
Abstract
Coronavirus disease 2019 (COVID-19) is an acute infection of the respiratory tract that emerged in late 20191,2. Initial outbreaks in China involved 13.8% of cases with severe courses, and 6.1% of cases with critical courses3. This severe presentation may result from the virus using a virus receptor that is expressed predominantly in the lung2,4; the same receptor tropism is thought to have determined the pathogenicity—but also aided in the control—of severe acute respiratory syndrome (SARS) in 20035. However, there are reports of cases of COVID-19 in which the patient shows mild upper respiratory tract symptoms, which suggests the potential for pre- or oligosymptomatic transmission6,7,8. There is an urgent need for information on virus replication, immunity and infectivity in specific sites of the body. Here we report a detailed virological analysis of nine cases of COVID-19 that provides proof of active virus replication in tissues of the upper respiratory tract. Pharyngeal virus shedding was very high during the first week of symptoms, with a peak at 7.11 × 108 RNA copies per throat swab on day 4. Infectious virus was readily isolated from samples derived from the throat or lung, but not from stool samples—in spite of high concentrations of virus RNA. Blood and urine samples never yielded virus. Active replication in the throat was confirmed by the presence of viral replicative RNA intermediates in the throat samples. We consistently detected sequence-distinct virus populations in throat and lung samples from one patient, proving independent replication. The shedding of viral RNA from sputum outlasted the end of symptoms. Seroconversion occurred after 7 days in 50% of patients (and by day 14 in all patients), but was not followed by a rapid decline in viral load. COVID-19 can present as a mild illness of the upper respiratory tract. The confirmation of active virus replication in the upper respiratory tract has implications for the containment of COVID-19.
Similar content being viewed by others

Main
There is a close genetic relationship between SARS coronavirus (SARS-CoV) and the causative agent of COVID-19, SARS-CoV-2. The predominant expression of ACE2 in the lower respiratory tract is believed to have determined the natural history of SARS as an infection of the lower respiratory tract5. Although the positive detection of SARS-CoV-2 in clinical specimens from the upper respiratory tract has previously been described9,10, these observations do not address the principal differences between SARS and COVID-19 in terms of clinical pathology. The patients who were studied here were enrolled because they acquired their infections upon known close contact to an index case, thereby avoiding representational biases owing to symptom-based case definitions. All patients were treated in a single hospital in Munich, Germany. Virological testing was done by two closely collaborating laboratories that used the same standards of technology for PCR with reverse transcription (RT–PCR) and virus isolation; these two laboratories confirmed each other’s results in almost all of the individual samples. Owing to the extremely high congruence of results, all data—except for the serological data (which are based on results from one laboratory only)—are presented together. The patients are part of a larger cluster of epidemiologically linked cases that occurred after 23 January 2020 in Munich, as discovered on 27 January (ref. 11). The present study uses samples taken during the clinical course in the hospital, as well as from initial diagnostic testing before admission. In cases in which this initial diagnostic testing was done by other laboratories, the original samples were retrieved and retested under the rigorous quality standards of the present study.
RT–PCR, replication sites and infectivity
To first understand whether the described clinical presentations are solely caused by infection with SARS-CoV-2, samples from all patients were tested against a panel of typical agents of respiratory viral infection, including human coronavirus (HCoV)-HKU1, HCoV-OC43, HCoV-NL63 and HCoV-229E, influenza virus A, influenza virus B, phinovirus, enterovirus, respiratory syncytial virus, human parainfluenza viruses 1–4, human metapneumovirus, adenovirus and human bocavirus. No coinfection was detected in any patient.
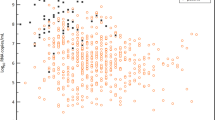
All patients were initially diagnosed by RT–PCR from oro- or nasopharyngeal swab specimens12. Both types of specimen were collected over the whole clinical course in all patients. There were no discernible differences in viral loads or detection rates when comparing naso- and oropharyngeal swabs (Fig. 1b). The earliest swabs were taken on day 1 of symptoms, which were often very mild or prodromal. All swabs from all patients taken between day 1 and day 5 tested positive. The average virus RNA load was 6.76 × 105 copies per whole swab until day 5, and the maximum load was 7.11 × 108 copies per swab. Swab samples taken after day 5 had an average viral load of 3.44 × 105 copies per swab and a detection rate of 39.93%. The last swab sample that tested positive was taken on day 28 after the onset of symptoms. The average viral load in sputum was 7.00 × 106 copies per ml, with a maximum of 2.35 × 109 copies per ml.

a, Samples and sample types per day. b, Viral RNA concentrations in samples from the upper respiratory tract. Neg., sample negative for RNA copies. c, Viral RNA concentrations in sputum and stool samples. d, Seroconversion and virus isolation success, dependent on day after the onset of symptoms. Top, fraction of seroconverted patients. Bottom, aggregated results of virus isolation trials. e, Virus isolation success, dependent on viral load. Viral loads were projected to RNA copies per ml (for sputum samples), per swab (for throat swab samples) or per g (for stool samples). f, g, Projected virus isolation success based on probit distributions. The inner lines are probit curves (dose–response rule). The outer dotted lines are 95% confidence interval. For a <5% isolation success, the estimated day was 9.78 (95% confidence interval 8.45–21.78) days after the onset of symptoms, and the estimated RNA concentration for <5% isolation success was estimated to be 5.40 log10(RNA copies per ml) (95% confidence interval −4.11–6.51). h, Subgenomic viral RNA transcripts in relation to viral genomic RNA. Dots represent mean values of RT–PCR data obtained from at least two independent experiments on samples from individual patients. Plots show median values with interquartile ranges.
Because swab samples had limited sensitivity for the initial diagnosis of cases of SARS13,14, we analysed the first paired swab and sputum samples taken on the same occasion from seven patients. All samples were taken between 2 and 4 days after the onset of symptoms. In two cases, swab samples had virus concentrations that were clearly higher than those in sputum samples, as indicated by a difference of >3 in the threshold cycle (Ct) value. The opposite was true in two other cases, and the remaining three cases had similar concentrations in both sample types.
None of 27 urine samples and none of 31 serum samples tested positive for RNA from SARS-CoV2.
To understand infectivity, we attempted live virus isolation on multiple occasions from clinical samples (Fig. 1d). Whereas the virus was readily isolated during the first week of symptoms from a considerable fraction of samples (16.66% of swabs and 83.33% of sputum samples), no isolates were obtained from samples taken after day 8 in spite of ongoing high viral loads.
Virus isolation from stool samples was never successful, irrespective of viral RNA concentration, on the basis of a total of 13 samples taken between day 6 and day 12 from 4 patients. The success of virus isolation also depended on viral load: samples that contained <106 copies per ml (or copies per sample) never yielded an isolate. For sputum samples, interpolation based on a probit model was done to obtain laboratory-based infectivity criteria for the discharge of patients (Fig. 1f, g).
High viral loads and successful isolation from early throat swabs suggested potential virus replication in tissues of the upper respiratory tract. To obtain proof of active virus replication in the absence of histopathology, we conducted RT–PCR tests to identify viral subgenomic mRNAs directly in clinical samples (Extended Data Fig. 1). Viral subgenomic mRNA is transcribed only in infected cells and is not packaged into virions, and therefore indicates the presence of actively infected cells in samples. Levels of viral subgenomic mRNA were compared against viral genomic RNA in the same sample. In sputum samples taken on day 4 to day 9, during which time active replication in sputum was obvious in all patients as per longitudinal viral load courses (as described in ‘Viral load, antibody response and clinical course’), the ratios of mean normalized subgenomic mRNA per genome were about 0.4% (Fig. 1g). A decline occurred from day 10 to day 11. In throat swabs, all samples taken up to day 5 were in the same range, whereas no subgenomic mRNA was detectable in swabs thereafter. Together, these data indicate the active replication of SARS-CoV-2 in the throat during the first five days after the onset of symptoms. No (or only minimal) indications of replication in stool were obtained by the same method (Fig. 1g).
During our study, we sequenced full virus genomes from all patients. A G6446A exchange was first detected in one patient, and later transmitted to other patients in the cluster11. In the first patient, this mutation was found in a throat swab while a sputum sample from the same day showed the original allele (G6446). The single-nucleotide polymorphism was analysed by RT–PCR and Sanger sequencing in all sequential samples available from that patient (Table 1). The presence of separate genotypes in throat swabs and sputum strongly supported our suspicion of independent virus replication in the throat, rather than passive shedding to the throat from the lung.
Viral load, antibody response and clinical course
Daily measurements of viral load in sputum, pharyngeal swabs and stool are summarized in Fig. 2. In general, the concentrations of viral RNA were very high in initial samples. In all patients except one, the concentration of viral RNA in throat swabs seemed to be already on the decline at the time of first presentation. Viral RNA concentrations in sputum declined more slowly, with a peak during the first week of symptoms in three out of eight patients. Viral RNA concentrations in stools were also high. In many cases, the course of viral RNA concentration in stools seemed to reflect the course in sputum (Fig. 2a–c). In only one case did independent replication in the intestinal tract seem obvious from the course of stool RNA excretion (Fig. 2d). Whereas symptoms mostly waned until the end of the first week (Table 2), viral RNA remained detectable in throat swabs well into the second week. Stool and sputum samples remained RNA-positive over three weeks in six of the nine patients, in spite of full resolution of symptoms.

a–i, The panels correspond to patients no. 1 (a), 2 (b), 3 (c), 4 (d), 7 (e), 8 (f), 10 (g), 14 (h) and 16 (i) in a previous publication11. Dotted lines, limit of quantification. Experiments were performed in duplicate and the data presented are the mean of results obtained by two laboratories independently. PRNT90, serum dilution that causes viral plaque reduction of 90%.
All cases had comparatively mild courses (Table 2). The two patients who showed some signs of lung infection were the only cases in which sputum viral loads showed a late and high peak around day 10 or 11, whereas sputum viral loads were on the decline by this time in all other patients (Fig. 2f, g). Of note, four out of nine patients showed a loss of taste and olfactory sensation, and described this loss to be stronger and more long-lasting than in common cold diseases.
Seroconversion was detected by IgG and IgM immunofluorescence using cells that express the spike protein of SARS-CoV-2 and a virus neutralization assay using SARS-CoV-2 (Table 3, Extended Data Fig. 2). Seroconversion in 50% of patients occurred by day 7, and in all patients by day 14 (Fig. 1d). No viruses were isolated after day 7. All patients showed detectable neutralizing antibodies, the titres of which did not suggest close correlation with clinical courses. Of note, patient no. 4, who showed the lowest virus neutralization titre at end of week 2, seemed to shed virus from stool over a prolonged time (Fig. 2d). Results from the differential recombinant immunofluorescence assay indicated cross-reactivity or cross-stimulation against the four endemic human coronaviruses in several patients (Extended Data Table 1).
Conclusions
The clinical courses in the patients under study—all of whom were young- to middle-aged professionals without notable underlying disease—were mild. Apart from one patient, all cases were first tested when symptoms were still mild or in the prodromal stage (a period in which most patients would present once there is general awareness of a circulating pandemic disease5). Diagnostic testing suggests that simple throat swabs will provide sufficient sensitivity at this stage of infection. This is in stark contrast to SARS; for instance, only 38 of 98 nasal or nasopharyngeal swab samples tested positive by RT–PCR in patients with SARS in Hong Kong15. Viral load also differs considerably between SARS and COVID-19. For SARS, it took 7 to 10 days after the onset of symptoms until peak RNA concentrations (of up to 5 × 105 copies per swab) were reached13,14. In the present study, peak concentrations were reached before day 5, and were more than 1,000 times higher. Successful isolation of live virus from throat swabs is another notable difference between COVID-19 and SARS, for which such isolation was rarely successful16,17,18. This suggests active virus replication in tissues of the upper respiratory tract, where SARS-CoV is not thought to replicate in spite of detectable ACE2 expression19,20. At the same time, the concurrent use of ACE2 as a receptor by SARS-CoV and SARS-CoV-2 corresponds to a highly similar excretion kinetic in sputum, with active replication in the lung. SARS-CoV was previously found13 in sputum at mean concentrations of 1.2–2.8 × 106 copies per ml, which corresponds to observations made here.
Whereas proof of replication by histopathology is awaited, extended tissue tropism of SARS-CoV-2 with replication in the throat is strongly supported by our studies of cells that transcribe subgenomic mRNA in throat swab samples, particularly during the first 5 days of symptoms. Notable additional evidence for independent replication in the throat is provided by sequence findings in one patient, who consistently showed a distinct virus in the throat as opposed to the lung. In addition, the disturbance of gustatory and olfactory senses points at an infection of the tissues of the upper respiratory tract.
Critically, the majority of patients in the present study seemed to be beyond their shedding peak in samples from the upper respiratory tract when they were first tested, whereas the shedding of infectious virus in sputum continued throughout the first week of symptoms. Together, these findings suggest a more efficient transmission of SARS-CoV-2 than SARS-CoV, through active pharyngeal viral shedding at a time at which symptoms are still mild and typical of infections of the upper respiratory tract. Later in the disease, COVID-19 resembles SARS in terms of replication in the lower respiratory tract. Of note, the two patients who showed some symptoms of the lungs being affected showed a prolonged viral load in sputum. Our study is limited, in that no severe cases were observed. Future studies that include severe cases should look at the prognostic value of an increase of viral load beyond the end of week 1, potentially indicating an aggravation of symptoms.
One of the most interesting hypotheses to explain the potential extension of tropism to the throat is the presence of a polybasic furin-type cleavage site at the S1–S2 junction in the SARS-CoV-2 spike protein that is not present in SARS-CoV17. The insertion of a polybasic cleavage site in the S1–S2 region in SARS-CoV has previously been shown to lead to a moderate, but discernible, gain-of-fusion activity that might result in increased viral entry in tissues with a low density of ACE2 expression21.
The combination of very high concentrations of virus RNA and the occasional detection of cells in stools that contain subgenomic mRNA indicate active replication in the gastrointestinal tract. Active replication is also suggested by a much higher detection rate compared to the Middle East respiratory system coronavirus (MERS-CoV), for which stool-associated RNA was found in only 14.6% of samples from 37 patients hospitalized in Riyadh (Saudi Arabia)22,23. If SARS-CoV-2 was only passively present in the stool (such as after swallowing respiratory secretions), similar detection rates as for MERS-CoV would be expected. Replication in the gastrointestinal tract is also supported by analogy with SARS-CoV, which was regularly excreted in stool (from which it could be isolated in cell culture24). Our failure to isolate live SARS-CoV-2 from stools may be due to the mild courses of cases, with only one case showing intermittent diarrhoea. In China, diarrhoea was seen in only 2 of 99 cases25. Further studies should therefore address whether SARS-CoV-2 shed in stools is rendered noninfectious though contact with the gut environment. Our initial results suggest that measures to contain viral spread should aim at droplet-, rather than fomite-, based transmission.
The prolonged viral shedding in sputum is relevant not only for the control of infections in hospitals, but also for discharge management. In a situation characterized by a limited capacity of hospital beds in infectious disease wards, there is pressure for early discharge after treatment. On the basis of the present findings, early discharge with ensuing home isolation could be chosen for patients who are beyond day 10 of symptoms and have less than 100,000 viral RNA copies per ml of sputum. Both criteria predict that there is little residual risk of infectivity, on the basis of cell culture.
The serological courses of all patients suggest a timing of seroconversion similar to, or slightly earlier than, in SARS-CoV infection18. Seroconversion in most cases of SARS occurred during the second week of symptoms. As in SARS and MERS, IgM was not detected considerably earlier than IgG in immunofluorescence; this might in part be due to technical reasons, as the higher avidity of IgG antibodies outcompetes IgM for viral epitopes in the assay. IgG depletion can only partially alleviate this effect. Because immunofluorescence assay is a labour-intensive method, enzyme-linked immunosorbent assay tests should be developed as a screening test. Neutralization testing is necessary to rule out cross-reactive antibodies directed against endemic human coronaviruses. On the basis of the frequently low neutralizing antibody titres observed in coronavirus infection26,27, we have here developed a particularly sensitive plaque-reduction neutralization assay. Considering the titres we observed, a simpler microneutralization test format is likely to provide sufficient sensitivity in routine application and population studies.
When aligned to viral load courses, it seems there is no abrupt virus elimination at the time of seroconversion. Rather, seroconversion early in week 2 coincides with a slow but steady decline of viral load in sputum. Whether properties such as the glycosylation pattern at critical sites of the glycoprotein have a role in the attenuation of the neutralizing antibody response needs further clarification. In any case, vaccine approaches targeting mainly the induction of antibody responses should aim to induce particularly strong antibody responses to be effective.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Clinical samples and viral load conversion
Sputum and stool samples were taken and shipped in native conditions. Oro- and nasopharyngeal throat swabs were preserved in 3 ml of viral transport medium. Viral loads in sputum samples were projected to RNA copies per ml, in stool samples to copies per g and in throat swabs to copies per 3 ml, assuming that all sample components were suspended in 3 ml viral transport medium. For swab samples suspended in less than 3 ml viral transport medium, this conversion was adapted to represent copies per whole swab. An aggregated overview of samples received per day after the onset of disease from all patients is shown in Fig. 1a.
RT–PCR for SARS-CoV-2 and other respiratory viruses
RT–PCR used targets in the E and RdRp genes as previously described12. Both laboratories used a pre-formulated oligonucleotide mixture (Tib-Molbiol) to make the laboratory procedures more reproducible. All patients were also tested for other respiratory viruses, including HCoV-HKU1, HCoV-OC43, HCoV-NL63 and HCoV-229E, influenza virus A, influenza virus B, rhinovirus, enterovirus, respiratory syncytial virus, human parainfluenza viruses 1–4, human metapneumovirus, adenovirus and human bocavirus using LightMix-Modular Assays (Roche). Additional technical details are provided in Supplementary Methods section 1.
Virus isolation
Virus isolation was done in two laboratories on Vero E6 cells. In brief, 100 μl of suspended, cleared and filtered clinical sample was mixed with an equal volume of cell culture medium. Supernatant was collected after 0, 1, 3 and 5 days and used in RT–PCR analysis. Additional technical details are provided in Supplementary Methods section 2a.
Serology
We performed recombinant immunofluorescence assays to determine the specific reactivity against recombinant spike proteins in VeroB4 cells, as previously described26,28. This assay used a cloned coronavirus spike protein from HCoV-229E, HCoV-NL63, HCoV-OC43, HCoV-HKU1 or SARS-CoV-2. The screening dilution was 1:10. Plaque reduction neutralization tests were done essentially as previously described for MERS-CoV26. Serum dilutions causing plaque reductions of 90% (PRNT90) and 50% (PRNT50) were recorded as titres. Additional technical details are provided in Supplementary Methods section 2b, c.
Statistical analyses
Statistical analyses were done using SPSS software (version 25) or GrapPad Prism (version 8).
Ethical approval statement
All patients provided informed consent for the use of their data and clinical samples for the purposes of the present study. Institutional review board clearance for the scientific use of patient data has been granted to the treating institution by the ethics committee at the Medical Faculty of the Ludwig Maximillians Universität Munich (vote 20-225 KB).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Sequence data are available in Gisaid under accession number EPI_ISL_406862. All other data are available from C.D. upon reasonable request.
Change history
11 December 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41586-020-2984-3
References
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020).
WHO. Report of the WHO–China Joint Mission on Coronavirus Disease 2019 (COVID-19) https://www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report.pdf (WHO, 2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Leung, G. M. et al. The epidemiology of severe acute respiratory syndrome in the 2003 Hong Kong epidemic: an analysis of all 1755 patients. Ann. Intern. Med. 141, 662–673 (2004).
Rothe, C. et al. Transmission of 2019-nCoV infection from an asymptomatic contact in Germany. N. Engl. J. Med. 382, 970–971 (2020).
Holshue, M. L. et al. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 382, 929–936 (2020).
Hoehl, S. et al. Evidence of SARS-CoV-2 infection in returning travelers from Wuhan, China. N. Engl. J. Med. 382, 1278–1280 (2020).
Zou, L. et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 382, 1177–1179 (2020).
Young, B. E. et al. Epidemiologic features and clinical course of patients infected with SARS-CoV-2 in Singapore. J. Am. Med. Assoc. 323, 1488–1494 (2020).
Böhmer, M. et al. Outbreak of COVID-19 in Germany resulting from a single travel-associated primary case. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(20)30314-5 (2020).
Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT–PCR. Euro Surveill. 25, 1–8 (2020).
Drosten, C. et al. Evaluation of advanced reverse transcription–PCR assays and an alternative PCR target region for detection of severe acute respiratory syndrome-associated coronavirus. J. Clin. Microbiol. 42, 2043–2047 (2004).
Peiris, J. S. et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361, 1767–1772 (2003).
Poon, L. L. et al. Detection of SARS coronavirus in patients with severe acute respiratory syndrome by conventional and real-time quantitative reverse transcription-PCR assays. Clin. Chem. 50, 67–72 (2004).
Ksiazek, T. G. et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966 (2003).
Drosten, C. et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976 (2003).
Peiris, J. S. et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361, 1319–1325 (2003).
Bertram, S. et al. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 7, e35876 (2012).
Xu, H. et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 12, 8 (2020).
Belouzard, S., Chu, V. C. & Whittaker, G. R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl Acad. Sci. USA 106, 5871–5876 (2009).
Corman, V. M. et al. Viral shedding and antibody response in 37 patients with Middle East respiratory syndrome coronavirus infection. Clin. Infect. Dis. 62, 477–483 (2016).
Zhou, J. et al. Human intestinal tract serves as an alternative infection route for Middle East respiratory syndrome coronavirus. Sci. Adv. 3, eaao4966 (2017).
Leung, W. K. et al. Enteric involvement of severe acute respiratory syndrome-associated coronavirus infection. Gastroenterology 125, 1011–1017 (2003).
Chen, N. et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395, 507–513 (2020).
Drosten, C. et al. Transmission of MERS-coronavirus in household contacts. N. Engl. J. Med. 371, 828–835 (2014).
Müller, M. A. et al. Presence of Middle East respiratory syndrome coronavirus antibodies in Saudi Arabia: a nationwide, cross-sectional, serological study. Lancet Infect. Dis. 15, 559–564 (2015).
Corman, V. M. et al. Assays for laboratory confirmation of novel human coronavirus (hCoV-EMC) infections. Euro Surveill. 17, 20334 (2012).
Acknowledgements
This work was funded by grants from the German Ministry of Research (01KI1723A) and the European Union (602525) to C.D. as well as by the German Bundeswehr Medical Service Biodefense Research Program. The funders had no role in study design, data collection and analysis or decision to publish. We thank P. Mackeldanz, E. Möncke-Buchner, A. Richter, M. Schmidt and J. Beheim-Schwarzbach for technical assistance.
Author information
Authors and Affiliations
Contributions
R.W. and V.M.C. planned and supervised laboratory testing, and evaluated data. W.G. and M.S. managed patients and evaluated clinical data. S.Z., T.B., S.B., J.S., R.E. and K.Z. performed laboratory testing. M.A.M. managed serological laboratory testing. D.N. managed and performed virus isolation studies. T.C.J. analysed sequences and population-specific polymorphisms. P.V. managed laboratory testing. C.R. managed initial patient contacts. M.H. managed initial patient contacts and evaluated clinical data. C.D. designed and supervised laboratory studies, and wrote the manuscript. C.W. designed and supervised clinical management and clinical data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Peter Openshaw and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Sequence analysis of E gene subgenomic mRNA.
The leader sequence (purple), putative transcription regulatory sequences (TRS) (grey) and nucleotides coding for the 5′-proximal part of the E gene (yellow box) are shown. PCR primer binding sites used for amplification and RT–PCR detection are shown as green arrows, and the 5′-nuclease PCR probe is shown as a red arrow.
Extended Data Fig. 2 Recombinant SARS-CoV-2-spike-based immunofluorescence test shows seroconversion of patient no. 4.
Representative outcome of a recombinant immunofluorescence test using serum dilutions 1:10, 1:100, 1:1,000 and 1:10,000 of patient no. 4 at 5 and 17 days after the onset of symptoms. Secondary detection was done by using a goat-anti human immunoglobulin labelled with Alexa Fluor 488 (shown in green). The experiment was performed in duplicate.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, which include a description of RNA extraction and RT-PCR methods, a description of cell culture and antibody detection methods and Supplementary References.
Rights and permissions
About this article

Cite this article
Wölfel, R., Corman, V.M., Guggemos, W. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020). https://doi.org/10.1038/s41586-020-2196-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2196-x
This article is cited by
-
Efficacy of povidone-iodine nasal rinse and mouth wash in COVID-19 management: a prospective, randomized pilot clinical trial (povidone-iodine in COVID-19 management)
BMC Infectious Diseases (2024)
-
The impact of delayed tracheostomy on critically ill patients receiving mechanical ventilation: a retrospective cohort study in a chinese tertiary hospital
BMC Anesthesiology (2024)
-
Cycle threshold of SARS-CoV-2 RT-PCR as a driver of retesting
Scientific Reports (2024)
-
An intranasal live-attenuated SARS-CoV-2 vaccine limits virus transmission
Nature Communications (2024)
-
Estimating actual SARS-CoV-2 infections from secondary data
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
