-type NSP5 and the epigenetic regulator histone deacetylase 2 (HDAC2), and predicted a cleavage site between the HDAC domain and the nuclear localization sequence of HDAC2 (Extended Data Fig. 6a–d), suggesting that NSP5 may inhibit the transport of HDAC2 into the nucleus and could potentially affect the ability of HDAC2 to mediate the inflammation and interferon response22,23. We also identified an interaction between catalytically dead NSP5(C145A) and tRNA methyltransferase 1 (TRMT1), which is responsible for the dimethylguanosine base modification (m2,2G) in both nuclear and mitochondrial tRNAs24. We predict that TRMT1 is also cleaved by NSP5 (Extended Data Fig. 6a–d), leading to the removal of its zinc finger and nuclear localization signal and probably resulting in an exclusively mitochondrial localization of TRMT1.
SARS-CoV-2 interacts with innate immune pathways
Several innate immune signalling proteins are targeted by SARS-CoV-2 viral proteins. The interferon pathway is targeted by NSP13 (TBK1 and TBKBP1), NSP15 (RNF41 (also known as NRDP1)) and ORF9b (TOMM70); and the NF-κB pathway is targeted by NSP13 (TLE1, TLE3 and TLE5) and ORF9c (NLRX1, F2RL1 and NDFIP2). Furthermore, two other E3 ubiquitin ligases that regulate antiviral innate immune signalling, TRIM59 and MIB1, are bound by ORF3a and NSP9, respectively25,26.
We also identified interactions between SARS-CoV-2 ORF6 and the NUP98–RAE1 complex (Fig. 4a), an interferon-inducible mRNA nuclear export complex27 that is hijacked or degraded by multiple viruses including vesicular stomatitis virus (VSV), influenza A, Kaposi’s sarcoma-associated herpesvirus and poliovirus, and is a restriction factor for influenza A infection28,29,30,31. The X-ray structure of the VSV M protein complexed with NUP98–RAE132 reveals key binding interactions, including a buried methionine residue on the M protein that packs into a hydrophobic pocket in RAE1, and neighbouring acidic residues that interact with a basic patch on the NUP98–RAE1 complex32. These features are also present in a conserved motif in the C-terminal region of SARS-CoV-2 ORF6 (Fig. 4b–d and Extended Data Fig. 7a, b), providing a structural hypothesis for the observed interaction. ORF6 of SARS-CoV antagonizes host interferon signalling by perturbing nuclear transport33, and the NUP98–RAE1 interaction with ORF6 may perform the same function for SARS-CoV-2.

a, ORF6 interacts with an mRNA nuclear export complex that can be targeted by selinexor. b, The C-terminal peptide of SARS-CoV-2 ORF6 (dark purple) is modelled into the binding site of the VSV M protein (yellow)–NUP98 (green)–RAE1 (light purple) complex (Protein Data Bank (PDB) ID: 4OWR). ORF6 and M protein residues are labelled. c, The C-terminal sequence of SARS-CoV-2 ORF6, highlighting the described trafficking motifs and putative NUP98–RAE1 binding sequence. The chemical properties of amino acids are shown as follows: polar, green; neutral, purple; basic, blue; acidic, red; and hydrophobic, black. d, Putative NUP98–RAE1 interaction motifs. Negatively charged residues (red) surround a conserved methionine (yellow) in several virus species. e, N targets stress granule proteins. f, Inhibition of casein kinase II (by silmitasertib or TMCB) disrupts stress granules. g, Inhibition of translation initiation. The MNK inhibitor tomivosertib prevents phosphorylation of eIF4E. 4ER1Cat blocks the interaction of eIF4E with eIF4G. Inhibition of eIF4A (zotatifin) may prevent the unwinding of the viral 5′ untranslated region to prevent its translation. h, Targeting the translation elongation factor-1A ternary complex (ternatin-4) or the Sec61 translocon (PS3061) may prevent viral protein production and membrane insertion, respectively. i, ORF10 interacts with the CUL2ZYG11B complex. j, Predicted secondary structure of ORF10. k, ORF10 possibly hijacks CUL2ZYG11B for the ubiquitination (Ub) of host proteins, which can be inhibited by pevonedistat. l, The protein E interacts with bromodomain proteins. j, Alignment of E proteins from SARS-CoV-2, SARS-CoV and bat-CoV with histone H3 and NS1 protein of influenza A H3N2. Identical and similar amino acids are highlighted. m, Bromodomain inhibitors (iBETs) may disrupt the interaction between E and BRDs. a, f–h, k, n, FDA-approved drugs are shown in green, clinical candidates are shown in orange and preclinical candidates are shown in purple.
SARS-CoV-2 interacts with host translation machinery
Nucleocapsid (N) of SARS-CoV-2 binds to the stress granule proteins G3BP1 and G3BP2, and to other host mRNA-binding proteins including the mTOR-regulated translational repressor LARP1, two subunits of casein kinase 2 (CK2), and mRNA decay factors UPF1 and MOV10 (Fig. 4e). Manipulation of the stress granule and related RNA biology is common among Coronaviridae34,35,36 and stress granule formation is thought to be a primarily antiviral response. The promotion of G3BP aggregation by eIF4A inhibitors28,37 may partially explain their antiviral activity (see ‘Antiviral activity of host-directed compounds’).
All coronavirus mRNAs rely on cap-dependent translation to produce their proteins, a process enhanced in trans by the SARS-CoV N protein38. Key eIF4F–cap binding complex constituents—the cap binding protein eIF4E, scaffold protein eIF4G and the DEAD-box helicase eIF4A—are candidates for therapeutic targeting of coronaviruses39,40. Therapeutic targeting (Fig. 4f, g) of viral translation by interfering with the eIF4F complex formation or the interactions between viral proteins N, NSP2 or NSP8 and the translational machinery may have therapeutic benefits (see ‘Antiviral activity of host-directed compounds’).
Cotranslational entry into the secretory pathway is a potential target for SARS-CoV-2 inhibition. Up to ten SARS-CoV-2 proteins are predicted to undergo insertion into the membrane of the endoplasmic reticulum mediated by the Sec61 translocon, which localizes to SARS-CoV replication complexes41. Furthermore, high-confidence interactions between NSP8 and three SRP components suggest that the virus hijacks the Sec61-mediated protein translocation pathway for entry into the endoplasmic reticulum. Sec61 inhibitors of protein biogenesis such as PS3061 (Fig. 4h), which has previously been shown to inhibit other enveloped RNA viruses42,43, may also block SARS-CoV-2 replication and assembly.
SARS-CoV-2 interacts with a Cullin ubiquitin ligase
Viruses commonly hijack ubiquitination pathways for replication and pathogenesis44. The ORF10 of SARS-CoV-2 interacts with members of a cullin-2 (CUL2) RING E3 ligase complex (Fig. 4i), specifically the CUL2ZYG11B complex. ZYG11B is the highest scoring protein in the ORF10 interactome, suggesting that there is a direct interaction between ORF10 and ZYG11B. Despite its small size (38 amino acids), ORF10 appears to contain an α-helical region (Fig. 4j) that may be adopted in complex with CUL2ZYG11B. The ubiquitin transfer to a substrate requires neddylation of CUL2 by NEDD8-activating enzyme (NAE), which is a druggable target45 (Fig. 4k). ORF10 may bind to the CUL2ZYG11B complex and hijack it for ubiquitination and degradation of restriction factors, or alternatively, ZYG11B may bind to the N-terminal glycine in ORF10 to target it for degradation31.
SARS-CoV-2 interacts with bromodomain proteins
We found that the transmembrane E protein, which is probably resident on the endoplasmic reticulum–Golgi intermediate compartment and Golgi membranes, binds to BRD2 and BRD4 (Fig. 4l), members of the bromodomain and extra-terminal (BET) domain family of epigenetic readers that bind to acetylated histones to regulate gene transcription46. The C-terminal region of E mimics the N-terminal segment of histone H3, which is a known interacting partner of bromodomains47. Notably, this region of E is highly conserved in SARS and bat coronaviruses, which suggests that it has a conserved function (Fig. 4m). A similar short peptide motif has also been identified in the NS1 protein of the influenza A H3N2 strain, in which it interferes with transcriptional processes that support an antiviral response47,48. Bromodomain inhibitors might disrupt the interaction between protein E and BRDs (Fig. 4n).
For a more comprehensive overview of virus–host interactions, see Supplementary Discussion and Supplementary Methods.
Identification of drugs that target host factors
To disrupt the SARS-CoV-2 interactome, we sought ligands of human proteins that interact with viral proteins (Methods). Molecules were prioritized by the MiST score of the interaction between the human and viral proteins; by their status as approved drugs, investigational drugs (drugs in clinical trials) or as preclinical candidates; by their selectivity; and by their availability (Supplementary Tables 4, 5). Chemoinformatics searches from the IUPHAR/BPS Guide to Pharmacology (2020-3-12) and the ChEMBL25 database on the human interactors yielded 16 approved drugs, 3 investigational drugs and 18 pre-clinical candidates (Supplementary Table 4); and target- and pathway-specific literature search revealed 13 approved drugs, 9 investigational drugs and 10 preclinical candidates (Supplementary Table 5). Of the 332 human targets that interact with the viral bait proteins with a high-confidence score (Fig. 3), 62 have 69 drugs, investigational drugs or preclinical molecules that modulate them and can be overlaid on our protein-interaction network (Fig. 5).

Protein–protein interactions of SARS-CoV-2 baits with approved drugs (green), clinical candidates (orange) and preclinical candidates (purple) with experimental activities against the host proteins (white background) or previously known host factors (grey background) are shown.
Antiviral activity of host-directed compounds
We next investigated the antiviral activity of these drugs and compounds, using two viral assays (Fig. 6a). First, at Mount Sinai Hospital in New York, we developed a medium-throughput immunofluorescence-based assay (which detects the viral NP protein) to screen 37 compounds for inhibition of SARS-CoV-2 infection in the Vero E6 cell line. Second, at the Institut Pasteur in Paris, viral RNA was monitored using quantitative PCR with reverse transcription (RT–qPCR) after treatment with 44 drugs and compounds. Together, both locations tested 47 of the 69 compounds that we identified, plus 13 to expand testing of the sigma-1 and sigma-2 receptors and mRNA translation targets, and 15 additional molecules that had been prioritized by other methods (Methods and Supplementary Table 6). Viral growth and cytotoxicity were monitored at both institutions (Extended Data Figs. 8, 9 and Supplementary Table 6). Two classes of molecules emerged as effectively reducing viral infectivity: protein biogenesis inhibitors (zotatifin, ternatin-4 and PS3061) (Fig. 6b and Extended Data Fig. 9) and ligands of the sigma-1 and sigma-2 receptors (haloperidol, PB28, PD-144418 and hydroxychloroquine, which is undergoing clinical trials in patients with COVID-19 (ClinicalTrials.gov, trial number NCT04332991)). We also subsequently found that the sigma-1- and sigma-2-receptor active drugs clemastine, cloperastine and progesterone, and the clinical molecule siramesine, were antiviral drugs (Fig. 6c and Extended Data Fig. 9). Median tissue culture infectious dose (TCID50) assays on supernatants from infected cells treated with PB28 (90% inhibitory concentration (IC90) = 0.278 μM) and zotatifin (IC90 = 0.037 μM) revealed a more potent inhibition than was observed in the NP-staining assay (Fig. 6d). Notably, in this assay, PB28 was around 20 times more potent than hydroxychloroquine (IC90 = 5.78 μM).

a, Schematic of viral infectivity assays. b, The mRNA translation inhibitors (zotatifin and ternatin-4) reduce viral infectivity in a concentration-dependent manner. Red, viral infectivity (anti-NP or plaque assays); blue, RT–qPCR; black, cell viability. The initial decrease in cell viability probably reflects cytostatic and not cytotoxic effects. Data are mean ± s.d.; n = 6 biologically independent samples for cell viability data and DMSO controls from Paris; all other data, n = 3. PFU, plaque-forming unit. c, Sigma receptor drugs and preclinical molecules inhibit viral infectivity (coloured as in b). Data are mean ± s.d.; n = 3 biologically independent samples. d, TCID50 assays using zotatifin, PB28 and hydroxychloroquine. e, Drugs added before or after high-titre virus (MOI = 2) addition had similar antiviral effects. Data are mean ± s.d.; n = 3 biologically independent samples. f, Sigma-1 and sigma-2 receptors are the common targets of the sigma ligands at the 1-μM activity threshold55. g, Dextromethorphan increases viral titres. Red, viral titre (plaque assay); black, cell viability. Data are mean ± s.d.; n = 3 biologically independent samples. h, On-target Kd values for the sigma-1 and sigma-2 receptor compared with the Kd values for the hERG ion channel. PB28 and PD-144418 show 500- to 5,000-fold selectivity, whereas chloroquine and hydroxychloroquine have about 30-fold selectivity for these targets. pKi values for hERG compared with sigma-1 receptor compared with sigma-2 receptor are: chloroquine (5.5 ± 0.1; 7.1 ± 0.1; 6.3 ± 0.1); hydroxychloroquine (5.6 ± 0.2; 6.9 ± 0.2; 6.0 ± 0.1); PB28 (6.0 ± 0.1; 8.7 ± 0.1; 8.6 ± 0.1); PD-144418 (5.0 ± 0.2; 8.7 ± 0.1; 6.1 ± 0.1); clemastine (6.8 ± 0.2; 8.0 ± 0.1; 7.6 ± 0.1). Data are mean ± s.d.; PB28, clemastine, PD-144418, n = 9 biologically independent samples for sigma-1 and sigma-2 receptors and hERG; chloroquine, hydroxychloroquine, n = 6 for sigma-1 and sigma-2 receptors and n = 4 for hERG.
To better understand the mechanism by which these inhibitors exert their antiviral effects, we performed a time course assay in which the drugs were added at different times before or after infection (Fig. 6e). Cells were infected during a single cycle of infection at high multiplicity of infection (MOI = 2) over the course of 8 h, and the drugs were added either 2 h before infection or at 0, 2 or 4 h after infection. PB28, zotatifin and hydroxychloroquine all decreased the detection of the viral NP protein even in this single cycle assay, indicating that the antiviral effect occurs before viral egress from the cell (Fig. 6e). Furthermore, all three molecules inhibited NP expression when added up to 4 h after infection, after viral entry has occurred. Thus, these molecules seem to exert their antiviral effect during viral replication.
Coronaviruses rely on cap-dependent mRNA translation through the translation machinery of the host. eIF4H, which interacts with NSP9, is a partner of eIF4A, and we observed a strong antiviral effect after treatment with the eIF4A inhibitor zotatifin (Fig. 6b), which is currently in a phase-I clinical trial for the treatment of cancer. We also observed potent antiviral effects of the elongation factor-1A (eEF1A) inhibitor ternatin-449 (Fig. 6b), which may suggest that the rate of translation elongation is critical for obtaining optimal levels of viral proteins. Of note, the eEF1A inhibitor aplidin/plitidepsin is used clinically in patients with multiple myeloma50. Multiple SARS-CoV-2 proteins are predicted to undergo SRP- and Sec61-mediated co-translational insertion into the endoplasmic reticulum, and SRP19, SRP54 and SRP72 were identified as NSP8-interacting proteins (Fig. 3). Consistent with previous studies of flaviviruses42, the Sec61 inhibitor PS3061 also blocked SARS-CoV-2 replication (Extended Data Fig. 9). The two translation inhibitors had cytostatic effects in uninfected Vero cells, which are immortalized cell lines with indefinite proliferative capacity that have mutations in key cell cycle inhibitors. These cells are more sensitive to anti-cancer compounds, which affect the cell cycle state of immortalized cells more strongly than non-immortalized cells. A critical question going forward is whether these or related inhibitors of viral protein biogenesis will show therapeutic benefits in patients with COVID-19. Plitidepsin is currently under consideration by the Spanish Medicines Agency for a phase-II trial in hospitalized patients with COVID-19.
Molecules that target the sigma-1 and sigma-2 receptors perturb the virus through different mechanisms than the translation inhibitors, which could include the cell stress response51. These molecules are also active against other aminergic receptors; however, the only targets shared among all of the tested molecules are the sigma-1 and sigma-2 receptors (Fig. 6f), into which these drugs can be readily modelled (Extended Data Fig. 10a). For instance, the antipsychotic drug haloperidol inhibits the dopamine D2 and histamine H1 receptors, whereas clemastine and cloperastine are antihistamines; each of these drugs is also a Sigma receptor ligand with antiviral activity (Fig. 6c). Conversely, the antipsychotic drug olanzapine, which also inhibits histamine H1 and dopamine D2 receptors, has little Sigma receptor activity and does not show antiviral activity (Extended Data Fig. 10b). Which of the Sigma receptors is responsible for the activity remains uncertain, as does the role of pharmacologically related targets, such as EBP and related sterol isomerases, the ligand recognition of which resembles those of the Sigma receptors. Notably, the sigma-1-receptor benzomorphan agonist dextromethorphan has proviral activity (Fig. 6g), further supporting the role of these receptors in viral infection. Overall, two features should be emphasized. First, several of the molecules that target the Sigma receptors, such as clemastine, cloperastine and progesterone, are approved drugs with a long history in human therapy. Many other widely used drugs, which show activity against the Sigma receptors, remain to be tested; and indeed, several drugs such as astemizole, which we show is a sigma-2 receptor ligand (with an Ki of 95 nM) (Extended Data Fig. 11), verapamil and amiodarone, have been reported by others to be active in viral replication assays, although this has not been linked to their Sigma receptor activity52,53. Second, the Sigma receptor ligands have a clear separation between antiviral and cytotoxic effects (Fig. 6b, c), and ligands such as PB28 have substantial selectivity for the Sigma receptors compared with side-effect targets, such as the hERG ion channel. Indeed, the lack of selectivity of chloroquine and hydroxychloroquine for hERG (Fig. 6h) and other off-targets (Extended Data Fig. 12) may be related to the adverse cardiac drug reactions54 that have limited their use.
Discussion
In this study, we have identified 332 high-confidence SARS-CoV-2 protein–human protein interactions that are connected with multiple biological processes, including protein trafficking, translation, transcription and regulation of ubiquitination. We found 69 ligands, including FDA-approved drugs, compounds in clinical trials and preclinical compounds, that target these interactions. Antiviral tests in two different laboratories reveal two broad sets of active drugs and compounds; those that affect translation and those modulate the sigma-1 and sigma-2 receptors. Within these sets are at least five targets and more than ten different chemotypes, providing a rich landscape for optimization.
The chemo-proteomic analysis that emerges from this study not only highlights clinically actionable drugs that target human proteins in the interactome, but also provides a context for interpreting their mechanism of action. The potent efficacy of the translation inhibitors on viral infectivity—in the 10 to 100 nM range—makes these molecules attractive as candidate antiviral agents, and also highlights this pathway as a point of intervention. Although the mechanism of action of the drugs that target the sigma-1 and sigma-2 receptors remains less defined, their activity as both anti- and proviral agents is mechanistically suggestive. The relatively strong efficacy of PB28, at an IC90 of 280 nM in the viral titre assay, and its high selectivity against off-target proteins, suggests that molecules of this class may be optimized as therapeutic agents. Although it is unclear whether approved drugs such as clemastine and cloperastine, which are used as antihistamines and antitussive drugs, have pharmacokinetics that are suitable for antiviral therapy, and although they are not free of binding to targets that cause side effects (Fig. 6f and Extended Data Fig. 12), these drugs have been used for decades. We caution against their use outside of controlled studies, because of their side-effect liabilities. By the same standard, we find that the widely used antitussive drug dextromethorphan has proviral activity and that therefore its use should merit caution and further study in the context of the treatment of COVID-19. More positively, there are dozens of approved drugs that show activity against the Sigma receptors, not all of which are generally recognized as Sigma receptor ligands. Many of these drugs remain to be tested as a treatment for COVID-19; although some have begun to appear in other studies52,53. This area of pharmacology has great promise for the repurposing and optimization of new agents in the fight against COVID-19.
Our approach of host-directed intervention as an antiviral strategy overcomes problems associated with drug resistance and may also provide panviral therapies as we prepare for the next pandemic. Furthermore, the possibilities for cotherapies are expanded—for example, with drugs that directly target the virus, including remdesivir—and, as we demonstrate in this study, there are numerous opportunities for the repurposing of FDA-approved drugs. More broadly, the pipeline described here represents an approach for drug discovery not only for panviral strategies, but also for the research of many diseases, and illustrates the speed with which science can be moved forward using a multi-disciplinary and collaborative approach.
Methods
Genome annotation
The GenBank sequence for SARS-CoV-2 isolate 2019-nCoV/USA-WA1/2020, accession MN985325, was downloaded on 24 January 2020. In total, we annotated 29 possible ORFs and proteolytically mature proteins encoded by SARS-CoV-21,12. Proteolytic products that result from NSP3- and NSP5-mediated cleavage of the ORF1a/ORF1ab polyprotein were predicted on the basis of the protease specificity of SARS-CoV proteases56, and 16 predicted nonstructural proteins (NSPs) were subsequently cloned (NSP1–NSP16). For the NSP5 protease (3Clike/3CLpro), we also designed the catalytically dead mutant NSP5(C145A)57,58. ORFs at the 3′ end of the viral genome annotated in the original GenBank file included 4 structural proteins: S, E, M, N and the additional ORFs ORF3a, ORF6, ORF7a, ORF8 and ORF10. On the basis of the analysis of ORFs in the genome and comparisons with other annotated SARS-CoV ORFs, we annotated a further four ORFs: ORF3b, ORF7b, ORF9b and ORF9c.
Cloning
ORFs and proteolytically mature NSPs annotated in the SARS-CoV-2 genome were human codon-optimized using the IDT codon-optimization tool (https://www.idtdna.com/codonopt) and internal EcoRI and BamHI sites were eliminated. Start and stop codons were added as necessary to NSPs 1–16, a Kozak sequence was added before each start codon, and a 2×Strep tag with linker was added to either the N or C terminus. To guide our tagging strategy, we used GPS-Lipid to predict protein lipid modification of the termini (http://lipid.biocuckoo.org/webserver.php)59,60, TMHMM Server v.2.0 to predict transmembrane/hydrophobic regions (http://www.cbs.dtu.dk/services/TMHMM/)61 and SignalP v.5.0 to predict signal peptides (http://www.cbs.dtu.dk/services/SignalP/)62. IDT gBlocks were ordered for all reading frames with 15-bp overlaps that corresponded to flanking sequences of the EcoRI and BamHI restriction sites in the lentiviral constitutive expression vector pLVX-EF1alpha-IRES-Puro (Takara). Vectors were digested and gel-purified, and gene fragments were cloned using InFusion (Takara). The S protein was synthesized and cloned into pTwist-EF1alpha-IRES-Puro (Twist Biosciences). NSP16 showed multiple mutations that could not be repaired before the time-sensitive preparation of this manuscript, and NSP3 was too large to be synthesized in time to be included in this study. Strep-tagged constructs encoding NSP3, NSP3(C857A) (catalytically dead mutant) and NSP16 will be used in future AP-MS experiments.
Cell culture
HEK-293T/17 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Corning) supplemented with 10% fetal bovine serum (FBS; Gibco, Life Technologies) and 1% penicillin–streptomycin (Corning) and maintained at 37 °C in a humidified atmosphere of 5% CO2. HEK-293T/17 cells were procured from the UCSF Cell Culture Facility, now available through UCSF’s Cell and Genome Engineering Core ((https://cgec.ucsf.edu/cell-culture-and-banking-services); cell line collection listed here: https://ucsf.app.box.com/s/6xkydeqhr8a2xes0mbo2333i3k1lndqv (CCLZR076)). STR analysis by the Berkeley Cell Culture Facility on 8 August 2017 authenticated HEK-293T/17 cells with 94% probability. Cells were tested on 3 July 2019 using the MycoAlert Mycoplasma Detection Kit (Lonza LT07-318) and were negative: B/A ratio < 1 (no detected mycoplasma).
Transfection
For each affinity purification (26 wild-type baits and one catalytically dead SARS-CoV-2 bait, one GFP control and one empty vector control), ten million HEK-293T/17 cells were plated per 15-cm dish and transfected with up to 15 μg of individual Strep-tagged expression constructs after 20–24 h. Total plasmid was normalized to 15 μg with empty vector and complexed with PolyJet Transfection Reagent (SignaGen Laboratories) at a 1:3 μg:μl ratio of plasmid:transfection reagent based on the manufacturer’s recommendations. After more than 38 h, cells were dissociated at room temperature using 10 ml Dulbecco’s phosphate-buffered saline without calcium and magnesium (DPBS) supplemented with 10 mM EDTA for at least 5 min and subsequently washed with 10 ml DPBS. Each step was followed by centrifugation at 200g, 4 °C for 5 min. Cell pellets were frozen on dry ice and stored at −80 °C. For each bait, n = 3 independent biological replicates were prepared for affinity purification.
Affinity purification
Frozen cell pellets were thawed on ice for 15–20 min and resuspended in 1 ml lysis buffer (IP buffer (50 mM Tris-HCl, pH 7.4 at 4 °C, 150 mM NaCl, 1 mM EDTA) supplemented with 0.5% Non-idet P40 substitute (NP40; Fluka Analytical) and cOmplete mini EDTA-free protease and PhosSTOP phosphatase inhibitor cocktails (Roche)). Samples were then frozen on dry ice for 10–20 min and partially thawed at 37 °C before incubation on a tube rotator for 30 min at 4 °C and centrifugation at 13,000g, 4 °C for 15 min to pellet debris. After reserving 50 μl lysate, up to 48 samples were arrayed into a 96-well Deepwell plate for affinity purification on the KingFisher Flex Purification System (Thermo Scientific) as follows: MagStrep ‘type3’ beads (30 μl; IBA Lifesciences) were equilibrated twice with 1 ml wash buffer (IP buffer supplemented with 0.05% NP40) and incubated with 0.95 ml lysate for 2 h. Beads were washed three times with 1 ml wash buffer and then once with 1 ml IP buffer. To directly digest bead-bound proteins as well as elute proteins with biotin, beads were manually suspended in IP buffer and divided in half before transferring to 50 μl denaturation–reduction buffer (2 M urea, 50 mM Tris-HCl pH 8.0, 1 mM DTT) and 50 μl 1× buffer BXT (IBA Lifesciences) dispensed into a single 96-well KF microtitre plate. Purified proteins were first eluted at room temperature for 30 min with constant shaking at 1,100 rpm on a ThermoMixer C incubator. After removing eluates, on-bead digestion proceeded (see ‘On-bead digestion’). Strep-tagged protein expression in lysates and enrichment in eluates were assessed by western blot and silver stain, respectively. The KingFisher Flex Purification System was placed in the cold room and allowed to equilibrate to 4 °C overnight before use. All automated protocol steps were performed using the slow mix speed and the following mix times: 30 s for equilibration and wash steps, 2 h for binding and 1 min for final bead release. Three 10-s bead collection times were used between all steps.
On-bead digestion
Bead-bound proteins were denatured and reduced at 37 °C for 30 min and after being brought to room temperature, alkylated in the dark with 3 mM iodoacetamide for 45 min and quenched with 3 mM DTT for 10 min. Proteins were then incubated at 37 °C, initially for 4 h with 1.5 μl trypsin (0.5 μg/μl; Promega) and then another 1–2 h with 0.5 μl additional trypsin. To offset evaporation, 15 μl 50 mM Tris-HCl, pH 8.0 were added before trypsin digestion. All steps were performed with constant shaking at 1,100 rpm on a ThermoMixer C incubator. Resulting peptides were combined with 50 μl 50 mM Tris-HCl, pH 8.0 used to rinse beads and acidified with trifluoroacetic acid (0.5% final, pH < 2.0). Acidified peptides were desalted for MS analysis using a BioPureSPE Mini 96-Well Plate (20 mg PROTO 300 C18; The Nest Group) according to standard protocols.
MS data acquisition and analysis
Samples were resuspended in 4% formic acid, 2% acetonitrile solution, and separated by a reversed-phase gradient over a Nanoflow C18 column (Dr Maisch). Each sample was directly injected via an Easy-nLC 1200 (Thermo Fisher Scientific) into a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific) and analysed with a 75 min acquisition, with all MS1 and MS2 spectra collected in the orbitrap; data were acquired using the Thermo software Xcalibur (4.2.47) and Tune (2.11 QF1 Build 3006). For all acquisitions, QCloud was used to control instrument longitudinal performance during the project63. All proteomic data were searched against the human proteome (Uniprot-reviewed sequences downloaded 28 February 2020), the eGFP sequence and the SARS-CoV-2 protein sequences using the default settings for MaxQuant (v.1.6.11.0)64,65. Detected peptides and proteins were filtered to 1% false-discovery rate in MaxQuant, and identified proteins were then subjected to protein–protein interaction scoring with both SAINTexpress (v.3.6.3)14 and MiST (https://github.com/kroganlab/mist)15,66. We applied a two-step filtering strategy to determine the final list of reported interactors, which relied on two different scoring stringency cut-offs. In the first step, we chose all protein interactions that had a MiST score ≥ 0.7, a SAINTexpress Bayesian false-discovery rate (BFDR) ≤ 0.05 and an average spectral count ≥ 2. For all proteins that fulfilled these criteria, we extracted information about the stable protein complexes that they participated in from the CORUM67 database of known protein complexes. In the second step, we then relaxed the stringency and recovered additional interactors that (1) formed complexes with interactors determined in filtering step 1 and (2) fulfilled the following criteria: MiST score ≥ 0.6, SAINTexpress BFDR ≤ 0.05 and average spectral counts ≥ 2. Proteins that fulfilled filtering criteria in either step 1 or step 2 were considered to be high-confidence protein–protein interactions (HC-PPIs) and visualized with Cytoscape (v.3.7.1)68. Using this filtering criteria, nearly all of our baits recovered a number of HC-PPIs in close alignment with previous datasets reporting an average of around 6 PPIs per bait69. However, for a subset of baits (ORF8, NSP8, NSP13 and ORF9c), we observed a much higher number of PPIs that passed these filtering criteria. For these four baits, the MiST scoring was instead performed using a larger in-house database of 87 baits that were prepared and processed in an analogous manner to this SARS-CoV-2 dataset. This was done to provide a more comprehensive collection of baits for comparison, to minimize the classification of non-specifically binding background proteins as HC-PPIs. All MS raw data and search results files have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD01811770,71. PPI networks have also been uploaded to NDEx.
GO overrepresentation analysis
The targets of each bait were tested for enrichment of GO biological process terms. The overrepresentation analysis was based on the hypergeometric distribution and performed using the enricher function of clusterProfiler package in R with default parameters. The GO terms were obtained from the c5 category of Molecular Signature Database (MSigDBv6.1). Significant GO terms (1% false-discovery rate) were identified and further refined to select non-redundant terms. To select non-redundant gene sets, we first constructed a GO term tree based on distances (1 − Jaccard similarity coefficients of shared genes) between the significant terms. The GO term tree was cut at a specific level (h = 0.99) to identify clusters of non-redundant gene sets. For a bait with multiple significant terms belonging to the same cluster, we selected the broadest term that is, largest gene set size.
Virus interactome similarity analysis
Interactome similarity was assessed by comparing the number of shared human-interacting proteins between pathogen pairs, using a hypergeometric test to calculate significance. The background gene set for the test consisted of all unique proteins detected by MS across all pathogens (n = 10,181 genes).
ORF6 peptide modelling
The proposed interaction between ORF6 and the NUP98–RAE1 complex was modelled in PyRosetta 4 (release v.2020.02-dev61090)72 using the crystal structure of VSV matrix (M) protein bound to NUP98–RAE1 as a template32 (PDB 4OWR; downloaded from the PDB-REDO server73). The M protein chain (C) was truncated after residue 54 to restrict the model to the putative interaction motif in ORF6 (M protein residues 49–54, sequence DEMDTH). These residues were mutated to the ORF6 sequence, QPMEID, using the mutate_residue function in the module pyrosetta.toolbox, without repacking at this initial step. After all six residues were mutated, the full model was relaxed to a low-energy conformation using the FastRelax protocol in the module pyrosetta.rosetta.protocols.relax. FastRelax was run with constraints to starting coordinates and scored with the ref2015 score function. The resulting model was inspected for any large energetic penalties associated with the modelled peptide residues or those NUP98 and RAE1 residues interacting with the peptide, and was found to have none. The model was visualized in PyMOL (The PyMOL Molecular Graphics System, v.3.4, Schrödinger).
ORF10 secondary structure prediction
The secondary structure of ORF10 was predicted using JPRED (https://www.compbio.dundee.ac.uk/jpred/index.html)74.
Protein E alignment
Protein E sequences from SARS-CoV-2 (YP_009724392.1), SARS-CoV (NP_828854.1) and bat SARS-like CoV (AGZ48809.1) were aligned using Clustal Omega75, and then manually aligned to the sequences of histone H3 (P68431) and influenza A H3N2 NS1 (YP_308845.1).
Chemoinformatic analysis of SARS-CoV-2-interacting partners
To identify drugs and reagents that modulate the 332 host factors that interact with SARS-CoV-2 and HEK-293T/17 cells (MiST ≥ 0.70), we used two approaches: (1) a chemoinformatic analysis of open-source chemical databases and (2) a target- and pathway-specific literature search, drawing on specialist knowledge within our group. Chemoinformatically, we retrieved 2,472 molecules from the IUPHAR/BPS Guide to Pharmacology (2020-3-12)55 (Supplementary Table 7) that interacted with 30 human ‘prey’ proteins (38 approved, 71 in clinical trials), and found 10,883 molecules (95 approved, 369 in clinical trials) from the ChEMBL25 database76 (Supplementary Table 8). For both approaches, molecules were prioritized on their FDA approval status, activity at the target of interest better than 1 μM and commercial availability, drawing on the ZINC database77. FDA-approved molecules were prioritized except when clinical candidates or preclinical research molecules had substantially better selectivity or potency on-target. In some cases, we considered molecules with indirect mechanisms of action on the general pathway of interest based solely on literature evidence (for example, captopril modulates ACE2 indirectly via its direct interaction with angiotensin-converting enzyme, ACE). Finally, we predicted 6 additional molecules (2 approved, 1 in clinical trials) for proteins with MIST scores between 0.7 and 0.6 to viral baits (Supplementary Tables 4, 5). Complete methods can be found at https://github.com/momeara/BioChemPantry/tree/master/vignette/COVID19.
Molecular docking
After their chemoinformatic assignment to the sigma-1 receptor, cloperastine and clemastine were docked into the agonist-bound state structure of the receptor (6DK1)78 using DOCK3.779. The best scoring configurations that ion pair with Glu172 are shown; both l-cloperastine and clemastine receive solvation-corrected docking scores between −42 and −43 kcal/mol, indicating high complementarity.
Viral growth and cytotoxicity assays in the presence of inhibitors
For studies carried out at Mount Sinai, SARS-CoV-2 (isolate USA-WA1/2020 from BEI RESOURCES NR-52281) was propagated in Vero E6 cells. Two thousand Vero E6 cells were seeded into 96-well plates in DMEM (10% FBS) and incubated for 24 h at 37 °C, 5% CO2. Vero E6 cells used were purchased from ATCC and thus authenticated (VERO C1008 (Vero 76, clone E6, Vero E6) (ATCC CRL-1586); tested negative for mycoplasma contamination before commencement). Then, 2 h before infection, the medium was replaced with 100 μl of DMEM (2% FBS) containing the compound of interest at concentrations 50% greater than those indicated, including a DMSO control. The Vero E6 cell line used in this study is a kidney cell line; therefore, we cannot exclude that lung cells yield different results for some inhibitors (see also ‘Cells and viruses’, ‘Antiviral activity assays’, ‘Cell viability assays’ and ‘Plaque-forming assays’ for studies carried out at Institut Pasteur). Plates were then transferred into the Biosafety Level 3 (BSL3) facility and 100 PFU (MOI = 0.025) was added in 50 μl of DMEM (2% FBS), bringing the final compound concentration to those indicated. Plates were then incubated for 48 h at 37 °C. After infection, supernatants were removed and cells were fixed with 4% formaldehyde for 24 h before being removed from the BSL3 facility. The cells were then immunostained for the viral NP protein (anti-sera produced in the García -Sastre laboratory; 1:10,000) with a DAPI counterstain. Infected cells (488 nM) and total cells (DAPI) were quantified using the Celigo (Nexcelcom) imaging cytometer. Infectivity is measured by the accumulation of viral NP protein in the nucleus of the Vero E6 cells (fluorescence accumulation). Percentage infection was quantified as ((infected cells/total cells) − background) × 100 and the DMSO control was then set to 100% infection for analysis. The IC50 and IC90 for each experiment were determined using the Prism (GraphPad) software. For some inhibitors, infected supernatants were assayed for infectious viral titres using the TCID50 method. For this, infectious supernatants were collected at 48 h after infection and frozen at −80 °C until later use. Infectious titres were quantified by limiting dilution titration on Vero E6 cells. In brief, Vero E6 cells were seeded in 96-well plates at 20,000 cells per well. The next day, SARS-CoV-2-containing supernatant was applied at serial tenfold dilutions ranging from 10−1 to 10−6 and, after 5 days, viral cytopathogenic effect was detected by staining cell monolayers with crystal violet. The TCID50/ml values were calculated using the previously described method80. Cytotoxicity was also performed using the MTT assay (Roche), according to the manufacturer’s instructions. Cytotoxicity was performed in uninfected VeroE6 cells with same compound dilutions and concurrent with viral replication assay. All assays were performed in biologically independent triplicates.
Cells and viruses
For studies at the Institut Pasteur, African green monkey kidney epithelial Vero E6 cells (ATCC, CRL-1586, authenticated by ATCC and tested negative for mycoplasma contamination before commencement (Vero 76, clone E6, Vero E6) (ATCC CRL-1586)) were maintained in a humidified atmosphere at 37 °C with 5% CO2, in DMEM containing 10% (v/v) FBS (Invitrogen) and 5 units/ml penicillin and 5 μg/ml streptomycin (Life Technologies). The Vero E6 cell line used in this study is a kidney cell line; therefore, we cannot exclude that lung cells yield different results for some inhibitors (see also ‘Viral growth and cytotoxicity assays in the presence of inhibitors’ for studies performed at Mount Sinai). SARS-CoV-2, isolate France/IDF0372/2020, was supplied by the National Reference Centre for Respiratory Viruses hosted by the Institut Pasteur and headed by S. van der Werf. The human sample from which strain BetaCoV/France/IDF0372/2020 was isolated has been provided by X. Lescure and Y. Yazdanpanah from the Bichat Hospital, Paris, France. The BetaCoV/France/IDF0372/2020 strain was supplied through the European Virus Archive goes Global (Evag) platform, a project that has received funding from the European Union’s Horizon 2020 research and innovation programme under the grant agreement no. 653316. Viral stocks were prepared by propagation in Vero E6 cells in DMEM supplemented with 2% FBS and 1 μg/ml TPCK-trypsin (Sigma-Aldrich). Viral titres were determined by plaque-forming assay in minimum essential medium supplemented with 2% (v/v) FBS (Invitrogen) and 0.05% agarose. All experiments involving live SARS-CoV-2 were performed at the Institut Pasteur in compliance with the guidelines of the Institut Pasteur following BSL3 containment procedures in approved laboratories. All experiments were performed in at least three biologically independent samples.
Antiviral activity assays
Vero E6 cells were seeded at 1.5 × 104 cells per well in 96-well plates 18 h before the experiment. Then, 2 h before infection, the cell-culture supernatant of triplicate wells was replaced with medium containing 10 μM, 2 μM, 500 nM, 200 nM, 100 nM or 10 nM of each compound or the equivalent volume of maximum DMSO vehicle used as a control. At the time of infection, the drug-containing medium was removed, and replaced with virus inoculum (MOI of 0.1 PFU per cell) containing TPCK-trypsin (Sigma-Aldrich). Following a 1-h adsorption incubation at 37 °C, the virus inoculum was removed and 200 μl of drug- or vehicle-containing medium was added. Then, 48 h after infection, the cell-culture supernatant was used to extract RNA using the Direct-zol-96 RNA extraction kit (Zymo) following the manufacturer’s instructions. Detection of viral genomes in the extracted RNA was performed by RT–qPCR, using previously published SARS-CoV-2-specific primers81. Specifically, the primers target the N gene region: 5′-TAATCAGACAAGGAACTGATTA-3′ (forward) and 5′-CGAAGGTGTGACTTCCATG-3′ (reverse). RT–qPCR was performed using the Luna Universal One-Step RT–qPCR Kit (NEB) in an Applied Biosystems QuantStudio 6 thermocycler, using the following cycling conditions: 55 °C for 10 min, 95 °C for 1 min, and 40 cycles of 95 °C for 10 s, followed by 60 °C for 1 min. The quantity of viral genomes is expressed as PFU equivalents, and was calculated by performing a standard curve with RNA derived from a viral stock with a known viral titre. In addition to measuring viral RNA in the supernatant derived from drug-treated cells, infectious virus was quantified by plaque-forming assay.
Cell viability assays
Cell viability in drug-treated cells was measured using Alamar blue reagent (ThermoFisher). In brief, 48 h after treatment, the drug-containing medium was removed and replaced with Alamar blue and incubated for 1 h at 37 °C and fluorescence measured in a Tecan Infinity 2000 plate reader. Percentage viability was calculated relative to untreated cells (100% viability) and cells lysed with 20% ethanol (0% viability), included in each plate.
Plaque-forming assays
Viruses were quantified by plaque-forming assays. For this, Vero E6 cells were seeded in 24-well plates at a concentration of 7.5 × 104 cells per well. The following day, tenfold serial dilutions of individual virus samples in serum-free MEM medium were added to infect the cells at 37 °C for 1 h. After the adsorption time, the overlay medium was added at final concentration of 2% FBS/MEM medium and 0.05% agarose to achieve a semi-solid overlay. Plaque-forming assays were incubated at 37 °C for 3 days before fixation with 4% formalin and visualization using crystal violet solution.
Off-target assays for Sigma receptor drugs and ligands
hERG binding assays were carried out as previously described82. In brief, compounds were incubated with hERG membranes, prepared from HEK-293 cells stably expressing hERG channels, and [3H]dofetilide (5 nM final) in a total of 150 μl for 90 min at room temperature in the dark. Reactions were stopped by filtering the mixture onto a glass fibre and were quickly washed three times to remove unbound [3H]dofetilide. The filter was dried in a microwave, melted with a scintillant cocktail and wrapped in a plastic film. Radioactivity was counted on a MicroBeta counter and results were analysed in Prism by fitting to the built-in one binding function to obtain affinity Ki. Radioligand binding assays for the muscarinic and alpha-adrenergic receptors were performed as previously described83. Detailed protocols are available on the NIMH PDSP website at https://pdspdb.unc.edu/html/tutorials/UNC-CH%20Protocol%20Book.pdf.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The AP-MS raw data and search results files generated during the current study are available in the ProteomeXchange Consortium via the PRIDE partner repository with dataset identifier PXD018117 (https://www.ebi.ac.uk/pride/archive/projects/PXD018117) and PPI networks have also been uploaded to NDEx (https://public.ndexbio.org/#/network/43803262-6d69-11ea-bfdc-0ac135e8bacf). An interactive version of these networks, including relevant drug and functional information, can be found at http://kroganlab.ucsf.edu/network-maps. All data generated or analysed during this study are included in the article and its Supplementary Information. Expression vectors used in this study are readily available from the authors to biomedical researchers and educators in the non-profit sector. Source data are provided with this paper.
Code availability
Complete methods for chemoinformatic analysis can be found on GitHub (https://github.com/momeara/BioChemPantry/tree/master/vignette/COVID19); details on MIST scoring can be found on GitHub (https://github.com/kroganlab/mist).
References
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
WHO. Coronavirus disease (COVID-2019) situation reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (2020).
Wang, C., Horby, P. W., Hayden, F. G. & Gao, G. F. A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Su, S. et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 24, 490–502 (2016).
Gates, B. Responding to Covid-19 — a once-in-a-century pandemic? N. Engl. J. Med. 382, 1677–1679 (2020).
Sheahan, T. P. et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 11, 222 (2020).
Sheahan, T. P. et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 12, eabb5883 (2020).
Paton, J. Moderna’s coronavirus vaccine trial set to begin this month. Bloomberg News (6 March 2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Prussia, A., Thepchatri, P., Snyder, J. P. & Plemper, R. K. Systematic approaches towards the development of host-directed antiviral therapeutics. Int. J. Mol. Sci. 12, 4027–4052 (2011).
Chan, J. F.-W. et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 9, 221–236 (2020).
Fehr, A. R. & Perlman, S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 1282, 1–23 (2015).
Teo, G. et al. SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics 100, 37–43 (2014).
Jäger, S. et al. Global landscape of HIV–human protein complexes. Nature 481, 365–370 (2012).
Bojkova, D. et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature https://doi.org/10.1038/s41586-020-2332-7 (2020).
Eckhardt, M., Hultquist, J. F., Kaake, R. M., Hüttenhain, R. & Krogan, N. J. A systems approach to infectious disease. Nat. Rev. Genet. 21, 339–354 (2020).
Harcourt, J. et al. Severe acute respiratory syndrome coronavirus 2 from patient with coronavirus disease, United States. Emerg. Infect. Dis. 26, 1266–1273 (2020).
Wang, D. et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15, e8503 (2019).
Li, M. et al. Identification of antiviral roles for the exon-junction complex and nonsense-mediated decay in flaviviral infection. Nat. Microbiol. 4, 985–995 (2019).
Penn, B. H. et al. An Mtb-human protein–protein interaction map identifies a switch between host antiviral and antibacterial responses. Mol. Cell 71, 637–648 (2018).
Barnes, P. J. Role of HDAC2 in the pathophysiology of COPD. Annu. Rev. Physiol. 71, 451–464 (2009).
Xu, P. et al. NOS1 inhibits the interferon response of cancer cells by S-nitrosylation of HDAC2. J. Exp. Clin. Cancer Res. 38, 483 (2019).
Dewe, J. M., Fuller, B. L., Lentini, J. M., Kellner, S. M. & Fu, D. TRMT1-catalyzed tRNA modifications are required for redox homeostasis to ensure proper cellular proliferation and oxidative stress survival. Mol. Cell. Biol. 37, e00214-17 (2017).
Kondo, T., Watanabe, M. & Hatakeyama, S. TRIM59 interacts with ECSIT and negatively regulates NF-κB and IRF-3/7-mediated signal pathways. Biochem. Biophys. Res. Commun. 422, 501–507 (2012).
Li, S., Wang, L., Berman, M., Kong, Y.-Y. & Dorf, M. E. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35, 426–440 (2011).
Faria, P. A. et al. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol. Cell 17, 93–102 (2005).
Slaine, P. D., Kleer, M., Smith, N. K., Khaperskyy, D. A. & McCormick, C. Stress granule-inducing eukaryotic translation initiation factor 4A inhibitors block influenza A virus replication. Viruses 9, 388 (2017).
Reineke, L. C. et al. Casein kinase 2 is linked to stress granule dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol. Cell. Biol. 37, e00596-16 (2017).
Kindrachuk, J. et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 59, 1088–1099 (2015).
Timms, R. T. et al. A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science 365, eaaw4912 (2019).
Quan, B., Seo, H.-S., Blobel, G. & Ren, Y. Vesiculoviral matrix (M) protein occupies nucleic acid binding site at nucleoporin pair (Rae1•Nup98). Proc. Natl Acad. Sci. USA 111, 9127–9132 (2014).
Frieman, M. et al. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81, 9812–9824 (2007).
Nakagawa, K., Narayanan, K., Wada, M. & Makino, S. Inhibition of stress granule formation by Middle East respiratory syndrome coronavirus 4a accessory protein facilitates viral translation, leading to efficient virus replication. J. Virol. 92, e00902-18 (2018).
Raaben, M., Groot Koerkamp, M. J. A., Rottier, P. J. M. & de Haan, C. A. M. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell. Microbiol. 9, 2218–2229 (2007).
Ivanov, P., Kedersha, N. & Anderson, P. Stress granules and processing bodies in translational control. Cold Spring Harb. Perspect. Biol. 11, a032813 (2019).
Thompson, P. A. et al. Abstract 2698: eFT226, a potent and selective inhibitor of eIF4A, is efficacious in preclinical models of lymphoma. Cancer Res. 79, 2698 (2019).
Nakagawa, K., Lokugamage, K. G. & Makino, S. Viral and cellular mRNA translation in coronavirus-infected cells. Adv. Virus Res. 96, 165–192 (2016).
Müller, C. et al. Broad-spectrum antiviral activity of the eIF4A inhibitor silvestrol against corona- and picornaviruses. Antiviral Res. 150, 123–129 (2018).
Cencic, R. et al. Blocking eIF4E–eIF4G interaction as a strategy to impair coronavirus replication. J. Virol. 85, 6381–6389 (2011).
Knoops, K. et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6, e226 (2008).
Shah, P. S. et al. Comparative flavivirus–host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell 175, 1931–1945 (2018).
Heaton, N. S. et al. Targeting viral proteostasis limits influenza virus, HIV, and dengue virus infection. Immunity 44, 46–58 (2016).
Mahon, C., Krogan, N. J., Craik, C. S. & Pick, E. Cullin E3 ligases and their rewiring by viral factors. Biomolecules 4, 897–930 (2014).
Soucy, T. A. et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009).
Faivre, E. J. et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 578, 306–310 (2020).
Filippakopoulos, P. et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231 (2012).
Marazzi, I. et al. Suppression of the antiviral response by an influenza histone mimic. Nature 483, 428–433 (2012).
Carelli, J. D. et al. Ternatin and improved synthetic variants kill cancer cells by targeting the elongation factor-1A ternary complex. eLife 4, e10222 (2015).
Spicka, I. et al. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Ann. Hematol. 98, 2139–2150 (2019).
Mitsuda, T. et al. Sigma-1Rs are upregulated via PERK/eIF2α/ATF4 pathway and execute protective function in ER stress. Biochem. Biophys. Res. Commun. 415, 519–525 (2011).
Si, L. et al. Human organs-on-chips as tools for repurposing approved drugs as potential influenza and COVID19 therapeutics in viral pandemics. Preprint at https://www.biorxiv.org/content/10.1101/2020.04.13.039917v1 (2020).
Riva, L. et al. A large-scale drug repositioning survey for SARS-CoV-2 antivirals. Preprint at https://www.biorxiv.org/content/10.1101/2020.04.16.044016v1 (2020).
White, N. J. Cardiotoxicity of antimalarial drugs. Lancet Infect. Dis. 7, 549–558 (2007).
Armstrong, J. F. et al. The IUPHAR/BPS Guide to pharmacology in 2020: extending immunopharmacology content and introducing the IUPHAR/MMV Guide to malaria pharmacology. Nucleic Acids Res. 48, D1006–D1021 (2020).
Yang, D. & Leibowitz, J. L. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 206, 120–133 (2015).
Yang, H. et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. USA 100, 13190–13195 (2003).
Thiel, V. et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 84, 2305–2315 (2003).
Xie, Y. et al. GPS-Lipid: a robust tool for the prediction of multiple lipid modification sites. Sci. Rep. 6, 28249 (2016).
Ren, J. et al. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng. Des. Sel. 21, 639–644 (2008).
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Almagro Armenteros, J. J. et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423 (2019).
Chiva, C. et al. QCloud: a cloud-based quality control system for mass spectrometry-based proteomics laboratories. PLoS ONE 13, e0189209 (2018).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 (2014).
Verschueren, E. et al. Scoring large-scale affinity purification mass spectrometry datasets with MiST. Curr. Protoc. Bioinformatics 49, 8.19.1–8.19.16 (2015).
Giurgiu, M. et al. CORUM: the comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 47, D559–D563 (2019).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Huttlin, E. L. et al. The BioPlex Network: a systematic exploration of the human interactome. Cell 162, 425–440 (2015).
Vizcaíno, J. A. et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 (2014).
Deutsch, E. W. et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 45, D1100–D1106 (2017).
Chaudhury, S., Lyskov, S. & Gray, J. J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 26, 689–691 (2010).
Joosten, R. P., Long, F., Murshudov, G. N. & Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220 (2014).
Drozdetskiy, A., Cole, C., Procter, J. & Barton, G. J. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394 (2015).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Gaulton, A. et al. The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954 (2017).
Sterling, T. & Irwin, J. J. ZINC 15—ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Schmidt, H. R., Betz, R. M., Dror, R. O. & Kruse, A. C. Structural basis for σ1 receptor ligand recognition. Nat. Struct. Mol. Biol. 25, 981–987 (2018).
Mysinger, M. M. & Shoichet, B. K. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 50, 1561–1573 (2010).
Reed, L. J. & Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Hyg. 27, 493–497 (1938).
Chu, D. K. W. et al. Molecular diagnosis of a novel coronavirus (2019-nCoV) causing an outbreak of pneumonia. Clin. Chem. 66, 549–555 (2020).
Huang, X.-P., Mangano, T., Hufeisen, S., Setola, V. & Roth, B. L. Identification of human Ether-à-go-go related gene modulators by three screening platforms in an academic drug-discovery setting. Assay Drug Dev. Technol. 8, 727–742 (2010).
Besnard, J. et al. Automated design of ligands to polypharmacological profiles. Nature 492, 215–220 (2012).
Jimenez-Morales, D., Rosa Campos, A., Von Dollen, J., Krogan, N. J. & Swaney, D. L. artMS: analytical R tools for mass spectrometry. R package version 1.6.5 http://bioconductor.org/packages/release/bioc/html/artMS.html (2020).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Acknowledgements
We thank J. Sarlo for his artistic contributions to the manuscript; A. Kruse for his advice on Sigma receptor functional activity and pharmacologically related enzymes; S. Worland and eFFECTOR Therapeutics for providing zotatifin; T. Haley from FedEx for his effort helping us to ship out drugs and SARS-CoV-2 expression constructs across the globe; R. Albrecht for support with BSL3 procedures and their partners and families for support in child care and other matters during this time. This research was funded by grants from the National Institutes of Health (P50AI150476, U19AI135990, U19AI135972, R01AI143292, R01AI120694, P01A1063302 and R01AI122747 to N.J.K.; R35GM122481 to B.K.S.; 1R01CA221969 and 1R01CA244550 to K.M.S.; K08HL124068 to J.S.C.; 1F32CA236347-01 to J.E.M., NIMH Psychoactive Drug Screening Contract to B.L.R.); from the Defense Advance Research Projects Agency HR0011-19-2-0020 (to B.K.S., N.J.K., A.G.-S., K.M.S., D.A.A. and K.M.V.); by CRIP (Center for Research for Influenza Pathogenesis), a NIAID supported Center of Excellence for Influenza Research and Surveillance (CEIRS, contract no. HHSN272201400008C) and by supplements to NIAID grant U19AI135972 and DoD grant W81XWH-19-PRMRP-FPA to A.G.-S.; by the Laboratoire d’Excellence ‘Integrative Biology of Emerging Infectious Diseases’ (grant ANR-10-LABX-62-IBEID) to M.V., O.S. and C.d’E.; funding from F. Hoffmann-La Roche and Vir Biotechnology and gifts from The Ron Conway Family. K.M.S. is an investigator of the Howard Hughes Medical Institute. The views, opinions, and findings contained in this study are those of the authors and do not represent the official views, policies, or endorsement of the Department of Defense or the US Government.
Author information
Authors and Affiliations
Contributions
The study was conceived by N.J.K. and D.E.G. Genome annotation was carried out by D.E.G., G.M.J. and B.J.P. Molecular cloning was performed by D.E.G., G.M.J. and J.Z.G. Cell culture, affinity purifications and peptide digestion was carried out by G.M.J. and J.X. Mass spectrometer operation and peptide search were performed by D.L.S. and proteomics data were processed by M. Bouhaddou, Y.Z., B.J.P., D.L.S and R.H. Network annotation was led by M. Bouhaddou with support from D.E.G., N.J.K., R.M.K. and the Supplementary Discussion literature review team. Interactome meta-analysis was performed by P.B., M. Bouhaddou, H.B., M.C., Z.Z.C.N., I.B.-H., D.M., C.H.-A., T.P., S.B.R., M.C.O., Y.C., J.C.J.C., D.J.B. and S.K. Drug selection and annotation was done by M.J.O., T.A.T., S.P., Y.S., Z.Z., W.S., I.T.K., J.E.M., J.S.C., K.L., S.A.D., J.L., L.C., S.V., J.L.-L., Y. Lin, X.-P.H., Y. Liu, P.P.S., N.A.W., D.K., H.-Y.W., K.M.S. and B.K.S. Structural modelling was performed by C.J.P.M., K.B.P., S.J.G., D.J.S., R.R., X.L., S.A.W., M. Bohn, F.S.U. and T.K. Live SARS-CoV-2 virus assays were led by K.M.W. and V.V.R. with support from F.R., T.V., A.M.K., L.M., E.M., C.K., N.S.S., Q.D.T., D.S., S.J.F. and M.H. Analysis of SARS-CoV-2 genomic diversity was done by M. Safari. Analysis of human gene positive selection was performed by J.M.Y., B.M. and H.S.M. NSP5 cleavage prediction and analysis was carried out by M. Bohn and C.S.C. with support from D.E.G. Figures were prepared by D.E.G., M. Bouhaddou, K.O., K.M.W., M.J.O., D.L.S., T.A.T., R.H., R.M.K., M.K., H.B., Y.Z., Z.Z., C.J.P.M., T.P., S.A.W., M. Bohn, M. Safari, F.S.U., N.A.W., D.G.F., S.N.F., J.D.G., D.R., T.K., P.B., K.M.S., B.K.S. and N.J.K. The Supplementary Information was assembled by R.M.K. with support from the literature review team: K.O., R.H., R.M.K., A.L.R., B.T., H.F., J.B., K.H., M.M., M.K., P.H., J.M.F., M. Eckhardt, M. Soucheray, M.J.B., M.C., M.J.M., Q.L., C.J.P.M., T.P., X.L., L.C., S.V., J.L.-L., Y. Lin, M. Bohn, R.T., D.A.C., J.H., T.L.R., U.R., A. Subramanian, J.N., N.J., S.M., S.N.F and J.D.G. The manuscript was prepared by D.E.G., M. Bouhaddou, K.O., M.J.O., D.L.S., T.A.T., R.H., R.M.K., M. Eckhardt, M. Soucheray, M.J.B., P.B., K.M.S., B.K.S. and N.J.K. The work was supervised by R.M.S., A.D.F., O.S.R., K.A.V., D.A.A., M.O., M. Emerman, N.J., M.v.Z., E.V., A.A., O.S., C.d’E., S.M., M.J., H.S.M., D.G.F., T.I., C.S.C., S.N.F., J.S.F., J.D.G., A. Sali, B.L.R., D.R., J.T., T.K., P.B., M.V., A.G.-S., K.M.S., B.K.S. and N.J.K.
Corresponding authors
Ethics declarations
Competing interests
The Krogan laboratory has received research support from Vir Biotechnology and F. Hoffmann-La Roche. K.M.S. has consulting agreements for the following companies, which involve monetary and/or stock compensation: Black Diamond Therapeutics, BridGene Biosciences, Denali Therapeutics, Dice Molecules, eFFECTOR Therapeutics (zotatifin and tomivosertib), Erasca, Genentech/Roche, Janssen Pharmaceuticals, Kumquat Biosciences, Kura Oncology, Merck, Mitokinin, Petra Pharma, Qulab, Revolution Medicines (WDB002), Type6 Therapeutics, Venthera, Wellspring Biosciences (Araxes Pharma). J.T. is a cofounder and shareholder of Global Blood Therapeutics, Principia Biopharma, Kezar Life Sciences and Cedilla Therapeutics. J.T. and P.P.S. are listed as inventors on a patent application that describes the cyclic depsipeptide, PS3061 (PCT/US2019/024731 by the Regents of the University of California and Kezar Life Sciences). D.R. is a shareholder of eFFECTOR Therapeutics and a member of its scientific advisory board (zotatifin and tomivosertib). C.S.C. is a co-founder of Catalyst Biosciences and Alaunus Biosciences, and receives grant/research support from ShangPharma/ChemPartners. A.A. is a co-founder of Tango Therapeutics, Azkarra Therapeutics, Ovibio Corporation; a consultant for SPARC, Bluestar, ProLynx, Earli, Cura, GenVivo and GSK; a member of the SAB of Genentech and GLAdiator; receives grant/research support from SPARC and AstraZeneca; holds patents on the use of PARP inhibitors held jointly with AstraZeneca, from which he has benefitted financially (and may do so in the future). The authors have not filed for patent protection on the SARS-CoV-2 host interactions or the use of predicted drugs for the treatment of COVID-19 to ensure all of the information is freely available to accelerate the discovery of a treatment.
Additional information
Peer review information Nature thanks Michael P. Weekes and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Mutations in overlapping coding regions result in premature termination of ORF3a and ORF9c.
a, Table of the SARS-CoV-2 proteins, including molecular mass, sequence similarity with the SARS-CoV homologue and inferred function based on the SARS-CoV homologue. b, Immunoblot detection of 2×Strep tag demonstrates expression of each bait in input samples, as indicated by the red arrowheads. For each bait, input from one of the three replicates prepared and affinity purified for mass spectrometry was used for western blot (n = 1). For gel source data, see Supplementary Fig. 1. c, Schematic of ORF3a (light green) and ORF3b (dark green) overlapping regions. A premature stop codon in ORF3b at position 14 (E14*) corresponds to a Q57H mutation in ORF3a. d, Schematic of the N (red), ORF9b (green) and ORF9c (green) overlapping regions. Two mutations in the N protein (S194L and S197L) correspond to premature stop codons at positions 41 and 44 in ORF9c. The analysis is based on 2,784 sequences obtained from GISAID on 4 April 2020.
Extended Data Fig. 2 Clustering analysis of the AP-MS dataset reveals good correlation between biological replicates of individual baits.
All AP-MS runs (n = 3 biologically independent samples) were compared and clustered using artMS84. All Pearson’s pairwise correlations between MS runs are shown and are clustered according to similar correlation patterns. Correlation between replicates for individual baits ranges from 0.46 to 0.72, and in most cases the experiments corresponding to each bait cluster together, with the exception of a few baits with lower numbers of specific host interactions (for example, E, NSP2, ORF6, ORF3a and ORF3b).
Extended Data Fig. 3 Enrichment of GO biological processes for SARS-CoV-2 host factors.
We performed GO biological process enrichment analyses (Methods) for the host factors identified as binding to each SARS-CoV-2 viral protein and the top five most significant terms for each viral protein are shown. The P values were calculated using a hypergeometric test and a false-discovery rate was used to account for multiple-hypothesis testing.
Extended Data Fig. 4 Enrichment of Pfam protein families for SARS-CoV-2 host factors.
The enrichments of individual protein family domains from the Pfam database85 was calculated using a hypergeometric test, for which success is defined as the number of domains, and the number of trials is the number of individual preys that were affinity purified with each viral bait. The population values were the numbers of individual Pfam domains in the human proteome. The P values were not adjusted for multiple testing. To make sure that the P values that indicated enrichment were meaningful, we only considered Pfam domains that have been affinity purified at least three times with any SARS-CoV-2 protein and that occurred in the human proteome at least five times. Here, we show Pfam domains with the lowest P value for a given viral bait protein.
Extended Data Fig. 5 Lung mRNA expression and specificity of SARS-CoV-2-interacting human proteins relative to other proteins.
a, Scatterplot of the lung mRNA expression (TPM) versus enrichment of lung mRNA expression (lung TPM/median of all tissue TPM) for human-interacting proteins. Red points denote drug targets that are labelled with their gene names. Points above the horizontal blue line represent interacting proteins that are enriched in lung expression and show how most SARS-CoV-2-interacting proteins tend to be enriched in the lung. b, Gene expression in the lung of the high-confidence human-interacting proteins was observed to be higher compared to all other proteins. Blue, interacting proteins (n = 332, median = 25.52 TPM); grey, all other proteins (n = 13,583, median = 3.198 TPM). P = 0.0007 using a Student’s t-test.
Extended Data Fig. 6 Candidate targets for the viral NSP5 protease.
a, Wild-type NSP5 and NSP5(C145A) (catalytic dead mutant) interactome. b, Domain maps of HDAC2 and TRMT1, illustrating predicted cleavage sites (using NetCorona 1.0). HDAC, histone deacetylase domain; NLS, nuclear localization sequence; MTS, mitochondrial targeting sequence; SAM-MT, S-adenosylmethionine-dependent methyltransferase domain. c, Peptide docking of predicted cleavage peptides into the crystal structure of SARS-CoV NSP5. d, NSP5-cleavage consensus site for SARS-CoV (left) and SARS-CoV-2 (right).
Extended Data Fig. 7 Consensus analysis of SARS-CoV-2 ORF6 homologues.
a, Sequence logo of SARS-CoV-2 ORF6 homologues, showing sequence conservation at each position computed from a multiple-sequence alignment of 35 sequences. The key methionine M58, and the acidic residues E55, E59 and D61 of the putative NUP98–RAE1-binding motif are shown to be highly conserved. Homology was determined from alignments to full-length sequences. Colours indicated chemical properties of amino acids: polar (G, S, T, Y, C; green), neutral (Q, N; purple), basic (K, R, H; blue), acidic (D, E; red) and hydrophobic (A, V, L, I, P, W, F, M; black). b, Multiple-sequence alignment of SARS-CoV-2 ORF6 homologues. The query sequence is shown at the top (sequence 1, ref|YP_009724394.1). Sequence coverage (cov) and percentage identity (pid) are shown for each homologous sequence.
Extended Data Fig. 8 Viral growth and cytotoxicity for compounds tested in New York.
Viral growth (percentage infection; red) and cytotoxicity (black) results for compounds tested at Mount Sinai in New York. TCID50 assay results (green) for zotatifin, hydroxychloroquine and PB28 are also shown. Zotafitin and midostaurin were tested in two independent experiments and data are shown in two individual panels. Data are mean ± s.d.; n = 3 biologically independent samples. The full dataset is available in Supplementary Table 6.
Extended Data Fig. 9 Virus plaque assays, qRT–PCR and cell viability for compounds tested in Paris.
Plaque assay (viral titre; red), qRT–PCR (viral RNA; blue) and cell viability (Alamar blue; black) results for compounds tested at the Pasteur Institute in Paris. PF-846 was tested in two independent experiments and data are shown in two individual panels. Data are mean ± s.d.; for virus plaque assay and RT–qPCR, n = 3 biologically independent samples for drug-treated cells, n = 5 for PS3061, n = 6 for DMSO controls; for cell viability, n = 6 biologically independent samples for drug-treated cells and DMSO controls. The full dataset is available in Supplementary Table 6.
Extended Data Fig. 10 Activity of sigma ligands.
a, The drugs cloperastine and clemastine can be readily fit into the agonist-bound structure of the sigma-1 receptor. b, Compounds tested for antiviral activity with annotated sigma-1 receptor and/or sigma-2 receptor (also known as TMEM97) activity are shown. Inhibition pIC50 values of SARS-CoV-2 infection are shown from blue to yellow, mode of functional activity at the sigma-1 receptor is shown by mark shape (upwards triangle, agonist; downwards triangle, antagonist; circle, binding), and pKi values for the sigma-1 receptor and sigma-2 receptor are shown along the x and y axes. We have not yet tested chloroquine for antiviral activity. Binding of E-52862 at the sigma-2 receptor is reported to be greater than 1 μM. Activities of pimozide and olanzapine at the sigma-2 receptor have not been reported. Activity of olanzapine at the sigma-1 receptor is reported to be greater than 5 μM.
Extended Data Fig. 11 Astemizole is a potent sigma-2 receptor ligand.
a, b, Concentration–response curves of astemizole from radioligand displacement assays for the sigma-2 (a; Ki = 95 nM) and the sigma-1 (Ki = 1.3 μM) receptors are shown. Data are mean ± s.e.m.; n = 4 independent assays for each receptor.
Extended Data Fig. 12 Off-target activities for characteristic Sigma receptor ligands.
Dose–response curves against a panel of eight targets that can confer adverse cardiac responses, respiratory difficulties and dry-mouth effects for chloroquine, hydroxychloroquine, PB28, PD-144418 and clemastine. These results are not meant to represent or replace a comprehensive test against off-target panels, as might commonly be assayed in drug progression for clinical use. The eight targets include the alpha-2A adrenergic receptors: alpha 2A (encoded by ADRA2A), alpha 2B (encoded by ADRA2B), and alpha 2C (encoded by ADRA2C); as well as the muscarinic acetylcholine receptors: M1 (encoded by CHRM1), M2 (encoded by CHRM2), M3 (encoded by CHRM3), M4 (encoded by CHRM4) and M5 (encoded by CHRM5). Data are mean ± s.d.; n = 3 biologically independent samples. The table summarizes the fitted pKi values for the five ligands at the eight targets.
Supplementary information
41586_2020_2286_MOESM1_ESM.pdf
Supplementary Information Supplementary Discussion and Methods. The Supplementary Discussion is a comprehensive literature review of all 27 wild-type and mutant SARS-CoV-2 bait protein-protein interaction (PPI) subnetworks. Individual bait subnetwork images are provided at the front of the document for reference (and are zoomed view representations from Fig. 3 of the main text). Supplementary Methods that were used to generate the data in Supplementary Tables and Extended Data Figures (when not referenced in the main text) are provided after the Supplementary Discussion. Supplementary References are included at the end of the document.
Supplementary Figure 1
Raw Western blot images with molecular weight ladder and black box outlining the image cropping used to generate Extended Data Figure 1b. Both blots were probed with primary antibody α-streptavidin, Qiagen #34850 (1:2500); and secondary α-mouse-HRP conjugate, BioRad #1706516 (1:20,000).
Supplementary Table 1
Scoring results for all baits and all proteins, showing spectral counts of experimental samples (columns AvgSpec and Spec) and empty controls (column Ctrl Counts).
Supplementary Table 2
SARS-CoV-2 high confidence interactors, showing spectral counts of experimental samples (columns AvgSpec and Spec) and empty controls (column Ctrl Counts).
Supplementary Table 3
Evolutionary Analysis for SARS-CoV-2 interactors. Phylogenetic Analysis by Maximum Likelihood (PAML) results for the 332 genes analysed, sorted in order of statistical significance. We used a likelihood ratio test to obtain a P value, by comparing twice the difference in log-likelihoods with the chi-squared distribution with 1 degree of freedom. After running all 332 analyses, we used the Benjamini-Hochberg procedure to control the false-discovery rate.
Supplementary Table 4
Literature-derived drugs and reagents that modulate SARS-CoV-2 interactors. Drug-target associations drawn from chemoinformatic searches of the literature, including information about purchasability.
Supplementary Table 5
Expert-identified drugs and reagents that modulate SARS-CoV-2 interactors. Drug-target associations drawn from expert knowledge of human protein interactors of SARS-CoV-2 and reagents and drugs that modulate them; not readily available from the chemoinformatically-searchable literature.
Supplementary Table 6
Summary of drugs/compounds and experimental results for SARS-CoV-2 viral inhibition and cell viability.
Supplementary Table 7
Raw chemical associations to prey proteins IUPHAR/BPS Guide to Pharmacology (2020-3-12).
Supplementary Table 8
Raw chemical associations to prey proteins ChEMBL25.
Supplementary Data
This zipped folder contains genome annotation of SARS-CoV-2 USA-WA1 (genbank format), and plasmid maps for all constructs generated for this study (genbank format).
Rights and permissions
About this article

Cite this article
Gordon, D.E., Jang, G.M., Bouhaddou, M. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020). https://doi.org/10.1038/s41586-020-2286-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2286-9
This article is cited by
-
Histopathological and clinical analysis of COVID-19-infected placentas
Surgical and Experimental Pathology (2024)
-
Graph embedding on mass spectrometry- and sequencing-based biomedical data
BMC Bioinformatics (2024)
-
Generic model to unravel the deeper insights of viral infections: an empirical application of evolutionary graph coloring in computational network biology
BMC Bioinformatics (2024)
-
SARS-CoV-2 outbreak: role of viral proteins and genomic diversity in virus infection and COVID-19 progression
Virology Journal (2024)
-
Integrated multi-omics analyses identify anti-viral host factors and pathways controlling SARS-CoV-2 infection
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
