
Abstract
Autophagy is a versatile degradation system for maintaining cellular homeostasis whereby cytosolic materials are sequestered in a double-membrane autophagosome and subsequently delivered to lysosomes, where they are broken down. In multicellular organisms, newly formed autophagosomes undergo a process called ‘maturation’, in which they fuse with vesicles originating from endolysosomal compartments, including early/late endosomes and lysosomes, to form amphisomes, which eventually become degradative autolysosomes. This fusion process requires the concerted actions of multiple regulators of membrane dynamics, including SNAREs, tethering proteins and RAB GTPases, and also transport of autophagosomes and late endosomes/lysosomes towards each other. Multiple mechanisms modulate autophagosome maturation, including post-translational modification of key components, spatial distribution of phosphoinositide lipid species on membranes, RAB protein dynamics, and biogenesis and function of lysosomes. Nutrient status and various stresses integrate into the autophagosome maturation machinery to coordinate the progression of autophagic flux. Impaired autophagosome maturation is linked to the pathogenesis of various human diseases, including neurodegenerative disorders, cancer and myopathies. Furthermore, invading pathogens exploit various strategies to block autophagosome maturation, thus evading destruction and even subverting autophagic vacuoles (autophagosomes, amphisomes and autolysosomes) for survival, growth and/or release. Here, we discuss the recent progress in our understanding of the machinery and regulation of autophagosome maturation, the relevance of these mechanisms to human pathophysiology and how they are harnessed by pathogens for their benefit. We also provide perspectives on targeting autophagosome maturation therapeutically.
Similar content being viewed by others

Introduction
Cells in multicellular organisms constantly experience diverse stresses, including protein misfolding, organelle damage, scarcity of nutrients/energy and invasion by pathogens. One mechanism exploited by cells to combat stresses is the lysosome-mediated degradation of intracellular components via autophagy. Autophagy involves the formation of an isolation membrane (or phagophore), which further expands and closes to form the double-membrane autophagosome1,2,3. The mechanism of autophagosome formation was excellently reviewed recently3. Various cellular materials can be sequestered in autophagosomes, including unselected cytosolic material or selected cargos such as protein aggregates, damaged organelles and pathogens. Following autophagosome closure via membrane abscission, cargos are delivered to the vacuole (yeasts and plants) or to lysosomes (animal cells) (for an overview of the autophagy process see4,5,6,7,8; Box 1). After degradation of the autophagic cargo, the digested content in the autolysosomes is released and lysosomes are re-formed to sustain the autophagic flux (Box 2). A series of proteins encoded by ATG (autophagy-related) genes and EPG (ectopic PGL granules) genes — identified mainly from genetic screens in Saccharomyces cerevisiae and Caenorhabditis elegans, respectively — act at different membrane remodelling steps for autophagosome formation and maturation3,4,9,10.
In yeast, autophagosomes are formed in the vicinity of and directly fuse with the much larger vacuole, whereas in multicellular organisms, newly formed autophagosomes fuse with different endolysosomal vesicles such as early/late endosomes and lysosomes to form non-degradative, single-membrane structures called ‘amphisomes’, which gradually acquire degradative properties through the accumulation of hydrolases. Maturation also involves autophagosome acidification mediated by V-ATPase, which is required for the activity of degradative enzymes9,10,11. Fusion of autophagosomes with endosomes/lysosomes requires the concerted actions of cognate SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complexes, tethers, phosphoinositides and RAB proteins11. Bidirectional microtubule-based transport of autophagic vacuoles (a term encompassing autophagosomes, amphisomes and autolysosomes) and late endosomes/lysosomes between the perinuclear space and periphery of cells maximizes the frequency of encounters and fusion efficiency12.
Recent studies revealed that the machinery mediating autophagosome maturation, such as SNARE complex assembly, tether recruitment and RAB protein dynamics, is tightly controlled and highly responsive to nutrient availability and stress conditions11. Disturbing these processes can cause accumulation of damaged organelles and toxic protein aggregates, and may also hamper endocytic trafficking. Accordingly, impaired autophagosome maturation is associated with the pathogenesis of various human disorders, including neurodegenerative diseases, cancer and myopathies11,13. Furthermore, invading viruses and bacteria also widely target maturation of autophagosomes to block their degradative capability. Viruses and bacteria thereby evade destruction and also accumulate autophagic vacuoles for their survival, replication and release14,15,16,17. Notably, unlike impairment of genes essential for autophagosome formation, loss of function of genes acting at autophagosome maturation usually causes a weaker defect and gradual accumulation of autophagic vacuoles. More importantly, in some cases, such defects can be suppressed by promoting the activities of partially redundant factors or mechanisms. For example, facilitating the recruitment of one tethering factor would alleviate the defect caused by depletion of another. This offers opportunities to ameliorate the defects of autophagosome maturation in relevant human diseases.
In this Review, we outline the most recent progress in our understanding of the molecular machinery that drives and regulates autophagosome maturation. We further discuss the causative link between impaired autophagosome maturation and the pathogenesis of neurodegeneration, muscle diseases and cancer. We also consider how viral proteins and bacterial effectors block autophagosome maturation for their own benefits. Finally, we offer insights into how autophagosome maturation can be therapeutically targeted to combat disease.

Machinery for autophagosome fusion
Maturation of autophagosomes requires their fusion with functional endolysosomal compartments such as early endosomes, late endosomes/multivesicular bodies and lysosomes. Accordingly, disturbances in biogenesis of these endolysosomal compartments — such as those caused by the loss of function of subunits of the ESCRT (endosomal sorting complexes required for transport) complex and COPI vesicles — cause accumulation of non-degradative autophagic vacuoles18,19. Fusion of autophagosomes with late endosomes/lysosomes involves the concerted action of RABs, tethers and the SNARE complex. Membrane fusion is driven by the assembly of a trans-SNARE complex composed of Qa, Qb, Qc and R SNAREs20. Tethering factors, which are recruited to target membranes and/or fusing vesicles by the active form of small RAB GTPases, phosphoinositides and SNARE proteins, facilitate the initial capture of vesicles and subsequent formation of the trans-SNARE complex21,22. Autophagic vacuoles are decorated with lipidated (phosphatidylethanolamine-conjugated) ubiquitin-like autophagy proteins related to yeast Atg8. These proteins, in the LC3 subfamily or the GABARAP subfamily, also help to recruit other factors such as tethers and RAB guanine nucleotide exchange factors (GEFs) for maturation23,24. LC3 is a widely used autophagic marker that labels autophagic structures at different stages of biogenesis, and, together with markers for late endosomes/lysosomes, it accumulates on amphisomes during maturation. Of note, in studies using LC3 to label autophagic structures after closure (which can be differentiated from isolation membranes by co-labelling with markers for other autophagy proteins or use of the HaloTag–LC3 method6,25), double-membrane autophagosomes cannot be distinguished from single-membrane amphisomes and autolysosomes. Hence, LC3-marked structures after closure should be referred to as ‘autophagic vacuoles’ unless the structure has been unambiguously demonstrated.
The role of SNARE complexes
SNARE complexes formed by autophagosomal membrane-localized STX17 (Qa), SNAP29 (Qbc) and late endosomal/lysosomal-localized VAMP8 (Vamp7 in flies and VAMP-7 in worms), or by the autophagosomal YKT6, SNAP29 and late endosomal/lysosomal-localized STX7, function partially redundantly to drive fusion of autophagosomes with late endosomes/lysosomes26,27,28 (Fig. 1a). Targeting of STX17 to forming autophagosomes accompanies autophagosome closure29. The small guanosine triphosphatase IRGM facilitates translocation of STX17 to autophagosomes by interacting with STX17 and ATG8-like proteins on the autophagosomal membrane30. SNAP29 is recruited by interacting with other SNAREs. STX17–SNAP29 complex assembly is promoted by the autophagosome-associated autophagy protein ATG14, which directly interacts with STX17 (ref.31) (Fig. 1a). Different SNARE complexes may act at different steps of autophagosome maturation or drive fusion of autophagic vacuoles with different sets of late endosomes/lysosomes27,32. Postfusion SNAREs disassemble and return to their donor compartments to maintain intracellular membrane identity and to prepare for new rounds of fusion20. The molecular mechanisms driving SNARE retrieval from autolysosomes have yet to be determined. It has been shown that release of STX17 from autolysosomes coincides with collapse of the autophagosomal inner membrane due to breakdown by lysosomal enzymes29.

a | Fusion between autophagosomes and late endosomes/lysosomes is mediated by two sets of SNARE complexes: the autophagosomal Qa SNARE STX17, the Qbc SNARE SNAP29 and the endolysosomal R SNARE VAMP8 (centre left); and the autophagosomal R SNARE YKT6, SNAP29 and the endolysosomal Qa SNARE STX7 (centre right). The unique hairpin structure formed by the two transmembrane domains of STX17 and the amino-terminal longin domain of YKT6 are involved in their autophagosomal recruitment26,27. The autophagosome-associated autophagy protein ATG14 interacts with STX17 to promote the assembly of the STX17–SNAP29 subcomplex (left). The assembly and function of both complexes are facilitated by tether proteins. b | Multiple tether proteins are involved in fusion of autophagosomes with late endosomes/lysosomes. EPG5 is targeted to autophagosomes and lysosomes by binding to LC3 and RAB7, respectively. In neural cells, WDR45 and WDR45B facilitate the lysosomal localization of EPG5. TECPR1 interacts with LC3C and ATG5–ATG12 on autophagosomes and phosphatidylinositol 4-phosphate (PtdIns4P) on lysosomes. RAB7 binds to the HOPS complex and PLEKHM1. On the autophagosome side, the HOPS complex binds to STX17 and LC3 proteins, and PLEKHM1 binds to GABARAPs. The active GTP-bound RAB7 associates with the membrane. Localization of RAB7 on autophagic vacuoles is facilitated by the LC3-binding MON1–CCZ1 guanine nucleotide exchange factor complex36,37.
The role of tethering factors
Both multisubunit tethering complexes and large individual tether proteins participate in the initial capture of autophagosomes and/or amphisomes and promote fusion efficiency and specificity with early/late endosomes and lysosomes but not with other vesicles such as recycling endosomes or secretory vesicles during autophagosome maturation (Fig. 1b).
The HOPS (homotypic fusion and protein sorting) complex is a prominent tethering complex that promotes assembly of the trans-SNARE complex, acts as a GEF for RAB7 and functions as a SNARE chaperone to facilitate fusion of autophagosomes with late endosomes and lysosomes22,33,34. The HOPS complex can be targeted to both autophagosomes and late endosomes/lysosomes to promote fusion33,34. Targeting of the HOPS complex to late endosomes/lysosomes is mediated by its binding to RAB7 and phosphoinositides such as phosphatidylinositol 3-phosphate (PtdIns3P)35, while multiple mechanisms facilitate its translocation from the cytosol to autophagosomes, including interaction with STX17 (refs33,34), active RAB7 (refs36,37), GABARAPs38 or large tether proteins (such as PLEKHM1) described below (Fig. 1b).
EPG5 is an evolutionarily conserved large tether protein for autophagosome maturation39,40. It is a RAB7 effector and translocates from the cytosol to late endosomes/lysosomes by interacting with RAB7 (ref.40) (Fig. 1b). During autophagosome maturation, EPG5 captures autophagosomes/amphisomes via binding to LC3 and promotes assembly of the STX17–SNAP29–VAMP8 complex40. EPG5 depletion causes accumulation of autophagosomes, amphisomes and non-degradative autolysosomes41. The function of EPG5 in autophagosome maturation appears to require its late endosomal/lysosomal localization. The β-propeller proteins WDR45 and WDR45B (mammalian homologues of the yeast PtdIns3P-binding autophagy protein Atg18; also known as WIPI4 and WIPI3, respectively) interact with and mediate late endosomal/lysosomal targeting of EPG5 (ref.25). In Wdr45–Wdr45b double-knockout cells, EPG5 is mislocalized on autophagosomes and the EPG5–STX17 interaction is enhanced. However, the assembly of STX17–SNAP29–VAMP8 is reduced, and consequently autophagosome maturation is impaired25.
PLEKHM1, another tethering protein and RAB7 effector, interacts preferentially with GABARAP subfamily members to promote autophagosome maturation23,42 (Fig. 1b). PLEKHM1 directly recruits the HOPS complex, ensuring fusion specificity and efficiency42,43. The lysosomal-localized pleckstrin homology (PH) domain-containing TECPR1 selectively binds to LC3C and promotes fusion of LC3C-decorated autophagosomes/amphisomes with phosphatidylinositol 4-phosphate (PtdIns4P)-enriched lysosomes44 (Fig. 1b). It has also been shown that TECPR1, upon binding to autophagosomal PtdIns3P, adopts a conformation that associates with the ATG12–ATG5 conjugate, thus facilitating tethering of autophagosomes with lysosomes45.
Vesicle transport during maturation
In non-polarized cells, autophagosomes are generated throughout the cytoplasm, while lysosomes locate mainly at perinuclear regions12,46. Autophagosome maturation requires retrograde transport of autophagic vacuoles and lysosomes driven by the microtubule motor protein dynein, as well as their anterograde transport mediated by kinesin motors12,46,47, which allow the two compartments to meet and fuse.
Machinery mediating transport of autophagic vacuoles and lysosomes
Anterograde transport of lysosomes enables them to fuse with peripheral autophagosomes. Anterograde transport is mediated by the late endosomal/lysosomal-associated multisubunit BORC complex, which recruits the small GTPase ARL8 to promote ARL8-dependent coupling to kinesin motors12 (Fig. 2). ARL8 directly interacts with kinesin 3, or interacts with its effector SKIP, which in turn couples to kinesin 1 (ref.48). Depletion of BORC complex decreases the fusion of peripheral autophagosomes with lysosomes49. The RAB7 effector FYCO1 also acts as an adaptor of kinesin 1 to mediate anterograde transport of late endosomes/lysosomes, and of autophagic vacuoles through its binding to LC3 and PtdIns3P50 (Fig. 2).

Autophagosomes, which form throughout the cytoplasm, and lysosomes, which are localized mainly in the perinuclear region, undergo bidirectional movement on microtubules. The late endosome/lysosome-localized BORC–ARL8 complex and the FYCO1–RAB7 pair recruit kinesin motors for anterograde transport of late endosomes/lysosomes. The RAB7 effectors RILP and ORP1L promote the membrane association of the dynein–dynactin motor machinery to mediate the retrograde transport of autophagic vacuoles (as well as late endosomes/lysosomes (not shown)). When cholesterol levels are low, ORP1L interacts with the endoplasmic reticulum (ER) protein VAPA to form ER–autophagosome membrane contacts, which releases dynein–dynactin and prevents retrograde trafficking, thereby interfering with autophagosome maturation. During transport, the tethering factors HOPS complex and PLEKHM1 are concomitantly recruited to promote fusion processes (see Fig. 1).
The recruitment of the dynein–dynactin motor complex to late endosomes/lysosomes and autophagic vacuoles for retrograde transport is mediated by RAB7 and its effectors RILP and ORP1L12 (Fig. 2). RILP interacts with the p150 subunit of the dynein–dynactin motor complex12, whereas ORP1L promotes the binding of the dynein complex with late endosomal/lysosomal membrane-associated βIII spectrin51. In addition, ORP1L can sense and couple cholesterol levels with transport of lysosomes and autophagic vacuoles (Fig. 2). Under low-cholesterol conditions, ORP1L interacts with the endoplasmic reticulum (ER) protein VAPA to form contacts between the ER and lysosomes/autophagic vacuoles; the dynein motor then disassociates from RAB7–RILP, causing dispersal of these organelles43,52. In Niemann–Pick type C disease and other lysosome storage diseases (LSDs), accumulation of cholesterol in the late endosomal/lysosomal compartments results in their perinuclear clustering and a defect in autophagosome maturation52.
Transport of autophagic vacuoles and late endosomes/lysosomes is tightly coordinated with recruitment of tethering factors. Specifically, the HOPS complex can be recruited by ARL8 (ref.49) or by the ORP1L–RAB7–RILP complex, which occurs either directly or via PLEKHM1 (ref.53). This allows coupling of anterograde and retrograde transport and fusion events53.
Directional transport of autophagic vacuoles is particularly prominent in polarized neuronal cells, where autophagosomes formed at the distal synaptic termini undergo long-range retrograde transport to the soma54,55. Maturation of autophagosomes is tightly linked with their dynein-driven transport to the soma, which requires coordinated actions of scaffolding proteins, including JNK-interacting protein 1 (JIP1), HTT-associated protein 1 (HAP1) and JIP3 (refs56,57). These factors bridge autophagic vacuoles with the dynein–dynactin complex, and their actions depend on location and autophagosomal maturity56,57. JIP1 acts in the distal portion of the axon, HAP1 functions in the mid-axon, while JIP3 primarily controls the motility of maturer autophagic vacuoles in the proximal portion of the axon57.
Regulation of autophagosome maturation
The machinery governing autophagosome maturation is dynamically regulated to allow adaptation of autophagic flux to the needs of the cell and to enable integration of autophagic degradation with external inputs. This involves regulation of the membrane recruitment of the key components mediating fusion events as well as transcriptional regulation, which occurs predominantly via the MiT/TFE family transcription factors TFEB and TFE3. During autophagosome biogenesis, various stresses and signalling pathways can modulate the initiation of autophagosome formation via multiple mechanisms, such as by regulating the kinase activity, stability and assembly of the Atg1 complex (ULK1 complex in mammals) and the Vps34 PtdIns3P kinase complex58,59,60,61,62. mTORC1, an evolutionarily conserved signalling hub that senses nutrient status and growth factor signals, plays a key role here. Nutrient status and other stresses also integrate into the autophagosome maturation machinery to add another level of control of autophagic flux. It is also noteworthy that the machinery governing autophagosome maturation is heterogeneous and demonstrates at least partial redundancy, which may allow flexible regulation of autophagic flux in different cell types.
Trafficking and post-translational modification of SNARE proteins
SNARE proteins dynamically move between distinct membrane compartments20. Upon starvation, RAB21, an endosomal RAB protein, is activated by its GEF MTMR13, which further promotes the translocation of plasma membrane-localized VAMP8 to late endosomes/lysosomes63 (Fig. 3a). SNARE protein dynamics is required for efficient fusion of the endolysosomal structures with autophagic vacuoles as evidenced by LSDs such as multiple sulfatase deficiency and mucopolysaccharidosis type IIIA, where SNAREs are sequestered in cholesterol-enriched regions of endolysosomal membranes and locked in assembled complexes64. Therefore, disassembly of postfusion SNARE complexes and their sorting and recycling back to target membranes is impaired, and, consequently, fusion of lysosomes with endocytic and autophagic vesicles is reduced64.

a | The SNARE domain of STX17 is modified by acetylation, a process controlled by the acetyltransferase CREBBP and the deacetylase HDAC2. Starvation inactivates CREBBP, resulting in deacetylation of STX17. Deacetylated STX17 interacts more strongly with the HOPS complex and SNAP29 and thus promotes autophagosome–lysosome fusion65. SNAP29 is O-GlcNAcylated by O-linked β-N-acetylglucosamine (O-GlcNAc) transferase (OGT). This modification attenuates the assembly of SNAP29-containing SNARE complexes. Under starvation conditions that decrease the intracellular UDP-GlcNAc level, or in OGT-knockdown cells, the O-GlcNAcylation of SNAP29 is reduced, which in turn facilitates the assembly of the trans-SNARE complex for autophagosome maturation66. MTMR13, the guanine nucleotide exchange factor for the endosomal protein RAB21, controls RAB21-dependent trafficking of plasma membrane-localized VAMP8 to late endosomes/lysosomes63. Upon starvation, MTMR13 activates RAB21, which subsequently promotes the translocation of VAMP8 R SNARE to late endosomes/lysosomes to promote fusion of endosomes/lysosomes with autophagic vacuoles (see Fig. 1). b | Phosphatidylinositol 3-phosphate (PtdIns3P) on autophagic vacuoles, generated by the UVRAG-containing VPS34 complex, facilitates autophagosome maturation by recruiting the tethering factors HOPS complex. Rubicon interacts with the UVRAG–VPS34 complex and negatively regulates its function. Pacer is targeted by autophagic vacuole-localized SNARE STX17 and phosphoinositides (PtdIns3P), and it antagonizes Rubicon and recruits the UVRAG–VPS34 complex to autophagic vacuoles. Pacer and UVRAG also recruit the HOPS complex. The RAB7 GTPase-activating protein Armus is targeted to autophagic vacuoles by interacting with LC3 and PtdIns3P, and promotes RAB7 dynamics, whereby it is recycled from the autophagic vacuole membranes. This generates a mobile pool of RAB7 that can be recruited to endosomes to drive their maturation to late endosomes/lysosomes. Depletion of the endoplasmic reticulum-localized transmembrane protein TMEM39A increases phosphatidylinositol 4-phosphate (PtdIns4P) levels on late endosomes/lysosomes (via inhibition of endoplasmic reticulum-to-Golgi apparatus trafficking of the PtdIns4P phosphatase SAC1, not shown), probably by increasing PtdIns4P levels in the trans-Golgi network (TGN), which promotes HOPS complex recruitment and enhances autophagosome–lysosome fusion. c | The transcription factor TFEB (as well as its homologue TFE3, not shown) activates the expression of genes involved in autophagy (including genes involved in autophagosome trafficking and fusion with lysosomes (UVRAG and VPS18)) and lysosomal biogenesis and function. The nuclear transport of TFEB is regulated by its phosphorylation levels. Various kinases, such as mTORC1 (downstream of nutrients) and glycogen synthase kinase 3β (GSK3β) (downstream of protein kinase C (PKC) signalling in response to various extracellular signals), phosphorylate TFEB to prevent its nuclear import, while protein phosphatase 2A (PP2A) and calcineurin (which is activated by calcium release from lysosomes) dephosphorylate TFEB to facilitate its translocation to the nucleus. The activity of TFEB is also controlled by liquid–liquid phase separation (LLPS), whereby TFEB forms condensates that promote gene transcription. The nuclear protein inositol polyphosphate multikinase (IPMK) directly binds to TFEB and inhibits the formation of TFEB condensates.
The SNARE activity of STX17 is regulated by acetylation of its SNARE domain, a modification controlled by the histone acetyltransferase CREBBP/CBP and the deacetylase HDAC2 (ref.65) (Fig. 3a). Starvation or mTORC1 inhibition inactivates CREBBP, while promoting the deacetylation of STX17. Deacetylation of STX17 facilitates the assembly of the STX17–SNAP29–VAMP8 complex and also enhances its binding to the HOPS complex, thus promoting autophagosome–lysosome fusion under stress conditions65 (Fig. 3a).
SNAP29 contains two antiparallel helix bundles and is the key SNARE for autophagosome maturation26,27. SNAP29 is post-translationally modified via O-GlcNAcylation at multiple serine/threonine residues, a process catalysed by O-linked β-N-acetylglucosamine (O-GlcNAc) transferase (OGT)66. O-GlcNAcylation of SNAP29 attenuates SNARE complex assembly (Fig. 3a). OGT knockdown or expression of O-GlcNAcylation-defective SNAP29 facilitates the formation of SNAP29-containing SNARE complexes and promotes autophagic flux66. Levels of UDP-GlcNAc, the donor for O-GlcNAc addition, are responsive to the availability of glucose, fatty acids, uridine and glutamine67. The SNAP29 O-GlcNAcylation level is reduced by starvation in mammalian cells and worms66, thus integrating nutrient status with autophagosome maturation.
Regulation of HOPS complex recruitment
During autophagosome biogenesis, the VPS34–beclin 1–ATG14 complex (PI3K complex I) generates PtdIns3P on the ER and/or isolation membrane to recruit effectors such as ATG18 for autophagosome initiation3,68. Steady levels of PtdIns3P on autophagic vacuoles also facilitate autophagosome maturation by recruiting the tethering factors HOPS complex (Fig. 3b). The VPS34–beclin 1–UVRAG complex (PI3K complex II), which is known for production of PtdIns3P on endosomes69, appears to mediate the generation of PtdIns3P on autophagic vacuoles as well70. Pacer (protein associated with UVRAG as autophagy enhancer), which is recruited to autophagic vacuoles by binding STX17 and phosphoinositides, is involved in targeting the VPS34–beclin 1–UVRAG complex71. Pacer and UVRAG also recruit the HOPS complex71,72 (Fig. 3b). Rubicon, a RAB7 effector, which interacts with beclin 1 and associates with VPS34 complexes, antagonizes UVRAG function and negatively regulates autophagosome maturation70,73,74. The UVRAG–Rubicon interaction is enhanced by mTORC1-mediated phosphorylation of UVRAG75, while active GTP-bound RAB7 and Pacer compete with Rubicon to release UVRAG, thus recruiting the HOPS complex and stimulating VPS34 activity under starvation71 (Fig. 3b). However, the role of UVRAG in autophagosome maturation is debated, as it is dispensable for autophagosome–lysosome fusion in fly fat cells and in certain mammalian cells33,34.
In addition to PtdIns3P, PtdIns4P also participates in multiple steps of the autophagy pathway. During autophagosome biogenesis, PtdIns4P contributes to recruitment of the ULK1 complex subunit ATG13 to the autophagosome initiation site76. During maturation, GABARAPs recruit phosphatidylinositol 4-kinase IIα (PI4KIIα) to autophagosomes to generate PtdIns4P, thus facilitating autophagosome–lysosome fusion77. Accumulation of PtdIns4P on the endosomes/lysosomes also promotes fusion events. The spatiotemporal distribution of PtdIns4P is regulated by PI4Ks and the PtdIns4P phosphatase SAC1 (ref.78). SAC1 is an integral membrane protein that cycles between the ER and Golgi compartments via trafficking mediated by COPI vesicles and COPII vesicles. The ER-localized transmembrane protein SUSR2 (also known as TMEM39A) acts as an adaptor by simultaneously interacting with SAC1 and the COPII coat proteins SEC23 and SEC24 to facilitate ER-to-Golgi apparatus transport of SAC1 (ref.79). Retention of SAC1 on the ER in TMEM39A-knockdown cells increases the late endosomal/lysosomal PtdIns4P level, which could result from an elevated PtdIns4P level in trans-Golgi network-derived anterograde vesicles and/or from an increased level of late endosomal/lysosomal PI4KIIα. Consequently, the recruitment of the HOPS complex, which binds strongly to PtdIns4P35, is greatly facilitated and subsequently autophagosome maturation is enhanced79 (Fig. 3b).
Activity and dynamics of RAB7 proteins
RAB7 activity and dynamics — involving its switch between an active GTP-bound state and an inactive GDP-bound form on autophagic vesicles — are essential for progression of autophagic flux. RAB7 activity is negatively regulated by a GTPase-activating protein (GAP) called ‘Armus’ (also known as TBC1D2A), whose targeting is mediated via binding to LC3 on autophagic vacuoles and PtdIns3P generated by VPS34 on endosomes. Armus finely tunes the nucleotide cycle of RAB7 to promote autolysosome formation and acidification80,81 (Fig. 3b). Armus was also demonstrated to be regulated by the nutritional status of the cell via starvation-induced inactivation of the small GTPase RAC1, which competes with Armus for binding to LC3 on autophagic vacuoles81. Limiting Armus recruitment to target membranes leads to persistence of active RAB7 on autophagic vacuoles and endosomes, which may have multiple inhibitory effects on autophagosome maturation: it reduces the availability of mobile RAB7 required for endosome maturation into late endosomes/lysosomes; it maintains high activity of RAB7 effectors, such as Rubicon, whose aberrant activity may impede autophagosome maturation as described earlier; and it also impedes formation of intraluminal vesicles in multivesicular bodies80 — a process that is important for the endocytic pathway and hence the formation of amphisomes as well as for endosome maturation into late endosomes/lysosomes11,18,82. In addition to regulation by Armus, the conversion of endosomal PtdIns4P generated by PI4KIIα to phosphatidylinositol 4,5-bisphosphate also inactivates RAB7 and causes its disassociation from late endosomes and from PLEKHM1. This step also facilitates autophagosome maturation83.
Regulation of TFEB/TFE3 activity by cytoplasm-to-nucleus transport
TFEB and TFE3 are transcription factors with key functions in autophagy as they control expression of a network of genes involved in autophagosome formation, autophagosome maturation and lysosomal biogenesis84,85,86,87. Their activity is extensively regulated by phosphorylation status, controlled by various kinases and phosphatases, with phosphorylation of TFEB and TFE3 inhibiting their cytoplasm-to-nucleus trafficking86,88 (Fig. 3c). This regulation is coupled to nutrient availability via mTORC1. Under nutrient-rich conditions, amino acids promote mTORC1 translocation to the lysosomal surface, where mTORC1 is activated. This lysosome surface-associated mTORC1 phosphorylates TFEB and TFE3, creating binding sites for the scaffold protein 14-3-3 for cytoplasmic retention, thereby preventing nuclear translocation and activity of these factors84,89. In addition to nutrients, various extracellular signals also regulate TFEB/TFE3. For example, it has been shown that glycogen synthase kinase 3β (GSK3β) — which is inactivated by protein kinase C (PKC), which responds to several signal transduction cascades — phosphorylates TFEB to prevent it from translocating into the nucleus90. In response to various stresses, TFEB/TFE3 phosphorylation is inhibited and nuclear transport is facilitated84. TFEB and TFE3 are also actively dephosphorylated by the ubiquitous protein phosphatase 2A (PP2A) and by calcineurin — a phosphatase activated by starvation-triggered lysosomal calcium release or ER stress91,92.
Regulation of TFEB activity via phase separation
Protein liquid–liquid phase separation (LLPS) is now an established mechanism for concentrating proteins in confined liquid-like compartments93,94,95. LLPS also compartmentalizes transcription factors, co-activators, the Mediator complex and RNA polymerase II into condensates for mediating gene expression96,97. It has been recently shown that TFEB undergoes LLPS and that phase-separated TFEB puncta colocalize with the Mediator complex and target mRNAs98. The nuclear protein inositol polyphosphate multikinase (IPMK) directly interacts with TFEB and chaperones it to inhibit its LLPS98 (Fig. 3c). IPMK knockout increases the formation of TFEB transcriptional condensates without altering TFEB phosphorylation or nuclear transport98. Consequently, IPMK knockout facilitates autophagosome maturation and promotes the maturation and degradation capability of lysosomes98.
Heterogeneity and redundancy of autophagosome maturation mechanisms
Overall, there is high variability in fusion events leading to mature, degradative autolysosomes. The process involves multiple, probably non-sequential, fusion processes, including fusion of autophagosomes with different endosomal and lysosomal structures, as well as various homotypic and heterotypic fusion events between the different types of autophagic vacuoles themselves11,46. This involves different tethers, which possess differential binding affinity for ATG8 members and thus mediate maturation of distinct populations of autophagosomes decorated with different ATG8-like proteins. Accordingly, different cell types, with distinct organization of endocytic vesicles, may require different tethers for autophagosome maturation. For example, PLEKHM1 depletion causes no autophagy defect in A549 lung adenocarcinoma cells and in C. elegans40,99. Loss of function of factors involved in autophagosome maturation may also result in accumulation of autophagic vacuoles at different stages of maturation in different cell types. Depletion of fly Rab2 causes accumulation of autophagosomes in muscle cells and amphisomes in larval fat cells100. To acquire degradative potential, autophagic vacuoles must be acidified efficiently, which is accomplished by further rounds of fusion with endosomes/lysosomes. Late endosomes/lysosomes also show heterogeneity, with different surface RAB proteins and distinct resident hydrolytic enzymes. Hence, efficient autophagic degradation requires fusion of autophagic vacuoles with multiple late endosomes/lysosomes11,46, which may require distinct SNAREs and tethers.
Notably, the functions of the multiple SNARE complexes and tethering factors are partially redundant, which, in some cases, means that impaired autophagosome maturation caused by depletion of one factor can be rescued by promotion of an alternative maturation mechanism. This is exemplified by characterization of suppressors of the autophagy defect associated with loss of EPG-5 activity in C. elegans. First, enhanced assembly of the STX17–SNAP29–VAMP8 complex achieved by reduced O-GlcNAcylation of SNAP29 suppresses the autophagy defect in epg-5-mutant worms, EPG5-deficient cells, cells depleted of VCP (also known as p97) — which is essential for maturation of ubiquitin cargo-containing autophagosomes (see also the next section) — and Wdr45–Wdr45b double-knockout cells25,66. Second, the autophagy defect caused by EPG5 deficiency is also suppressed by enhancement of late endosomal/lysosomal recruitment of the HOPS complex in TMEM39A-knockdown cells or by elevation of lysosomal function and biogenesis by promoting TFEB activity via IPMK knockout79,98. Finally, defective autophagosome maturation in epg-5-mutant worms was rescued by promotion of RAB-7 dynamics on non-degradative autophagic vacuoles by expression of the GDP-bound form of RAB-7 or by promotion of lysosome biogenesis and function by depletion of the RBG-1–RBG-2 complex — a GEF and GAP complex known for promoting RAB protein dynamics for synaptic transmission and lipid droplet biogenesis. RBG-1–RBG-2 has emerged as a new regulator of RAB-7 activity, which may act by targeting the RAB-7 GEF to lysosomes, or by promoting its activity on lysosomes101. These mechanisms, however, fail to suppress the autophagy defect caused by knockout of genes involved in autophagosome formation66,79,98,101.
Autophagosome maturation is intricately connected with endocytic trafficking pathways that are highly responsive to growth conditions. The heterogeneity and redundancy of the mechanisms governing autophagosome maturation confer flexibility and robustness on dynamic endocytic trafficking to maintain cellular homeostasis.
Autophagosome maturation and disease
Impaired formation of degradative autolysosomes is associated with the pathogenesis of various human diseases, including neurodegenerative disorders, cancer and muscle diseases (myopathies). Defective autophagosome maturation causes accumulation of toxic protein aggregates and damaged organelles that disrupt cellular homeostasis. Non-functional amphisomes, autolysosomes and even hybrid vesicles resulting from promiscuous fusion with other compartments, such as recycling endosomes, may impede endocytic trafficking and recycling, which also contribute to disease pathogenesis.
Dysfunction in neurodegenerative disorders
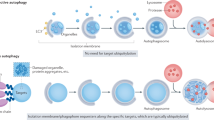
Autophagy is critical for neuronal function. Its key role in the maintenance of axon homeostasis relies on robust degradation-mediated recycling of synaptic proteins, protein aggregates and damaged mitochondria. Various neurodegenerative diseases have been linked to the dysfunction of the autophagy–endolysosomal system, including Alzheimer disease, Parkinson disease, Huntington disease, amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (FTLD). Notably, in these diseases, autophagosomes are formed but then accumulate as non-degradative autophagic vacuoles102,103. Various mutations have been implicated in these neurodegenerative disorders (Fig. 4). Some of these mutations affect endocytic trafficking at the intersection with autophagosome maturation102. For example, mutations affecting the p150 subunit of the dynein–dynactin motor cause motor neuron disease104, while mutations in the ESCRT-III complex subunit CHMP2B cause familial ALS and FTLD105. Impaired lysosomal function is also related to the pathogenesis of neurodegenerative diseases. When lysosomal acidification is defective, fusion of lysosomes with autophagic vacuoles can still occur, but the resulting autolysosomes are non-degradative106. Mutant presenilin 1, one of the major causes of familial Alzheimer disease, impairs lysosomal acidification107. Also, LSDs featuring dysfunctional, non-degradative autolysosomes, such as Gaucher disease, multiple sulfatase deficiency, mucopolysaccharidosis type IIIA and mucolipidosis type IV, are associated with neurodegeneration108,109. Impaired lysosomal biogenesis is also a feature of neurodegenerative disorders. In this case, disease proteins, such as accumulated α-synuclein in Parkinson disease or androgen receptor with polyglutamine expansion in X-linked spinal and bulbar muscular atrophy sequester TFEB in the cytoplasm, impairing lysosomal function and autophagosome maturation110,111. Lysosomal function can also be impaired by physical damage instigated by disease proteins. Amyloid assemblies of disease-associated proteins, including Aβ and tau in Alzheimer disease, α-synuclein in Parkinson disease and huntingtin (HTT) with a pathological polyglutamine expansion in Huntington disease, can be secreted and transported between cells, and, following endocytosis, can induce endolysosome rupture in recipient cells112,113.

Illustration of endolysosome trafficking and the progression of autophagosomes into autolysosomes. Impairments of several steps in the process have been linked to the pathogenesis of neurodegenerative diseases, and examples of the related diseases are shown. AD, Alzheimer disease; ALS, amyotrophic lateral sclerosis; BPAN, β-propeller protein-associated neurodegeneration; FTD, frontotemporal dementia; HD, Huntington disease; ID, intellectual disability; LSD, lysosomal storage disease; MLIV, mucolipidosis type IV; MPSIIIA, mucopolysaccharidosis type IIIA; MSD, multiple sulfatase deficiency; PD, Parkinson disease; poly(Q)-AR, androgen receptor with polyglutamine expansion; poly(Q)-HTT, huntingtin with polyglutamine expansion; SBMA, spinal and bulbar muscular atrophy.
Mutations in genes directly involved in autophagosome maturation can also cause selective damage of certain populations of neurons, resulting in neurodegenerative features (Fig. 4). Mice deficient in Epg5 show selective loss of motor neurons and display key characteristics of ALS41. Recessive mutations in human EPG5 are associated with the multisystem disorder Vici syndrome114. Patients with Vici syndrome exhibit neurodevelopmental and neurodegenerative features that are recapitulated in Epg5-knockout mice115,116. Patients with Vici syndrome also display muscle abnormalities (skeletal muscle myopathy and cardiomyopathy)115, which, as discussed later, have been linked to autophagosome maturation defects. In neural cells, WDR45 and WDR45B act redundantly during autophagosome maturation to target EPG5 to late endosomes/lysosomes25. De novo mutations in WDR45 cause β-propeller protein-associated neurodegeneration, previously known as static encephalopathy of childhood with neurodegeneration in adulthood117,118. Wdr45-knockout mice exhibit extensive swollen axons and impaired learning and memory, reminiscent of β-propeller protein-associated neurodegeneration119. A potential causative role of WDR45B in intellectual disability recently emerged120. Wdr45b-knockout mice also display abnormal motor behaviour and cognitive impairment, and pathologically exhibit cerebellar atrophy and accumulation of autophagosomes in swollen axons121.
Of note, mutations in different genes acting in autophagosome maturation result in distinct neuropathological deficits. This could be because the genes are differentially expressed in different neuronal subpopulations or are required in different types of neural cells or because their mutation results in accumulation of different types of autophagic cargo that may differ in different cells. The differential function of these genes in endocytic trafficking and recycling may also contribute to the distinct pathological defects.
Pathogenesis of muscle diseases
Autophagy is essential for preserving muscle mass and maintaining myofibre integrity122. Defects in lysosomal function and their degradative potential have been associated with a group of autophagic vacuolar myopathies, including Pompe disease, Danon disease and X-linked myopathy with excessive autophagy, which all present with accumulation of non-functional autophagic vacuoles123. Pompe disease is caused by deficiency of the lysosomal acid α-glucosidase (GAA), which hydrolyses glycogen to glucose. It is characterized by massive accumulation of autophagic vacuoles, especially in skeletal muscles124. Patients with Danon disease, which is linked to deficiency in the lysosomal membrane protein LAMP2, display cardiomyopathy, myopathy and variable mental retardation125. Massive autophagic vacuoles accumulate in cardiomyocytes and skeletal muscle cells of patients with Danon disease and LAMP2-deficient mice125,126. X-linked myopathy with excessive autophagy, a childhood-onset disease with progressive vacuolation and atrophy of skeletal muscle, is caused by compromised lysosomal acidification due to deficiency in the vacuolar ATPase assembly factor VMA21 (ref.127).
Muscle cells from patients with inclusion body myopathy, Paget disease of bone and frontotemporal dementia (IBMPFD)-related myopathy accumulate large, ubiquitin-positive rimmed vacuoles, which are non-digested autophagic vacuoles128. The causative gene for IBMPFD is VCP. VCP is primarily known for its roles in extraction of ubiquitylated proteins — which are often misfolded and prone to aggregation — from membranes or protein complexes for degradation or recycling. VCP knockdown or expression of IBMPFD mutant VCP results in accumulation of autophagic vacuoles containing ubiquitin-positive contents, and VCP was found to be essential for autophagosome maturation at late stages (following initial acidification), but the exact mechanisms through which VCP promotes further maturation are elusive128,129. VCP mutations are also associated with familial ALS103.
Deregulation in cancer
Autophagy functions as a tumour suppressor and promoter in a context-dependent manner130,131. Autophagy impedes tumour initiation and early stages of tumour progression by acting in quality control processes such as removal of damaged mitochondria and maintenance of genomic stability. By contrast, in advanced tumours or during cancer therapy, autophagy enables tumour cells to survive harsh conditions such as hypoxia and metabolic stresses. In line with this, elevated lysosomal activity resulting from increased activity of TFEB and TFE3 — which may be caused by chromosomal rearrangements, gene amplification or upregulation, and enhanced nuclear transport — is associated with the development or metastasis of various tumours, such as renal cell carcinomas, prostate cancer, pancreatic cancer, non-small-cell lung cancer (NSCLC) and breast cancer88,132. It is important to note that TFE3 and TFEB may also promote cancer progression, at least in part independently of their function in autophagy, by activating signalling pathways implicated in tumorigenesis, such as WNT and TGFβ signalling132. However, increased expression and nuclear import of TFE3 and TFEB have been demonstrated to maintain a high autophagy level to sustain intracellular amino acid pools in the pathogenesis of pancreatic ductal adenocarcinoma133. Enhanced TFEB activity can also promote lysosome exocytosis, releasing proteolytic enzymes, such as cathepsins, into the cell microenvironment, which fuels extracellular matrix remodelling, thereby stimulating cancer cell invasion and metastasis134.
Perturbation of autophagosome maturation machinery may also contribute to the occurrence and development of tumours. Alterations in EPG5 have been suggested to be involved in breast and prostate cancers135,136. EPG5 expression is significantly lower in NSCLC clinical samples, and EPG5 knockdown promotes NSCLC cell proliferation and tumorigenesis137. WDR45 is genetically altered in patients with uterine corpus endometrial carcinoma and downregulated in cervical cancer development138,139. However, owing to the heterogeneity and redundancy in the autophagosome maturation machinery highlighted above, changes in individual components are probably not sufficient to drive tumorigenesis and tumorigenic progression, and it is likely that alterations in autophagosome maturation coexist with other oncogenic lesions, further aggravating pathology.
Interplay with pathogen life cycles
Autophagy responds to invading pathogens by capturing them and delivering them to lysosomes for degradation (a process known as xenophagy); this facilitates antigen presentation for activation of innate and adaptive immune responses15,16,17,140. Pathogens have evolved diverse strategies to inhibit autophagy at the initiation and/or maturation steps to escape destruction. Certain pathogens even block autophagosome maturation and subvert the resulting vesicles for their own benefit.
Harnessing of autophagosomes/amphisomes for viral replication and release
Positive-strand RNA viruses belonging to the family Picornaviridae, such as poliovirus, rhinovirus, coxsackievirus B3 and enterovirus D68, utilize double-membrane vesicles (DMVs) as membrane scaffolds for replication and transcription141,142,143. DMVs formed in virus-infected cells are smaller than regular autophagosomes. They exhibit hallmarks of autophagosomes/amphisomes such as positivity for LC3 and the late endosomal/lysosomal marker LAMP1, but their further maturation into degradative autolysosomes is blocked14,144,145,146.
The betacoronaviruses, including mouse hepatitis virus (MHV), Middle East respiratory syndrome coronavirus, severe acute respiratory syndrome coronavirus (SARS-CoV) and SARS-CoV-2, also induce the formation of DMVs for anchoring the viral replication and transcription complexes32,147,148,149. The viral RNA products are localized in the DMV lumen and transported to the cytosol for translation and virion assembly via double-membrane-spanning molecular pores150. However, the canonical autophagic machinery, such as the LC3 lipidation system, is not required for DMV formation or coronavirus replication151,152. Instead, the DMVs in MHV-infected cells are related to the ER-derived small vesicles, called ‘EDEMosomes’, that deliver short-lived regulators of ER–associated degradation (ERAD) to late endosomes/lysosomes153. Certain autophagy proteins, however, are required for coronavirus infection. LC3 decorates the DMVs and is required for MHV replication153. Unlike on autophagic structures, where LC3 is conjugated with phosphatidylethanolamine, non-lipidated LC3 is present on the DMVs in MHV-infected cells153. Autophagy proteins involved in the generation of PtdIns3P are required for SARS-CoV-2 infection154,155. The ER-localized transmembrane autophagy proteins EPG3 (also known as VMP1) and TMEM41B are essential for autophagosome formation39,156,157,158,159. EPG3 and TMEM41B are also essential for replication of coronaviruses such as SARS-CoV-2 (refs151,160), but the step at which these proteins act during the viral life cycle has yet to be identified. In betacoronavirus-infected cells, the replicated viruses are transported inside lysosomes and released through the exocytic pathway161. Deacidification of lysosomes by loading with too many viral particles and/or by viral proteins such as ORF3a of SARS-CoV-2 promotes lysosomal exocytosis and thus viral egress from the infected cell32,161. Autophagosomes also can sequestrate and mediate non-lytic extracellular release of poliovirus, coxsackievirus B3 and enterovirus D68 (refs141,145,162).
Bacterial survival and proliferation in autophagic vacuoles
Bacteria invade host cells via phagocytosis and reside in bacterium-containing vacuoles. If the vacuolar membrane is damaged, bacteria can escape into the cytosol. Autophagy captures bacteria in the cytosol or in damaged vacuoles and delivers them to lysosomes for destruction (via xenophagy)16,140. Bacteria use different secretion systems to deliver effectors or toxins to evade autophagy surveillance and even to exploit autophagic vacuoles for intracellular survival and growth. Bacterial virulence factors can block autophagy by inhibiting the autophagy induction signal, impairing autophagy recognition, or directly attenuating the function of autophagy proteins16,140. For example, the Legionella pneumophila effector protein RavZ inhibits host autophagy by functioning as a cysteine protease that uncouples lipid-conjugated ATG8 proteins163. RavZ cleaves ATG8 proteins between the carboxy-terminal glycine and the penultimate aromatic residue, producing ATG8 proteins that cannot be reconjugated163. SopF, the effector of Salmonella enterica subsp. enterica serovar Typhimurium, impairs initiation of xenophagy by interfering with the binding of the V-ATPase on damaged bacterium-containing vacuoles to ATG16L164. Maturation of bacterium-containing autophagic vacuoles (autophagosomes or fused vesicles of bacterium-containing vacuoles and autophagosomes) into degradative autolysosomes by deposition of lytic enzymes can also be inhibited. In macrophages, autophagic vacuoles containing the virulent strain of Mycobacterium tuberculosis (H37Rv) fail to recruit RAB7 for maturation into autolysosomes165,166. Vacuoles containing Mycobacterium marinum and Yersinia pestis exhibit features of non-degradative autolysosomes that are devoid of lysosomal enzymes167,168. Some bacteria (for example, Serratia marcescens, Staphylococcus aureus, Anaplasma phagocytophilum and Coxiella burnetii) even exploit autophagic vacuoles as replication niches for intracellular growth and proliferation169,170,171,172,173,174. Fusion with autophagosomes facilitates the formation of vacuoles where bacteria reside and replicate — by promoting fusion of individual bacterium-containing vacuoles and/or providing a membrane for vacuole expansion — and also supplies nutrients to the pathogen171,172,174,175. In line with this, replication of these pathogens is promoted by autophagy induction and is blocked by autophagy inhibition172,173,174.
Mechanisms used by pathogens to interfere with autophagosome maturation
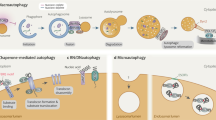
Inhibition of the assembly of STX17–SNAP29–VAMP8 is widely used by viruses to prevent the formation of degradative autolysosomes and/or to accumulate autophagosomes/amphisomes for their replication or release (Fig. 5a). Viral proteinase 3C of coxsackievirus B3 and enterovirus D68 mediates cleavage of SNAP29, separating the two SNARE motifs and thus impairing the formation of the SNARE complex144,145. The phosphoprotein (P) of human parainfluenza virus type 3 binds to SNAP29 to inhibit its interaction with STX17 (ref.176), whereas cells in which hepatitis C virus is replicating exhibit reduced levels of STX17 due to its decreased expression and increased turnover177. Tethering factors can also be targeted by viral effectors. PLEKHM1 is proteolytically targeted by proteinase 3C of coxsackievirus B3 to separate the HOPS complex-binding and LC3-binding amino terminus from the RAB7-interacting carboxy terminus, thus abolishing its tethering function145 (Fig. 5a). Cells infected with SARS-CoV-2, or expressing the viral accessory protein ORF3a, sequester components of the HOPS complex on late endosomes32. This prevents functional HOPS complex from interacting with STX17 and consequently inhibits assembly of the STX17–SNAP29–VAMP8 complex32 (Fig. 5a). M2 protein of influenza virus A inhibits autophagosome maturation by interfering with the beclin 1-containing and UVRAG-containing VPS34 complex178 (Fig. 5a). Bacterial virulence factors also block the maturation and elimination of bacterium-containing autophagic vacuoles, but the mechanisms are less well understood. Examples are shown in Fig. 5b.

Autophagy can capture invading pathogens and deliver them to lysosomes for destruction. Pathogens (including viruses and bacteria) therefore use various mechanisms to block autophagosome–lysosome fusion to escape autophagy clearance. Pathogens can also modulate autophagic structures — including induction of accumulation of autophagic vacuoles or autophagy-independent double-membrane vesicles marked by certain autophagosomal proteins and modulation of endolysosomal degradative enzymes and pH — to promote their own survival, replication and/or release (not shown). a | Viral proteinase 3C of coxsackievirus B3 (CVB3) and enterovirus D68 (EVD68) cleaves SNAP29 to reduce SNARE assembly144,145. Proteinase 3C of CVB3 also cleaves the tether protein PLEKHM1. Phosphoprotein (P) of human parainfluenza virus type 3 (HPIV3) competes with STX17 for SNAP29 binding176. ORF3a of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) sequestrates the HOPS complex on late endosomes, thus impairing SNARE complex assembly. M2 protein of influenza virus A (IVA) dampens the activity of the VPS34 complex to prevent autophagosome maturation. b | Streptolysin O (SLO) damages the membrane of group A Streptococcus (GAS)-containing endosomes to trigger their engulfment by autophagosomes. Translocation of the co-toxin NAD-glycohydrolase (NADase) into the cytoplasm blocks the fusion of GAS-containing autophagic vacuoles with lysosomes213. The virulence factor IsaB of methicillin-resistant Staphylococcus aureus (MRSA) blocks lysosomal acidification to suppress the function of autolysosomes214. The virulence factor VacA of Helicobacter pylori prevents TRPML1-mediated calcium efflux from endosomes to disrupt endolysosomal trafficking and thus autophagosome maturation215.
Therapeutic interventions
Different steps of the autophagy pathway are potential targets for therapeutic interventions179,180. Inhibitors and activators of autophagosome formation, including VPS34 and ULK1 inhibitors or the activating peptide Tat–beclin 1, are potent modulators of autophagy, but are not yet available for clinical use181,182. By contrast, lysosomotropic agents, which inhibit the activity of lysosomes and block their fusion with autophagic vesicles, have been used in several clinical trials183,184,185. Modulation of autophagosome maturation through targeting key components of the maturation machinery or through controlling autolysosomal activity by targeting the transcriptional programme of lysosome and autophagosome biogenesis is also a potential therapeutic option to regulate autophagic flux, but such approaches require further optimization if they are to be used in a clinical setting. Inhibiting and stimulating autophagosome maturation are important therapeutic avenues to explore. In many neurodegenerative diseases, autophagosome maturation is blocked186,187, so restoring the autophagy flux would be an important therapeutic option. When inhibition of autophagy is required, as, for example, in cancer cells, it is important to consider the step at which the autophagic pathway must be interrupted. Different outcomes can be observed when autophagy is blocked at autophagosome formation or autophagosome maturation. In line with this, it has been shown that blocking maturation of autophagosomes favours cell death via necroptosis in human cancer cells in response to TNF-related apoptosis-inducing ligand (TRAIL), whereas blocking the formation of autophagosomes triggers TRAIL-dependent apoptosis in the same cells188.
Targeting the autophagosome maturation machinery
Targeting the machinery that controls autophagosome maturation (such as ESCRT, SNAREs and the HOPS complex) or targeting post-translational modifications of these proteins would be ideal to either increase or decrease autophagy flux189,190,191. Inhibitors of O-GlcNAc transferase and those of the opposing enzyme, O-GlcNAcase, can stimulate or inhibit autophagosome maturation, respectively, by modulating the O-GlcNAcylation of SNAP29 (refs66,192,193) (Table 1). As many cellular proteins are O-GlcNAcylated, blocking O-GlcNAc sites on a specific protein would require the development of methods to increase the selectivity of targeting, such as the use of aptamers or nanobodies194. Another strategy to inhibit autophagosome maturation would be to use the small molecule TCH-165 to specifically activate the proteasomal degradation of SNAP29 and STX17 (ref.195). Inhibiting Rubicon is a potential approach to promote autophagy; however, pharmacological Rubicon inhibitors have yet to be developed. This strategy must be carefully evaluated because Rubicon is a positive modulator of LC3-associated phagocytosis, a process known to have a protective effect in many inflammatory diseases196,197,198.
Modulation of the transcription programme of autophagosome maturation
Modulation of TFEB/TFE3 is an obvious strategy to control autophagy on a transcriptional level. It has been shown that TFEB overexpression has beneficial effects in ameliorating LSDs and obesity by stimulating lipophagy199,200,201. However, chronic overexpression of TFEB favours the progression of pancreatic tumours and NSCLCs200. Thus, developing small molecules to acutely stimulate TFEB and harness the autophagy–lysosomal pathway would be beneficial in ameliorating diseases in which autophagy has a defensive role201. Small molecules can activate TFEB indirectly by modulating its upstream kinases or phosphatases (Table 1). For example, TFEB nuclear transport is promoted by rapamycin via inhibiting mTORC1 activity or by compounds isolated from the herb Euphorbia peplus Linn via the PKC–GSK3β cascade90.
Blocking autophagosome maturation with lysosomotropic agents
Chloroquine (CQ), hydroxychloroquine (HCQ) and their derivatives are the only clinically approved drugs that act on autophagosome maturation (Table 1). They are used alone or in combination with other drugs, mostly in ongoing oncology trials, in general with the goal of optimizing therapies by blocking autophagy induced by cancer treatments183,184,185. The new-generation dimeric CQ derivatives Ly05 and DQ661 are active at lower concentrations than CQ and HCQ130,183,202.
CQ and HCQ block autophagy flux by inhibiting the hydrolytic capacity of autolysosomes. They increase the pH in autolysosomal compartments and hence block the activity of acidic proteases and other enzymes203. Beyond their capacity for H+ trapping, monomeric and dimeric CQ derivatives also block lysosomal function by inhibiting the activity of the lysosomal enzyme palmitoyl-protein thioesterase 1 (PPT1), which is involved in stabilizing the lysosomal localization of V-ATPase subunits202. Thus, inhibition of PPT1 results in autophagy inhibition. In addition, lysosomal function and/or fusion with autophagosomes could be targeted by other means of pH regulation (such as via modulation of V-ATPase), inhibition of the cation channel TRPML1, inhibition of lysosomal enzymes or modulation of lysosomal membrane dynamics — fusion and fission — which impact on lysosomal number and function179,180,183,204 (Table 1).
Of note, lysosomotropic agents target all acidic compartments and also other pathways203,205, and thus in some cases the beneficial effects of lysosomotropic agents can be attributed to mechanisms other than a blockade of autophagy130,203. For example, these drugs inhibit tumour progression, independently of the autophagy blockade, by altering the trafficking of signalling molecules (that is, NOTCH1) in the endocytic pathway206 and by other mechanisms184,203. It is also worth mentioning that the activity of CQ and HCQ observed in vitro may not be the same in vivo due to different parameters. For example, during viral infection, the cell type used in vitro may not reflect the tropism of the virus in vivo and/or the sensitivity to CQ207. Moreover, the acidic environment in tumours can protonate the lysosomotropic agent and greatly reduce its cellular uptake208.
Conclusions and perspectives
Autophagosome maturation is an essential step in the autophagy pathway that ensures the formation of degradative autolysosomes. It adds another layer of complexity and provides an extra node to integrate nutrient status and stresses for regulation of autophagic degradation. The distinct organization and trafficking of the endolysosomal compartment in different cell types and growth conditions add complexity at the intersection of the autophagy and endocytic pathways. Thus, the trans-SNARE complexes and tethering factors act coordinately with context-specific factors to mediate fusion of autophagosomes with endocytic vesicles and lysosomes. Further investigations are needed to elucidate how different signalling pathways and stresses coordinate autophagosome initiation and maturation to ensure efficient progression of autophagic flux and how these processes are adapted in different cell types or pathophysiological contexts.
Autophagosome maturation is widely manipulated by pathogens to escape from destruction and for replication and growth. Pathogens that use autophagic vacuoles for replication can both activate autophagosome initiation and block maturation to achieve their maximal accumulation. Understanding how viral proteins and bacterial virulence factors modulate host autophagy will help us to develop strategies to interfere with the pathogen–host interaction and even to restore autophagy as a defence mechanism. Such strategies are urgently required with the evolution of multidrug-resistant bacteria. Elucidating the underlying mechanisms for autophagosome maturation defects and deregulation of the function of the autophagosome–lysosome system is also key for us to understand the pathogenesis of various human diseases. Targeting autophagosome maturation — via modulation of SNAREs, tethers and their regulators as well as lysosome biogenesis and function — offers an effective strategy for the treatment of these diseases.
Biomarkers and methods that reliably monitor autophagy flux in vivo are needed to examine temporal changes of autophagy activity and to evaluate interventions that target autophagosome maturation. A combination of assays has been used to measure autophagy flux and to monitor autophagosome maturation25. However, many of these assays are difficult to implement in humans. Several methods have recently been developed to serve as reliable autophagy biomarkers in humans209. Analysis of autophagy flux in isolated peripheral blood mononuclear cells is used to measure autophagy activity in human blood samples210,211. Positron emission tomography can be used with hypoxia tracers to correlate hypoxia and autophagy in tumours and also to gauge the level of specific autophagy substrates in tissues by the use of positron emission tomography ligands that bind to autophagy substrates184,209. The levels of specific molecules in biological fluids can also be used to determine autophagy flux in tissues. For instance, the blood level of arginase 1 reflects autophagy activity in the liver212. Nevertheless, these methods are of low throughout and/or can be applied only to selected cells or tissues. Thus, to screen drugs targeting autophagy, there is an urgent need for reliable, high-throughput clinical biomarkers to measure autophagic activity by the identification of tissue-specific circulating autophagy by-products and the development of flux probes for use in imaging techniques.
References
Feng, Y. C., He, D., Yao, Z. Y. & Klionsky, D. J. The machinery of macroautophagy. Cell Res. 24, 24–41 (2014).
Lamb, C. A., Yoshimori, T. & Tooze, S. A. The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 14, 759–774 (2013).
Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell. Biol. 21, 439–458 (2020).
Mizushima, N., Yoshimori, T. & Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132 (2011).
Stolz, A., Ernst, A. & Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 16, 495–501 (2014).
Takahashi, Y. et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 9, 2855 (2018).
Takahashi, Y. et al. VPS37A directs ESCRT recruitment for phagophore closure. J. Cell Biol. 218, 3336–3354 (2019).
Zhen, Y. et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy 16, 826–841 (2020).
Ktistakis, N. T. & Tooze, S. A. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 26, 624–635 (2016).
Zhao, Y. G. & Zhang, H. Formation and maturation of autophagosomes in higher eukaryotes: a social network. Curr. Opin. Cell Biol. 53, 29–36 (2018).
Zhao, Y. G. & Zhang, H. Autophagosome maturation: an epic journey from the ER to lysosomes. J. Cell Biol. 218, 757–770 (2019).
Pu, J., Guardia, C. M., Keren-Kaplan, T. & Bonifacino, J. S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016).
Jiang, P. D. & Mizushima, N. Autophagy and human diseases. Cell Res. 24, 69–79 (2014).
Choi, Y., Bowman, J. W. & Jung, J. U. Autophagy during viral infection — a double-edged sword. Nat. Rev. Microbiol. 16, 341–354 (2018).
Deretic, V., Saitoh, T. & Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737 (2013).
Kimmey, J. M. & Stallings, C. L. Bacterial pathogens versus autophagy: implications for therapeutic interventions. Trends Mol. Med. 22, 1060–1076 (2016).
Levine, B., Mizushima, N. & Virgin, H. W. Autophagy in immunity and inflammation. Nature 469, 323–335 (2011).
Filimonenko, M. et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 179, 485–500 (2007).
Razi, M., Chan, E. Y. W. & Tooze, S. A. Early endosomes and endosomal coatomer are required for autophagy. J. Cell Biol. 185, 305–321 (2009).
Jahn, R. & Scheller, R. H. SNAREs — engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643 (2006).
Langemeyer, L., Frohlich, F. & Ungermann, C. Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol. 28, 957–970 (2018).
Yu, I. M. & Hughson, F. M. Tethering factors as organizers of intracellular vesicular traffic. Annu. Rev. Cell Dev. Biol. 26, 137–156 (2010).
Nguyen, T. N. et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 215, 857–874 (2016).
Vaites, L. P., Paulo, J. A., Huttlin, E. L. & Harper, J. W. Systematic analysis of human cells lacking ATG8 proteins uncovers roles for GABARAPs and the CCZ1/MON1 regulator C18orf8/RMC1 in macroautophagic and selective autophagic flux. Mol. Cell Biol. 38, e00392-17 (2018).
Ji, C. C., Zhao, H. Y., Chen, D., Zhang, H. & Zhao, Y. G. β-Propeller proteins WDR45 and WDR45B control autophagosome maturation into autolysosomes in neural cells. Curr. Biol. 31, 1666–1677.e6 (2021).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269 (2012). This study reveals that the SNARE complex composed of autophagosomal Qa SNARE STX17, Qbc SNARE SNAP29 and Qc SNARE VAMP8 mediates autophagosome–late endosome/lysosome fusion.
Matsui, T. et al. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 217, 2633–2645 (2018).
Takats, S. et al. Autophagosomal syntaxin 17-dependent lysosomal degradation maintains neuronal function in Drosophila. J. Cell Biol. 201, 531–539 (2013).
Tsuboyama, K. et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354, 1036–1041 (2016).
Kumar, S. et al. Mechanism of Stx17 recruitment to autophagosomes via IRGM and mammalian Atg8 proteins. J. Cell Biol. 217, 997–1013 (2018).
Diao, J. J. et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520, 563–566 (2015).
Miao, G. et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 56, 427–442.e425 (2021). In this work, the authors demonstrate that the late endosome/lysosome-localized ORF3a protein of SARS-CoV-2 prevents fusion of autophagic vacuoles with lysosomes by sequestrating the HOPS complex component VPS39 and hence blocking the assembly of the SNARE complex required for the formation of degradative autolysosomes.
Takats, S. et al. Interaction of the HOPS complex with syntaxin 17 mediates autophagosome clearance in Drosophila. Mol. Biol. Cell 25, 1338–1354 (2014).
Jiang, P. et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 25, 1327–1337 (2014).
Stroupe, C., Collins, K. M., Fratti, R. A. & Wickner, W. Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J. 25, 1579–1589 (2006).
Gao, J. Q., Langemeyer, L., Kummel, D., Reggiori, F. & Ungermann, C. Molecular mechanism to target the endosomal Mon1-Ccz1 GEF complex to the pre-autophagosomal structure. eLife 7, e31145 (2018).
Hegedus, K. et al. The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Mol. Biol. Cell 27, 3132–3142 (2016).
Manil-Segalen, M. et al. The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev. Cell 30, 110–110 (2014).
Tian, Y. et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 141, 1042–1055 (2010).
Wang, Z. et al. The Vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol. Cell 63, 781–795 (2016). This study shows that EPG5, the gene causing Vici syndrome, encodes a tether protein that is recruited to late endosomes/lysosomes by binding to RAB7 and facilitates SNARE complex formation to mediate autophagosome maturation.
Zhao, H. Y. et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J. Cell Biol. 200, 731–741 (2013).
McEwan, D. G. et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 57, 39–54 (2015). The authors demonstrate that PLEKHM1 tethers autophagosomes and lysosomes by simultaneously binding to autophagosomal LC3 and lysosome-localized RAB7, and also promotes autophagosome–lysosome fusion by recruiting the HOPS complex.
Wijdeven, R. H. et al. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat. Commun. 7, 11808 (2016).
Wetzel, L. et al. TECPR1 promotes aggrephagy by direct recruitment of LC3C autophagosomes to lysosomes. Nat. Commun. 11, 2993 (2020).
Chen, D. D. et al. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol. Cell 45, 629–641 (2012).
Jahreiss, L., Menzies, F. M. & Rubinsztein, D. C. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic 9, 574–587 (2008).
Korolchuk, V. I. et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 13, 453–460 (2011).
Bonifacino, J. S. & Neefjes, J. Moving and positioning the endolysosomal system. Curr. Opin. Cell Biol. 47, 1–8 (2017).
Jia, R., Guardia, C. M., Pu, J., Chen, Y. & Bonifacino, J. S. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy 13, 1648–1663 (2017).
Pankiv, S. et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 188, 253–269 (2010).
Johansson, M. et al. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J. Cell Biol. 176, 459–471 (2007).
Rocha, N. et al. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150(Glued) and late endosome positioning. J. Cell Biol. 185, 1209–1225 (2009).
van der Kant, R. et al. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J. Cell Sci. 126, 3462–3474 (2013).
Lee, S., Sato, Y. & Nixon, R. A. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 31, 7817–7830 (2011).
Maday, S., Wallace, K. E. & Holzbaur, E. L. F. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 196, 407–417 (2012). This work reveals that in primary neurons, autophagosomes formed at the distal tip of the axon undergo retrograde transport to the cell soma, accompanied by gradual maturation and acidification.
Fu, M. M., Nirschl, J. J. & Holzbaur, E. L. F. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev. Cell 29, 577–590 (2014).
Cason, S. E. et al. Sequential dynein effectors regulate axonal autophagosome motility in a maturation-dependent pathway. Preprint at bioRxiv https://doi.org/10.1101/2020.11.01.363333 (2020).
Russell, R. C., Yuan, H. X. & Guan, K. L. Autophagy regulation by nutrient signaling. Cell Res. 24, 42–57 (2014).
Furuya, T. et al. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol. Cell 38, 500–511 (2010).
Nazio, F. et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 15, 406–416 (2013).
Nazio, F. et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J. Cell Biol. 215, 841–856 (2016).
Liu, C. C. et al. Cul3-KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol. Cell 61, 84–97 (2016).
Jean, S., Cox, S., Nassari, S. & Kiger, A. A. Starvation-induced MTMR13 and RAB21 activity regulates VAMP8 to promote autophagosome-lysosome fusion. EMBO Rep. 16, 297–311 (2015).
Fraldi, A. et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 29, 3607–3620 (2010).
Shen, Q. H. et al. Acetylation of STX17 (syntaxin 17) controls autophagosome maturation. Autophagy 17, 1157–1169 (2021).
Guo, B. et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat. Cell Biol. 16, 1215–1226 (2014).
Hanover, J. A., Krause, M. W. & Love, D. C. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim. Biophys. Acta 1800, 80–95 (2010).
Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 20, 521–527 (2018).
Funderburk, S. F., Wang, Q. J. & Yue, Z. Y. The beclin 1-VPS34 complex — at the crossroads of autophagy and beyond. Trends Cell Biol. 20, 355–362 (2010).
Matsunaga, K. et al. Two beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11, 385–396 (2009).
Cheng, X. W. et al. Pacer mediates the function of class III PI3K and HOPS complexes in autophagosome maturation by engaging Stx17. Mol. Cell 65, 1029–1043 (2017).
Liang, C. Y. et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 10, 776–787 (2008).
Sun, Q. M., Westphal, W., Wong, K. N., Tan, I. & Zhong, Q. Rubicon controls endosome maturation as a Rab7 effector. Proc. Natl Acad. Sci. USA 107, 19338–19343 (2010).
Zhong, Y. et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 11, 468–476 (2009).
Kim, Y. M. et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 57, 207–218 (2015).
Judith, D. et al. ATG9A shapes the forming autophagosome through arfaptin 2 and phosphatidylinositol 4-kinase III beta. J. Cell Biol. 218, 1634–1652 (2019).
Wang, H. Z. et al. GABARAPs regulate PI4P-dependent autophagosome: lysosome fusion. Proc. Natl Acad. Sci. USA 112, 7015–7020 (2015).
Del Bel, L. M. & Brill, J. A. Sac1, a lipid phosphatase at the interface of vesicular and nonvesicular transport. Traffic 19, 301–318 (2018).
Miao, G., Zhang, Y., Chen, D. & Zhang, H. The ER-localized transmembrane protein TMEM39A/SUSR2 regulates autophagy by controlling the trafficking of the PtdIns(4)P phosphatase SAC1. Mol. Cell 77, 618–632.e5 (2020).
Jaber, N. et al. Vps34 regulates Rab7 and late endocytic trafficking through recruitment of the GTPase-activating protein Armus. J. Cell Sci. 129, 4424–4435 (2016).
Carroll, B. et al. The TBC/RabGAP armus coordinates Rac1 and Rab7 functions during autophagy. Dev. Cell 25, 15–28 (2013).
Piper, R. C. & Luzio, J. P. Late endosomes: sorting and partitioning in multivesicular bodies. Traffic 2, 612–621 (2001).
Baba, T., Toth, D. J., Sengupta, N., Kim, Y. J. & Balla, T. Phosphatidylinositol 4,5-bisphosphate controls Rab7 and PLEKHM1 membrane cycling during autophagosome-lysosome fusion. EMBO J. 38, e100312 (2019).
Raben, N. & Puertollano, R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol. 32, 255–278 (2016).
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009).
Settembre, C. et al. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011).
Ballabio, A. & Bonifacino, J. S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118 (2020).
Puertollano, R., Ferguson, S. M., Brugarolas, J. & Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 37, e98804 (2018).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012).
Li, Y. et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 18, 1065–1077 (2016).
Medina, D. L. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015). This study shows that upon activation by lysosomal calcium release through MCOLN1, the phosphatase calcineurin dephosphorylates TFEB and promotes its nuclear translocation to induce autophagy and lysosomal biogenesis.
Martina, J. A. & Puertollano, R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem. 293, 12525–12534 (2018).
Banani, S. F., Lee, H. O., Hyman, A. A. & Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017).
Shin, Y. & Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017).
Zhang, H. et al. Liquid-liquid phase separation in biology: mechanisms, physiological functions and human diseases. Sci. China Life Sci 63, 953–985 (2020).
Boija, A. et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855 (2018).
Sabari, B. R. Biomolecular condensates and gene activation in development and disease. Dev. Cell 55, 84–96 (2020).
Chen, D. et al. Inositol polyphosphate multikinase inhibits liquid-liquid phase separation of TFEB to negatively regulate autophagy activity. Dev. Cell 55, 588–602 (2020). This study demonstrates that the TFEB condensate formed via LLPS is involved in transcription and its formation is negatively regulated by the nuclear-localized protein IPMK.
Tabata, K. et al. Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway via a novel Rab7-binding domain. Mol. Biol. Cell. 21, 4162–4172 (2010).
Fujita, N. et al. Genetic screen in Drosophila muscle identifies autophagy-mediated T-tubule remodeling and a Rab2 role in autophagy. eLife 6, e23367 (2017).
Wang, Z. Y. et al. The RBG-1-RBG-2 complex modulates autophagy activity by regulating lysosomal biogenesis and function in C. elegans. J. Cell Sci. 132, jcs234195 (2019).
Nixon, R. A., Yang, D. S. & Lee, J. H. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4, 590–599 (2008).
Otomo, A., Pan, L. & Hadano, S. Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol. Res. Int. 2012, 498428 (2012).
Puls, I. et al. Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456 (2003).
Parkinson, N. et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67, 1074–1077 (2006).
Mauvezin, C., Nagy, P., Juhasz, G. & Neufeld, T. P. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 6, 7007 (2015).
McBrayer, M. & Nixon, R. A. Lysosome and calcium dysregulation in Alzheimer’s disease: partners in crime. Biochem. Soc. Trans. 41, 1495–1502 (2013).
Settembre, C. et al. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 17, 119–129 (2008).
Vergarajauregui, S., Connelly, P. S., Daniels, M. P. & Puertollano, R. Autophagic dysfunction in mucolipidosis type IV patients. Hum. Mol. Genet. 17, 2723–2737 (2008).
Decressac, M. et al. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl Acad. Sci. USA 110, E1817–E1826 (2013).
Cortes, C. J. et al. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci. 17, 1180–1189 (2014). This work reveals that the polyglutamine-expanded androgen receptor physically binds to and inactivates TFEB, thereby impairing autophagy function and contributing to the pathogenesis of spinal and bulbar muscular atrophy.
Flavin, W. P. et al. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta. Neuropathol. 134, 629–653 (2017).
Jiang, P. Z., Gan, M., Yen, S. H., McLean, P. J. & Dickson, D. W. Impaired endo-lysosomal membrane integrity accelerates the seeding progression of alpha-synuclein aggregates. Sci. Rep. 7, 28794446 (2017).
Cullup, T. et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 45, 83–87 (2013).
Byrne, S., Dionisi-Vici, C., Smith, L., Gautel, M. & Jungbluth, H. Vici syndrome: a review. Orphanet J. Rare Dis. 11, 21 (2016).
Zhao, Y. G., Zhao, H. Y., Sun, H. Y. & Zhang, H. Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy 9, 1258–1262 (2013).
Haack, T. B. et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 91, 1144–1149 (2012).
Saitsu, H. et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449 (2013).
Zhao, Y. G. et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 11, 881–890 (2015).
Suleiman, J. et al. WDR45B-related intellectual disability, spastic quadriplegia, epilepsy, and cerebral hypoplasia: a consistent neurodevelopmental syndrome. Clin. Genet. 93, 360–364 (2018).
Ji, C. C. et al. Role of Wdr45b in maintaining neural autophagy and cognitive function. Autophagy 16, 615–625 (2020).
Jiao, J. Q. & Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharmacol. 34, 1–6 (2017).
Dowling, J. J., Moore, S. A., Kalimo, H. & Minassian, B. A. X-linked myopathy with excessive autophagy: a failure of self-eating. Acta. Neuropathol. 129, 383–390 (2015).
Myerowitz, R., Puertollano, R. & Raben, N. Impaired autophagy: the collateral damage of lysosomal storage disorders. EBioMedicine 63, 103166 (2021).
Nishino, I. et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Am. J. Hum. Genet. 67, 390–390 (2000).
Tanaka, Y. et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906 (2000).
Saraste, A. et al. No cardiomyopathy in X-linked myopathy with excessive autophagy. Neuromuscul. Disord. 25, 485–487 (2015).
Tresse, E. et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227 (2010).
Ju, J. S. et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888 (2009).
Towers, C. G., Wodetzki, D. & Thorburn, A. Autophagy and cancer: modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 219, e201909033 (2020).
Dikic, I. & Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364 (2018).
Astanina, E., Bussolino, F. & Doronzo, G. Multifaceted activities of transcription factor EB in cancer onset and progression. Mol. Oncol. 15, 327–346 (2021).
Perera, R. M. et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 (2015). The authors demonstrate that pancreatic ductal adenocarcinoma cells contain higher levels of nuclear MITF, TFE3 and TFEB than normal cells, which in turn activate autophagy to promote tumour maligancy.
Kundu, S. T. et al. TMEM106B drives lung cancer metastasis by inducing TFEB-dependent lysosome synthesis and secretion of cathepsins. Nat. Commun. 9, 2731 (2018).
Sjoblom, T. et al. The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 (2006).
Wu, B. et al. Intratumoral heterogeneity and genetic characteristics of prostate cancer. Int. J. Cancer 146, 3369–3378 (2020).
Li, H. et al. C-myc/miR-150/EPG5 axis mediated dysfunction of autophagy promotes development of non-small cell lung cancer. Theranostics 9, 5134–5148 (2019).
Bai, M. X., Che, Y. Y., Lu, K. & Fu, L. Analysis of deubiquitinase OTUD5 as a biomarker and therapeutic target for cervical cancer by bioinformatic analysis. PeerJ 8, e9146 (2020).
Lebovitz, C. B. et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 11, 1668–1687 (2015).
Huang, J. & Brumell, J. H. Bacteria-autophagy interplay: a battle for survival. Nat. Rev. Microbiol. 12, 101–114 (2014).
Jackson, W. T. et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3, 861–871 (2005).
Wong, H. H. & Sanyal, S. Manipulation of autophagy by (+) RNA viruses. Semin. Cell Dev. Biol. 101, 3–11 (2020).
Wong, J. et al. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82, 9143–9153 (2008).
Corona, A. K., Saulsbery, H. M., Velazquez, A. F. C. & Jackson, W. T. Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit. Cell Rep. 22, 3304–3314 (2018).
Mohamud, Y. et al. Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication. Cell Rep. 22, 3292–3303 (2018).
Kemball, C. C. et al. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 84, 12110–12124 (2010).
Knoops, K. et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6, 1957–1974 (2008).
Miller, K. et al. Coronavirus interactions with the cellular autophagy machinery. Autophagy 16, 2131–2139 (2020).
Snijder, E. J. et al. A unifying structural and functional model of the coronavirus replication organelle: tracking down RNA synthesis. PLoS Biol. 18, e3000715 (2020).
Wolff, G. et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 369, 1395–1398 (2020).
Schneider, W. M. et al. Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell 184, 120–132.e14 (2021).
Zhao, Z. J. et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 3, 581–585 (2007).
Reggiori, F. et al. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 7, 500–508 (2010). Using MHV infection as a model, the authors show that coronaviruses hijack LC3-labelled EDEMsomes as DMVs for their replication.
Dniloski, Z. et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 184, 92–105.e16 (2021).
Wang, R. et al. Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell 184, 106–119.e14 (2021).
Morita, K. et al. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J. Cell Biol. 217, 3817–3828 (2018).
Zhao, Y. G. et al. The ER-localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol. Cell 67, 974–989 (2017).
Moretti, F. et al. TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 19, e45889 (2018).
Shoemaker, C. J. et al. CRISPR screening using an expanded toolkit of autophagy reporters identifies TMEM41B as a novel autophagy factor. PLoS Biol. 17, e2007044 (2019).
Hoffmann, H. H. et al. TMEM41B is a pan-flavivirus host factor. Cell 184, 133–148 (2021).
Ghosh, S. et al. β-Coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 183, 1520–1535 (2020). This is the first demonstration that betacoronaviruses exploit lysosomal exocytosis for egress, accompanied by blockage of lysosomal acidification, inactivation of lysosomal degradation and impaired antigen presentation.
Bird, S. W., Maynard, N. D., Covert, M. W. & Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl Acad. Sci. USA 111, 13081–13086 (2014).
Choy, A. et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338, 1072–1076 (2012).
Xu, Y. et al. A bacterial effector reveals the V-ATPase-ATG16L1 axis that initiates xenophagy. Cell 178, 552–566 (2019). This study demonstrates that the Salmonella Typhimurium T3SS effector SopF blocks V-ATPase on damaged bacterium-containing vacuoles from recruiting ATG16L1 to initiate xenophagy.
Chandra, P. et al. Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Sci. Rep. 5, 16320 (2015).
Romagnoli, A. et al. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 8, 1357–1370 (2012).
Lerena, M. C. & Colombo, M. I. Mycobacterium marinum induces a marked LC3 recruitment to its containing phagosome that depends on a functional ESX-1 secretion system. Cell Microbiol. 13, 814–835 (2011).
Pujol, C. et al. Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect. Immun. 77, 2251–2261 (2009).
Beron, W., Gutierrez, M. G., Rabinovitch, M. & Colombo, M. I. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect. Immun. 70, 5816–5821 (2002).
Fedrigo, G. V., Campoy, E. M., Di Venanzio, G., Colombo, M. I. & Vescovi, E. G. Serratia marcescens is able to survive and proliferate in autophagic-like vacuoles inside non-phagocytic cells. PLoS ONE 6, e24054 (2011).
Niu, H., Xiong, Q. M., Yamamoto, A., Hayashi-Nishino, M. & Rikihisa, Y. Autophagosomes induced by a bacterial beclin 1 binding protein facilitate obligatory intracellular infection. Proc. Natl Acad. Sci. USA 109, 20800–20807 (2012).
Niu, H., Yamaguchi, M. & Rikihisa, Y. Subversion of cellular autophagy by Anaplasma phagocytophilum. Cell Microbiol. 10, 593–605 (2008).
Schnaith, A. et al. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J. Biol. Chem. 282, 2695–2706 (2007).
Winchell, C. G., Graham, J. G., Kurten, R. C. & Voth, D. E. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect. Immun. 82, 2229–2238 (2014).
Newton, H. J. et al. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 10, e1004286 (2014).
Ding, B. B. et al. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 15, 564–577 (2014). This study demonstrates that the viral phosphoprotein (P) of human parainfluenza virus type 3 interacts with SNAP29 to prevent its binding to STX17, thereby blocking fusion of autophagosomes with lysosomes to avoid autophagic degradation.
Ren, H. et al. The autophagosomal SNARE protein syntaxin 17 is an essential factor for the hepatitis C virus life cycle. J. Virol. 90, 5989–6000 (2016).
Gannage, M. et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380 (2009).
Galluzzi, L., Bravo-San Pedro, J. M., Levine, B., Green, D. R. & Kroemer, G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 16, 487–511 (2017).
Morel, E. et al. Autophagy: a druggable process. Annu. Rev. Pharmacol. Toxicol. 57, 375–398 (2017).
Levine, B., Packer, M. & Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Invest. 125, 14–24 (2015).
Vakifahmetoglu-Norberg, H., Xia, H. G. & Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Invest. 125, 5–13 (2015).
Amaravadi, R. K., Kimmelman, A. C. & Debnath, J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 9, 1167–1181 (2019).
Levy, J. M. M., Towers, C. G. & Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542 (2017).
Marsh, T., Tolani, B. & Debnath, J. The pleiotropic functions of autophagy in metastasis. J. Cell Sci. 134, jcs247056 (2021).
Djajadikerta, A. et al. Autophagy induction as a therapeutic strategy for neurodegenerative diseases. J. Mol. Biol. 432, 2799–2821 (2020).
Ganesan, D. & Cai, Q. Understanding amphisomes. Biochem. J. 478, 1959–1976 (2021).
Goodall, M. L. et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev. Cell 37, 337–349 (2016).
Corona, A. K. & Jackson, W. T. Finding the middle ground for autophagic fusion requirements. Trends Cell Biol. 28, 869–881 (2018).
van der Beek, J., Jonker, C., van der Welle, R., Liv, N. & Klumperman, J. CORVET, CHEVI and HOPS — multisubunit tethers of the endo-lysosomal system in health and disease. J. Cell Sci. 132, jcs189134 (2019).
Vietri, M., Radulovic, M. & Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 21, 25–42 (2020).
Dodson, M. et al. Increased O-GlcNAcylation of SNAP29 drives arsenic-induced autophagic dysfunction. Mol. Cell Biol. 38, e00595-17 (2018).
Zhou, F. X. et al. Down-regulation of OGT promotes cisplatin resistance by inducing autophagy in ovarian cancer. Theranostics 8, 5200–5212 (2018).
Zhu, Y. & Hart, G. W. Targeting O-GlcNAcylation to develop novel therapeutics. Mol. Aspects Med. 79, 100885 (2020).
Njomen, E. & Tepe, J. J. Regulation of autophagic flux by the 20S proteasome. Cell Chem. Biol. 26, 1283–1294.e5 (2019).
Galluzzi, L. & Green, D. R. Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699 (2019).
Wan, J. H. et al. LC3-associated phagocytosis protects against inflammation and liver fibrosis via immunoreceptor inhibitory signaling. Sci. Transl. Med. 12, eaaw8523 (2020).
Wong, S. W., Sil, P. & Martinez, J. Rubicon: LC3-associated phagocytosis and beyond. FEBS J. 285, 1379–1388 (2018).
Settembre, C. et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 15, 1016–1016 (2013).
Napolitano, G. & Ballabio, A. TFEB at a glance. J. Cell Sci. 129, 2475–2481 (2016).
Parenti, G., Medina, D. L. & Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 13, e12836 (2021).
Rebecca, V. W. et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. 9, 220–229 (2019).
Nirk, E. L., Reggiori, F. & Mauthe, M. Hydroxychloroquine in rheumatic autoimmune disorders and beyond. EMBO Mol. Med. 12, e12476 (2020).
Whitmarsh-Everiss, T. & Laraia, L. Small molecule probes for targeting autophagy. Nat. Chem. Biol. 17, 653–664 (2021).
Chude, C. I. & Amaravadi, R. K. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 18, 1279 (2017).
Maes, H. et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26, 190–206 (2014).
Hoffmann, M. et al. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature 585, 588–590 (2020).
Pellegrini, P. et al. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy 10, 562–571 (2014).
Mizushima, N. & Murphy, L. O. Autophagy assays for biological discovery and therapeutic development. Trends Biochem. Sci. 45, 1080–1093 (2020).
Pietrocola, F. et al. Metabolic effects of fasting on human and mouse blood in vivo. Autophagy 13, 567–578 (2017).
Bensalem, J. et al. Measurement of autophagic flux in humans: an optimized method for blood samples. Autophagy https://doi.org/10.1080/15548627.2020.1846302 (2020).
Poillet-Perez, L. et al. Autophagy maintains tumour growth through circulating arginine. Nature 565, 569–573 (2018).
O’Seaghdha, M. & Wessels, M. R. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A streptococcus from xenophagic killing. PLoS Pathog. 9, e1003394 (2013). The authors show that during group A Streptococcus infection, the pore-forming toxin streptolysin O damages the bacterium-containing vacuole, resulting in release of active NADase into the cytosol, which prevents the fusion of group A Streptococcus-containing autophagosomes with lysosomes.
Liu, P. F. et al. IsaB inhibits autophagic flux to promote host transmission of methicillin-resistant Staphylococcus aureus. J. Invest. Dermatol. 135, 2714–2722 (2015).
Capurro, M. I. et al. VacA generates a protective intracellular reservoir for Helicobacter pylori that is eliminated by activation of the lysosomal calcium channel TRPML1. Nat. Microbiol. 4, 1411–1423 (2019).
Zhang, H. Lipid transfer at ER-isolation membrane contacts. Nat. Rev. Mol. Cell Biol. 21, 121 (2020).
Schutter, M., Giavalisco, P., Brodesser, S. & Graef, M. Local fatty acid channeling into phospholipid synthesis drives phagophore expansion during autophagy. Cell 180, 135–149.e14 (2020).
Yang, C. & Wang, X. Lysosome biogenesis: regulation and functions. J. Cell Biol. 220, e202102001 (2021).
Teter, S. A. et al. Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J. Biol. Chem. 276, 2083–2087 (2001).
Liu, Y. B. et al. Autophagy-dependent ribosomal RNA degradation is essential for maintaining nucleotide homeostasis during C. elegans development. eLife 7, e36588 (2018).
Liu, B., Du, H. W., Rutkowski, R., Gartner, A. & Wang, X. C. LAAT-1 is the lysosomal lysine/arginine transporter that maintains amino acid homeostasis. Science 337, 351–354 (2012).
Rong, Y. G. et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl Acad. Sci. USA 108, 7826–7831 (2011).
Yu, L. et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946 (2010).
Chen, Y. & Yu, L. Development of research into autophagic lysosome reformation. Mol. Cell 41, 45–49 (2018).
Sun, T., Wang, X. W., Lu, Q., Ren, H. Y. & Zhang, H. CUP-5, the C. elegans ortholog of the mammalian lysosomal channel protein MLN1/TRPML1, is required for proteolytic degradation in autolysosomes. Autophagy 7, 1308–1315 (2011).
Acknowledgements
The authors are grateful to I. Hanson for editing work. This work was supported by the following grants to H.Z.: 92054301, 31630048 and 31790403 from the National Natural Science Foundation of China, 2017YFA0503401 from the Chinese Ministry of Science and Technology, Z181100001318003 from the Beijing Municipal Science and Technology Committee, XDB19000000 from the Strategic Priority Research Program of the Chinese Academy of Sciences, and QYZDY-SSW-SMC006 from the Key Research Program of Frontier Sciences, Chinese Academy of Sciences. P.C. is supported by grants from the French Agence Nationale pour la Recherche (R18004KK, R16167KK and R18158KK) and Fondation pour la Recherche Médicale.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Molecular Cell Biology thanks E. Eskelinen, N. Mizushima and H. Nakatogawa for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- V-ATPase
-
A proton pump, driven by ATP hydrolysis, which transports protons into the lumen of membrane compartments for their acidification. V-ATPases are multisubunit complexes, containing V1 and V0 sectors.
- SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complexes
-
Complexes consisting of the Qa, Qb, Qc and R SNARE proteins in opposing membranes. The SNARE motifs of these proteins are assembled into a hetero-oligomeric, four-helix bundle to drive membrane fusion.
- Tethers
-
Proteins or protein complexes that link the transported vesicles to target membranes and also promote the assembly of the trans-SNARE complex.
- Phosphoinositides
-
Phosphorylated derivatives of phosphatidylinositol that define membrane identity and participate in various signalling and membrane trafficking processes.
- RAB proteins
-
A subgroup of small GTPases that dynamically associate with membrane-bound compartments driven by GTP–GDP cycling to regulate multiple aspects of membrane dynamics, such as vesicle transport, fusion and positioning.
- Multivesicular bodies
-
Endocytic vesicles formed by invagination of the endosomal membrane to form membrane-bound intraluminal vesicles. They subsequently undergo lysosomal degradation or extracellular release.
- COPI vesicles
-
Coat protein I (COPI)-coated vesicles mediating the retrieval of proteins and lipids from the Golgi apparatus to the endoplasmic reticulum and also transport between Golgi apparatus cisternae.
- LC3 subfamily
-
A subfamily of Atg8 homologues containing LC3A, LC3B and LC3C.
- GABARAP subfamily
-
A subfamily of Atg8 homologues containing GABARAP, GABARAPL1 and GABARAPL2.
- Guanine nucleotide exchange factors
-
(GEFs). Factors that catalyse the conversion of the GDP-bound inactive form of small GTPase proteins to their GTP-bound active form.
- Recycling endosomes
-
Endosomal compartments for recycling materials internalized by endocytosis back to the cell surface to maintain the composition of the plasma membrane.
- HOPS (homotypic fusion and protein sorting) complex
-
A multisubunit seahorse-shaped complex consisting of VPS41, VPS39, VPS18, VPS33A, VPS11 and VPS16. It facilitates fusion events involving late endosomes and lysosomes.
- SNARE chaperone
-
A protein chaperone such as Sec1/Munc18 (SM) proteins that binds to individual SNAREs or assembly intermediates. It ensures the fast and accurate assembly of trans-SNARE complexes to drive efficient membrane fusion.
- β-Propeller proteins
-
A family of proteins with 4–12 repeats of a β-stranded blade that function as structural scaffolds for ligand binding, enzymatic activity and assembly of multiple protein complexes.
- Pleckstrin homology (PH) domain
-
A protein domain that binds phosphatidylinositol lipids (such as phosphatidylinositol 4,5-bisphosphate) and proteins with high affinity and specificity. It functions in vesicle trafficking, cellular signalling and cytoskeletal remodelling.
- Retrograde transport
-
Intracellular movement from the cell periphery towards the nucleus mediated by the dynein–dynactin complex. Also known as centripetal movement or minus-end transport.
- Anterograde transport
-
Intracellular movement from the nucleus towards the cell periphery mediated by kinesin motors. Also known as centrifugal movement or plus-end transport.
- BORC complex
-
A multisubunit complex that associates peripherally with the lysosomal membrane to regulate lysosomal positioning by recruiting ARL8. It comprises eight subunits: BLOS1, BLOS2, snapin, KXD1, myrlysin, lyspersin, diaskedin and MEF2BNB.
- ARL8
-
A small ADP-ribosylation factor-like RAS family GTPase that mediates kinesin-driven lysosome transport, and also regulates lysosome fusion by recruiting the HOPS complex.
- Lysosome storage diseases
-
(LSDs). A group of inherited metabolic disorders characterized by abnormal storage of toxic materials. They result from deficiencies of lysosomal enzymes or transporters.
- Atg1 complex
-
The Atg1 complex consists of the protein kinase Atg1 together with Atg13, Atg17, Atg31 and Atg29. Activation of this complex triggers the initiation of autophagy.
- Vps34 PtdIns3P kinase complex
-
A complex, containing the phosphatidylinositol 3-phosphate (PtdIns3P) kinase Vps34 together with Vps15, Atg6 and Atg14, that generates PtdIns3P at the autophagosome formation site to recruit downstream effectors for autophagosome formation.
- O-GlcNAcylation
-
A reversible post-translational modification in which a single O-linked N-acetylglucosamine (O-GlcNAc) moiety is linked to serine/threonine residues of proteins. It is catalysed by O-GlcNAc transferase (OGT) and reversed by O-GlcNAcase (OGA).
- COPII vesicles
-
Vesicles whose formation is driven by COPII coat proteins. COPII vesicles transport proteins from the endoplasmic reticulum to the Golgi apparatus.
- GTPase-activating protein
-
(GAP). A protein that facilitates the GTPase activity of small GTPases resulting in hydrolysis of GTP to GDP and phosphate.
- ER stress
-
A response elicited by accumulation of unfolded proteins in the endoplasmic reticulum (ER). It promotes several intracellular signal transduction pathways, collectively known as the unfolded protein response, to restore ER homeostasis.
- Liquid–liquid phase separation
-
(LLPS). A process triggered by multivalent interactions of constituents, resulting in their concentration in a confined liquid-like compartment that stably coexists with the surrounding liquid milieu.
- Mediator complex
-
A multiprotein complex that interacts with transcription factors and RNA polymerase II and functions as a transcriptional co-activator.
- α-Synuclein
-
An aggregation-prone protein that accumulates in Lewy bodies and plays a central role in the pathogenesis of Parkinson disease and other synucleinopathies.
- Polyglutamine expansion
-
An expansion of the CAG-trinucleotide repeat in disease-related genes resulting in polyglutamine expansion disorders such as Huntington disease and spinocerebellar ataxias.
- X-linked spinal and bulbar muscular atrophy
-
An X-linked, recessive lower motor neuron disease caused by a CAG polyglutamine repeat expansion within the first exon of the androgen receptor gene.
- Aβ
-
A mixture of small peptides (37–43 amino acids) derived from sequential secretase-mediated proteolytic processing of amyloid precursor protein (APP). Aβ aggregates into insoluble neurotoxic plaques in patients with Alzheimer disease.
- Tau
-
A major component of the neurofibrillary tangles found in a number of neurodegenerative diseases, including Alzheimer disease.
- Vici syndrome
-
A multisystem disorder (agenesis of the corpus callosum, progressive microcephaly, cataracts, hypopigmentation, combined immunodeficiency and recurrent infections, and cardiomyopathy) caused by mutations in the autophagy gene EPG5.
- β-Propeller protein-associated neurodegeneration
-
A disease caused by WDR45 mutations. Patients show static psychomotor retardation in early childhood, then develop sudden-onset progressive parkinsonism, dystonia and cognitive impairment in early adulthood.
- LAMP2
-
The receptor for chaperone-mediated autophagy. Mutations in the gene encoding LAMP2 are causatively linked with Danon disease.
- EDEMosomes
-
Endoplasmic reticulum-derived vesicles that tune endoplasmic reticulum-associated degradation activity by targeting regulators of endoplasmic reticulum-associated degradation (such as EDEM1) to lysosomes for degradation.
- ER-associated degradation
-
(ERAD). A process involving extraction of misfolded proteins from the endoplasmic reticulum (ER) for subsequent proteasomal degradation.
- Secretion systems
-
Apparatuses used by bacteria to secrete virulence factors from the bacterial cytosol into host cells or the host cell environment.
- Tat–beclin 1
-
A cell-penetrating peptide that induces autophagy. Eighteen amino acids from the evolutionarily conserved domain of beclin 1 are covalently coupled with the HIV-1 Tat protein transduction domain.
- Lysosomotropic agents
-
Substances that are taken up selectively into lysosomes irrespective of their chemical nature or uptake mechanism.
- Aptamers
-
Short, single-stranded DNA or RNA molecules or peptides that non-covalently bind to a target molecule with high affinity and specificity.
- Nanobodies
-
Antibodies consisting of a single monomeric heavy chain variable domain used in pharmaceutical applications such as diagnosis and therapy due to their small size, solubility and stability.
- Chloroquine
-
(CQ). An antimalarial drug also commonly used as a lysosomotropic agent that changes the lysosomal pH.
- Positron emission tomography
-
An imaging technique that visualizes metabolic changes by three-dimensional mapping of administered positron-emitting radiopharmaceuticals.
Rights and permissions
About this article

Cite this article
Zhao, Y.G., Codogno, P. & Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol 22, 733–750 (2021). https://doi.org/10.1038/s41580-021-00392-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41580-021-00392-4
This article is cited by
-
PLEKHM2 deficiency induces impaired mitochondrial clearance and elevated ROS levels in human iPSC-derived cardiomyocytes
Cell Death Discovery (2024)
-
The STX17-SNAP47-VAMP7/VAMP8 complex is the default SNARE complex mediating autophagosome–lysosome fusion
Cell Research (2024)
-
Viruses and autophagy: bend, but don’t break
Nature Reviews Microbiology (2024)
-
High-voltage pulsed radiofrequency improves ultrastructure of DRG and enhances spinal microglial autophagy to ameliorate neuropathic pain induced by SNI
Scientific Reports (2024)
-
Mechanism and role of mitophagy in the development of severe infection
Cell Death Discovery (2024)
