
Abstract
The many forms of vasculitis are characterized by inflammation of blood vessels, leading to potentially long-term sequelae including vision loss, aneurysm formation and kidney failure. Accurate estimation of the incidence and prevalence has been hampered by the absence of reliable diagnostic criteria and the rarity of these conditions; however, much progress has been made over the past two decades, although data are still lacking from many parts of the world including the Indian subcontinent, China, Africa and South America. Giant cell arteritis occurs in those aged 50 years and over and seems to mainly affect persons of northern European ancestry, whereas Takayasu arteritis occurs mainly in those aged under 40 years. By contrast, Kawasaki disease mainly occurs in children aged under 5 years and is most common in children of Asian ancestry, and IgA vasculitis occurs in children and adolescents. Although much less common than giant cell arteritis, the different forms of antineutrophil cytoplasmic antibody-associated vasculitis are being increasingly recognized in most populations and occur more frequently with increasing age. Behçet syndrome occurs most commonly along the ancient silk road between Europe and China. Much work needs to be done to better understand the influence of ethnicity, geographical location, environment and social factors on the development of vasculitis.
Key points
-
Vasculitis is a group of generally uncommon diseases of generally unknown aetiology.
-
Vasculitis mainly occurs at the ends of the age spectrum, in young children and older adults.
-
Kawasaki disease is most common in children aged 5 years or less and occurs most commonly in Japan and in children of Southeast Asian ancestry.
-
Giant cell arteritis is the most common form of vasculitis in elderly individuals, particularly in those of Northern European ancestry.
-
The three types of antineutrophil cytoplasmic antibody-associated vasculitis are rare; of the three types, granulomatosis with polyangiitis is the most prevalent.
-
Behçet syndrome has the highest prevalence along the ancient silk road, which stretches from the Mediterranean through the Middle East to East Asia.
Similar content being viewed by others
Introduction
Systemic vasculitis is a term that covers a group of rare conditions characterized by inflammation of blood vessels, which leads to organ ischaemia and damage. Vasculitis can affect individuals of all ages, but certain conditions show a marked age tropism; for example, Kawasaki disease affects young children, whereas giant cell arteritis (GCA) predominantly affects those aged >50 years. The reasons for these differences are in general unknown but might reflect interactions between a widespread environmental trigger and a genetically predisposed child in Kawasaki disease and interactions between infection and ‘immunosenescence’ in the ageing immune system in GCA1,2. Although much is now known about the immunopathogenesis of vasculitis, relatively little is known about the events leading up to the development of clinical disease. Classic descriptive epidemiology can provide some clues, for example, by demonstrating evidence of clustering or a cyclical pattern of occurrence that might indicate a possible infectious aetiology. However, the rarity of the conditions has posed a number of methodological problems, together with uncertainty as to case definition.
Reliable classification criteria are an absolute requirement for conducting accurate epidemiology, yet there has been no universal system of classification for vasculitis. Vasculitis is defined according to the predominant size of the vessel involved3. Large vessels include the aorta and its major branches, medium vessels include the main visceral arteries and their branches, and small vessels include intraparenchymal arteries, arterioles, capillaries and venules. The ACR promulgated classification criteria for seven types of vasculitis in 1990, but these criteria were subsequently shown to be unreliable following developments in imaging and autoantibody serology4,5. The criteria were developed using patients from North America without representation from Europe or Asia, which is clearly not appropriate for conditions that have geographical variation in the pattern of disease. The Chapel Hill Consensus conferences of 1994 and 2012 produced definitions that are widely used in clinical studies, but these were not designed for use as classification criteria3,6. New classification criteria for antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and large vessel vasculitis have been developed over the past decade7. Those for AAV will be published during 2022. It is hoped that these will provide a secure basis from which to conduct future studies8,9,10.
Two main types of study have been used to assess the incidence and prevalence of vasculitis: cohort studies and registry studies. Cohort studies have the benefit that case classification can be confirmed, the downside being that it can take a long time to accumulate enough cases to conduct an accurate study. The geographical location from which the study population is drawn also needs to be tightly defined to ensure complete case capture. By contrast, registry studies have the advantage that a large number of cases can be identified rapidly from large populations, but confirmation of case classification is often impossible. Statistical methods such as capture–recapture analysis enable the identification of cases that might have been missed, but are only possible where potential cases can be identified from multiple independent sources11. Differences in study design are therefore considered to be responsible for some of the variation in prevalence seen between geographical regions. For example, a study in which the effect of methodology on the geographic variation in Behçet syndrome prevalence was explored concluded that studies that used a sample survey design reported strikingly greater prevalence rates than studies with a census design12. Furthermore, the structure of many health-care systems does not lend itself to epidemiological studies. Regions with well-developed comprehensive state insurance-based systems are good locations for registry studies, whereas those with more fragmented systems can often only produce centre-based studies or registries based on private insurance systems. Reliable data are therefore lacking from many regions, particularly Africa, the Indian subcontinent, China and Latin America. Importantly, this lack of data does not mean that the conditions do not occur in these locations, just that the tools for measuring disease occurrence and burden are not available.
In this Review, we examine the occurrence of types of vasculitis that are of particular relevance to rheumatologists (Table 1), focusing on large vessel vasculitis (Takayasu arteritis and GCA), medium vessel vasculitis (polyarteritis nodosa and Kawasaki disease), small vessel vasculitis (AAV, IgA vasculitis and hypocomplementaemic urticarial vasculitis) and Behçet syndrome, across the globe to develop an overview of the current burden of disease.
Large vessel vasculitis
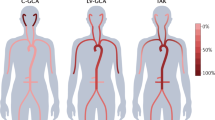
Large vessel vasculitis encompasses Takayasu arteritis and GCA, which are conditions that cause inflammation of the aorta and its major branches3. There has been considerable debate as to whether these two conditions are distinct or represent a spectrum13. The 1990 ACR classification criteria included strict age criteria of ≤40 years for Takayasu arteritis and ≥50 years for GCA14,15. However, many patients with Takayasu arteritis present after a considerable delay after the age of 40 years, making application of the criteria challenging. The patterns of arterial involvement also overlap, making distinction on clinical grounds difficult16.
Takayasu arteritis
Compared with GCA, there are relatively few data on the occurrence of Takayasu arteritis. In addition, there is often a long diagnostic delay, which makes incidence studies unreliable. The annual incidence rate is in the range 0.4–3.4 per million people with a female predominance, although the majority of studies have been in white populations from Northern Europe17,18,19,20,21. Takayasu arteritis was originally described in Japan and it has long been considered that the occurrence is greatest in individuals from Southeast Asia. Data from the 1980s from Japan suggest that the incidence rate there is similar to that seen in Europe (1–2 per million people per year)22. A 2021 systematic meta-analysis estimated the incidence rate to be 1.11 per million person years (95% CI 0.70–1.76), although considerable heterogeneity in data was noted, suggesting substantial variation in incidence rates across different populations23.
Studies from the past 10 years suggest prevalence figures of 8.4 per million in the USA24, 9.0 per million in Italy25, 13.2 per million in Sweden19, 14.5 per million in Switzerland26 and 25.2 per million in Norway18, which are approaching those seen in Japan (40.0 per million), South Korea (28.2 per million) and Turkey (33.0 per million)21,27,28. Studies from Scandinavia report a higher prevalence in people of non-Northern European ancestry than in those with Scandinavian ancestry18,19. In Norway, there was a 3.5-fold to 5.0-fold difference in prevalence between individuals of Northern European ancestry (22.0 per million) and those of Asian ancestry (78.1 per million) or African ancestry (108.3 per million)18. However, interpretation of these studies is limited by the small numbers of patients in the non-Scandinavian populations included.
Giant cell arteritis
GCA is a common systemic vasculitis in adults aged >50 years29. Cranial GCA is characterized by acute onset headache, scalp tenderness, jaw claudication and visual loss. Patients with extracranial GCA have a more severe systemic illness with fever, weight loss, aortitis and involvement of peripheral arteries. Permanent blindness is the most feared long-term sequela of untreated GCA, occurring in up to 30% of patients30. Modern imaging techniques, including positron emission tomography, computed tomography and vascular ultrasonography, have demonstrated that extra-cranial large vessel involvement is much more common than was previously recognized, occurring in up to 83% of patients31. Furthermore, a subset of patients with GCA present with systemic illness without classic features of cranial GCA32. The epidemiological characteristics of this phenotype are not well described and might overlap with cranial GCA and with Takayasu arteritis. In some regions, vascular imaging (particularly ultrasonography of temporal arteries) is replacing traditional temporal artery biopsy (TAB) for GCA diagnosis, but vascular imaging is not routinely undertaken in all regions and centres. The 1990 ACR criteria for GCA do not include vascular imaging15, although the performance of the criteria can be improved by including imaging33. New joint ACR–EULAR criteria are currently being developed that will address this issue7.
GCA is well known as a disease that is more common in populations with Northern European ancestry (Fig. 1a, Supplementary Table 1). Comparable epidemiological features have been reported in Scandinavian and Northern American populations, particularly in areas of North America with immigrants from Scandinavia34,35. However, globally, the majority of studies have reported on populations with Northern European ancestry, and it is possible that the apparently higher frequency of GCA in Northern Europe is an artefact of the populations studied. A 2021 meta-analysis of studies that included more than 50 cases and that were published before 2019 reported an overall pooled incidence of GCA of 10.0 per 100,000 in people aged ≥50 years, with the highest incidence in Scandinavia (21.6 per 100,000) followed by North and South America (10.89 per 100,000), Oceania (7.8 per 100,000) and Europe (7.3 per 100,000)29. This meta-analysis confirmed the previously reported association of latitude with incidence but not with prevalence or mortality36. Notably, the association of incidence with latitude is independent of the increased frequency of HLA-DRB1*04 (a GCA susceptibility allele) in Northern European populations36.

a | Giant cell arteritis occurs most frequently in populations of Northern European ancestry. See Supplementary Table 1 for data used. Grey indicates no data available. b | Changing incidence of giant cell arteritis. Giant cell arteritis is gradually becoming less common, having peaked in incidence around 1990. Data from refs34,38,39,40.
The reported epidemiological characteristics of GCA vary worldwide depending on the case definition used, as some studies only include patients with TAB-confirmed GCA, whereas others rely on clinical manifestations and include patients who fulfil the 1990 ACR classification criteria, which do not mandate confirmatory histology15, or patients with evidence of large vessel vasculitis from imaging studies. As expected, studies that use clinical and histological data yield slightly higher incidence figures than those that do not; in a systematic review, the mean incidence rate of GCA in European studies using only patients with a positive TAB was 14.6 per 100,000 persons aged ≥50 years (range 6.0–43.6) compared with 15.2 per 100,000 in studies that used broader clinical criteria37. However, the incidence of TAB-confirmed GCA might be decreasing. Several long-term studies have now reported data from the same population over 40 years34,38,39,40. Figure 1b illustrates changes in the incidence rate of TAB-confirmed GCA over 40 years in two Swedish regions with comparable populations that used similar case identification. The incidence rate of TAB-proven GCA in Sweden during 1976–1995 was 22.2 per 100,000 inhabitants aged ≥50, decreasing to 13.3 per 100,000 during 1997–2019, and there was a simultaneous decrease in the rate at which TAB was performed, from 74.1 per 100,000 (95% CI 70.7–77.5) during 1997–2002 to 50.2 per 100,000 (95% CI 47.4–53.0) during 2015–2019 (refs34,40). The decreases in the rate of performing TABs and in the incidence of TAB-proven GCA might be explained by the increasing use of imaging studies in place of TAB and the changing demographics of Sweden, as immigration from areas with a lower prevalence of GCA has increased.
A striking feature of the epidemiology of GCA is the strong effect of age. For example, in a well-documented population in Sweden, the incidence rate was 2.0 per 100,000 in those aged 50–60 years and 31.3 per 100,000 in those aged 71–80 years41. Data suggest that there might be a decline in incidence in those aged >90 years, which could reflect underdiagnosis in this age group41. The mean age at diagnosis of GCA increased by 4.5 years between 1997 and 2010, mainly as a result of increased incidence in people aged 70 years and older, findings that might indicate interactions between environmental factors and an ageing immune system42. In addition, numerous studies have shown that GCA is 2–3 times more common in women than men in all age groups37 (Supplementary Table 1).
The aetiology of GCA is poorly understood, but associations with host and environmental factors have been investigated. Obesity and diabetes mellitus are negatively associated with GCA43,44,45, whereas exposure to different infections, particularly those that affect the respiratory system, is associated with an increased risk of GCA46,47. Studies on smoking have shown variable effects that range from a protective effect on GCA development in men to an association with increased risk of GCA in women48,49. The effect of seasonality on GCA onset has also produced conflicting data: some studies have shown increasing occurrence during summer50,51,52 or spring and summer40, whereas a large study that included >2,200 patients with GCA from Australia, New Zealand, Germany and the Netherlands showed an even distribution of GCA diagnosis throughout the year53.
Compared with incidence studies, few studies on the prevalence of GCA have been published. This paucity of data is probably due to the nature of the disease, as many patients achieve long-term remission off treatment and are therefore not included in prevalence estimates. Similar to incidence estimates, different criteria and case definitions, as well as different epidemiological definitions, are used for prevalence studies, which either report point prevalence or cumulative prevalence. In general, more recent studies have reported higher prevalence figures than older studies. For example, in the UK, a cumulative prevalence of 250 per 100,000 was reported in 2016 using data from a primary care setting54; however, these results were based on a single general practice, and a UK study from the 1990s reported a prevalence of 84 per 100,000 (ref.55). In North America, data from Olmsted County, MN, USA suggest a prevalence of 204 per 100,000 in 2015 (ref.56), whereas in Ontario, Canada, the prevalence in 2018 was 235 per 100,000, compared with 125 per 100,000 in 2000 (ref.57). A Swedish study from 2021, in which treatment with glucocorticoids on the date of prevalence calculation was required for inclusion, reported an overall prevalence of 75.5 per 100,000 (107.8 per 100,000 in women and 40.1 per 100,000 in men)40. The prevalence of GCA in the same population irrespective of steroid treatment was 127.1 per 100,000. The higher figure reflects inclusion of patients with polymyalgia rheumatica who had ever been treated with steroids. By contrast, a very low prevalence rate of 1.47 per 100,000 was reported in a study from Japan in the 1990s58. Interestingly, unlike the association between incidence of GCA and latitude, a 2021 meta-analysis did not demonstrate an association between latitude and GCA prevalence29. This discrepancy between incidence and prevalence in the meta-analysis probably reflects the relative paucity of prevalence studies.
Medium vessel vasculitis
Polyarteritis nodosa
Polyarteritis nodosa is a systemic necrotizing vasculitis that predominantly affects medium-sized muscular arteries3. Hepatitis B virus (HBV) is a well-recognized trigger infection for polyarteritis nodosa, and HBV-associated polyarteritis nodosa is now classified as a separate disease. With the introduction of widespread vaccination against HBV, there has been a reduction in the proportion of patients with polyarteritis nodosa who are infected with HBV59. Polyarteritis nodosa is estimated to have an annual incidence rate of 0.9–8.0 per million in European countries60,61, and a prevalence of 31 per million59,62. Polyarteritis nodosa can affect patients of all ethnicities, typically occurs in patients aged 40–60 years and, unlike most other types of vasculitis, has a male preponderance (male-to-female ratio of 1.5:1)59. In addition, polyarteritis nodosa can occur in association with familial Mediterranean fever63.
Kawasaki disease
Kawasaki disease differs in important ways from the other forms of vasculitis covered in this Review. Kawasaki disease almost exclusively affects children (85% of affected patients are under the age of 5 years64), and the vasculitis is self-limited, with all patients seeming to completely recover. Another difference is that the systemic inflammation during the acute phase of the illness is not confined to the arterial wall but also affects the myocardium; myocarditis by histological criteria is a universal finding during acute illness65,66. Decades later, autopsy studies of patients with giant aneurysms who had previously had Kawasaki disease show diffuse, bridging fibrosis beyond the territories supplied by the affected arteries67,68. The genesis of this fibrosis is uncertain but it could be the result of subclinical, smouldering microvascular inflammation or of chronic myocardial inflammation. Another unique feature of Kawasaki disease is its emergence as a new condition in Asia after World War II69,70. The existence of Kawasaki disease in North America in the nineteenth and early twentieth centuries as a rare, uniformly fatal condition called infantile polyarteritis nodosa is supported by comparison of autopsies of patients with infantile polyarteritis nodosa and those with Kawasaki disease71.
The epidemiology of Kawasaki disease is an imprecise science as there is no specific diagnostic test for the condition. Kawasaki disease is diagnosed on the basis of clinical criteria coupled with laboratory evidence of acute inflammation with or without dilation of the coronary arteries by transthoracic echocardiography. Infants and children present with fever and mucocutaneous manifestations including conjunctival injection, rash, erythema of the oral mucosa and vermillion border, cervical lymphadenopathy and swelling of the dorsa of the hands and feet72. Without treatment, 25% of children will develop coronary artery aneurysms that can lead to thrombosis, myocardial infarction, heart failure or death73. Treatment with intravenous immunoglobulin as an immunomodulatory agent reduces the incidence of aneurysms to 3–5%74.
Kawasaki disease has been reported in more than 60 regions spanning five continents75. The epidemiology of Kawasaki disease has been most clearly defined for Japan, the location with the highest known incidence in the world, where questionnaire surveys of hospitals with at least 100 beds have been conducted every 2 years since 1970 (ref.76). Nationwide epidemics of Kawasaki disease in Japan in 1979, 1982 and 1986 were followed by steadily increasing numbers of cases, leading to the current incidence rate of 359 per 100,000 children <5 years of age. According to the latest estimates from Japan, approximately 1 in every 64 boys and 1 in every 80 girls will develop Kawasaki disease during the first 10 years of their life77. A genetic predisposition to Kawasaki disease is supported by its occurrence in first-degree relatives of patients with the disease, the increased incidence of Kawasaki disease in children of Asian descent living outside of Asia, and the discovery of genetic variants that influence susceptibility78. The incidence of Kawasaki disease is high throughout Northeast Asia, with incidence rates per 100,000 children <5 years of 195 in South Korea and 60 in Taiwan79,80 (Fig. 2, Supplementary Table 2). Both of these regions have national health systems and nationwide databases that permit accurate tracking of disease incidence. In other parts of Asia, the lack of accurate population statistics and a centralized database has resulted in uncertainty regarding the incidence of Kawasaki disease. For example, in India and China, the two most populous regions of the world, there is no centralized collection of data to aid in disease epidemiology. The epidemiology of Kawasaki disease in China and India has been fully reviewed elsewhere81. Briefly, questionnaire-based studies from Beijing and Shanghai have reported incidence rates of 46.3–55.1 per 100,000 children aged <5 years. The emergence of Kawasaki disease in India is thought to be a recent occurrence and case numbers have been steadily rising, although there is a debate as to whether this is due to increased case ascertainment or a true rising incidence82. Increased recognition of Kawasaki disease might also be due to a reduction in vaccine-preventable diseases that manifest with rash and fever, and which can mimic the clinical features of Kawasaki disease83. Only regional incidence rates are available for India. In Chandigarh in northern India, incidence rates from 2009–2014 have varied between 1 per 100,000 and 9 per 100,000 children aged <5 years84.

Kawasaki disease occurs most frequently in East Asia, especially Japan, South Korea and China, with a relatively equal distribution elsewhere. See Supplementary Table 2 for data used. Grey indicates no data available.
European and North American locations with national health services and centralized data collection have reported incidence rates per 100,000 in children aged <5 years that range from 19.6 in Canada to 4.5–9.0 in the UK and most of Europe85,86,87,88. Low rates have also been reported in Finland, Sweden and Norway (11.4, 5.4 and 7.4 per 100,000 children aged <5 years, respectively)89. In the USA, administrative databases have been used to estimate the burden of disease. Using such databases, estimated incidence rates for Kawasaki disease are approximately 20 per 100,000 children aged <5 years90. Prospective epidemiological investigation over a 10-year period from a region in southern California with a large Hispanic population revealed incidence rates per 100,000 children aged <5 years of 14.9, 20 and 50 for children of European, Hispanic and Asian or Pacific Islander descent, respectively64.
Intriguing aspects of Kawasaki disease epidemiology include the distinct global seasonality, with peaks in winter, spring and mid-summer, and the temporal and spatial clustering of cases91,92,93,94. No evidence exists to support person-to-person transmission of Kawasaki disease, and clinical features, including cervical lymphadenopathy, hoarseness and retropharyngeal oedema, all suggest a respiratory point of entry95,96,97. Potential clues to the aetiology of Kawasaki disease can be gleaned from the analysis of temporal clusters that share distinct clinical and laboratory features. The existence of these sub-phenotypes suggests that Kawasaki disease might be triggered by diverse agents that each yield slight variations in clinical presentation98. A surprising observation from the past year is the large decrease in the incidence of Kawasaki disease in Japan and the USA during the SARS-CoV-2 pandemic, which supports the hypothesis that Kawasaki disease is triggered by inhaled aerosols that can be blocked or reduced by the use of facial masking99,100. The possibility of long-range transport of aerosols linked to fluctuations in Kawasaki disease incidence has been studied in both Northern and Southern Hemispheres93,101,102. Atmospheric circulation patterns have been reported that link winds from northeastern China with fluctuations in incidence rates of Kawasaki disease in Japan and winds from the Atacama Desert with Kawasaki disease incidence in southern Chile102,103. At least in Northeast Asia, exposure to the causative agents of Kawasaki disease continues to increase and case numbers continue to steadily climb104.
Small vessel vasculitis
ANCA-associated vasculitis
AAV is a group of three conditions predominantly affecting small vessels, the main types being granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA)3 (Table 1). The incidence of AAV has increased during the past 40 years. The combined incidence rate for GPA, MPA and EGPA from Norway (1999–2013) and the USA (1996–2015) was around 24.7 per million in Norway105 and 33.0 per million in the USA106, compared with a rate of only 1.5 per million for GPA alone in the UK in the 1980s107. Figure 3 and Supplementary Table 3 summarize the incidence rates of AAV around the globe since 2000. The increased reported incidence is attributable to improved case recognition following the widespread introduction of ANCA testing in the 1990s and the application of uniform, albeit imperfect, classification criteria. The widespread adoption of the 1994 Chapel Hill Consensus definitions for vasculitis resulted in the recognition of MPA as an entity distinct from polyarteritis nodosa6. With the acknowledged imperfections of the ACR and Chapel Hill Consensus criteria for classification, there has been debate as to whether it is preferable to classify patients by ANCA serotype (proteinase 3 and myeloperoxidase (MPO)) alone rather than by the clinical phenotype108. It is hoped that the new EULAR–ACR classification system, which incorporates ANCA status, will help to resolve this debate8,9,10.

Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis subtypes show different patterns of occurrence across populations. For example, in Southern Europe and Japan, microscopic polyangiitis is more common than granulomatosis with polyangiitis, whereas as in most other populations, granulomatosis with polyangiitis is the more common form. Eosinophilic granulomatosis with polyangiitis is the least common of the three conditions. See Supplementary Table 3 for data used. Grey indicates no data available. a | The global incidence of granulomatosis with polyangiitis. b | The global incidence of microscopic polyangiitis. c | The global incidence of eosinophilic granulomatosis with polyangiitis.
In the most recent study from northern Norway, the incidence rate for MPA substantially increased from 2.7 per million in 1999–2003, to 10.4 per million in 2009–2013 (ref.105). A study using data from the 1980s and 1990s in which the incidences of GPA and MPA were compared across Europe suggested that GPA was more common than MPA in Northern Europe, whereas the opposite was true in Southern Europe109. However, data from the late 1990s and 2000s from several European countries have suggested that this might no longer be true61,110,111. Data from epidemiological studies, serological studies and large case series suggest that MPA is much more common than GPA in China and Japan112,113. To confirm these observations, it will be critical to rigorously classify cases using the same criteria. Moreover, separating the influence of genetic and environmental factors in such studies will be difficult, as people with different genetic backgrounds have been studied in different geographical locations, limiting comparison. A global study reported that MPO-ANCA was much more common in Japanese, Chinese and Southern European individuals than in Northern European individuals114. In the same study, ophthalmological and ear, nose and throat involvement was less common in Japanese and Chinese patients with AAV than in Northern European patients with AAV. In a multi-ethnic series from Chapel Hill in the USA, GPA was less common in African American individuals than in those with European ancestry115. By contrast, in the UK, there was no evidence of an effect of ethnicity on the occurrence of GPA in a combined primary and secondary database study116. In all countries studied so far, EGPA is the rarest of the three forms of AAV, with an incidence rate in the range 0.14–4.0 per million and no evidence of a change over time (Supplementary Table 3).
In addition to the increasing incidence of AAV, there has also been an increase in the peak age at diagnosis. In studies looking at data from the late twentieth century, the peak age-specific incidence was reported in those aged 65–74 years in the UK and Finland60,117. In data from the early twenty-first century in the UK, the peak age for AAV incidence had increased to >80 years116. Again, the reasons for the increased diagnosis of AAV in older individuals are not clear, but could be related to the increasing ease of ANCA testing, which might lead to the identification of patients who would not previously have been considered to have AAV. At the other end of the age spectrum, AAV is very uncommon. The incidence rate of AAV in those aged 0–17 years is 0.45 per million in France118 and 3.2 per million in Sweden119. Overall, AAV is slightly more common in men than in women (male-to-female ratio of between 1.07:1 and 1.48:1)110,111,120,121.
The trigger for development of AAV is unknown, and environmental factors have been extensively investigated122. Some studies have reported a cyclical pattern of incidence suggestive of an infectious trigger but, so far, no clear infectious trigger has been identified for AAV123,124. Seasonality has also been investigated as a clue to infection; however, no clear pattern has been observed. European studies from the 1990s suggested a trend towards an increase in GPA in the winter123,125, whereas more recent studies have suggested a summer link or no association126,127, which might reflect differences in case definition by date of symptom onset or date of diagnosis. A study looking at health-care events occurring prior to a diagnosis of GPA in a UK general practice database did not report any health events that predicted subsequent development of vasculitis128. Silica has also been proposed as a possible trigger in a number of small studies129 and, in 2021, a geospatial association was reported between GPA and quarries in the Alsace region of France130. Other reported risk factors include rural living and farming131,132. Social factors have also been investigated, but no clear association with socio-economic status has so far been demonstrated, possibly reflecting differences in case definition (such as renal versus non-renal vasculitis) and assessment of socioeconomic status127.
Compared with incidence studies, there are relatively fewer studies on the prevalence of AAV. Prevalence rates for GPA as high as 261.0 per million have been reported in Norway, with rates of 58.2 per million for MPA and 32.9 per million for EGPA105. The prevalence of AAV has increased owing to a combination of increasing incidence, improved case definitions, the emergence of a number of local or national vasculitis registries in different locations around the world and improved survival as a result of improvements in treatment.
IgA vasculitis
IgA vasculitis (previously known as Henoch–Schönlein purpura) is an immune complex vasculitis that predominantly affects small vessels3. In children and adolescents, IgA vasculitis is the most common type of vasculitis, the main features being cutaneous palpable purpura, arthralgia or arthritis, bowel angina, and haematuria or proteinuria119,133. The annual incidence rate of IgA vasculitis is between 3.5 (in Japan) and 26.7 (in Scotland) per 100,000 persons aged <15 years134,135, and the highest rate reported is between the ages of 4 and 6 years (70.3 per 100,000 in the UK)119,133. European studies have reported an incidence in Sweden (2004–2014) of 17.5 per 100,000 persons aged <15 years and in France (2012–2014) of 18.6 per 100,000 persons aged <15 years11,119. A study in a multi-ethnic cohort from Birmingham, UK, reported that IgA vasculitis was more common in those of Indian subcontinent ancestry (24.0 per 100,000 persons aged <17 years) than in white individuals (17.8 per 100,000 persons <17 years) or Black individuals (predominantly those of Afro-Caribbean ancestry; 6.2 per 100,000 persons aged <17 years)133. A 2017 study from France similarly noted an increased occurrence of IgA vasculitis in children of North African ancestry11.
In adults, IgA vasculitis is much less common than in children, with a broad range of incidence estimates. Studies suggest that the incidence in Spain between 1992 and 2010 was 1.5 per million and in Finland (2010) it was 15 per million, although the latter figure is based on two cases136,137. The mean age of onset for IgA vasculitis in adults is 50 years, and the condition is more common in men than in women138. IgA vasculitis is typically a disease with a relatively short duration and there are few data on prevalence, although a study from Spain has reported a prevalence of 7.9 per million136.
Hypocomplementaemic urticarial vasculitis
Hypocomplementaemic urticarial vasculitis is a form of vasculitis characterized by urticaria and hypocomplementaemia and is associated with anti-C1q antibodies3. Only a single study exists that describes the epidemiology of hypocomplementaemic urticarial vasculitis. A small Swedish study reported an annual incidence rate of 0.7 per million persons with a point prevalence on 31 December 2015 of 9.5 per million139. The median age of onset was 51 years and 87.5% of patients were women.
Behçet syndrome
Behçet syndrome is a variable vessel vasculitis that runs a remitting and relapsing course with oral and genital ulcers, erythema nodosum-like and papulopustular skin lesions, arthritis, uveitis, arterial aneurysms, arterial and venous thrombosis, parenchymal nervous system lesions and intestinal ulcers3. Formal incidence studies are rare for Behçet syndrome. Annual incidence rates have been reported from South Korea (3.9 per 100,000), Taiwan (2.4 per 100,000), Poland (0.05 per 100,000) and Sweden (0.2 per 100,000)140,141,142,143. By contrast, the prevalence of Behçet syndrome has been widely studied and shows substantial variability between countries12,140,141,142,144,145,146,147,148,149,150,151,152,153,154 (Fig. 4 and Supplementary Table 4). The highest burden is documented along the ancient silk road, which runs from the eastern Mediterranean and the Middle East to East Asia, with estimated prevalence rates of 421 per 100,000 in Turkey, 660 per 100,000 in Jordan, 100 per 100,000 in Iran, 35.0 per 100,000 in South Korea, 13.5 per 100,000 in Japan and 10.0 per 100,000 in China147,149,150,151,152,154. In the USA, studies have reported estimated prevalence rates of between 0.33 and 10.6 per 100,000 and in South America, rates of 0.3 per 100,000 have been reported in Brazil and 1.1 per 100,000 in Colombia144,145,146,153. A 2018 meta-analysis showed that pooled estimates of prevalence proportions were 10.3 per 100,000 for all studies included, and 31.8 per 100,000 for the Middle East, 4.5 per 100,000 for Asia and 3.3 per 100,000 for Europe12. The prevalence tends to decrease towards the north in Europe and towards the south in Africa. Differences in study design, calculating prevalence rates versus prevalence odds ratios and the criteria used for case ascertainment make it difficult to compare prevalence studies from different countries. Notably, in the meta-analysis, population-based studies showed a 12-fold higher prevalence than hospital or registry-based studies in the same geographic area12.

Behçet syndrome occurs most commonly along the ancient silk road between the Mediterranean and China. See Supplementary Table 4 for data used. Grey indicates no data available.
Similar to disease prevalence, disease severity and the occurrence of specific manifestations in Behçet syndrome, as well as the association with HLA-B51 positivity, also show geographical variation. The risk of developing Behçet syndrome is increased by a factor of 5.78 for carriers of HLA-B51 or HLA-B5 (ref.155), and the prevalence of Behçet syndrome in a population is generally positively associated with the prevalence of HLA-B51. For example, a study from Turkey showed that 30% of the general population were positive for HLA-B51 and Behçet syndrome had a prevalence of 421.0 per 100,000 (ref.149). By contrast, a study showed that 8.9% of the general population was HLA-B51 positive in the UK156, where the prevalence of Behçet syndrome is 14.6 per 100,000 (ref.157). An exception to this rule is some Native American populations in southwestern USA, in which HLA-B51 is common but the prevalence of Behçet syndrome is low144.
Behçet syndrome occurs at a similar rate in both men and women, but a more severe prognosis, more organ involvement and higher mortality rates are reported among men158. Behçet syndrome usually starts during the second or third decade of life, and an early age of disease onset seems to be a poor prognostic factor in addition to male sex159. Despite differences between cohorts, in general, women more commonly have isolated skin, mucosal and musculoskeletal involvement, whereas men more commonly have uveitis, vascular and, in some series, central nervous system involvement158. Differences have also been observed in how common certain manifestations are among geographical locations. Gastrointestinal involvement was reported in up to 30% of patients in East Asia and the USA, compared with around 1% in Turkey, Tunisia and Spain148. A positive reaction to the pathergy test (hyperreactivity of the skin to a needle prick) is more common in the Mediterranean region and East Asia, whereas it is much less common in Northern Europe148,160. A number of studies that compared patients with Behçet syndrome in the USA with those in other countries showed more women, more gastrointestinal and nervous system involvement, higher disease activity and worse quality of life in patients in the USA than patients in Turkey161; more genital ulcers, less epididymitis and pulmonary involvement than patients in Japan162; and more women, earlier age of onset, more genital ulcers, skin lesions, arthritis, nervous system and cardiovascular involvement than patients in Iran163. The general contention has been that Behçet syndrome runs a more severe course in countries with a high prevalence. Although the results of these studies run contrary to that presumption, it is thought that the reports of more severe disease in the USA might be associated with the fact that only those patients with more severe disease are referred and diagnosed in the USA.
A number of studies have looked at the change in the prevalence, severity and occurrence of disease manifestations of Behçet syndrome over time, with varying results. A study from Japan reported a decrease in Behçet syndrome prevalence over decades, whereas a South Korean study reported a small, but steady increase152,164. By contrast, a more recent study from South Korea suggested that there had been a substantial decline in the incidence of Behçet syndrome165. Overall, a milder disease course has been reported in different studies, with fewer skin lesions, genital ulcers and positive pathergy tests, and a decrease in the occurrence and severity of uveitis164,166,167. One of the hypotheses for explaining the milder disease course was the possibility of improved living conditions and hygiene, as an association with infections has been proposed in the pathogenesis of Behçet syndrome. Although infections have been considered to be the main environmental trigger, no solid evidence exists to support a link with any specific microorganism168. Some dysbiosis has been noted in all microbiota studies in Behçet syndrome, but which microorganisms were abundant or decreased differed in studies from different regions. Interestingly, an increase in the occurrence of gastrointestinal involvement has been reported in both Japan and South Korea164,167. Notably, an increase in the prevalence of inflammatory bowel diseases has also been reported in East Asia, which was attributed to the adoption of a Western diet169,170. Therefore, it is not clear whether a similar association might also be true for gastrointestinal involvement of Behçet syndrome, which highly resembles Crohn’s disease.
By contrast, an increase in the prevalence of Behçet syndrome over time has been reported in Europe, including in the UK, Ireland and Sweden143,157,171. Increased awareness among physicians and changes in the disease criteria that were used have been considered as possible explanations for this increase, as well as immigration from regions with a high prevalence of Behçet syndrome. Studies comparing the prevalence and disease characteristics of Behçet syndrome among immigrants and locals in Europe have provided important clues to our understanding of Behçet syndrome regarding the roles of genetic and environmental factors. Such studies from Germany, France, Italy, Switzerland, Sweden and the Netherlands reported that the prevalence of Behçet syndrome among immigrants was higher than the prevalence among locals, and was somewhat lower than or similar to the prevalence in the immigrant’s region of origin143,172,173,174,175,176. These results were thought to support the role of both genetics and the environment in the pathogenesis of Behçet syndrome, but could be biased by differences in living conditions and health-care access between people of different ethnicities within the same geographic area. Moreover, the association with age at immigration and prevalence among second-generation and third-generation immigrants have not been adequately studied.
Conclusions
In conclusion, knowledge of the epidemiology of vasculitis has increased over the past two decades, and it has become apparent that vasculitis occurs most commonly at the ends of the age spectrum. Kawasaki disease is predominantly a disease of those aged under 5 years and IgA vasculitis is predominantly a disease of childhood and adolescence, suggesting a role for infection in the initiation of vascular inflammation in these conditions. By contrast, GCA is a disease of older individuals, being the most common type of vasculitis in those aged over 65 years, perhaps pointing to an interaction between infection and the ageing immune system. Although much less common than GCA, AAV also shows an increasing incidence with age. The underlying triggering factors for most forms of vasculitis remain broadly unknown, although some clues are emerging in relation to infection, the role of the environment (such as links between certain minerals and AAV) and genetics. However, considerable lacunae remain in our knowledge of the basic descriptive epidemiology of vasculitis. Very few high-quality data exist from the two most populous regions of the planet, and the effects of ethnic background, together with other social factors including deprivation, are largely unknown. Future research should address the urgent need for a global dataset to describe the occurrence of disease, variations in which both temporally and geographically might provide insights into pathogenesis. Better global prevalence data will enable the disease burden to be estimated, which is important as vasculitis is associated with substantial long-term morbidity, including vision loss in GCA, coronary artery aneurysms in Kawasaki disease and renal failure in AAV. These long-term morbidities are associated with considerable costs, both financial to health services and society, and to individuals.
References
Newburger, J. W., Takahashi, M. & Burns, J. C. Kawasaki disease. J. Am. Coll. Cardiol. 67, 1738–1749 (2016).
Akiyama, M. et al. Innate and adaptive immunity in giant cell arteritis. Front. Immunol. 11, 621098 (2020).
Jennette, J. et al. 2012 revised international Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 65, 1–11 (2013).
Fries, J. F. et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis. Summary. Arthritis Rheum. 33, 1135–1136 (1990).
Seeliger, B. et al. Are the 1990 American College of Rheumatology vasculitis classification criteria still valid? Rheumatology 56, 1154–1161 (2017).
Jennette, J. C. et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 37, 187–192 (1994).
Craven, A. et al. ACR/EULAR-endorsed study to develop diagnostic and classification criteria for vasculitis (DCVAS). Clin. Exp. Nephrol. 17, 619–621 (2013).
Grayson, P. et al. 2021 American College of Rheumatology/European League Against Rheumatism classification criteria for eosinophilic granulomatosis with polyangiitis. Ann. Rheum. Dis. (in the press) (2021).
Suppiah, R. et al. 2021 American College of Rheumatology/European League Against Rheumatism classification criteria for microscopic polyangiitis. Ann. Rheum. Dis. (in the press) (2021).
Robson, J. et al. 2021 American College of Rheumatology/European League Against Rheumatism classification criteria for granulomatosis with polyangiitis. Ann. Rheum. Dis. (in the press) (2021).
Piram, M. et al. Incidence of IgA vasculitis in children estimated by four-source capture — recapture analysis: a population-based study. Rheumatology 56, 1358–1366 (2017).
Maldini, C., Druce, K., Basu, N., LaValley, M. P. & Mahr, A. Exploring the variability in Behçet’s disease prevalence: a meta-analytical approach. Rheumatology 57, 185–195 (2018).
Maksimowicz-McKinnon, K., Clark, T. M. & Hoffman, G. S. Takayasu arteritis and giant cell arteritis: a spectrum within the same disease? Medicine 88, 221–226 (2009).
Arend, W. P. et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 33, 1129–1134 (1990).
Hunder, G. G. et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 33, 1122–1128 (1990).
Gribbons, K. B. et al. Patterns of arterial disease in Takayasu arteritis and giant cell arteritis. Arthritis Care Res. 72, 1615–1624 (2020).
Watts, R., Al-Taiar, A., Mooney, J., Scott, D. & Macgregor, A. The epidemiology of Takayasu arteritis in the UK. Rheumatology 48, 1008–1011 (2009).
Gudbrandsson, B., Molberg, Ø., Garen, T. & Palm, Ø. Prevalence, incidence and disease characteristics of Takayasu arteritis differ by ethnic background; data from a large, population based cohort resident in southern Norway. Arthritis Care Res. 69, 278–285 (2017).
Mohammad, A. J. & Mandl, T. Takayasu arteritis in southern Sweden. J. Rheumatol. 42, 853–858 (2015).
Kanecki, K. et al. Takayasu’s arteritis: a rare disease in Poland. Ann. Agric. Environ. Med. 25, 469–472 (2018).
Park, S. J. et al. Incidence, prevalence, mortality and causes of death in Takayasu Arteritis in Korea — a nationwide, population-based study. Int. J. Cardiol. 235, 100–104 (2017).
Koide, K. Takayasu arteritis in Japan. Heart Vessels Suppl. 7, 48–54 (1992).
Rutter, M., Bowley, J., Lanyon, P. C., Grainge, M. J. & Pearce, F. A. A systematic review and meta-analysis of the incidence rate of Takayasu arteritis. Rheumatology 60, 4982–4990 (2021).
Sanchez-Alvarez, C., Crowson, C. S., Koster, M. J. & Warrington, K. J. Prevalence of Takayasu arteritis: a population-based study. J. Rheumatol. 48, 952 (2021).
Muratore, F. et al. Incidence and prevalence of large vessel vasculitis (giant cell arteritis and Takayasu arteritis) in northern Italy: a population-based study. Semin. Arthritis Rheum. 51, 786–792 (2021).
Gloor, A. D. et al. Takayasu arteritis: prevalence and clinical presentation in Switzerland. PLoS ONE 16, e0250025 (2021).
Terao, C., Yoshifuji, H. & Mimori, T. Recent advances in Takayasu arteritis. Int. J. Rheum. Dis. 17, 238–247 (2014).
Saritas, F., Donmez, S., Direskeneli, H. & Pamuk, O. N. The epidemiology of Takayasu arteritis: a hospital-based study from northwestern part of Turkey. Rheumatol. Int. 36, 911–916 (2016).
Li, K. J., Semenov, D., Turk, M. & Pope, J. A meta-analysis of the epidemiology of giant cell arteritis across time and space. Arthritis Res. Ther. 23, 82 (2021).
Donaldson, L. & Margolin, E. Vision loss in giant cell arteritis. Pract. Neurol. https://doi.org/10.1136/practneurol-2021-002972 (2021).
Blockmans, D. et al. Repetitive 18F-fluorodeoxyglucose positron emission tomography in giant cell arteritis: a prospective study of 35 patients. Arthritis Rheum. 55, 131–137 (2006).
Gribbons, K. B. et al. Diagnostic assessment strategies and disease subsets in giant cell arteritis: data from an international observational cohort. Arthritis Rheumatol. 72, 667–676 (2020).
Wiberg, F., Naderi, N., Mohammad, A. J. & Turesson, C. Evaluation of revised classification criteria for giant cell arteritis and its clinical phenotypes. Rheumatology https://doi.org/10.1093/rheumatology/keab353 (2021).
Petursdottir, V., Johansson, H., Nordborg, E. & Nordborg, C. The epidemiology of biopsy-positive giant cell arteritis: special reference to cyclic fluctuations. Rheumatology 38, 1208–1212 (1999).
Salvarani, C., Crowson, C. S., O’Fallon, W. M., Hunder, G. G. & Gabriel, S. E. Reappraisal of the epidemiology of giant cell arteritis in Olmsted County, Minnesota, over a fifty-year period. Arthritis Rheum. 51, 264–268 (2004).
Mackie, S. L. et al. Association of HLA-DRB1 amino acid residues with giant cell arteritis: genetic association study, meta-analysis and geo-epidemiological investigation. Arthritis Res. Ther. 17, 195 (2015).
Sharma, A., Mohammad, A. J. & Turesson, C. Incidence and prevalence of giant cell arteritis and polymyalgia rheumatica: a systematic literature review. Semin. Arthritis Rheum. 50, 1040–1048 (2020).
Bengtsson, B. A. & Malmvall, B. E. The epidemiology of giant cell arteritis including temporal arteritis and polymyalgia rheumatica. Incidences of different clinical presentations and eye complications. Arthritis Rheum. 24, 899–904 (1981).
Nordborg, E. & Bengtsson, B. A. Epidemiology of biopsy-proven giant cell arteritis (GCA). J. Intern. Med. 227, 233–236 (1990).
Stamatis, P., Turkiewicz, A., Englund, M., Turesson, C. & Mohammad, A. J. Epidemiology of biopsy-confirmed giant cell arteritis in southern Sweden — an update on incidence and first prevalence estimate. Rheumatology https://doi.org/10.1093/rheumatology/keab269 (2021).
Mohammad, A. J., Nilsson, J.-Å., Jacobsson, L. T., Merkel, P. A. & Turesson, C. Incidence and mortality rates of biopsy-proven giant cell arteritis in southern Sweden. Ann. Rheum. Dis. 50, 1040–1048 (2015).
Kermani, T. A. et al. Increase in age at onset of giant cell arteritis: a population-based study. Ann. Rheum. Dis. 69, 780–781 (2010).
Ungprasert, P., Thongprayoon, C. & Warrington, K. J. Lower body mass index is associated with a higher risk of giant cell arteritis: a systematic review and meta-analysis. Ann. Transl. Med. 3, 232 (2015).
Tomasson, G., Bjornsson, J., Zhang, Y., Gudnason, V. & Merkel, P. A. Cardiovascular risk factors and incident giant cell arteritis: a population-based cohort study. Scand. J. Rheumatol. 48, 213–217 (2019).
Wadström, K. et al. Negative associations for fasting blood glucose, cholesterol and triglyceride levels with the development of giant cell arteritis. Rheumatology 59, 3229–3236 (2020).
Rhee, R. L., Grayson, P. C., Merkel, P. A. & Tomasson, G. Infections and the risk of incident giant cell arteritis: a population-based, case-control study. Ann. Rheum. Dis. 76, 1031–1035 (2017).
Stamatis, P. et al. Infections are associated with increased risk of giant cell arteritis: a population-based case-control study from southern Sweden. J. Rheumatol. 48, 251–257 (2021).
Larsson, K., Mellström, D., Nordborg, E., Nordborg, C. & Odén, A. Early menopause, low body mass index, and smoking are independent risk factors for developing giant cell arteritis. Ann. Rheum. Dis. 65, 529–532 (2006).
Duhaut, P. et al. Giant cell arteritis and cardiovascular risk factors: a multicenter, prospective case-control study. Arthritis Rheum. 41, 1960–1965 (1998).
Bas-Lando, M. et al. The incidence of giant cell arteritis in Jerusalem over a 25-year period: annual and seasonal fluctuations. Clin. Exp. Rheumatol. 25, S15–S17 (2007).
Kønig, E. B. et al. Seasonal variation in biopsy-proven giant cell arteritis in eastern Denmark from 1990–2018. Acta Ophthalmol. 99, 527–532 (2021).
Gokoffski, K. K., Chatterjee, A. & Khaderi, S. K. Seasonal incidence of biopsy-proven giant cell arteritis: a 20-year retrospective study of the University of California Davis Medical System. Clin. Exp. Rheumatol. 37 (Suppl. 117), 90–97 (2019).
De Smit, E. et al. Geo-epidemiology of temporal artery biopsy-positive giant cell arteritis in Australia and New Zealand: is there a seasonal influence? RMD Open 3, e000531 (2017).
Yates, M., Graham, K., Watts, R. A. & MacGregor, A. J. The prevalence of giant cell arteritis and polymyalgia rheumatica in a UK primary care population. BMC Musculoskelet. Disord. 17, 285 (2016).
Smeeth, L., Cook, C. & Hall, A. J. Incidence of diagnosed polymyalgia rheumatica and temporal arteritis in the United Kingdom, 1990–2001. Ann. Rheum. Dis. 65, 1093–1098 (2006).
Crowson, C. S. & Matteson, E. L. Contemporary prevalence estimates for giant cell arteritis and polymyalgia rheumatica, 2015. Semin. Arthritis Rheum. 47, 253–256 (2017).
Barra, L. et al. Incidence and prevalence of giant cell arteritis in Ontario, Canada. Rheumatology 59, 3250–3258 (2020).
Kobayashi, S. et al. Clinical and epidemiologic analysis of giant cell (temporal) arteritis from a nationwide survey in 1998 in Japan: the first government-supported nationwide survey. Arthritis Rheum. 49, 594–598 (2003).
Mahr, A., Guillevin, L., Poissonnet, M. & Aymé, S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum. 51, 92–99 (2004).
Watts, R. A., Lane, S. E., Bentham, G. & Scott, D. G. Epidemiology of systemic vasculitis: a ten-year study in the United Kingdom. Arthritis Rheum. 43, 414–419 (2000).
Mohammad, A. J., Jacobsson, L. T. H., Westman, K. W. A., Sturfelt, G. & Segelmark, M. Incidence and survival rates in Wegener’s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology 48, 1560–1565 (2009).
Mohammad, A. J., Jacobsson, L. T. H., Mahr, A. D., Sturfelt, G. & Segelmark, M. Prevalence of Wegener’s granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology 46, 1329–1337 (2007).
Balcı-Peynircioğlu, B. et al. Comorbidities in familial Mediterranean fever: analysis of 2000 genetically confirmed patients. Rheumatology 59, 1372–1380 (2020).
Skochko, S. M. et al. Kawasaki disease outcomes and response to therapy in a multiethnic community: a 10-year experience. J. Pediatr. 203, 408–415.e3 (2018).
Yonesaka, S. et al. Biopsy-proven myocardial sequels in Kawasaki disease with giant coronary aneurysms. Cardiol. Young 20, 602–609 (2010).
Yonesaka, S. et al. Endomyocardial biopsy in children with Kawasaki disease. Acta Paediatr. Jpn. Overseas Ed. 31, 706–711 (1989).
Orenstein, J. M. et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLoS ONE 7, e38998 (2012).
Numano, F. et al. Galectin-3 is a marker of myocardial and vascular fibrosis in Kawasaki disease patients with giant aneurysms. Int. J. Cardiol. 201, 429–437 (2015).
Shibuya, N., Shibuya, K., Kato, H. & Yanagisawa, M. Kawasaki disease before Kawasaki at Tokyo university hospital. Pediatrics 110, e17 (2002).
Burns, J. C. History of the worldwide emergence of Kawasaki disease. Int. J. Rheum. Dis. 21, 13–15 (2018).
Landing, B. H. & Larson, E. J. Are infantile periarteritis nodosa with coronary artery involvement and fatal mucocutaneous lymph node syndrome the same? Comparison of 20 patients from North America with patients from Hawaii and Japan. Pediatrics 59, 651–662 (1977).
Burns, J. C. & Glode, M. P. Kawasaki syndrome. Lancet 364, 533–544 (2004).
Kato, H. et al. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation 94, 1379–1385 (1996).
Newburger, J. W. et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N. Engl. J. Med. 315, 341–347 (1986).
Lin, M.-T. & Wu, M.-H. The global epidemiology of Kawasaki disease: review and future perspectives. Glob. Cardiol. Sci. Pract. 2017, e201720 (2017).
Ae, R. et al. Epidemiology, treatments, and cardiac complications in patients with Kawasaki disease: the nationwide survey in Japan, 2017-2018. J. Pediatr. 225, 23–29.e2 (2020).
Matsubara, Y. et al. Cumulative incidence of Kawasaki disease with cardiac sequelae in Japan. Pediatr. Int. 62, 444–450 (2020).
Onouchi, Y. The genetics of Kawasaki disease. Int. J. Rheum. Dis. 21, 26–30 (2018).
Huang, Y.-H. et al. Increased incidence of Kawasaki disease in Taiwan in recent years: a 15 years nationwide population-based cohort study. Front. Pediatr. 7, 121 (2019).
Kim, G. B. et al. Epidemiology and clinical features of Kawasaki disease in South Korea, 2012–2014. Pediatr. Infect. Dis. J. 36, 482–485 (2017).
Jiao, F. et al. The emergence of Kawasaki disease in India and China. Glob. Cardiol. Sci. Pract. 2017, e201721 (2017).
Kushner, H. I., Macnee, R. P. & Burns, J. C. Kawasaki disease in India: increasing awareness or increased incidence? Perspect. Biol. Med. 52, 17–29 (2009).
Singh, S. et al. Is Kawasaki disease incidence rising in Chandigarh, North India? Arch. Dis. Child. 96, 137–140 (2011).
Singh, S. & Bhattad, S. Kawasaki disease incidence at Chandigarh, North India, during 2009–2014. Rheumatol. Int. 36, 1391–1397 (2016).
Singh, S., Vignesh, P. & Burgner, D. The epidemiology of Kawasaki disease: a global update. Arch. Dis. Child. 100, 1084–1088 (2015).
Manlhiot, C. et al. Epidemiology of Kawasaki disease in Canada 2004 to 2014: comparison of surveillance using administrative data vs periodic medical record review. Can. J. Cardiol. 34, 303–309 (2018).
Tulloh, R. M. R. et al. Kawasaki disease: a prospective population survey in the UK and Ireland from 2013 to 2015. Arch. Dis. Child. 104, 640–646 (2019).
Heuclin, T. et al. Increased detection rate of Kawasaki disease using new diagnostic algorithm, including early use of echocardiography. J. Pediatr. 155, 695–699.e1 (2009).
Salo, E. et al. Incidence of Kawasaki disease in northern European countries. Pediatr. Int. 54, 770–772 (2012).
Maddox, R. A. et al. Kawasaki disease and Kawasaki disease shock syndrome hospitalization rates in the United States, 2006–2018. Pediatr. Infect. Dis. J. 40, 284–288 (2021).
Burns, J. C. et al. Seasonality and temporal clustering of Kawasaki syndrome. Epidemiology 16, 220–225 (2005).
Burns, J. C. et al. Seasonality of Kawasaki disease: a global perspective. PLoS ONE 8, e74529 (2013).
Manlhiot, C. et al. Environmental epidemiology of Kawasaki disease: linking disease etiology, pathogenesis and global distribution. PLoS ONE 13, e0191087 (2018).
Rypdal, M. et al. Clustering and climate associations of Kawasaki disease in San Diego County suggest environmental triggers. Sci. Rep. 8, 16140 (2018).
Kanegaye, J. T. et al. Lymph-node-first presentation of Kawasaki disease compared with bacterial cervical adenitis and typical Kawasaki disease. J. Pediatr. 162, 1259–1263 (2013).
Leuin, S. C. et al. Hoarseness as a presenting sign in children with Kawasaki disease. Pediatr. Infect. Dis. J. 32, 1392–1394 (2013).
Gámez-González, L. B. et al. Kawasaki disease presenting with hoarseness: a multinational study of the REKAMLATINA network. Pediatr. Int. 63, 643–648 (2021).
Burns, J. C. et al. Temporal clusters of Kawasaki disease cases share distinct phenotypes that suggest response to diverse triggers. J. Pediatr. 229, 48–53.e1 (2021).
Hara, T. et al. Assessment of pediatric admissions for Kawasaki disease or infectious disease during the COVID-19 state of emergency in Japan. JAMA Netw. Open 4, e214475 (2021).
Shulman, S., Geevarghese, B., Kim, K.-Y. & Rowley, A. The impact of social distancing for COVID-19 upon diagnosis of Kawasaki disease. J. Pediatr. Infect. Dis. Soc. 10, 742–744 (2021).
Rodó, X. et al. Revisiting the role of environmental and climate factors on the epidemiology of Kawasaki disease. Ann. N. Y. Acad. Sci. 1382, 84–98 (2016).
Jorquera, H., Borzutzky, A., Hoyos-Bachiloglu, R. & García, A. Association of Kawasaki disease with tropospheric winds in central Chile: is wind-borne desert dust a risk factor? Environ. Int. 78, 32–38 (2015).
Rodó, X. et al. Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan. Proc. Natl Acad. Sci. USA 111, 7952–7957 (2014).
Makino, N. et al. Nationwide epidemiologic survey of Kawasaki disease in Japan, 2015–2016. Pediatr. Int. 61, 397–403 (2019).
Nilsen, A. T. et al. Increasing incidence and prevalence of ANCA-associated vasculitis in northern Norway. Rheumatology 59, 2316–2324 (2020).
Berti, A., Cornec, D., Crowson, C. S., Specks, U. & Matteson, E. L. The epidemiology of antineutrophil cytoplasmic autoantibody-associated vasculitis in Olmsted County, Minnesota (USA): a 20 year population-based study. Arthritis Rheumatol. 69, 2338–2350 (2017).
Andrews, M., Edmunds, M., Campbell, A., Walls, J. & Feehally, J. Systemic vasculitis in the 1980s — is there an increasing incidence of Wegener’s granulomatosis and microscopic polyarteritis? J. R. Coll. Physicians 24, 284–288 (1990).
Windpessl, M. et al. ANCA status or clinical phenotype — what counts more? Curr. Rheumatol. Rep. 23, 37 (2021).
Watts, R. A. et al. Epidemiology of vasculitis in Europe. Ann. Rheum. Dis. 60, 1156–1157 (2001).
Pearce, F. A. et al. Incidence of ANCA-associated vasculitis in a UK mixed ethnicity population. Rheumatology 55, 1656–1663 (2016).
Pamuk, Ö. N., Dönmez, S., Calayır, G. B. & Pamuk, G. E. The epidemiology of antineutrophil cytoplasmic antibody-associated vasculitis in northwestern Turkey. Clin. Rheumatol. 35, 2063–2071 (2016).
Fujimoto, S. et al. Comparison of the epidemiology of anti-neutrophil cytoplasmic antibody-associated vasculitis between Japan and the U.K. Rheumatology 50, 1916–1920 (2011).
Li, J. et al. The frequency of ANCA-associated vasculitis in a national database of hospitalized patients in China. Arthritis Res. Ther. 20, 226 (2018).
Pearce, F. A., Craven, A., Merkel, P. A., Luqmani, R. A. & Watts, R. A. Global ethnic and geographic differences in the clinical presentations of anti-neutrophil cytoplasm antibody–associated vasculitis. Rheumatology 56, 1962–1969 (2017).
Cao, Y. et al. DRB1*15 allele is a risk factor for PR3-ANCA disease in African Americans. J. Am. Soc. Nephrol. 22, 1161–1167 (2011).
Pearce, F. A., Grainge, M. J., Lanyon, P. C., Watts, R. A. & Hubbard, R. B. The incidence, prevalence and mortality of granulomatosis with polyangiitis in the UK Clinical Practice Research Datalink. Rheumatology 56, 589–596 (2017).
Takala, J. H., Kautiainen, H., Malmberg, H. & Leirisalo-Repo, M. Incidence of Wegener’s granulomatosis in Finland 1981–2000. Clin. Exp. Rheumatol. 26, S81–S85 (2008).
Sacri, A.-S. et al. Clinical characteristics and outcomes of childhood-onset ANCA-associated vasculitis: a French nationwide study. Nephrol. Dial. Transpl. 30 (Suppl. 1), i104–i112 (2015).
Mossberg, M., Segelmark, M., Kahn, R., Englund, M. & Mohammad, A. Epidemiology of primary systemic vasculitis in children: a population-based study from southern Sweden. Scand. J. Rheumatol. 47, 295–302 (2018).
Gonzalez-Gay, M. A., Garcia-Porrua, C., Guerrero, J., Rodriguez-Ledo, P. & Llorca, J. The epidemiology of the primary systemic vasculitides in northwest Spain: implications of the Chapel Hill Consensus Conference definitions. Arthritis Rheum. 49, 388–393 (2003).
Berti, A., Cornec, D., Crowson, C. S., Specks, U. & Matteson, E. L. The epidemiology of antineutrophil cytoplasmic autoantibody-associated vasculitis in Olmsted County, Minnesota (USA): a 20 year population-based study. Arthritis Rheumatol. 69, 2338–2350 (2017).
Scott, J., Hartnett, J., Mockler, D. & Little, M. A. Environmental risk factors associated with ANCA associated vasculitis: a systematic mapping review. Autoimmun. Rev. 19, 102660 (2020).
Tidman, M., Olander, R., Svalander, C. & Danielsson, D. Patients hospitalized because of small vessel vasculitides with renal involvement in the period 1975-95: organ involvement, anti-neutrophil cytoplasmic antibodies patterns, seasonal attack rates and fluctuation of annual frequencies. J. Intern. Med. 244, 133–141 (1998).
Watts, R. A., Mooney, J., Skinner, J., Scott, D. G. I. & Macgregor, A. J. The contrasting epidemiology of granulomatosis with polyangiitis (Wegener’s) and microscopic polyangiitis. Rheumatology 51, 926–931 (2012).
Carruthers, D. M., Watts, R. A., Symmons, D. P. M. & Scott, D. G. I. Wegener’s granulomatosis — increased incidence or increased recognition? Br. J. Rheumatol. 35, 142–145 (1996).
Mahr, A. et al. Seasonal variations in onset of Wegener’s granulomatosis: increased in summer? J. Rheumatol. 33, 1615–1622 (2006).
Aiyegbusi, O. et al. ANCA-associated renal vasculitis is associated with rurality but not seasonality or deprivation in a complete national cohort study. RMD Open 7, e001555 (2021).
Pearce, F. A. et al. Can granulomatosis with polyangiitis be diagnosed earlier in primary care? A case–control study. QJM . Int. J. Med. 111, 39–45 (2018).
Gómez-Puerta, J. A., Gedmintas, L. & Costenbader, K. H. The association between silica exposure and development of ANCA-associated vasculitis: systematic review and meta-analysis. Autoimmun. Rev. 12, 1129–1135 (2013).
Giorgiutti, S. et al. Prevalence of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis and spatial association with quarries in a French northeast region. Arthritis Rheumatol. 73, 2078–2085 (2021).
Lane, S. E., Watts, R. A. & Scott, D. G. I. Seasonal variations in onset of Wegener’s granulomatosis: Increased in summer? J. Rheumatol. 34, 889–890 (2007).
Stamp, L. K. et al. Association between environmental exposures and granulomatosis with polyangiitis in Canterbury, New Zealand. Arthritis Res. Ther. 17, 333 (2015).
Gardner-Medwin, J. M. M., Dolezalova, P., Cummins, C. & Southwood, T. R. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 360, 1197–1202 (2002).
Kawasaki, Y. et al. The incidence and severity of Henoch-Schönlein purpura nephritis over a 22-year period in Fukushima Prefecture, Japan. Int. Urol. Nephrol. 42, 1023–1029 (2010).
Penny, K., Fleming, M., Kazmierczak, D. & Thomas, A. An epidemiological study of Henoch-Schönlein purpura. Paediatr. Nurs. 22, 30–35 (2010).
Romero-Gómez, C. et al. Epidemiological study of primary systemic vasculitides among adults in southern Spain and review of the main epidemiological studies. Clin. Exp. Rheumatol. 33 (2 Suppl. 89), S11–S18 (2014).
Elfving, P. et al. Estimating the incidence of connective tissue diseases and vasculitides in a defined population in northern Savo area in 2010. Rheumatol. Int. 36, 917–924 (2016).
Audemard-Verger, A., Pillebout, E., Guillevin, L., Thervet, E. & Terrier, B. IgA vasculitis (Henoch-Shönlein purpura) in adults: diagnostic and therapeutic aspects. Autoimmun. Rev. 14, 579–585 (2015).
Sjöwall, C., Mandl, T., Skattum, L., Olsson, M. & Mohammad, A. J. Epidemiology of hypocomplementaemic urticarial vasculitis (anti-C1q vasculitis). Rheumatology 57, 1400–1407 (2018).
Lin, Y.-H. et al. Epidemiology of Behcet’s disease in Taiwan: a population-based study. Ophthalmic Epidemiol. 25, 323–329 (2018).
Lee, Y. B. et al. Incidence, prevalence, and mortality of Adamantiades-Behçet’s disease in Korea: a nationwide, population-based study (2006–2015). J. Eur. Acad. Dermatol. Venereol. 32, 999–1003 (2018).
Kanecki, K. et al. Behçet disease: a rare systemic vasculitis in Poland. Pol. Arch. Intern. Med. 127, 652–656 (2017).
Mohammad, A., Mandl, T., Sturfelt, G. & Segelmark, M. Incidence, prevalence and clinical characteristics of Behcet’s disease in southern Sweden. Rheumatology 52, 304–310 (2013).
Muruganandam, M. et al. Characteristics of Behcet’s disease in the American Southwest. Semin. Arthritis Rheum. 49, 296–302 (2019).
Tunes, R. S., Anjos, T. C., Martins, G. B., Barreto, E. R. M. & Santiago, M. B. Prevalence of Behcet’s syndrome in patients with recurrent aphthous ulcerations in Brazil. Rheumatol. Int. 29, 875–878 (2009).
Fernández-Ávila, D. G., Rincón-Riaño, D. N., Bernal-Macías, S., Dávila, J. M. G. & Rosselli, D. Prevalence and demographic characteristics of Behcet disease in Colombia: data from the national health registry 2012–2016. Rheumatol. Int. 40, 17–20 (2020).
Nakae, K. et al. Recent epidemiological features of Behçet’s disease in Japan. in Behçet’s Disease (eds Wechsler, B. & Godeau, P.) 145–151 (Excerpta Medica, 1993).
Yurdakul, S. Epidemiology of Behçet syndrome and regional differences in disease expression. in Behçet’s Syndrome (eds Yazici, Y., Hatemi, G., Seyahi, E. & Yazici, H.) 21–35 (Springer, 2020).
Azizlerli, G. et al. Prevalence of Behçet’s disease in Istanbul, Turkey. Int. J. Dermatol. 42, 803–806 (2003).
Madanat, W. Y. et al. The prevalence of Behçet’s disease in the north of Jordan: a hospital-based epidemiological survey. Clin. Exp. Rheumatol. 35 (Suppl. 108), 51–54 (2017).
Moghimi, N. et al. WHO-ILAR COPCORD study (stage 1, urban study) in Sanandaj, Iran. Clin. Rheumatol. 34, 535–543 (2015).
Kim, J. N., Kwak, S. G., Choe, J.-Y. & Kim, S.-K. The prevalence of Behçet’s disease in Korea: data from Health Insurance Review and Assessment Service from 2011 to 2015. Clin. Exp. Rheumatol. 35 (Suppl. 1), 38–42 (2017).
O’Duffy, J. D. Summary of International Symposium on Behçet’s Disease. Istanbul, September 29–30, 1977. J. Rheumatol. 5, 229–233 (1978).
Li, R. et al. Epidemiology of eight common rheumatic diseases in China: a large-scale cross-sectional survey in Beijing. Rheumatology 51, 721–729 (2012).
de Menthon, M., Lavalley, M. P., Maldini, C., Guillevin, L. & Mahr, A. HLA-B51/B5 and the risk of Behçet’s disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. 61, 1287–1296 (2009).
Ahmad, T. et al. CARD15 polymorphisms in Behçet’s disease. Scand. J. Rheumatol. 34, 233–237 (2005).
Thomas, T. et al. Epidemiology, morbidity and mortality in Behçet’s disease: a cohort study using The Health Improvement Network (THIN). Rheumatology 59, 2785–2795 (2020).
Bonitsis, N. G. et al. Gender-specific differences in Adamantiades-Behçet’s disease manifestations: an analysis of the German registry and meta-analysis of data from the literature. Rheumatology 54, 121–133 (2015).
Yazici, H. et al. Influence of age of onset and patient’s sex on the prevalence and severity of manifestations of Behçet’s syndrome. Ann. Rheum. Dis. 43, 783–789 (1984).
Yazici, H., Chamberlain, M. A., Tüzün, Y., Yurdakul, S. & Müftüoglu, A. A comparative study of the pathergy reaction among Turkish and British patients with Behçet’s disease. Ann. Rheum. Dis. 43, 74–75 (1984).
Sibley, C. et al. Behçet syndrome manifestations and activity in the United States versus Turkey — a cross-sectional cohort comparison. J. Rheumatol. 41, 1379–1384 (2014).
Kobayashi, T. et al. Differences in clinical manifestations, treatment, and concordance rates with two major sets of criteria for Behçet’s syndrome for patients in the US and Japan: data from a large, three-center cohort study. Mod. Rheumatol. 23, 547–553 (2013).
Shahram, F. et al. Geographical variations in ocular and extra-ocular manifestations in Behçet’s disease. Eur. J. Rheumatol. 6, 199–206 (2019).
Kirino, Y. et al. Continuous evolution of clinical phenotype in 578 Japanese patients with Behçet’s disease: a retrospective observational study. Arthritis Res. Ther. 18, 217 (2016).
Jun, J.-B. et al. Significant decline in the incidence of Behcet’s disease in South Korea: a nationwide population-based study (2004–2017). Arthritis Care Res. 73, 1689–1696 (2020).
Davatchi, F. et al. Behcet’s disease in Iran: analysis of 7641 cases. Mod. Rheumatol. 29, 1023–1030 (2019).
Kim, D. Y., Choi, M. J., Cho, S., Kim, D. W. & Bang, D. Changing clinical expression of Behçet disease in Korea during three decades (1983–2012): chronological analysis of 3674 hospital-based patients. Br. J. Dermatol. 170, 458–461 (2014).
Hatemi, G. & Yazici, H. Behçet’s syndrome and micro-organisms. Best. Pract. Res. Clin. Rheumatol. 25, 389–406 (2011).
Mehmood, N., Low, L. & Wallace, G. R. Behçet’s disease-do microbiomes and genetics collaborate in pathogenesis? Front. Immunol. 12, 648341 (2021).
Park, J. & Cheon, J. H. Incidence and prevalence of inflammatory bowel disease across Asia. Yonsei Med. J. 62, 99–108 (2021).
Adeeb, F., Stack, A. G. & Fraser, A. D. New insights into Behçet’s disease in Ireland: the Midwest cohort study. Clin. Exp. Rheumatol. 36, 33–39 (2018).
Papoutsis, N. G. et al. Prevalence of Adamantiades-Behçet’s disease in Germany and the municipality of Berlin: results of a nationwide survey. Clin. Exp. Rheumatol. 24, S125 (2006).
Mahr, A. et al. Population-based prevalence study of Behçet’s disease: differences by ethnic origin and low variation by age at immigration. Arthritis Rheum. 58, 3951–3959 (2008).
Kappen, J. H. et al. Behçet’s disease, hospital-based prevalence and manifestations in the Rotterdam area. Neth. J. Med. 73, 471–477 (2015).
Villiger, R. A., Stefanski, A.-L., Grobéty, V., Adler, S. & Villiger, P. M. Behçet’s syndrome: clinical presentation and prevalence in Switzerland. Swiss Med. Wkly. 149, w20072 (2019).
Cartella, S. Prevalence of Behçet’s disease in the province of Brescia in northern Italy. Clin. Exp. Rheumatol. 32 (4 Suppl. 84), S176 (2014).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of this manuscript.
Corresponding author
Ethics declarations
Competing interests
R.A.W. declares sponsorship of less than $10,000 from Gilead. G.H. declares payments of less than $10,000 from Abbvie, Amgen, Bayer, Celgene, Johnson & Johnson, Lilly, Novartis and UCB Pharma. A.J.M. declares payments of less than $10,000 from Amgen Sweden, Lilly Sweden and Roche Sweden. J.C.B. declares no competing interests.
Additional information
Peer review information
Nature Reviews Rheumatology thanks H. Direskeneli and K. Warrington for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Watts, R.A., Hatemi, G., Burns, J.C. et al. Global epidemiology of vasculitis. Nat Rev Rheumatol 18, 22–34 (2022). https://doi.org/10.1038/s41584-021-00718-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41584-021-00718-8
This article is cited by
-
Risk factors and scores for prediction of coronary artery aneurysms in Kawasaki disease: a European monocentric study
BMC Pediatrics (2024)
-
Sex-specific circulating unconventional neutrophils determine immunological outcome of auto-inflammatory Behçet’s uveitis
Cell Discovery (2024)
-
Clinical and anamnestic features of patients with systemic vasculitis: a single-center retrospective study
Rheumatology International (2024)
-
Immunglobulin-A-Vaskulitis
Die Innere Medizin (2024)
-
Renal artery involvement is associated with increased morbidity but not mortality in Takayasu arteritis: a matched cohort study of 215 patients
Clinical Rheumatology (2024)
