
Abstract
The ability of cells to deal with different types of stressful situations in a precise and coordinated manner is key for survival and involves various signalling networks. Over the past 25 years, p38 kinases — in particular, p38α — have been implicated in the cellular response to stress at many levels. These span from environmental and intracellular stresses, such as hyperosmolarity, oxidative stress or DNA damage, to physiological situations that involve important cellular changes such as differentiation. Given that p38α controls a plethora of functions, dysregulation of this pathway has been linked to diseases such as inflammation, immune disorders or cancer, suggesting the possibility that targeting p38α could be of therapeutic interest. In this Review, we discuss the organization of this signalling pathway focusing on the diversity of p38α substrates, their mechanisms and their links to particular cellular functions. We then address how the different cellular responses can be generated depending on the signal received and the cell type, and highlight the roles of this kinase in human physiology and in pathological contexts.
Similar content being viewed by others
Introduction
Most cells are subjected to different stresses during their lifetime, in both homeostatic and pathological conditions, and effective management of stress is essential for cell and organismal survival. In their response to stress (Supplementary Box 1), cells rely on several signalling networks, among which p38 kinases are of central importance.
p38 kinases are proline-directed serine/threonine kinases of the mitogen-activated protein kinase (MAPK) family, which are found in all eukaryotes and whose structural and regulatory characteristics are conserved from yeast to human. Unlike the prototypic MAPKs ERK1 and ERK2, p38 kinases do not typically respond to mitogens but are activated by environmental stresses and inflammatory signals. For this reason, they are referred to as stress-activated protein kinases.
As of today, thousands of reports have implicated p38 kinases in cellular responses to virtually all types of stresses, from environmental and intracellular insults to pathologies such as infection or tumorigenesis, and including processes such as cell differentiation that are not harmful but involve a certain stress1,2,3,4,5,6.
The general idea is that p38 kinases are highly versatile and can integrate many types of signals, contributing to various biological responses. Given all of the processes that p38 kinases can potentially control, dysregulation of this pathway has been linked to several diseases, suggesting that pharmacological targeting of p38 signalling could be therapeutically useful.
In this Review, we focus on p38α (also known as MAPK14 and SAPK2a), the best characterized member of the family. The regulation and functions of other members, p38γ and p38δ, have been recently reviewed7. We will discuss mechanisms that control the p38α pathway activity, and the biochemical and cellular processes involved in the particular cellular responses regulated by p38α activation. We also address how the pleiotropic character of this pathway can be reconciled with the specificity of its responses depending on the context. Finally, we describe recent findings using animal models that implicate p38α signalling in pathophysiological functions, and the prospects of using chemical inhibitors of this pathway in the clinic.
p38 kinase family members
The first mammalian p38 protein was independently reported in four studies: as a 38-kDa protein that was tyrosine phosphorylated following lipopolysaccharide (LPS) stimulation8; as RK, a protein kinase activated in response to arsenite, heat shock or osmotic stress9; as p40, activated in response to IL-1 (ref.10); and as CSBP2, the target of pyridinyl imidazole compounds such as SB203580 with anti-inflammatory properties11. p38, RK, p40 and CSBP2 all refer to the same protein, which showed high homology to Saccharomyces cerevisiae HOG1, a MAPK involved in protection from osmotic stress. This protein is now known as p38α, and proteins with high homology were subsequently identified and named p38β, p38γ and p38δ3,6 (Fig. 1a). Several spliced variants of p38α have been reported, including CSPB1 (ref.11), EXIP12 and MXI2 (ref.13), but their roles in and contribution to cell pathophysiology remain unclear.

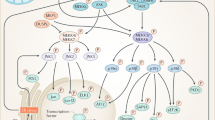
a | Schematic representation of the four human p38 kinases, indicating gene names (in parentheses), amino acid numbers and the different domains. The kinase domain is 90% identical in amino acid sequence among the four members. The CD domain is a negatively charged region involved in high-affinity docking interactions with substrates and regulators that contain positively charged docking (D) motifs. The ED domain contributes to substrate docking and specificity, being particularly important for interactions with mitogen-activated protein kinase (MAPK)-activated protein (MAPKAP) kinase 2 (MK2) and MK3. The ATP binding site and the phosphorylated Thr and Tyr residues of the activation loop are also indicated. p38γ has an additional carboxy-terminal region that binds to PDZ domain-containing proteins (serving as scaffolding proteins for various signalling pathways). p38 kinases are also referred to as stress-activated protein kinases: SAPK2a (p38α), SAPK2b (p38β), SAPK3 (p38γ) and SAPK4 (p38δ). b | Schematic representation of the three human MAP2Ks involved in p38 kinase activation, indicating gene names (in parentheses), amino acid numbers and highlighting the kinase domain, the ATP binding site, the D site involved in docking to p38 kinases, the DVD site that mediates interaction with MAP3Ks and the phosphorylated Ser and Thr residues of the activation loop. c | Canonical and non-canonical p38 kinase activation pathways. The colour of the phosphates (P) indicates the kinase responsible for the phosphorylation. In the canonical pathway, the first step is activation of MAP3Ks, which is triggered by various stimuli, encompassing cytokines acting via their receptors, ligands of G protein-coupled receptors (GPCRs; which include hormones, metabolites, cytokines and neurotransmitters) and stress signals. Mechanistically, MAP3Ks can be activated by multiple mechanisms, including binding to RHO, CDC45 and RAC small GTPases, phosphorylation by STE20 kinases and ubiquitylation by TRAF ubiquitin ligases, triggering phosphorylation of MAP2K, which in turn phosphorylate and activate p38 kinases. In the non-canonical pathways, activation is triggered by autophosphorylation of p38α either by binding to proteins such as transforming growth factor-β-activated protein 1 (TAB1) (observed in various cell types, but the signals responsible for activating this pathway are not well defined (question mark)) or by ZAP70 phosphorylation (specific to T cells) downstream of T cell receptor (TCR) activation. d | Scheme showing the main mechanisms leading to p38α signal termination, including phosphatases that target the activation loop phosphorylated residues, and p38α-triggered negative feedback loops (dotted lines). e | Scheme indicating human p38α protein sequence with post-translational modifications known to regulate the p38α activity. Interestingly, most modifications occur in amino acids involved in ATP binding and in the Thr-Gly-Tyr (TGY) sequence of the activation loop or near these regions. DUSP, dual-specificity phosphatase; MKP, MAPK phosphatase; PPase, protein phosphatase.
Mammalian p38 kinases share more than 60% amino acid sequence identity, with p38α being 75% identical to p38β and p38γ being 75% identical to p38δ. In spite of their structural similarities, p38 kinases differ with respect to their downstream targets and sensitivity to chemical inhibitors such as the widely used SB203580 (refs14,15). Moreover, p38α and p38β are ubiquitously expressed, p38α usually at higher levels than p38β except in some brain regions, whereas p38γ and p38δ expression tends to be more tissue-specific.
The p38 kinases serve a plethora of cellular functions, in both development and tissue homeostasis, but there are clear functional differences between the family members. Notably, p38α is the only p38 kinase that is essential for mouse embryo development owing to its key function in placental morphogenesis16,17,18, whereas p38β is mostly redundant in the presence of p38α19,20. This could be due to the higher p38α expression in most cell types, but might also reflect that p38α can perform particular functions, as suggested by the inability of p38β expressed under control of the p38α endogenous promoter to rescue p38α phenotypes in mice21. Nevertheless, experiments with cell cultures have identified some functions that can be mostly performed by p38β22. In addition, some cells may rely on p38β as a backup for p38α, as shown by the additional phenotypes observed in mice in which genes encoding both p38α and p38β were knocked out, compared with the single knockouts. Overall, p38α and p38β cooperate in heart development21, sex determination23, mitotic entry inhibition24 and regulatory T cell induction25. Along the same line, p38γ and p38δ can often perform overlapping functions, for example in tissue regeneration and immune responses26, but we are not aware of genetic evidence supporting that p38γ or p38δ can perform p38α functions. Interestingly, p38α downregulation sometimes leads to the enhanced activation of p38γ and/or p38δ27 (B.C and A.R.N., unpublished observations), suggesting that p38α might negatively regulate other p38 kinases28 or may be reflecting intrinsic differences in the affinity of p38α and other family members for upstream pathway regulators29. Therefore, a deeper understanding of both individual behaviour and functional interactions of the four p38 kinases is needed to fully understand the biological roles of this signalling pathway.
Signal transduction by p38 kinases
The activity of p38 kinases is tightly regulated, and involves activation by dedicated kinases that integrate multiple inputs, inactivation by several types of phosphatases, and the possibility of modulation by feedback loops and various post-translational modifications acting on different components of the pathway.
Activation mechanisms
p38 kinases are activated through dual phosphorylation by an MAP2K, which in turn is phosphorylated by an MAP3K (Fig. 1b,c). Up to ten MAP3Ks have been reported to contribute to the activation of p38 kinases, although some of them can also trigger activation of other MAPKs, mostly JNKs. Because different MAP3Ks are activated by different signals, this diversity in the upstream components of the p38 kinase cascade allows the pathway to integrate a wide range of stimuli, providing versatility to the response. Once activated, MAP3Ks phosphorylate the MAP2Ks MKK3 and MKK6, which share 80% amino acid sequence homology and are highly specific for p38 kinases, or MKK4 that normally activates JNKs but can also activate p38α18 (Fig. 1b). The contribution of each MAP2K to p38 kinase activation depends on the cell type and the stimulus3. MAP2K-catalysed phosphorylation of Thr and Tyr residues in the activation loop (Thr180 and Tyr182 in p38α) is important for full kinase activity (Box 1). This phosphorylation cascade is typical of most MAPKs, and is known as the canonical activation pathway (Fig. 1c).
Besides the MAP2K-based phosphorylation cascade, p38α can be activated by two non-canonical pathways (Fig. 1c). One involves binding to transforming growth factor-β-activated kinase 1-binding protein 1 (TAB1), which induces p38α autophosphorylation30. This mechanism has been intensively studied in cardiomyocytes under myocardial ischaemia31,32,33, and has also been implicated in T cell senescence34, skin inflammation35, triiodothyronine-mediated browning of white adipose tissue36 and endothelial inflammation triggered by G protein-coupled receptor (GPCR) agonists37. It should be noted that TAB1 can also induce p38α activation through the canonical pathway, by binding to the MAP3K TAK1. The other non-canonical mechanism of p38α activation seems to operate exclusively in T cells stimulated through the T cell receptor (TCR), and involves phosphorylation on Tyr323 by ZAP70, which leads to autophosphorylation of both p38α and p38β38. In contrast to the canonical pathway where p38α is dually phosphorylated by MAP2K, Tyr323-induced autophosphorylation of p38α occurs preferentially on Thr180, and this mono-phosphorylated p38α shows altered substrate specificity in vitro39.
The existence of different activation mechanisms may provide higher versatility to modulate the pathway activity and greater selectivity in defining relevant targets, helping to fine-tune the response in different cell types and contexts.

Signal termination
p38α hyperactivation is usually deleterious for the cell, and mechanisms that ensure signal termination are essential for homeostasis. Several phosphatases can inactivate p38α by targeting the activation loop phosphorylation including serine/threonine phosphatases, tyrosine phosphatases and dual-specificity phosphatases of the DUSP/MKP family (Fig. 1d). Interestingly, some phosphatases such as DUSP1 can be induced by p38α signalling, generating a negative feedback loop that may lead to asynchronous oscillations and cell-to-cell heterogeneity in p38 activity. This has been shown to be important for pro-inflammatory gene expression40 and stress-induced cell death41,42.
In addition, p38α can trigger other negative regulatory loops by limiting the expression of MKK6 (ref.43), by phosphorylating TAB1 (potentially affecting both the non-canonical activation and the TAK1-mediated canonical activation)44 or by phosphorylating ZAP70, which shortens the association of ZAP70 with TCR and decreases p38α activation in T cells45 (Fig. 1d).
Taken together, dephosphorylation of the p38α activation loop is key for pathway downregulation, but negative feedback loops can also shape the extent of p38α signalling, providing ample means for fine-tuning p38α activity in different contexts (Fig. 1d).
Regulatory mechanisms
The activity of p38α can also be fine-tuned by several mechanisms that are independent of the activation loop phosphorylation. These mechanisms include: Thr123 phosphorylation by GRK2, which impairs the binding of p38α to both MKK6 and substrates46; binding to the protein GADD45α that inhibits Tyr323 phosphorylation by ZAP70 (ref.47); and binding to DAPK1 that enhances p38α phosphorylation by MKK3 (ref.48). Post-translational modifications other than phosphorylation further modulate p38α activity, including acetylation of Lys53 that enhances activity by promoting ATP binding49, isomerization of Pro224 that facilitates MAP2K-mediated phosphorylation50 or arginine methylations that promote particular p38α functions51,52 (Fig. 1e). In addition, p38α signalling can be modulated by scaffold proteins, such as JIP4, OSM and DLGH1, which simultaneously interact with several pathway components, tethering them into complexes and helping to localize the complexes to a specific area of the cell for site-specific signalling53,54. p38α can also be regulated by importin-mediated nuclear translocation55 and by caspase-mediated protein degradation56.
Other components of the p38α pathway may also be affected by post-translational modifications resulting in either signal upregulation or downregulation. Thus, ubiquitylation can stabilize upstream activators such as TAK1 or MKK6 (ref.57), or induce the degradation of MKK6 (ref.58), the MAP3Ks DLK1 and MLK3 or the phosphatases PP2A, WIP1 and MKP1 (ref.59). Moreover, MKK6 can be inactivated by oxidation of specific cysteines60, and the MKK6 mRNA can be targeted by miR-625-3p, which in both cases downregulates p38 signalling61. Bacterial and protozoan proteins can also modulate p38α signalling, such as Bacillus anthracis lethal factor that cleaves and inactivates MKK3 and MKK6 (ref.62), Yersinia species YopP/YopJ that acetylates MKK6 and TAK1 in the activation loop residues blocking their activation63,64 or Toxoplasma gondii GRA24 that induces p38α autophosphorylation65.
Overall, the existence of multiple regulatory mechanisms combined with modulation of the enzymes responsible for particular post-translational modifications offer additional opportunities to adjust p38α signalling in specific contexts. As many of these mechanisms have been described in very specific contexts, it is likely that additional p38α-regulating modifications remain to be discovered.
Cues activating p38 kinases
p38 kinases are activated by essentially all environmental stresses, including oxidative stress and osmotic stress, ultraviolet radiation or gamma radiation, as well as by cytokines and inflammatory signals. Moreover, ligands that activate GPCRs, such as thrombin, glutamate or endothelin, can also activate p38α signalling to regulate various cellular responses. By contrast, mitogens — typically associated with the activation of ERK1/2 and other MAPK signalling pathways — are usually poor activators of p38 kinase signalling compared with stress and cytokines66,67. We discuss below examples of pathways involved in p38α activation by different types of signals.
Oxidative stress
The stress-induced activation of p38 kinases occurs in all eukaryotic cells, but little is known of how different stresses lead to activation of MAP3Ks. One of the best-known examples is the response to reactive oxygen species (ROS) mediated by the MAP3K ASK1, which normally binds to the inhibitory protein thioredoxin; upon thioredoxin oxidation, both proteins dissociate allowing ASK1 homo-oligomerization and activation68. Recently, the MAP3K MTK1 (also known as MEKK4) was also shown to function as a redox sensor, which is activated by coupled oxidation–reduction modifications of specific cysteine residues69. Thus, the coordinated activity of MTK1 and ASK1 likely mediates the ROS-induced activation of p38α signalling, recurrently observed in multiple contexts with largely different effects on cells69,70,71,72,73,74,75,76 (Fig. 2). Taken together, p38α activation by ROS seems to be of extraordinary importance for the regulation of cellular viability, but how ROS can lead to such different context-dependent responses is not yet clear.

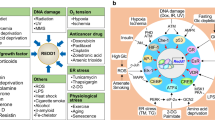
Reactive oxygen species (ROS) have been reported to activate p38α in various homeostatic and pathological contexts. Importantly, ROS play essential signalling roles and their levels are known to impact cell biology in various ways251. Despite the vast amount of literature linking ROS production with p38α activation, the actual levels of ROS are rarely experimentally determined. The signals reported to induce ROS and to activate p38α in different contexts are on the left, and the biological responses observed on the right. The signals and responses are organized according to the expected ROS levels in the cell, increasing from bottom to top. Lower ROS levels tend to be linked to physiological processes and homeostatic responses such as cell proliferation and differentiation or cytokine production, whereas higher ROS levels are usually generated in pathological contexts and in response to persistent stresses, eventually leading to severe cell dysfunction and death. However, how different signals trigger different ROS levels, and how diverse ROS amounts can differentially modulate p38α activation and particular biological responses remain to be fully understood.
Cytokines
Inflammatory cytokines such as TNF, IL-6 and IL-1β are prototypic activators of p38α. Cytokines can bind different types of surface receptors, which in turn determine the pathway leading to p38α phosphorylation. For example, signalling downstream of IL-1β and TNF usually engages TRAF ubiquitin ligases and TAK1, as well as other MAP3Ks77.
Cytokines that are not primarily related to inflammatory processes also activate p38α, many of which can modulate cell differentiation. For example, receptor activator of nuclear factor-κB ligand (RANKL) regulates several processes from immune responses to bone metabolism, and can trigger the differentiation of bone marrow cells into osteoclasts by activating p38α through the scaffold protein RACK1, TRAF6 and TAK1 (ref.78). The multifunctional cytokine TGFβ also induces p38α, which frequently involves TAK1 (ref.79), although other MAP3Ks have been implicated in some cell types80.
Infection
Bacterial and microbial infection triggers p38α activation usually through Toll-like receptors (TLRs), which through the adaptor MyD88 engage IRAK kinases, TRAFs and TAK1, similarly to IL-1β, but some signalling elements differentially contribute depending on the context4,81,82. For instance, upon persistent Salmonella typhimurium infection, TLR4 can mediate p38α activation through the adaptor TRIF and ROS, ignoring MyD88 (ref.83), whereas Mycobacterium tuberculosis infection induces p38α activation through the receptor MINCLE, probably involving the tyrosine protein kinase SYK84.
Activation of p38α has also been observed in response to viral infections and may regulate viral replication. This activation is usually mediated by the canonical pathway, although inactivation of phosphatases might contribute in some cases85. Recent work has shown that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection induces late p38α activation, suggesting its implication in advanced stages of viral infection. Enhanced MKK3/6 phosphorylation was detected in SARS-CoV-2-infected cells but upstream pathway components were not characterized86.
Mechanical forces
Stretching, compression and pressure overload can trigger p38α activation in different cell types. Interpreting these cues allow cells to recognize their position and shape, as well as environmental perturbations, which is essential in tissues such as muscles, and can drive diverse cellular responses from cell proliferation and differentiation to cell death. The cytoskeleton plays a pivotal role in cellular mechanosensing, which probably involves different elements to elaborate the response for each force. There is evidence that p38α activation occurs downstream of cytoskeleton-modulating kinases, such as ROCK87 and PKC88, or the small GTPase CDC42 (ref.89), depending on the mechanical stimuli, suggesting that the activation pathway may impact on the biological response.
Chemical drugs
Virtually every chemical compound that induces cellular stress will activate p38 kinases. Anisomycin, tunicamycin, hydroxyurea or cycloheximide are examples of compounds that affect different cellular processes and all induce p38α activation. It is important to highlight drugs used for chemotherapy treatments. The pathway involved usually depends on the mechanism of action of the drug, but in many cases implicates TAO family MAP3Ks that act downstream of DNA damage90. As drugs can induce DNA damage by different mechanisms, the BRAF–TAK1 pathway91 or ROS production92 has also been implicated, and it is possible that more than one MAP3K is involved in each case. Moreover, the microtubule poison paclitaxel can induce p38α activation through downregulation of the phosphatase DUSP16 (ref.93).
p38α substrates and functions
A multitude of proteins potentially phosphorylated by p38α have been identified using chemical inhibitors and genetic downregulation of pathway components (but the actual list of substrates requires validation involving in vitro phosphorylation assays). There is evidence that p38α directly phosphorylates more than 100 proteins6,94, which can be located throughout the cell and can regulate transcription and chromatin remodelling, mRNA stability and translation, protein degradation and localization, cell cycle, endocytosis, metabolism and cytoskeleton dynamics3,6. Some p38α substrates are protein kinases, which in turn phosphorylate additional proteins, expanding the versatility of the pathway to regulate diverse processes. Of special relevance are MAPK-activated protein (MAPKAP) kinase 2 (MK2) and MK3. Unphosphorylated p38α and MK2 interact, forming a complex, which coordinates the activation of both kinases to regulate particular cellular functions. MK2 plays an important role in post-transcriptional regulation of gene expression, by phosphorylating adenylate–uridylate-rich element (ARE)-binding proteins such as tristetraprolin (TTP) and HuR, and in actin filament remodelling through Hps27 phosphorylation95. Other kinases that can be regulated by p38α (activated also by ERK1/2 downstream of mitogens) are MSK1 and MSK2, which control gene expression by phosphorylating transcription factors or nucleosome components such as histone H3 (ref.96), and MNK1 and MNK2, which regulate protein synthesis through phosphorylation of the initiation factor eIF4E97. Below, we highlight some p38α substrates and key targets implicated in various cellular processes (Fig. 3).

p38α directly phosphorylates more than 100 proteins and can indirectly modulate a wider network of targets, explaining the versatility of this pathway. The top bar shows the relative distribution of p38α substrates according to their biological function. The panels illustrate key substrates and targets in three main p38α-regulated processes. In the stress response, p38α has been connected to many protein phosphorylation changes, which probably reflects the suitability of this mechanism for cellular adaptation by facilitating a rapid control of processes such as cell cycle progression, DNA damage repair or mRNA processing. In the immune response, p38α controls the phosphorylation of kinases, transcription factors and regulators of mRNA stability, which collectively regulate the expression of cytokines and other factors involved in inflammatory processes. In addition, the p38α pathway controls the phosphorylation of RIPK1 and the IFNα/β receptor IFNAR1, which are important in the response to pathogens and inflammation, as well as GSK3, which upon p38α phosphorylation regulates lymphocyte fitness and the adaptive immune response. In cell differentiation, and in agreement with the irreversible character of this process, p38α phosphorylates many transcription factors and chromatin modulators that will directly or indirectly control the gene expression programmes driving cell differentiation in different tissues. Dashed arrows represent indirect regulation by p38α. MK2, mitogen-activated protein kinase (MAPK)-activated protein (MAPKAP) kinase 2; RB, retinoblastoma protein; STAT, signal transducer and activator of transcription.
Stress response
The p38α pathway plays a key role in the regulation of cell survival in response to stress, which usually involves halting cell proliferation to allow for the repair of any stress-induced damage, thereby promoting cell survival. p38α induces cell cycle arrest through the upregulation of cyclin-dependent kinase (CDK) inhibitors, p53 or GADD45α, or the downregulation of cyclin D or CDC25 via several mechanisms98,99. Moreover, p38α can prevent cancer cell proliferation by phosphorylating the amino terminus of retinoblastoma protein (RB)100 or by inhibiting the transcription regulators CREB, YAP and MYC101. Alternatively, p38α can control the apoptotic machinery through regulation of BCL-2 family proteins102.
Other pro-survival mechanisms engaged by p38α involve the modulation of alternative splicing through MNK1/2-mediated phosphorylation of hnRNPA1 (ref.103) or the phosphorylation of SKIIP by p38α104. The p38α pathway can also facilitate the survival of stressed cells through the MK2-mediated phosphorylation of NELFE105 or RBM7 (ref.106), which enables a RNA polymerase II transcriptional response including genes required for telomere maintenance or DNA repair.
Autophagy is another process linked to cell survival, which can be regulated by p38α through the phosphorylation of lysosomal LAMP2A, a key activator of chaperone-mediated autophagy107. The macroautophagy regulators Beclin-1 and ULK1 can also be phosphorylated by MK2 and p38α, respectively108,109. In addition, p38α may facilitate cell viability in response to metabolic stresses by reducing mitochondrial oxidative phosphorylation through phosphorylation of the transcription co-adaptor KAP1 (also known as TRIM28)75, by promoting fatty acid β-oxidation through phosphorylation of PPARγ110 and by restricting endoplasmic reticulum stress111.
In contrast to the generally pro-survival functions, stress-induced p38α activation can sometimes induce cell death, which tends to correlate with higher/sustained levels of pathway activity. This may be mediated by the p38α phosphorylation and inhibition of Drosha, a key enzyme in miRNA biogenesis whose downregulation sensitizes cells to stress112, or by phosphorylation and cytoplasmic translocation of the transcription factor TEAD, which impairs YAP activity, potentially reducing the expression of anti-apoptotic genes113. Moreover, p38α activation can induce other stress-induced deleterious effects, although substrates are not characterized, including mitochondrial malfunction114, decreased proteasome activity115 and postmitotic apoptosis mediated by HIF1α inhibition and metabolic stress116.
In summary, p38α can regulate stress responses via different mechanisms but not all of them operate simultaneously. On the contrary, selected signalling branches are engaged depending on the context, causing different outcomes that usually support cell survival but sometimes drive cell death.
DNA repair
DNA damage can happen in both homeostatic and pathological situations, and p38α signalling is emerging as a particularly important regulator of cancer cell survival in this context. As an example, once activated by p38α, MK2 phosphorylates hnRNPA0, which controls the stability of CDKN1B and GADD45A mRNAs, regulating cell cycle progression and cell survival in response to DNA damage-inducing drugs such as cisplatin99. Moreover, p38α can directly phosphorylate DNA repair regulators such as CtIP, coordinating the DNA damage response and limiting replication stress and chromosome instability117. In addition, p38α facilitates activation of the DNA damage response kinase ATM, by controlling the phosphorylation of p45-IKKα, a nuclear form of the NF-κB regulatory kinase IKKα that shows distinct functions91. Likewise, p38α phosphorylation of the transcription factor BRN2 induces its association with DNA damage response proteins to promote error-prone DNA repair via non-homologous end-joining, and suppresses apoptosis-associated gene expression118. Collectively, these studies illustrate how inhibiting p38α signalling may be useful to curb DNA repair mechanisms, for example to enhance the cytotoxicity of chemotherapy drugs, which are key exogenous inducers of DNA damage.
Inflammation
p38α signalling regulates the production of inflammatory cytokines in different immune cell types, as well as in epithelial cells, fibroblasts and endothelial cells. Various inflammatory mediators can be regulated by p38α, which occurs through modulation of pro-inflammatory transcription factors, such as NF-κB, or by regulating the stability or the translation of the corresponding mRNAs, which often involves MK2 (refs1,2,95). Experiments using genetically modified mice support that p38α signalling in several cell types can promote inflammation in vivo119,120,121,122,123,124,125,126. However, p38α also has anti-inflammatory roles in innate immune cells, which are mediated by the kinases MSK1/2 and involve phosphorylation of the transcription factor CREB and histone H3, leading to the expression of anti-inflammatory genes such as IL10 (ref.96).
Besides regulating the production of inflammatory mediators, p38α can control the expression of cytokine receptors as well as modulate the receptor-initiated intracellular signals. For example, p38α can phosphorylate and induce the ubiquitin-dependent degradation of the IFNα/β receptor IFNAR1, independently of ligand binding127, which supports viral infections128 and has implications in diseases such as cancer129,130,131.
Recent findings indicate that p38α can also control the resolution of inflammation by impairing the engulfment of apoptotic bodies (efferocytosis) in macrophages. This is probably mediated by inhibition of the histone acetyltransferase p300 through p38α phosphorylation and subsequent reduced expression of the receptor TIM4, which recognizes phosphatidylserine on apoptotic cells132.
Moreover, p38α–MK2 signalling has emerged as a crucial regulator of the balance between cytokine production and cell death in response to inflammation and infection. Inflammatory signals, such as TNF or LPS, or Yersinia enterocolitica infection can induce cell death through the kinase RIPK1, which is repressed by MK2-mediated phosphorylation. Accordingly, MK2 inhibition boosts TNF-induced death in several cell types and sensitizes mice to the cytotoxic effects of TNF133,134,135.
Cell differentiation
In addition to acute or persistent stress, p38α regulates situations of mild stress such as cell differentiation, which frequently involves substantial morphological changes. Differentiation takes place during normal physiology, such as in embryo development or adult tissue cell turnover, as well as in response to certain tissue injuries. The process is often initiated by cytokines, which together with ROS are major cues activating p38α that, in turn, controls transcriptional programmes implicated in the differentiation of several cell types.
As one example, p38α can regulate muscle gene expression by phosphorylating myogenic transcription regulators such as MEF2 and E47, or by inducing chromatin remodelling through the phosphorylation of BAF60c and p18Hamlet (ref.136). In addition, myogenic differentiation requires degradation of EZH2, the catalytic subunit of the epigenetic repressor complex PRC2, which involves EZH2 phosphorylation by p38α followed by ubiquitylation by the E3 ubiquitin ligase Praja1 (ref.137). Similarly, osteoblast differentiation can be induced by p38α phosphorylation and activation of specific transcription factors such as Osterix, RUNX2 and DLX5 (ref.138).
In contrast to its well-established functions in muscle and bone, the role of p38α in adipocyte differentiation is much less precisely defined138,139. However, p38α has a well-defined role in the thermogenic programme of brown adipocytes and has been implicated in the browning process that transforms white adipocytes into brown-like adipocytes. Here, the PKA–ASK1–p38α kinase axis triggers, through several transcription factors, the expression of brown adipocyte genes including UCP1, which uncouples mitochondrial respiration from ATP synthesis to produce heat, as well as FGF21, a systemic regulator of energy homeostasis. Moreover, p38α upregulates the receptor GPR120, which feeds back on p38α to boost the transcription of FGF21 (refs140,141,142,143,144) (Fig. 4a).

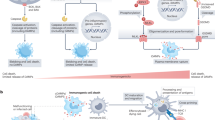
The multifactorial nature of p38α signalling is illustrated by showing the diversity of functions that can be regulated by p38α depending on the cell type and the signals received. In every panel, the cell type, the extracellular stimuli (top), the signalling elements involved (when known) and the biological outcome (bottom) are indicated. Green boxes indicate homeostatic responses and red boxes pathological or deleterious events. a | p38α has a well-established role in the activation of thermogenesis in brown adipocytes. The different signals and mediators leading to p38α activation, the direct substrates and the effector targets that drive the p38α-orchestrated thermogenic programme are indicated. p38α can also regulate adipocyte differentiation, involving C/EBP and PPARγ transcription regulators of adipogenesis. Of note, contrary effects of p38α on adipogenesis have been reported depending on the model used (indicated by the split, dashed arrow), which can be linked to high dependency of p38α-mediated responses on the context as highlighted in this Review. b | The functions of p38α signalling in haematopoietic stem cells (HSCs) can be classified according to the stimuli. Upon severe or persistent stress such as infection, radiation or ageing, p38α activation correlates with elevated reactive oxygen species (ROS) levels and usually leads to detrimental responses that impair HSC function. However, in response to mild stresses, such as acute inflammation, regenerative stress or differentiation, which often involve cytokine exposure, p38α coordinates a pro-survival programme aimed to recover homeostasis. c | p38α activation has been traditionally linked to neurodegenerative diseases, especially Alzheimer disease, due to its implication in β-amyloid (Aβ) plaque formation and cytotoxicity, at least in part via its contribution to Tau hyperphosphorylation. But recent work describes additional p38α functions in different cells of the central nervous system, both in homeostasis and pathological situations. Known substrates are indicated, but targets involved in p38α-regulated synaptic plasticity, myelination or neuroinflammation remain largely unknown. There is also evidence that the role of p38α in myelination may depend on both the cause of nerve injury and the cell type252. d | In hepatocytes, p38α can promote cell death or support cell viability depending on the strength of the stress: high levels of stress (such as combination of a high-fat diet (HFD) with infection/inflammation) results in cell death, whereas milder stress (such as HFD alone) generally promotes hepatocyte function in metabolizing fatty acids, by increasing their trafficking and β-oxidation, thereby reducing triglyceride (TG) storage and load in the liver. The hepatic function can be further modulated by p38α-regulated production of pro-inflammatory cytokines in macrophages that links to hepatocyte cell death.
However, p38α does not always function as a positive regulator of cell differentiation. For example, its role in adipogenesis seems to depend on the cellular system analysed, with reports of both stimulating and inhibitory effects27,138,139. p38α also restrains erythropoiesis through the repression of MEKK4 to keep JNK activity low, resulting in erythroblast apoptosis145. Likewise, the TGFβ-induced differentiation of mesenchymal stem cells into endothelial-like cells is negatively regulated by p38α via TAK1 and JNK inhibition146.
Stemness
p38α plays various roles in stem cell physiology, depending on the tissue and context147. For example, in hepatic stellate cells treated with TGFβ, p38α induces, probably through ATF2 transcription factor, the expression of the RNA binding protein CUGBP1, which reduces IFNγ production, a requisite for stellate cell fibrotic activation148. In keratinocytes, p38α phosphorylates and induces the degradation of the transcription factor p63, limiting the expression of stem cell-associated genes149. Also, the p38α-induced upregulation of p53 and p16 has been implicated in the decreased number and activity of intestinal stem cells observed in geriatric mice, which is driven by the increase in MKK6 protein synthesis downstream of nutrient signalling150.
In addition, p38α can regulate the recovery of haematopoietic stem cells (HSCs) from stresses such as bone marrow transplantation through the induction of inosine-5′-monophosphate dehydrogenase 2 (IMPDH2) — mediated by p38α activation of transcription factors CREB and MITF — to regulate the purine metabolism promoting HSC proliferation151. However, p38α activation limits the lifespan of HSCs in response to persistent infection, radiation or ageing, driving their ROS-induced exhaustion in vivo83,152. This dichotomy probably reflects the different ROS levels generated in ageing or infection versus bone marrow transplantation assays, which might translate to differential choice of substrates by p38α (see below) (Fig. 4b).
Other functions
p38α is an important inducer of the senescence-associated secretory phenotype (SASP). This function has been ascribed to p38α-mediated induction of NF-κB transcriptional activation153, and to stabilization of SASP factor transcripts mediated by the inhibition of RNA-binding proteins that destabilize mRNAs, such as AUF1 and ZFP36L1, either by p38α-induced displacement of AUF1 (ref.154) or by MK2 phosphorylation of ZFP36L1 (ref.155).
Some neuronal functions have been linked to specific p38α phosphorylation, including phosphorylation of the A-type K+ channel subunit Kv4.2, affecting neuronal excitability156, or the proteasome-binding protein PI31, which regulates axonal proteasome motility and synaptic proteostasis157.
In pathogenic contexts, p38α can phosphorylate the microtubule-associated protein Tau, promoting its aggregation to form neurofibrillary tangles — a hallmark of Alzheimer disease158 — and Parkinson disease-associated protein Parkin, resulting in defects in mitochondrial clearance via mitophagy and, in consequence, mitochondrial abnormalities and neuronal degeneration159 (Fig. 4c).
Context-dependence of p38α signalling
As outlined above, p38α controls many processes in a cell type and context-specific manner, but the molecular basis of this versatility remains elusive. Phosphorylation of many proteins can be regulated by p38α either directly or through downstream kinases and the crosstalk with other signalling pathways (Box 2). However, the p38α substrates whose phosphorylation leads to particular cellular responses are often unknown, and when specific phosphorylation events are linked to a phenotype, it is difficult to demonstrate that they are the exclusive mediators of p38α activity. We discuss below two main factors that may affect the response to p38α activation: signal and cell context.
Signal
The stimulus received by the cell is bound to play an important role in defining the response to p38α activation at several levels. First, different stimuli are likely to modulate the activity of various signalling pathways, which in turn may alter gene expression programmes affecting the availability of potential p38α substrates and regulators. In addition, every signal probably engages different combinations of p38α adaptors, MAP3Ks and MAP2Ks in the case of the canonical cascade, and may trigger non-canonical activation pathways depending on the cell type, which can affect the phosphorylation of specific substrates.
The activation of p38α through two mechanisms that cooperate to balance CD4+ T cell responses provides an example of how p38α in a particular cell type can perform different functions depending on the signal160,161. On the one hand, non-canonical activation of p38α upregulates the transcription factor NFATc1, which is required for T cell proliferation and cytokine production. On the other, stress-induced activation of p38α by the canonical pathway results in phosphorylation and cytoplasmic retention of NFATc1, antagonizing T cell function162. This may be explained by the ability of MAP2K-activated p38α to phosphorylate NFATc1, which is a poor substrate for ZAP70-phoshorylated p38α, perhaps due to the latter being mono-phosphorylated in the activation loop. It is tempting to postulate that other post-translational modifications of p38α, such as methylation or acetylation, can be differentially modulated by the type of signal and structurally impact the selection of substrates.
In addition, different signals may induce different levels of p38α activation, which is likely to affect the substrates that are phosphorylated. In this regard, it is important to distinguish between homeostatic functions and stress responses, as the latter are usually associated with higher levels of p38α activity67,163. The molecular basis for the differential phosphorylation of particular substrates depending on p38α activity levels may rely on both the substrate expression levels and the existence of particular sequences, such as so-called docking sites, which facilitate the interaction between the substrate and p38α (ref.164). MK2 and MEF2a are prototypic docking site-containing substrates, which show robust interaction with p38α, but this is not observed in many p38α substrates (our unpublished data). The exact number of substrates that contain docking sites is unknown as the motifs are rather degenerated and the docking ability should be determined empirically using protein–protein interaction assays165. Thus, a protein that is highly expressed or with a docking site may have a higher chance of being phosphorylated by a small number of active p38α molecules. On the contrary, substrates that are actively targeted by phosphatases may require a higher number of active p38α molecules to achieve a substantial fraction of substrate phosphorylation.
An example of how p38α activity levels may affect different functions can be found in HSCs, where p38α supports viability upon differentiation cues or bone marrow transplantation, but drives apoptosis and other deleterious responses following persistent infection or irradiation (Fig. 4b). This seems to correlate with different ROS levels produced in each context83,151,152,166. Similarly, hepatic p38α protects from high-fat diet-induced liver steatosis and liver injury by limiting the accumulation of fatty acids, but the combination of a high-fat diet with inflammatory signals boosts the levels of hepatic p38α activation leading to hepatocyte death (Fig. 4d). Although the p38α substrates responsible for the different hepatocyte responses were not elucidated, ROS were speculated to be involved110.
The ability of p38α to regulate a particular process at different levels and with opposite effects also offers the possibility for the integration of different inputs to modulate the signalling output balancing the response in a way most appropriate for the context. For example, p38α induces the degradation of the IFNα/β receptor IFNAR1, but positively modulates the transcription of interferon-stimulated genes. This allows cells to fine-tune the extent and duration of IFNα/β signalling and antiviral defences128,167. Likewise, the p38α–MK2 pathway modulates TNF expression and mediates TNF-induced production of pro-inflammatory cytokines, while restricting TNF-induced cell death. This provides a way to link the regulation of cell death and inflammation so that cells can support the inflammatory response and control TNF-induced cytotoxicity133,134,135.
Finally, although most p38α substrates appear to be cytoplasmic, about 30% can be detected in the nucleus168, and some in other locations such as lysosomes or the cytoskeleton94. It is therefore conceivable that p38α pools might exist in different subcellular locations, and that activation of particular pools would impact on the specificity of p38α-regulated processes. Supporting this idea, p38α phosphorylates GSK3β in the nucleus, where it associates with the double-strand break marker γH2AX in response to DNA damage169. Thus, substrate specificity could be linked to particular subcellular locations.
Cell context
Besides the signal received, different cell types express different sets of proteins, including the substrates that can be potentially phosphorylated by p38α. The ability of p38α to regulate specific functions depending on the cell type was nicely illustrated by a report showing a sex reversal phenotype — where an XY genotype has female gonads — in mice with p38α and p38β deletion23. This phenotype was mediated by p38α and p38β controlling expression of the Sry gene, a master regulator of sex determination. Interestingly, p38 signalling is activated in both male and female gonads, but as the Sry gene is located on the Y chromosome, it is only expressed in males. This example shows how availability of the target rather than activation of the pathway per se determines a specific response. Of course, target availability is not the only cell-specific factor that determines p38α function. For instance, p38α induces apoptosis when aneuploidy is induced in near-diploid HCT116 cancer cells that have robust mechanisms of genomic stability control116, whereas aneuploid cancer cells rely on p38α to engage DNA repair and facilitate survival117. It is therefore tempting to speculate that mechanisms linking aneuploidy with p38α-induced apoptosis might be non-operative in some cancer cells, and that the final fate of aneuploid cells may depend on the status of other DNA damage response proteins.
Post-translational modifications that target p38α or its regulators may also affect the cell type-specific responses orchestrated by the pathway, as some of the enzymes responsible for the modifications are expressed in particular cell types. For example, the arginine methylase PRMT7 is upregulated during myoblast differentiation and through p38α methylation facilitates muscle gene expression51. How methylation facilitates p38α activation is not yet clear.
The outcome of p38α signalling can be also regulated by the status of other signalling pathways due to cell type intrinsic characteristics or mutations in particular genes, which may change the balance among different players in the signalling network, potentially making p38α-regulated functions more obvious. The pro-survival roles of p38α in cancer cells with mutated p53 (refs99,102,170) or that express low levels of the phosphatase PP2AC171 illustrate this point (Box 2).
In summary, the diversity of functions performed by p38α depends on several factors that are engaged mostly by the nature of the signal and the cell context (Fig. 5a).

a | p38α is known to modulate many cellular processes, but not all of these functions are performed simultaneously. The gears illustrate the required coordination among several key factors that can influence the diversity of p38α-driven cellular responses. b | Genetic and pharmacological targeting of p38α in mouse models has revealed the implication of this signalling pathway in several physiological functions, and its dysregulation has been linked to a plethora of diseases. The homeostatic functions (left) and the diseases (right) in which the p38α pathway has been reported to play a role are indicated. c | Results from animal and cell-based preclinical models have supported the interest of p38α as a potential target in some of these pathologies, and several clinical trials have been developed using pharmacological p38α inhibitors, alone or in combination with other drugs. The boxes show the proportion of different pathologies targeted in clinical trials (ClinicalTrials.gov database) with p38α inhibitors (upper) and their evolution over the past 20 years (bottom). The disappointing results obtained in most clinical trials performed so far have led to a decline in the number of studies testing p38α inhibitors in patients in recent years, probably reflecting the decision of pharmaceutical companies to pursue novel targets. However, encouraging preclinical results have stimulated ongoing phase II clinical trials that either use mitogen-activated protein kinase (MAPK)-activated protein (MAPKAP) kinase 2 (MK2) inhibitors or target new diseases, as shown in Table 1. COPD, chronic obstructive pulmonary disease.
Physiopathological functions of p38α
Signalling by p38α can regulate many biological responses, and has been linked to several human pathologies (Fig. 5b). However, it is not clear that diseases are always caused by increased p38α signalling, as enhanced p38α phosphorylation (as a surrogate for activity) is not consistently detected in pathologies and, when detected, p38α activation might be a consequence rather than a driver of the pathogenesis. Alternatively, during disease development p38α might acquire new functions that favour pathogenesis without concomitant upregulation of the pathway activity. Nevertheless, the mechanisms of pathological p38α activation are not well understood in most cases. Below, we address various functions of p38α in physiological and pathological situations.
Roles in immune cell function, inflammatory responses and inflammatory diseases
The p38α pathway plays an important and evolutionary conserved role in innate immune responses and the defence against bacterial and viral pathogens82,172. Accordingly, many bacteria have evolved mechanisms to increase virulence by inhibiting p38α signalling in the cellular host. However, p38α can also facilitate the replication of some viruses, indicating that this pathway can regulate viral pathogenesis at different levels.
There is ample evidence linking p38α to pro-inflammatory functions, both by mediating signalling in response to cytokine exposure and by controlling the production of inflammatory modulators. In agreement, mouse experiments support that p38α signalling contributes to the pathogenesis of several inflammatory diseases including rheumatoid arthritis and chronic obstructive pulmonary disease173. However, p38α can also have cell type-specific anti-inflammatory functions that are thought to be important for turning off the inflammatory responses to avoid tissue damage82.
Beyond controlling innate immunity and cytokine production, p38α can regulate the function of several cell types implicated in adaptive immunity82,160,174. Thus, p38α inhibition improves T cell expansion and expression of stemness markers, and promotes redox balance and genomic stability in T cells, enhancing the antitumour efficacy of T cell-based immunotherapy175, but seems to be counterproductive during V(D)J recombination or class switch recombination, impairing B cell survival169. In addition, p38α inhibition has been reported to boost HSC renewal and repopulation activity in response to persistent stress, suggesting a strategy to tackle infection83 and ageing166.
Inflammation has been linked to diseases such as neurodegenerative conditions (see next subsection) and cancer, suggesting potential therapeutic uses for p38α inhibitors. In the context of cancer, p38α inhibition in macrophages or dendritic cells reduces colon inflammation and the associated tumorigenesis in mice120,121. Moreover, inhibition of p38α stabilizes IFNAR1, which improves the viability of cytotoxic T lymphocytes potentiating antitumour immune responses in colorectal tumours129, and induces chemokine expression that enables neutrophil infiltration into lungs130,131, in both cases suppressing tumour growth.
Overall, p38α is a crucial coordinator of immune cell function and inflammatory responses in various cell types, which in turn impinge on diverse pathologies such as infection, neurodegenerative diseases and cancer. However, the ability of p38α to control both pro-inflammatory and anti-inflammatory functions complicates the clinical use of pharmacological inhibitors of this pathway, which would benefit from a greater understanding of the mechanisms that underlie contextual p38α signalling.
Neuronal regulation and roles in neurodegenerative diseases
p38α has been implicated in several neuronal or glial-specific functions that are relevant for brain physiology, such as neuronal excitability156, synaptic plasticity157,176,177,178 or myelination179,180,181 (Fig. 4c). However, p38α has also been associated with neuronal pathologies. For example, neuropathic pain has been ascribed to p38α activation in microglia and the production of pro-inflammatory cytokines, which in turn induce neuron hyperactivity and pain hypersensitivity. The therapeutic potential of inhibiting p38α in pain transduction has been investigated in clinical trials, but no drug is approved yet and the interest seems to have faded182.
By contrast, the potential clinical use of p38α inhibitors to treat neurodegenerative diseases is on the rise183. p38α phosphorylation is detected in the early stages of Alzheimer disease184,185,186. Consistently, the phosphatase DUSP1 is downregulated in brains of individuals diagnosed with Alzheimer disease, and its upregulation ameliorates cognitive impairment in mouse models187. Alzheimer disease is a multifactorial disease characterized by the accumulation of hyperphosphorylated Tau proteins and β-amyloid plaques, as well as increased neuroinflammation, and p38α has been involved in all of these processes188. In fact, inhibition of p38α attenuates neuroinflammation, which correlates with improved spatial memory in mouse models of Alzheimer disease189,190. Altogether, preclinical studies in various animal models have increased expectations of p38α as a potential therapeutic target for Alzheimer disease189.
Similarly, downregulation of p38α in a mouse model of Parkinson disease that expresses mutated α-synuclein alleviates the synaptic loss in dopaminergic neurons in vitro159. Moreover, MK2-deficient mice treated with neurotoxin showed decreased loss of dopaminergic neurons and lower neuroinflammation, supporting the potential benefits of inhibiting p38α signalling in Parkinson disease191.
In contrast to Alzheimer disease and Parkinson disease, amyotrophic lateral sclerosis (ALS) has an earlier onset, which is caused by the degeneration of motor neurons ultimately leading to cell death. p38α activation has been detected both in motor neurons and microglia of the ALS mouse model and in patients with ALS192,193, and p38α inhibition improves ALS-associated defects such as axonal retrograde transport in mice194 or loss of survival in human motor neurons cultured in vitro195.
Moreover, p38α inhibition normalizes both physiological and behavioural perturbations in a mouse model of autism spectrum disorder, suggesting p38α inhibition as a potential therapy for this disease196. However, as in Parkinson disease or ALS, further work is needed to validate p38α as a therapeutic target.
Regulation of cardiomyocytes and roles in cardiovascular diseases
p38α can modulate several functions in cardiomyocytes, including hypertrophy, contractibility, fibrosis and apoptosis, which may impact on heart failure197. The activation of this pathway often correlates with cardiac pathologies such as atherosclerosis or myocardial ischaemia198, supporting the efforts to use p38α inhibitors in the clinic. Although p38α inhibitors are well tolerated and attenuate some inflammatory components199, a phase III clinical trial showed no effect of these inhibitors on lowering the risk of major ischaemic cardiovascular events200. However, given the consistent preclinical data supporting the benefits of inhibiting p38α signalling, alternative strategies have been proposed, such as inhibition of MK2 (ref.201) or targeting the TAB1-induced activation of p38α, which has been implicated in cardiomyocyte death upon ischaemia–reperfusion32,33,202.
Roles in metabolism and metabolic diseases
p38α has been implicated in the regulation of cellular bioenergetics at different levels, including the ability to phosphorylate proteins such as PGC1α, CREB or C/EBPβ that regulate glucose or lipid metabolism203. Recently, a brain cell type named tanycyte has been shown to produce in a p38α-dependent manner the hormone FGF21, which regulates body lipid homeostasis, suggesting that p38α can regulate energy expenditure at a systemic level too76.
One of the best characterized roles of p38α in metabolism is the activation of the thermogenic programme in adipocytes, a calorie-burning process that avoids body weight accumulation (Fig. 4a). There is evidence that p38α is required for both brown adipocyte thermogenesis and the browning process. Unexpectedly, mice lacking p38α in adipose tissue were recently shown to display resistance to diet-induced obesity and increased energy expenditure, in contrast to the pro-thermogenic role of p38α. This may be explained by regulatory differences among various adipose depots — uniformly affected by the knock out — and/or the impact of other p38 family members that becomes visible upon loss of p38α27,204. Moreover, p38α inhibition can revert the decline in browning capacity observed with age, by avoiding entry into senescence of the aged adipocyte progenitors205. These results indicate that p38α plays different roles depending on the adipocyte type and status, providing a more complex picture of how this pathway can regulate adipose tissue functions.
There is also evidence that p38α can play a dual role in non-alcoholic fatty liver disease by performing different functions in hepatocytes exposed to a high-fat diet depending on the disease stage, which might be linked to different p38α activation levels. Thus, at early stages, p38α is weakly activated and protects from steatosis, but as the disease progresses and becomes associated with inflammation, p38α activation is stronger and exacerbates steatohepatitis110 (Fig. 4d). Moreover, p38α in macrophages can further impact on liver diseases by controlling pro-inflammatory cytokine production, which boosts steatohepatitis206.
Although not strictly a metabolic disease, cachexia is a muscle-wasting syndrome, often associated with chronic diseases such as cancer, characterized by an enhanced metabolic rate that is not compensated by increased caloric or protein intake, leading to skeletal muscle loss. p38α was proposed to boost this process by promoting increased mitochondrial respiration in the muscle207, which is linked to muscle catabolism and, subsequently, muscle mass loss. In addition, cachectic cells secrete inflammatory factors that induce fatty acid oxidation and enhance oxidative stress, leading to p38α activation in neighbouring cells fuelling muscle atrophy in this feedforward loop. Accordingly, inhibition of p38α has emerged as a potential strategy to slow down cachexia74,208.
Roles in cancer
p38α was initially described as a tumour suppressor in normal epithelial cells, based on its ability to inhibit oncogene-induced malignant cell transformation in cell cultures, which can be mediated by inhibiting cell proliferation, triggering cell death or promoting cell differentiation2,209,210. Further experiments confirmed that genetic downregulation of p38α enhances tumour growth in mouse models of liver, lung, colon and skin cancer2,149,211,212,213. These data collectively indicate that p38α can suppress tumour initiation both in vitro and in vivo. However, there is also evidence from diverse experimental systems showing that this pathway is often harnessed by malignant cells to support tumour progression214. Thus, studies in mouse models of colon, breast and lung cancer indicate that p38α can engage different mechanisms in cancer cells to support primary tumour growth in vivo, including the modulation of intracellular signalling pathways that control cell survival and proliferation, the regulation of DNA repair or the production of extracellular factors that support cancer cell proliferation117,213,215,216. Moreover, p38α may promote metastasis of breast cancer, ovarian cancer and melanoma cells by targeting various proteins involved in the regulation of epithelial–mesenchymal transition, cell migration and extravasation217,218,219,220,221. However, similar to other roles of p38α, its effects on cancer cell spreading are context-dependent, as p38α was reported to prevent early dissemination of HER2 (also known as ERBB2)-positive mammary cancer cells222 and the ability of colon cancer cells to colonize the lung from liver metastasis223. In addition, the p38α–MSK1 axis controls dormancy of disseminated ER+ breast cancer cells224. The different environments to which cancer cells are exposed during metastasis and in the primary tumour probably affect the functions regulated by p38α.
Recent work has also highlighted the importance of p38α in the crosstalk between cancer cells and non-malignant cells of the tumour stroma. In fibroblasts, p38α signalling can support tumour growth in different ways that include triggering the production of pro-tumorigenic SASP factors154, remodelling the extracellular matrix through hyaluronan synthesis to prepare the tumour niche225, fuelling cancer cell metabolism by inducing cytokines that mobilize glycogen in cancer cells to release glucose226 or inducing the expression of chemokines that enable infiltration by neutrophils into the lung to facilitate lung metastasis131. Pro-tumorigenic roles of p38α signalling have been also reported to be conferred by immune cells, such as macrophages and dendritic cells, where the p38α pathway facilitates inflammation, which was associated with colon tumorigenesis in mouse models120,121,227. Furthermore, non-canonical p38α activation in T cells promotes an inflammatory state that facilitates pancreatic ductal carcinoma development228. In addition, cancer cells also rely on p38α to produce cytokines and chemokines that recruit pro-tumorigenic myeloid cells to the tumour niche229. Similarly, the p38α–MK2 axis was implicated in the upregulation of T cell inhibitory protein PDL1 in cancer cells favouring immune suppression signalling71.
Besides its role in tumour initiation and progression, p38α activity has been linked to chemotherapy response, by either promoting or antagonizing cytotoxicity depending on the chemotherapeutic drug and the tumour model. Targeting p38α generally impairs cell death induced by oxaliplatin61,230, or the nucleoside analogues gemcitabine and cytarabine, but has a less clear effect on chemotherapeutic drugs such as cisplatin or 5-fluoracil214,231. Given the variability in p38α function observed in established cancer cell lines232, and considering both the role of p38α in the tumour stroma and the contribution of stromal cells to the chemotherapy response, it is particularly important to use in vivo models to better predict the response of patients to p38α-inhibiting drugs. In this regard, a few studies that combine p38α inhibitors and chemotherapy treatments in vivo have reported promising results. Thus, pharmacological inhibition of p38α synergizes with cisplatin in a breast cancer model233, decreases resistance to the multikinase inhibitor sorafenib in a hepatocarcinoma model234 and boosts the cytotoxic effect of taxanes in a breast cancer mouse model and human-derived xenografts117. Moreover, p38α and MK2 inhibitors potentiate the effect of targeted drugs such as checkpoint kinase 1 (CHK1) inhibitors in KRAS or BRAF mutant tumours235 or Smac mimetics in leukaemia236, supporting the interest of combining p38α inhibitors with clinically used anticancer agents. Of note, inhibition of the p38α–MK2 pathway can also prevent chemotherapy-induced bone loss in mice237.
In summary, there is overwhelming support for the idea that p38α functions as a non-oncogene addiction factor in malignant cells, which enables the survival and proliferation of many cancer cell types through various mechanisms, perhaps explaining why p38α is not commonly mutated in tumours, despite displaying tumour suppressor functions in normal epithelial cells. However, the role of p38α is not restricted to malignant cells and it may also function in tumour stromal cells to promote tumour growth and dissemination (in part via pro-inflammatory signals). Overall, inhibition of p38α signalling may produce antitumoural effects by targeting this pathway in different cell types of the tumour and its niche, by enhancing the efficacy of immunotherapies and by potentiating chemotherapy treatments. However, therapeutic interference with p38α signalling may also result in unexpected adverse effects, so p38α inhibitors should be used with care.
p38α as a therapeutic target
The discovery of p38α as an inflammation and immune response modulator supported the efforts to develop chemical inhibitors for diseases such as rheumatoid arthritis, chronic obstructive pulmonary disease or asthma. The initial impetus to target inflammatory diseases led to the generation of a series of potent p38α inhibitors with good pharmacokinetic properties that are being repurposed for other pathologies. These include neflamapimod (VX-745) for patients with early Alzheimer disease, Lewy body dementia and early Huntington disease, talmapimod (SCIO-469) for multiple myeloma, ARRY-371797 for cardiomyopathy, and losmapimod (GW856553) for muscular dystrophy and, more recently, the treatment of patients affected by COVID-19 (information from ClinicalTrial.gov database). These efforts are further supported by the encouraging results observed in preclinical studies that pharmacologically target the p38α pathway in various disease models (Table 1).
However, application of p38α inhibitors in the clinical setting has proven difficult so far. For example, a phase II clinical trial with a small-molecule inhibitor of p38α, PH-797804, showed improved lung function parameters in patients with chronic obstructive pulmonary disease238, but further studies generated disappointing results, as did many other clinical trials performed over the past 20 years6 (Fig. 5c). The reasons for the lack of success in applying p38α inhibition in the clinic may range from the widespread use in initial studies of SB203580, a potent inhibitor of the kinase but with clear non-specific effects239, to the use of models that did not faithfully recapitulate the actual disease environment, thereby generating preclinical results that did not align with the drug application in the patient setting240. In addition, the diversity of p38α-regulated functions, sometimes resulting in opposite responses, could contribute to the failure of clinical trials using p38α inhibitors. In this regard, inhibition of the downstream target MK2 has been proposed as an alternative to decrease undesired effects. Accordingly, the MK2 inhibitor ATI-450 (also known as CDD-450) was shown to inhibit pro-inflammatory cytokine production with no serious adverse effects in a phase I trial, supporting its further clinical development241. Overall, it is likely that more tailored approaches focusing on a specific cell type or p38α-directed signalling branch could be more successful in correcting this pathway in disease as compared with generalized inhibition.
Given that p38α signalling can potentially regulate many functions, the long-term treatments required for chronic pathologies such as autoimmune or neurodegenerative diseases are more likely to result in undesirable effects, as well as in adaptation to the inhibition and lack of efficacy. Thus, pathologies such as cancer where p38α inhibitors can be used acutely and in combination with other drugs might provide better responses242,243. Novel approaches to target p38α signalling are now emerging. Besides the inhibition of particular p38α downstream branches, it is appealing to consider compounds that target p38α for degradation244, or the possibility of tissue-specific targeting of p38α to improve efficacy and reduce the undesired effects of systemic administration245. Moreover, it has been reported that inhibiting the nuclear translocation of p38α, which may interfere only with p38α functions in the nucleus, reduces inflammation in mouse models246. It would be also interesting to explore the possibility of generating drugs that mimic the effect of p38α phosphorylation on substrates that function as tumour suppressors247.
In summary, a better understanding of the p38α contribution to diseases at the cellular and organism levels should help develop new compounds to target specific p38α activation mechanisms or particular effectors, which could increase selectivity and reduce clinical drawbacks of the current inhibitors.
Conclusions and perspectives
Over the past 25 years, p38 kinases — in particular, p38α — have gone from being known as regulators of environmental stress and inflammation to being recognized as key players in homeostasis maintenance at the cellular, tissue and organism levels. Recent work using genetically modified mice and improved disease models supports the implication of p38α in the development of several pathologies, including cancer, autoimmune disorders, neurodegenerative diseases and malfunction of the cardiovascular system (Fig. 5b). However, attempts to translate these findings into therapies have not yet been successful.
The versatility of the p38α pathway allows it to regulate a wide range of biological processes, raising the question of how specific responses are orchestrated depending on the cell context. Detailed accounts of the underlying mechanisms for context-specific signalling are largely lacking, but some common themes are starting to emerge. Thus, high levels of p38α activity are often linked to deleterious effects and cell death, whereas cell survival and homeostatic functions tend to rely on milder or transient p38α activation levels. In fact, cell survival regulation in response to stress is a key function of p38α, which has been hijacked by cancer cells to thrive under conditions, such as high DNA damage, that would be detrimental for normal cells. It is important to keep in mind that cell type or disease-specific genetic alterations may affect the wiring of signalling networks that operate during homeostasis, which combined with p38α signalling versatility may result in the acquisition of new p38α functions, explaining the difficulty in foreseeing the effects of targeting this pathway in different scenarios.
Although thousands of papers have been published on p38α, we still know very little on the collection of p38α phosphorylated proteins induced by each signal, the regulation and function of different p38α pools inside the cell and how p38α signals are integrated with other pathways to regulate particular processes. This information is critical to understand how p38α activation may result in different, sometimes even opposite, outcomes depending on the context.
Future work should build on techniques to study kinase activity and protein modifications at the single-cell level, and the use of affordable and reliable omics technologies to identify substrates and protein interactors as well as gene expression changes associated with p38α activation in different cell types and upon exposure to different inputs. This should inform on where, when and how p38α is activated, and which targets are engaged depending on the context. In addition, genome-wide screening analysis should provide valuable information on pathways that collaborate with or antagonize p38α signalling.
Altogether, further characterization of the molecular basis of p38α regulation and function in different contexts is expected to provide important insights into the biology of human health and disease. Further understanding of the diversity and context-specificity of p38α signalling will also open up a path towards the design of improved drugs and therapeutic strategies to target this pathway more effectively.
References
Cuenda, A. & Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 1773, 1358–1375 (2007).
Wagner, E. F. & Nebreda, A. R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9, 537–549 (2009).
Cuadrado, A. & Nebreda, A. R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417 (2010).
Kyriakis, J. M. & Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 92, 689–737 (2012).
Hotamisligil, G. S. & Davis, R. J. Cell signaling and stress responses. Cold Spring Harb. Perspect. Biol. 8, a006072 (2016).
Han, J., Wu, J. & Silke, J. An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling. F1000Res. 9, 653 (2020).
Cuenda, A. & Sanz-Ezquerro, J. J. p38γ and p38δ: from spectators to key physiological players. Trends Biochem. Sci. 42, 431–442 (2017). This extensive review addresses the functions of p38γ and p38δ.
Han, J., Lee, J. D., Bibbs, L. & Ulevitch, R. J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265, 808–811 (1994).
Rouse, J. et al. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 78, 1027–1037 (1994).
Freshney, N. W. et al. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell 78, 1039–1049 (1994).
Lee, J. C. et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746 (1994). Together with Han et al. (1994), Rouse et al. (1994) and Freshney et al. (1994), this paper reports the identification of the first mammalian p38 kinase using different experimental systems and approaches.
Sudo, T., Yagasaki, Y., Hama, H., Watanabe, N. & Osada, H. Exip, a new alternative splicing variant of p38α, can induce an earlier onset of apoptosis in HeLa cells. Biochem. Biophys. Res. Commun. 291, 838–843 (2002).
Sanz, V., Arozarena, I. & Crespo, P. Distinct carboxy-termini confer divergent characteristics to the mitogen-activated protein kinase p38α and its splice isoform Mxi2. FEBS Lett. 474, 169–174 (2000).
Eyers, P. A., Craxton, M., Morrice, N., Cohen, P. & Goedert, M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem. Biol. 5, 321–328 (1998).
Gum, R. J. et al. Acquisition of sensitivity of stress-activated protein kinases to the p38 inhibitor, SB 203580, by alteration of one or more amino acids within the ATP binding pocket. J. Biol. Chem. 273, 15605–15610 (1998).
Adams, R. H. et al. Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell 6, 109–116 (2000).
Mudgett, J. S. et al. Essential role for p38α mitogen-activated protein kinase in placental angiogenesis. Proc. Natl Acad. Sci. USA 97, 10454–10459 (2000). Together with Adams et al. (2000), this paper provides in vivo evidence for a critical role of p38α in the regulation of a process unrelated to the acute stress response.
Brancho, D. et al. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 17, 1969–1978 (2003).
Beardmore, V. A. et al. Generation and characterization of p38β (MAPK11) gene-targeted mice. Mol. Cell Biol. 25, 10454–104645 (2005).
Greenblatt, M. B. et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Invest. 120, 2457–2473 (2010).
del Barco Barrantes, I., Coya, J. M., Maina, F., Arthur, J. S. & Nebreda, A. R. Genetic analysis of specific and redundant roles for p38α and p38β MAPKs during mouse development. Proc. Natl Acad. Sci. USA 108, 12764–12769 (2011).
Arriazu, E. et al. A new regulatory mechanism of protein phosphatase 2A activity via SET in acute myeloid leukemia. Blood Cancer J. 10, 3 (2020).
Warr, N. et al. Gadd45γ and Map3k4 interactions regulate mouse testis determination via p38 MAPK-mediated control of Sry expression. Dev. Cell 23, 1020–1031 (2012).
Llopis, A. et al. The stress-activated protein kinases p38α/β and JNK1/2 cooperate with Chk1 to inhibit mitotic entry upon DNA replication arrest. Cell Cycle 11, 3627–3637 (2012).
Hayakawa, M. et al. Loss of functionally redundant p38 isoforms in T cells enhances regulatory T cell induction. J. Biol. Chem. 292, 1762–1772 (2017).
Escos, A., Risco, A., Alsina-Beauchamp, D. & Cuenda, A. p38γ and p38δ mitogen activated protein kinases (MAPKs), new stars in the MAPK galaxy. Front. Cell Dev. Biol. 4, 31 (2016).
Matesanz, N. et al. p38α blocks brown adipose tissue thermogenesis through p38δ inhibition. PLoS Biol. 16, e2004455 (2018).
Qi, X. et al. p38α antagonizes p38γ activity through c-Jun-dependent ubiquitin–proteasome pathways in regulating Ras transformation and stress response. J. Biol. Chem. 282, 31398–31408 (2007).
Alonso, G., Ambrosino, C., Jones, M. & Nebreda, A. R. Differential activation of p38 mitogen-activated protein kinase isoforms depending on signal strength. J. Biol. Chem. 275, 40641–40648 (2000).
Ge, B. et al. MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α. Science 295, 1291–1294 (2002). This paper presents evidence for a non-canonical mechanism of p38α activation based on TAB1-induced autophosphorylation.
Tanno, M. et al. Diverse mechanisms of myocardial p38 mitogen-activated protein kinase activation: evidence for MKK-independent activation by a TAB1-associated mechanism contributing to injury during myocardial ischemia. Circ. Res. 93, 254–261 (2003).
DeNicola, G. F. et al. Mechanism and consequence of the autoactivation of p38α mitogen-activated protein kinase promoted by TAB1. Nat. Struct. Mol. Biol. 20, 1182–1190 (2013). This paper provides important structural information on the mechanism by which TAB1 binding triggers p38α autophosphorylation.
De Nicola, G. F. et al. The TAB1–p38α complex aggravates myocardial injury and can be targeted by small molecules. JCI Insight 3, e121144 (2018).
Lanna, A., Henson, S. M., Escors, D. & Akbar, A. N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 15, 965–972 (2014).
Theivanthiran, B. et al. The E3 ubiquitin ligase Itch inhibits p38α signaling and skin inflammation through the ubiquitylation of Tab1. Sci. Signal. 8, ra22 (2015).
Matesanz, N. et al. MKK6 controls T3-mediated browning of white adipose tissue. Nat. Commun. 8, 856 (2017).
Grimsey, N. J. et al. G protein-coupled receptors activate p38 MAPK via a non-canonical TAB1–TAB2- and TAB1–TAB3-dependent pathway in endothelial cells. J. Biol. Chem. 294, 5867–5878 (2019).
Salvador, J. M. et al. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 6, 390–395 (2005). This paper describes an alternative mechanism of p38α activation that involves tyrosine phosphorylation and is specific for T cells.
Mittelstadt, P. R., Yamaguchi, H., Appella, E. & Ashwell, J. D. T cell receptor-mediated activation of p38α by mono-phosphorylation of the activation loop results in altered substrate specificity. J. Biol. Chem. 284, 15469–15474 (2009).
Tomida, T., Takekawa, M. & Saito, H. Oscillation of p38 activity controls efficient pro-inflammatory gene expression. Nat. Commun. 6, 8350 (2015).
Staples, C. J., Owens, D. M., Maier, J. V., Cato, A. C. & Keyse, S. M. Cross-talk between the p38α and JNK MAPK pathways mediated by MAP kinase phosphatase-1 determines cellular sensitivity to UV radiation. J. Biol. Chem. 285, 25928–25940 (2010).
Miura, H., Kondo, Y., Matsuda, M. & Aoki, K. Cell-to-cell heterogeneity in p38-mediated cross-inhibition of JNK causes stochastic cell death. Cell Rep. 24, 2658–2668 (2018).
Ambrosino, C. et al. Negative feedback regulation of MKK6 mRNA stability by p38α mitogen-activated protein kinase. Mol. Cell Biol. 23, 370–381 (2003).
Cheung, P. C., Campbell, D. G., Nebreda, A. R. & Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2a/p38α. EMBO J. 22, 5793–5805 (2003).
Giardino Torchia, M. L. et al. Intensity and duration of TCR signaling is limited by p38 phosphorylation of ZAP-70T293 and destabilization of the signalosome. Proc. Natl Acad. Sci. USA 115, 2174–2179 (2018).
Peregrin, S. et al. Phosphorylation of p38 by GRK2 at the docking groove unveils a novel mechanism for inactivating p38MAPK. Curr. Biol. 16, 2042–2047 (2006).
Salvador, J. M., Mittelstadt, P. R., Belova, G. I., Fornace, A. J. Jr. & Ashwell, J. D. The autoimmune suppressor Gadd45α inhibits the T cell alternative p38 activation pathway. Nat. Immunol. 6, 396–402 (2005).
Wu, Y. H. et al. Tumor suppressor death-associated protein kinase 1 inhibits necroptosis by p38 MAPK activation. Cell Death Dis. 11, 305 (2020).
Pillai, V. B. et al. Acetylation of a conserved lysine residue in the ATP binding pocket of p38 augments its kinase activity during hypertrophy of cardiomyocytes. Mol. Cell Biol. 31, 2349–2363 (2011).
Brichkina, A. et al. Proline isomerisation as a novel regulatory mechanism for p38MAPK activation and functions. Cell Death Differ. 23, 1592–1601 (2016).
Jeong, H. J. et al. Prmt7 promotes myoblast differentiation via methylation of p38MAPK on arginine residue 70. Cell Death Differ. 27, 573–586 (2020).
Liu, M. Y., Hua, W. K., Chen, C. J. & Lin, W. J. The MKK-dependent phosphorylation of p38α is augmented by arginine methylation on Arg49/Arg149 during erythroid differentiation. Int. J. Mol. Sci. 21, 3546 (2020).
Round, J. L. et al. Scaffold protein Dlgh1 coordinates alternative p38 kinase activation, directing T cell receptor signals toward NFAT but not NF-κB transcription factors. Nat. Immunol. 8, 154–161 (2007).
Uhlik, M. T. et al. Rac–MEKK3–MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 5, 1104–1110 (2003).
Zehorai, E. & Seger, R. Beta-like importins mediate the nuclear translocation of mitogen-activated protein kinases. Mol. Cell Biol. 34, 259–270 (2014).
Weaver, B. P. et al. Non-canonical caspase activity antagonizes p38 MAPK stress-priming function to support development. Dev. Cell 53, 358–369.e6 (2020).
Liu, K. et al. Mutual stabilization between TRIM9 short isoform and MKK6 potentiates p38 signaling to synergistically suppress glioblastoma progression. Cell Rep. 23, 838–851 (2018).
Liu, J. et al. F-box only protein 31 (FBXO31) negatively regulates p38 mitogen-activated protein kinase (MAPK) signaling by mediating lysine 48-linked ubiquitination and degradation of mitogen-activated protein kinase kinase 6 (MKK6). J. Biol. Chem. 289, 21508–21518 (2014).
Zou, X. & Blank, M. Targeting p38 MAP kinase signaling in cancer through post-translational modifications. Cancer Lett. 384, 19–26 (2017).
Diao, Y. et al. Oxidation-induced intramolecular disulfide bond inactivates mitogen-activated protein kinase kinase 6 by inhibiting ATP binding. Proc. Natl Acad. Sci. USA 107, 20974–20979 (2010).
Rasmussen, M. H. et al. miR-625-3p regulates oxaliplatin resistance by targeting MAP2K6-p38 signalling in human colorectal adenocarcinoma cells. Nat. Commun. 7, 12436 (2016).
Turk, B. E. Manipulation of host signalling pathways by anthrax toxins. Biochem. J. 402, 405–417 (2007).
Mukherjee, S. et al. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214 (2006).
Paquette, N. et al. Serine/threonine acetylation of TGFβ-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc. Natl Acad. Sci. USA 109, 12710–12715 (2012).
Pellegrini, E. et al. Structural basis for the subversion of MAP kinase signaling by an intrinsically disordered parasite secreted agonist. Structure 25, 16–26 (2017).
Katz, M., Amit, I. & Yarden, Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta 1773, 1161–1176 (2007).
Faust, D. et al. Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun. Signal. 10, 6 (2012).
Sakauchi, C., Wakatsuki, H., Ichijo, H. & Hattori, K. Pleiotropic properties of ASK1. Biochim. Biophys. Acta Gen. Subj. 1861, 3030–3038 (2017).
Matsushita, M., Nakamura, T., Moriizumi, H., Miki, H. & Takekawa, M. Stress-responsive MTK1 SAPKKK serves as a redox sensor that mediates delayed and sustained activation of SAPKs by oxidative stress. Sci. Adv. 6, eaay9778 (2020).
Dolado, I. et al. p38α MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 11, 191–205 (2007).
Coelho, M. A. et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 47, 1083–1099.e6 (2017).
Rodriguez-Colman, M. J. et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 543, 424–427 (2017). This article shows that physiological levels of ROS produced by mitochondrial OXPHOS activate p38α in intestinal stem cells to ensure intestinal crypt homeostasis.
L’Honore, A. et al. The role of Pitx2 and Pitx3 in muscle stem cells gives new insights into p38α MAP kinase and redox regulation of muscle regeneration. eLife 7, e32991 (2018).
Fukawa, T. et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 22, 666–671 (2016).
Cheng, C. T. et al. Metabolic stress-induced phosphorylation of KAP1 Ser473 blocks mitochondrial fusion in breast cancer cells. Cancer Res. 76, 5006–5018 (2016).
Geller, S. et al. Tanycytes regulate lipid homeostasis by sensing free fatty acids and signaling to key hypothalamic neuronal populations via FGF21 secretion. Cell Metab. 30, 833–844.e7 (2019).
Sabio, G. & Davis, R. J. TNF and MAP kinase signalling pathways. Semin. Immunol. 26, 237–245 (2014).
Lin, J., Lee, D., Choi, Y. & Lee, S. Y. The scaffold protein RACK1 mediates the RANKL-dependent activation of p38 MAPK in osteoclast precursors. Sci. Signal. 8, ra54 (2015).
Tzavlaki, K. & Moustakas, A. TGF-β signaling. Biomolecules 10, 487 (2020).
Sapkota, G. P. The TGFβ-induced phosphorylation and activation of p38 mitogen-activated protein kinase is mediated by MAP3K4 and MAP3K10 but not TAK1. Open. Biol. 3, 130067 (2013).
Gaestel, M., Kotlyarov, A. & Kracht, M. Targeting innate immunity protein kinase signalling in inflammation. Nat. Rev. Drug Discov. 8, 480–499 (2009).
Arthur, J. S. & Ley, S. C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692 (2013).
Takizawa, H. et al. Pathogen-induced TLR4–TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell 21, 225–240.e5 (2017).
Lee, W. B. et al. Mincle-mediated translational regulation is required for strong nitric oxide production and inflammation resolution. Nat. Commun. 7, 11322 (2016).
Johnson, R. A., Huong, S. M. & Huang, E. S. Activation of the mitogen-activated protein kinase p38 by human cytomegalovirus infection through two distinct pathways: a novel mechanism for activation of p38. J. Virol. 74, 1158–1167 (2000).
Bouhaddou, M. et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182, 685–712.e19 (2020).
Wagstaff, L. et al. Mechanical cell competition kills cells via induction of lethal p53 levels. Nat. Commun. 7, 11373 (2016).
Hofmann, M. et al. Mechanical pressure-induced phosphorylation of p38 mitogen-activated protein kinase in epithelial cells via Src and protein kinase C. Biochem. Biophys. Res. Commun. 316, 673–679 (2004).
Liu, Z. et al. MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep. 16, 1810–1819 (2016).
Raman, M., Earnest, S., Zhang, K., Zhao, Y. & Cobb, M. H. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J. 26, 2005–2014 (2007).
Colomer, C. et al. IKKα kinase regulates the DNA damage response and drives chemo-resistance in cancer. Mol. Cell 75, 669–682.e5 (2019).
Bent, E. H., Gilbert, L. A. & Hemann, M. T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 30, 1811–1821 (2016).
Lu, H. et al. Reciprocal regulation of DUSP9 and DUSP16 expression by HIF1 controls ERK and p38 MAP kinase activity and mediates chemotherapy-induced breast cancer stem cell enrichment. Cancer Res. 78, 4191–4202 (2018).
Trempolec, N., Dave-Coll, N. & Nebreda, A. R. SnapShot: p38 MAPK substrates. Cell 152, 924–924.e1 (2013).
Gaestel, M. MAPK-activated protein kinases (MKs): novel insights and challenges. Front. Cell Dev. Biol. 3, 88 (2015).
Reyskens, K. M. & Arthur, J. S. Emerging roles of the mitogen and stress activated kinases MSK1 and MSK2. Front. Cell Dev. Biol. 4, 56 (2016).
Joshi, S. & Platanias, L. C. Mnk kinase pathway: cellular functions and biological outcomes. World J. Biol. Chem. 5, 321–333 (2014).
Dolado, I. & Nebreda, A. R. Regulation of tumorigenesis by p38α MAP kinase. Top. Curr. 20, 99–128 (2008).
Cannell, I. G. et al. A pleiotropic RNA-binding protein controls distinct cell cycle checkpoints to drive resistance of p53-defective tumors to chemotherapy. Cancer Cell 28, 623–637 (2015).
Gubern, A. et al. The N-terminal phosphorylation of RB by p38 bypasses its inactivation by CDKs and prevents proliferation in cancer cells. Mol. Cell 64, 25–36 (2016).
Muranen, T. et al. ERK and p38 MAPK activities determine sensitivity to PI3K/mTOR inhibition via regulation of MYC and YAP. Cancer Res. 76, 7168–7180 (2016).
Phong, M. S. et al. p38 mitogen-activated protein kinase promotes cell survival in response to DNA damage but is not required for the G2 DNA damage checkpoint in human cancer cells. Mol. Cell Biol. 30, 3816–3826 (2010).
Guil, S., Long, J. C. & Caceres, J. F. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell Biol. 26, 5744–5758 (2006).
Carbonell, C. et al. Functional network analysis reveals the relevance of SKIIP in the regulation of alternative splicing by p38 SAPK. Cell Rep. 27, 847–859.e6 (2019).
Borisova, M. E. et al. p38–MK2 signaling axis regulates RNA metabolism after UV-light-induced DNA damage. Nat. Commun. 9, 1017 (2018).
Bugai, A. et al. P-TEFb activation by RBM7 shapes a pro-survival transcriptional response to genotoxic stress. Mol. Cell 74, 254–267.e10 (2019).
Li, W. et al. Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat. Commun. 8, 1763 (2017).
Wei, Y. et al. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. eLife 4, e05289 (2015).
Slobodnyuk, K. et al. Autophagy-induced senescence is regulated by p38α signaling. Cell Death Dis. 10, 376 (2019).
Hwang, S. et al. Protective and detrimental roles of p38α MAPK in different stages of nonalcoholic fatty liver disease. Hepatology 72, 873–891 (2020).
Soustek, M. S. et al. Inhibition of the ER stress IRE1α inflammatory pathway protects against cell death in mitochondrial complex I mutant cells. Cell Death Dis. 9, 658 (2018).
Yang, Q. et al. Stress induces p38 MAPK-mediated phosphorylation and inhibition of Drosha-dependent cell survival. Mol. Cell 57, 721–734 (2015).
Lin, K. C. et al. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat. Cell Biol. 19, 996–1002 (2017).
Trempolec, N. et al. Induction of oxidative metabolism by the p38α/MK2 pathway. Sci. Rep. 7, 11367 (2017).
Leestemaker, Y. et al. Proteasome activation by small molecules. Cell Chem. Biol. 24, 725–736.e7 (2017).
Simoes-Sousa, S. et al. The p38α stress kinase suppresses aneuploidy tolerance by inhibiting Hif-1α. Cell Rep. 25, 749–760.e6 (2018).
Canovas, B. et al. Targeting p38α increases DNA damage, chromosome instability, and the anti-tumoral response to taxanes in breast cancer cells. Cancer Cell 33, 1094–1110.e8 (2018). This paper describes a p38α-mediated mechanism that regulates DNA repair in cancer cells, and provides evidence that pharmacological inhibitors of p38α potentiate the cytotoxic effect of taxanes in breast cancer mouse models and patient-derived xenografts.
Herbert, K. et al. BRN2 suppresses apoptosis, reprograms DNA damage repair, and is associated with a high somatic mutation burden in melanoma. Genes Dev. 33, 310–332 (2019).
Kang, Y. J. et al. Macrophage deletion of p38α partially impairs lipopolysaccharide-induced cellular activation. J. Immunol. 180, 5075–5082 (2008).
Youssif, C. et al. Myeloid p38α signaling promotes intestinal IGF-1 production and inflammation-associated tumorigenesis. EMBO Mol. Med. 10, e8403 (2018).
Zheng, T. et al. Protein kinase p38α signaling in dendritic cells regulates colon inflammation and tumorigenesis. Proc. Natl Acad. Sci. USA 115, E12313–E12322 (2018).
Li, C. et al. Dendritic cell MST1 inhibits TH17 differentiation. Nat. Commun. 8, 14275 (2017).
Li, J. et al. Activation of DR3 signaling causes loss of ILC3s and exacerbates intestinal inflammation. Nat. Commun. 10, 3371 (2019).
Petrova, T., Pesic, J., Pardali, K., Gaestel, M. & Arthur, J. S. C. p38 MAPK signalling regulates cytokine production in IL-33 stimulated type 2 innate lymphoid cells. Sci. Rep. 10, 3479 (2020).
Gopfert, C. et al. The p38-MK2/3 module is critical for IL-33-induced signaling and cytokine production in dendritic cells. J. Immunol. 200, 1198–1206 (2018).
McCarthy, P. C. et al. IL-33 regulates cytokine production and neutrophil recruitment via the p38 MAPK-activated kinases MK2/3. Immunol. Cell Biol. 97, 54–71 (2019).
Bhattacharya, S. et al. Role of p38 protein kinase in the ligand-independent ubiquitination and down-regulation of the IFNAR1 chain of type I interferon receptor. J. Biol. Chem. 286, 22069–22076 (2011).
Fuchs, S. Y. Ubiquitination-mediated regulation of interferon responses. Growth Factors 30, 141–148 (2012).
Katlinski, K. V. et al. Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 31, 194–207 (2017).
Ortiz, A. et al. An interferon-driven oxysterol-based defense against tumor-derived extracellular vesicles. Cancer Cell 35, 33–45.e6 (2019).
Gui, J. et al. Activation of p38α stress-activated protein kinase drives the formation of the pre-metastatic niche in the lungs. Nat. Cancer 1, 603–619 (2020). This extensive study analyses how p38α signalling in fibroblasts facilitates lung tumour growth.
De Maeyer, R. P. H. et al. Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat. Immunol. 21, 615–625 (2020). This paper shows that pharmacological inhibition of p38α restores the macrophage-mediated resolution of dermal inflammation in older humans, supporting that p38α inhibitors might be useful to treat particular inflammatory diseases.
Jaco, I. et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 66, 698–710.e5 (2017).
Dondelinger, Y. et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19, 1237–1247 (2017).
Menon, M. B. et al. p38MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 19, 1248–1259 (2017). Together with Jaco et al. (2017) and Dondelinger et al. (2017), this paper demonstrates an important role for the p38/MK2 pathway restraining TNF-induced cell death through the phosphorylation of RIPK1.
Segales, J., Perdiguero, E. & Munoz-Canoves, P. Regulation of muscle stem cell functions: a focus on the p38 MAPK signaling pathway. Front. Cell Dev. Biol. 4, 91 (2016).
Consalvi, S., Brancaccio, A., Dall’Agnese, A., Puri, P. L. & Palacios, D. Praja1 E3 ubiquitin ligase promotes skeletal myogenesis through degradation of EZH2 upon p38α activation. Nat. Commun. 8, 13956 (2017).
Rodriguez-Carballo, E., Gamez, B. & Ventura, F. p38 MAPK signaling in osteoblast differentiation. Front. Cell Dev. Biol. 4, 40 (2016).
Bost, F., Aouadi, M., Caron, L. & Binetruy, B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie 87, 51–56 (2005).
Cao, W. et al. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell Biol. 24, 3057–3067 (2004). This paper describes p38 kinase signalling as a central regulator of thermogenesis in brown adipocytes.
Hattori, K. et al. ASK1 signalling regulates brown and beige adipocyte function. Nat. Commun. 7, 11158 (2016).
Yi, D. et al. Zc3h10 acts as a transcription factor and is phosphorylated to activate the thermogenic program. Cell Rep. 29, 2621–2633.e4 (2019).
Ng, R. et al. miRNA-32 drives brown fat thermogenesis and trans-activates subcutaneous white fat browning in mice. Cell Rep. 19, 1229–1246 (2017).
Quesada-Lopez, T. et al. The lipid sensor GPR120 promotes brown fat activation and FGF21 release from adipocytes. Nat. Commun. 7, 13479 (2016).
Hu, P. et al. p38α/JNK signaling restrains erythropoiesis by suppressing Ezh2-mediated epigenetic silencing of Bim. Nat. Commun. 9, 3518 (2018).
Batlle, R. et al. Regulation of tumor angiogenesis and mesenchymal–endothelial transition by p38α through TGF-β and JNK signaling. Nat. Commun. 10, 3071 (2019).
Oeztuerk-Winder, F. & Ventura, J. J. The many faces of p38 mitogen-activated protein kinase in progenitor/stem cell differentiation. Biochem. J. 445, 1–10 (2012).
Wu, X. et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat. Commun. 7, 13498 (2016).
Choo, M. K., Kraft, S., Missero, C. & Park, J. M. The protein kinase p38α destabilizes p63 to limit epidermal stem cell frequency and tumorigenic potential. Sci. Signal. 11, eaau0727 (2018).
He, D. et al. Gut stem cell aging is driven by mTORC1 via a p38 MAPK–p53 pathway. Nat. Commun. 11, 37 (2020).
Karigane, D. et al. p38α activates purine metabolism to initiate hematopoietic stem/progenitor cell cycling in response to stress. Cell Stem Cell 19, 192–204 (2016).
Ito, K. et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 12, 446–451 (2006).
Freund, A., Patil, C. K. & Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548 (2011).
Alspach, E. et al. p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. 4, 716–729 (2014).
Herranz, N. et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 17, 1205–1217 (2015).
Hu, J. H. et al. Activity-dependent isomerization of Kv4.2 by Pin1 regulates cognitive flexibility. Nat. Commun. 11, 1567 (2020).
Liu, K. et al. PI31 is an adaptor protein for proteasome transport in axons and required for synaptic development. Dev. Cell 50, 509–524.e10 (2019).
Lloret, A., Fuchsberger, T., Giraldo, E. & Vina, J. Molecular mechanisms linking amyloid β toxicity and Tau hyperphosphorylation in Alzheimers disease. Free Radic. Biol. Med. 83, 186–191 (2015).
Chen, J. et al. Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T α-synuclein model of Parkinson’s disease. Cell Death Dis. 9, 700 (2018).
Ashwell, J. D. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat. Rev. Immunol. 6, 532–540 (2006).
Jun, J. E., Kulhanek, K. R., Chen, H., Chakraborty, A. & Roose, J. P. Alternative ZAP70–p38 signals prime a classical p38 pathway through LAT and SOS to support regulatory T cell differentiation. Sci. Signal. 12, eaao0736 (2019).
Alam, M. S. et al. Counter-regulation of T cell effector function by differentially activated p38. J. Exp. Med. 211, 1257–1270 (2014).
Mace, G., Miaczynska, M., Zerial, M. & Nebreda, A. R. Phosphorylation of EEA1 by p38 MAP kinase regulates μ opioid receptor endocytosis. EMBO J. 24, 3235–3246 (2005).
Biondi, R. M. & Nebreda, A. R. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem. J. 372, 1–13 (2003).
Zeke, A. et al. Systematic discovery of linear binding motifs targeting an ancient protein interaction surface on MAP kinases. Mol. Syst. Biol. 11, 837 (2015).
Jung, H. et al. Thioredoxin-interacting protein regulates haematopoietic stem cell ageing and rejuvenation by inhibiting p38 kinase activity. Nat. Commun. 7, 13674 (2016).
Joshi, S., Kaur, S., Kroczynska, B. & Platanias, L. C. Mechanisms of mRNA translation of interferon stimulated genes. Cytokine 52, 123–127 (2010).
Maik-Rachline, G., Lifshits, L. & Seger, R. Nuclear P38: roles in physiological and pathological processes and regulation of nuclear translocation. Int. J. Mol. Sci. 21, 6102 (2020).
Thornton, T. M. et al. Inactivation of nuclear GSK3β by Ser389 phosphorylation promotes lymphocyte fitness during DNA double-strand break response. Nat. Commun. 7, 10553 (2016).
Reinhardt, H. C., Aslanian, A. S., Lees, J. A. & Yaffe, M. B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11, 175–189 (2007).
Zhang, Y. et al. PP2AC level determines differential programming of p38–TSC–mTOR signaling and therapeutic response to p38-targeted therapy in colorectal cancer. EBioMedicine 2, 1944–1956 (2015).
Kim, D. H. et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297, 623–626 (2002).
Gupta, J. & Nebreda, A. R. Roles of p38α mitogen-activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J. 282, 1841–1857 (2015).
Rincon, M. & Davis, R. J. Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 228, 212–224 (2009).
Gurusamy, D. et al. Multi-phenotype CRISPR–Cas9 screen identifies p38 kinase as a target for adoptive immunotherapies. Cancer Cell 37, 818–833.e9 (2020). This paper identifies the p38 kinase pathway as a central regulator of several phenotypes associated with the antitumour efficacy of T cells.
Navarrete, M. et al. Astrocytic p38α MAPK drives NMDA receptor-dependent long-term depression and modulates long-term memory. Nat. Commun. 10, 2968 (2019).
Bolshakov, V. Y. et al. Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3–CA1 synapses. Nat. Neurosci. 3, 1107–1112 (2000).
Zhu, J. J., Qin, Y., Zhao, M., Van Aelst, L. & Malinow, R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110, 443–455 (2002).
Chung, S. H. et al. The p38α mitogen-activated protein kinase is a key regulator of myelination and remyelination in the CNS. Cell Death Dis. 6, e1748 (2015).
Haines, J. D., Fragoso, G., Hossain, S., Mushynski, W. E. & Almazan, G. p38 mitogen-activated protein kinase regulates myelination. J. Mol. Neurosci. 35, 23–33 (2008).
Liu, X. et al. BMP7 retards peripheral myelination by activating p38 MAPK in Schwann cells. Sci. Rep. 6, 31049 (2016).
Lin, X., Wang, M., Zhang, J. & Xu, R. p38 MAPK: a potential target of chronic pain. Curr. Med. Chem. 21, 4405–4418 (2014).
Scheltens, P. et al. An exploratory clinical study of p38α kinase inhibition in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 5, 464–473 (2018).
Hensley, K. et al. p38 kinase is activated in the Alzheimer’s disease brain. J. Neurochem. 72, 2053–2058 (1999).
Zhu, X. et al. Activation of MKK6, an upstream activator of p38, in Alzheimer’s disease. J. Neurochem. 79, 311–318 (2001).
Sun, A., Liu, M., Nguyen, X. V. & Bing, G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 183, 394–405 (2003).
Du, Y. et al. MKP-1 reduces Aβ generation and alleviates cognitive impairments in Alzheimer’s disease models. Signal. Transduct. Target. Ther. 4, 58 (2019).
Kheiri, G., Dolatshahi, M., Rahmani, F. & Rezaei, N. Role of p38/MAPKs in Alzheimer’s disease: implications for amyloid β toxicity targeted therapy. Rev. Neurosci. 30, 9–30 (2018).
Lee, J. K. & Kim, N. J. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 22, 1287 (2017).
Colie, S. et al. Neuronal p38α mediates synaptic and cognitive dysfunction in an Alzheimer’s mouse model by controlling β-amyloid production. Sci. Rep. 7, 45306 (2017).
Thomas, T. et al. MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing neuroinflammation—relevance in a mouse model of Parkinson’s disease. J. Neurochem. 105, 2039–2052 (2008).
Dewil, M., dela Cruz, V. F., Van Den Bosch, L. & Robberecht, W. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1(G93A)-induced motor neuron death. Neurobiol. Dis. 26, 332–341 (2007).
Bendotti, C. et al. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J. Neuropathol. Exp. Neurol. 63, 113–119 (2004).
Gibbs, K. L. et al. Inhibiting p38 MAPKα rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis. 9, 596 (2018).
Bhinge, A., Namboori, S. C., Zhang, X., VanDongen, A. M. J. & Stanton, L. W. Genetic correction of SOD1 mutant iPSCs reveals ERK and JNK activated AP1 as a driver of neurodegeneration in amyotrophic lateral sclerosis. Stem Cell Rep. 8, 856–869 (2017).
Robson, M. J. et al. p38α MAPK signaling drives pharmacologically reversible brain and gastrointestinal phenotypes in the SERT Ala56 mouse. Proc. Natl Acad. Sci. USA 115, E10245–E10254 (2018).
Arabacilar, P. & Marber, M. The case for inhibiting p38 mitogen-activated protein kinase in heart failure. Front. Pharmacol. 6, 102 (2015).
Martin, E. D., Bassi, R. & Marber, M. S. p38 MAPK in cardioprotection—are we there yet? Br. J. Pharmacol. 172, 2101–2113 (2015).
Newby, L. K. et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet 384, 1187–1195 (2014).
O’Donoghue, M. L. et al. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: a randomized clinical trial. JAMA 315, 1591–1599 (2016).
Meng, Q. et al. MMI-0100 inhibits cardiac fibrosis in a mouse model overexpressing cardiac myosin binding protein C. J. Am. Heart Assoc. 6, e006590 (2017).
Wang, Q. et al. Disruption of TAB1/p38α interaction using a cell-permeable peptide limits myocardial ischemia/reperfusion injury. Mol. Ther. 21, 1668–1677 (2013).
Manieri, E. & Sabio, G. Stress kinases in the modulation of metabolism and energy balance. J. Mol. Endocrinol. 55, R11–R22 (2015).
Zhang, S. et al. Metabolic benefits of inhibition of p38α in white adipose tissue in obesity. PLoS Biol. 16, e2004225 (2018).
Berry, D. C. et al. Cellular aging contributes to failure of cold-induced beige adipocyte formation in old mice and humans. Cell Metab. 25, 166–181 (2017).
Zhang, X. et al. Macrophage p38α promotes nutritional steatohepatitis through M1 polarization. J. Hepatol. 71, 163–174 (2019).
Puigserver, P. et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 8, 971–982 (2001).
Brown, J. L. et al. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 9, 987–1002 (2018).
Bulavin, D. V. & Fornace, A. J. Jr. p38 MAP kinase’s emerging role as a tumor suppressor. Adv. Cancer Res. 92, 95–118 (2004).
Loesch, M. & Chen, G. The p38 MAPK stress pathway as a tumor suppressor or more? Front. Biosci. 13, 3581–3593 (2008).
Hui, L. et al. p38α suppresses normal and cancer cell proliferation by antagonizing the JNK–c-Jun pathway. Nat. Genet. 39, 741–749 (2007).
Ventura, J. J. et al. p38α MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat. Genet. 39, 750–758 (2007).
Gupta, J. et al. Dual function of p38α MAPK in colon cancer: suppression of colitis-associated tumor initiation but requirement for cancer cell survival. Cancer Cell 25, 484–500 (2014). This study describes pro-tumorigenic and anti-tumorigenic functions of p38α in the same mouse model of cancer depending on the tumorigenesis stage.
Igea, A. & Nebreda, A. R. The stress kinase p38α as a target for cancer therapy. Cancer Res. 75, 3997–4002 (2015). This paper presents an overview of the cell autonomous roles of p38α in cancer cells.
Vitos-Faleato, J. et al. Requirement for epithelial p38α in KRAS-driven lung tumor progression. Proc. Natl Acad. Sci. USA 117, 2588–2596 (2020).
Saad, M. I. et al. ADAM17 selectively activates the IL-6 trans-signaling/ERK MAPK axis in KRAS-addicted lung cancer. EMBO Mol. Med. 11, e9976 (2019).
Wu, X. et al. Ubiquitin-conjugating enzyme Ubc13 controls breast cancer metastasis through a TAK1–p38 MAP kinase cascade. Proc. Natl Acad. Sci. USA 111, 13870–13875 (2014).
Tichet, M. et al. Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat. Commun. 6, 6993 (2015).
Anwar, T. et al. p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat. Commun. 9, 2801 (2018).
Ryu, K. J. et al. p38 stabilizes Snail by suppressing DYRK2-mediated phosphorylation that is required for GSK3β–βTrCP-induced Snail degradation. Cancer Res. 79, 4135–4148 (2019).
Naffa, R. et al. p38 MAPK promotes migration and metastatic activity of BRAF mutant melanoma cells by inducing degradation of PMCA4b. Cells 9, 1209 (2020).
Harper, K. L. et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 540, 588–592 (2016).
Urosevic, J. et al. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat. Cell Biol. 16, 685–694 (2014).
Gawrzak, S. et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+ breast cancer. Nat. Cell Biol. 20, 211–221 (2018).
Brichkina, A. et al. p38MAPK builds a hyaluronan cancer niche to drive lung tumorigenesis. Genes Dev. 30, 2623–2636 (2016).
Curtis, M. et al. Fibroblasts mobilize tumor cell glycogen to promote proliferation and metastasis. Cell Metab. 29, 141–155.e9 (2019). This interesting study on the interplay between fibroblasts and cancer cells shows how p38α signalling in fibroblasts can modulate the metabolism of cancer cells.
Suarez-Lopez, L. et al. MK2 contributes to tumor progression by promoting M2 macrophage polarization and tumor angiogenesis. Proc. Natl Acad. Sci. USA 115, E4236–E4244 (2018).
Alam, M. S. et al. Selective inhibition of the p38 alternative activation pathway in infiltrating T cells inhibits pancreatic cancer progression. Nat. Med. 21, 1337–1343 (2015).
Zonneville, J. et al. Blockade of p38 kinase impedes the mobilization of protumorigenic myeloid populations to impact breast cancer metastasis. Int. J. Cancer 147, 2279–2292 (2020).
Santoro, V. et al. Role of reactive oxygen species in the abrogation of oxaliplatin activity by cetuximab in colorectal cancer. J. Natl Cancer Inst. 108, djv394 (2016).
Garcia-Cano, J. et al. p38MAPK and chemotherapy: we always need to hear both sides of the story. Front. Cell Dev. Biol. 4, 69 (2016).
Lin, A. et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl Med. 11, eaaw841 (2019).
Pereira, L., Igea, A., Canovas, B., Dolado, I. & Nebreda, A. R. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol. Med. 5, 1759–1774 (2013).
Rudalska, R. et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat. Med. 20, 1138–1146 (2014).
Dietlein, F. et al. A synergistic interaction between Chk1- and MK2 inhibitors in KRAS-mutant cancer. Cell 162, 146–159 (2015).
Lalaoui, N. et al. Targeting p38 or MK2 enhances the anti-leukemic activity of smac-mimetics. Cancer Cell 29, 145–158 (2016). This article shows an interesting cooperation between the inhibitors of p38 or MK2 and Smac-mimetic drugs in leukaemia cells, and paves the way for the discovery of an important link between p38/MK2 and RIPK in the inflammatory response.
Murali, B. et al. Inhibition of the stromal p38MAPK/MK2 pathway limits breast cancer metastases and chemotherapy-induced bone loss. Cancer Res. 78, 5618–5630 (2018).
MacNee, W., Allan, R. J., Jones, I., De Salvo, M. C. & Tan, L. F. Efficacy and safety of the oral p38 inhibitor PH-797804 in chronic obstructive pulmonary disease: a randomised clinical trial. Thorax 68, 738–745 (2013).
Godl, K. et al. An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc. Natl Acad. Sci. USA 100, 15434–15439 (2003).
Jones, D. S., Jenney, A. P., Joughin, B. A., Sorger, P. K. & Lauffenburger, D. A. Inflammatory but not mitogenic contexts prime synovial fibroblasts for compensatory signaling responses to p38 inhibition. Sci. Signal. 11, eaal1601 (2018).
Wang, C. et al. Selective inhibition of the p38α MAPK–MK2 axis inhibits inflammatory cues including inflammasome priming signals. J. Exp. Med. 215, 1315–1325 (2018).
Patnaik, A. et al. A first-in-human phase I study of the oral p38 MAPK inhibitor, ralimetinib (LY2228820 dimesylate), in patients with advanced cancer. Clin. Cancer Res. 22, 1095–1102 (2016).
Vergote, I. et al. A randomized, double-blind, placebo-controlled phase 1b/2 study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine and carboplatin versus gemcitabine and carboplatin for women with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 156, 23–31 (2020).
Donoghue, C. et al. Optimal linker length for small molecule PROTACs that selectively target p38α and p38β for degradation. Eur. J. Med. Chem. 201, 112451 (2020).
Casadome-Perales, A. et al. Inhibition of p38 MAPK in the brain through nasal administration of p38 inhibitor loaded in chitosan nanocapsules. Nanomedicine 14, 2409–2422 (2019).
Maik-Rachline, G., Zehorai, E., Hanoch, T., Blenis, J. & Seger, R. The nuclear translocation of the kinases p38 and JNK promotes inflammation-induced cancer. Sci. Signal. 11, eaao3428 (2018).
Martinez-Limon, A., Joaquin, M., Caballero, M., Posas, F. & de Nadal, E. The p38 pathway: from biology to cancer therapy. Int. J. Mol. Sci. 21, 1913 (2020).
Gee, M. S. et al. A selective p38α/β MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse. Alzheimers Res. Ther. 12, 45 (2020).
Roy, S. M. et al. Targeting human central nervous system protein kinases: an isoform selective p38αMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. ACS Chem. Neurosci. 6, 666–680 (2015).
Evans, B. C. et al. MK2 inhibitory peptide delivered in nanopolyplexes prevents vascular graft intimal hyperplasia. Sci. Transl Med. 7, 291ra295 (2015).
Sies, H. & Jones, D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383 (2020).
Asih, P. R. et al. Functions of p38 MAP kinases in the central nervous system. Front. Mol. Neurosci. 13, 570586 (2020).
Kuzmanic, A. et al. Changes in the free-energy landscape of p38α MAP kinase through its canonical activation and binding events as studied by enhanced molecular dynamics simulations. eLife 6, e22175 (2017).
Tokunaga, Y., Takeuchi, K., Takahashi, H. & Shimada, I. Allosteric enhancement of MAP kinase p38α’s activity and substrate selectivity by docking interactions. Nat. Struct. Mol. Biol. 21, 704–711 (2014).
Kumar, G. S. et al. Dynamic activation and regulation of the mitogen-activated protein kinase p38. Proc. Natl Acad. Sci. USA 115, 4655–4660 (2018). Together with Kuzmanic et al. (2017) and Tokunaga et al. (2014), this paper provides detailed insights into the mechanism of p38α kinase activation using NMR and molecular modelling.
Haller, V., Nahidino, P., Forster, M. & Laufer, S. A. An updated patent review of p38 MAP kinase inhibitors (2014–2019). Expert Opin. Ther. Pat. 30, 453–466 (2020).
Yang, L. et al. p38α mitogen-activated protein kinase is a druggable target in pancreatic adenocarcinoma. Front. Oncol. 9, 1294 (2019).
Shah, N. G. et al. Novel noncatalytic substrate-selective p38α-specific MAPK inhibitors with endothelial-stabilizing and anti-inflammatory activity. J. Immunol. 198, 3296–3306 (2017).
Nichols, C. et al. Mining the PDB for tractable cases where X-ray crystallography combined with fragment screens can be used to systematically design protein–protein inhibitors: two test cases illustrated by IL1β–IL1R and p38α–TAB1 complexes. J. Med. Chem. 63, 7559–7568 (2020).
Lemmens, B. et al. DNA replication determines timing of mitosis by restricting CDK1 and PLK1 activation. Mol. Cell 71, 117–128.e3 (2018).
Thornton, T. M. et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3β inactivation. Science 320, 667–670 (2008).
Cully, M. et al. A role for p38 stress-activated protein kinase in regulation of cell growth via TORC1. Mol. Cell Biol. 30, 481–495 (2010).
Li, L. et al. TLR8-mediated metabolic control of human Treg function: a mechanistic target for cancer immunotherapy. Cell Metab. 29, 103–123.e5 (2019).
Hernandez, G. et al. A novel cardioprotective p38-MAPK/mTOR pathway. Exp. Cell Res. 317, 2938–2949 (2011).
Acknowledgements
The authors thank members of the Nebreda laboratory for critical reading of the manuscript. The authors’ work is supported by grants from the Spanish Ministerio de Ciencia e Innovación (MICINN; SAF2016-81043-R and PID2019-109521RB-I00), Worldwide Cancer Research (18-0666), Asociación Española Contra el Cancer (AECC), AGAUR (2017 SRG-557) and the European Research Council (ERC). The group is part of the network of excellence iDIFFER of MICINN (RED2018-102723-T). IRB Barcelona is the recipient of institutional funding from MICINN through the Centres of Excellence Severo Ochoa award and from the Centres de Recerca de Catalunya (CERCA) Program of the Catalan Government. The authors apologize to those colleagues whose research articles could not be directly cited due to space limitations.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
IRB Barcelona and ICREA have filed a patent application on compounds that modulate p38α autophosphorylation (WO2020120576). A.R.N. is a named inventor on this application. B.C. declares no competing interests.
Additional information
Peer review information
Nature Reviews Molecular Cell Biology thanks A. Cuenda, J. Han and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ClinicalTrials.gov database: https://clinicaltrials.gov
Supplementary information
Glossary
- Mitogen
-
A molecule of peptidic or non-peptidic nature that stimulates cell division triggering mitosis.
- Pleiotropic
-
In the context of cell regulation, the ability of one protein to control several unrelated functions or processes.
- Lipopolysaccharide
-
(LPS). A major component of the outer membrane of Gram-negative bacteria that induces a strong immune response and is used as a prototypical endotoxin.
- Osmotic stress
-
A sudden variation in the solute concentration around cells that induces a movement of water through the cell membrane.
- Triiodothyronine
-
A thyroid hormone that controls a wide range of physiological processes in the body, including the metabolic rate and maintenance of bones, as well as brain, heart, muscle and digestive functions.
- G protein-coupled receptor
-
(GPCR). The largest and most diverse group of membrane receptors in eukaryotes, which can receive multiple signals such as light energy, peptides, lipids, sugars or proteins.
- Phosphotransfer reaction
-
Chemical process in which a phosphate group is transferred from a donor to an acceptor molecule. In the case of protein kinases, this transfer usually takes place between ATP and a specific amino acid of the protein substrate.
- Isomerization
-
A chemical process by which a molecule is transformed into a different form (isomer) with the same composition but a different chemical configuration, which usually involves different chemical properties.
- Scaffold proteins
-
Proteins that simultaneously bind two or more proteins creating functional complexes that enhance the efficacy and fidelity of a signalling pathway.
- Importin
-
A protein that binds to other proteins containing nuclear localization signals, and transports them from the cytoplasm to the nucleus of the cell.
- Caspase
-
A cysteine protease with essential functions in programmed cell death.
- Oxidative stress
-
An excess of reactive oxygen species due to an imbalance between the production of oxygen derivatives and antioxidants, which occurs naturally during ageing or due to environmental factors, and can lead to cellular damage.
- Reactive oxygen species
-
(ROS). Highly reactive non-radical and free radical derivatives of molecular oxygen, including hydrogen peroxide (H2O2) and superoxide (O2–), which are produced mainly by the mitochondrial transport chain and NADPH oxidases, and are important in signal transduction.
- Thioredoxin
-
A protein involved in redox regulation that can act as a signalling molecule by interacting with and facilitating the reduction of oxidized cysteine residues in other proteins, functioning as an antioxidant.
- Redox sensor
-
A molecule that detects and signals redox imbalances in the cell.
- Receptor activator of nuclear factor-κB ligand
-
(RANKL). A membrane-bound protein that can also be found in soluble form, which through binding to the receptor RANK regulates osteoblast differentiation, bone remodelling and immune responses.
- Toll-like receptors
-
(TLRs). A family of membrane proteins that recognize pathogen-associated molecular patterns and play key roles in the innate immune response.
- MINCLE
-
A macrophage inducible Ca2+-dependent lectin receptor that regulates innate immunity by recognizing bacteria, fungi and other molecules.
- SYK
-
A non-receptor cytoplasmic tyrosine protein kinase that functions downstream of several receptors involved in innate and adaptive immunity, and has been implicated in haematopoietic malignancies.
- BRAF
-
A serine/threonine protein kinase involved in RAS signalling, which functions as an MAP3K for the ERK1/2 pathway and has been found mutated in cancer cells, especially in melanoma but also in lung and colorectal tumours.
- Adenylate–uridylate-rich element
-
(ARE). Sequences found in the 3′ untranslated region of many mRNAs, which are key determinants of mRNA stability in mammalian cells.
- Retinoblastoma protein
-
(RB). A tumour suppressor protein that is usually mutated in cancer and negatively controls cell cycle progression through binding to E2F family transcription factors.
- YAP
-
A transcription regulator that functions as an effector of the Hippo signalling pathway and has been implicated in organ development and tumour progression through the modulation of cell proliferation, apoptosis and other processes.
- BCL-2 family proteins
-
An evolutionarily conserved family of proteins that share BCL-2 homology (BH) domains and can interact with a complex network of proteins. Their primary function is to control cell death by regulating the permeabilization of the mitochondrial outer membrane and caspase activation.
- hnRNPA1
-
An RNA-binding protein that controls pre-mRNA processing and transport from the nucleus to the cytoplasm, and facilitates cell viability in response to stress, probably mediated by its recruitment to stress granules.
- SKIIP
-
A component of the spliceosome that controls mRNA splicing and can also regulate transcription, and may modulate cell adaptation to stress through its phosphorylation by p38α and the generation of the GADD45α alternatively spliced isoform.
- NELFE
-
An RNA-binding protein that is a component of the negative elongation factor (NELF) complex, which represses RNA polymerase II transcript elongation.
- RBM7
-
An RNA-binding subunit of the trimeric nuclear exosome targeting (NEXT) complex, initially described to direct non-coding short-lived RNAs for exosomal degradation.
- Chaperone-mediated autophagy
-
A process that allows the degradation of selected intracellular proteins through the recognition of a degradation tag by chaperones, which then translocate across the lysosomal membrane in a receptor-mediator manner for protein degradation.
- Macroautophagy
-
A process in which cytoplasmic proteins and organelles are enclosed in vesicles called autophagosomes that then fuse with lysosomes, leading to the degradation of their content.
- β-Oxidation
-
A multistep catabolic process that takes place in the mitochondria and breaks down fatty acids to produce energy.
- Endoplasmic reticulum stress
-
Saturation of the capacity of endoplasmic reticulum-resident chaperones to fold proteins, which gives rise to a stress response referred to as the unfolded protein response.
- HIF1α
-
A subunit of the HIF1 heterodimeric transcription factor, which is induced by low-oxygen conditions and is considered a master regulator of the response to hypoxia.
- Brown adipocytes
-
The main cells in the brown adipose tissue characterized by the presence of small lipid droplets, a high number of mitochondria and the expression of uncoupling protein 1 (UCP1), which uncouples oxidative phosphorylation from ATP production to release heat.
- Hepatic stellate cells
-
Liver-specific mesenchymal cells located between the hepatocytes and the endothelial cells of small blood vessels that play key roles in hepatic physiology and fibrogenesis.
- Senescence-associated secretory phenotype
-
(SASP). The ability of senescent cells to secrete various proteins such as cytokines, growth factors and proteases that act in a paracrine fashion on other cells.
- Rheumatoid arthritis
-
An inflammatory disease that causes pain, swelling and stiffness mainly in the joints, and is the most common form of autoimmune arthritis.
- Chronic obstructive pulmonary disease
-
A progressive lung condition characterized by chronic inflammation and tissue damage that causes airflow blockage and breathing difficulties, including emphysema and chronic bronchitis.
- V(D)J recombination
-
An essential process in the adaptive immune system by which developing T lymphocytes and B lymphocytes randomly assemble different gene segments in order to generate a diverse repertoire of antigen receptors that will allow the recognition of pathogens.
- Class switch recombination
-
Intrachromosomal DNA rearrangement of the immunoglobulin heavy-chain locus that allows proliferating B cells to change the class of the immunoglobulin expressed, maintaining the antigen specificity but modifying the antibody properties.
- β-Amyloid plaques
-
Insoluble aggregates of misfolded protein that form in the space between nerve cells and that are thought to contribute to pathogenesis of Alzheimer disease.
- α-Synuclein
-
A neuronal protein mainly localized at synapses that constitutes the major component of Lewy bodies and Lewy neurites, which are the hallmarks of a group of neurodegenerative diseases that include Parkinson disease.
- Tanycyte
-
A nutrient-sensing cell integrated in the hypothalamic neural network of the brain, which is involved in the control of energy homeostasis.
- Tumour stroma
-
Non-cancer cells in the tumour including fibroblasts, endothelial cells and immune cells, as well as extracellular matrix components such as collagen or hyaluronan.
- PDL1
-
A transmembrane protein that is involved in immunosuppression during physiological processes but has been also implicated in some immune diseases and is often upregulated in cancer cells.
- Checkpoint kinase 1
-
(CHK1). A serine/threonine protein kinase involved in checkpoint signalling and coordination of the DNA damage response, which plays a key role in maintenance of genomic integrity.
- Smac mimetics
-
Targeted drugs that induce apoptosis in cancer cells by interfering with the pro-survival function of inhibitor of apoptosis proteins (IAP).
- Non-oncogene addiction factor
-
The cancer cell dependency on the cellular functions of proteins that themselves do not trigger malignant cell transformation, that is, do not have oncogenic activity.
- Lewy body dementia
-
A disease driven by the accumulation of abnormal deposits of α-synuclein in the brain, which causes problems in behaviour, memory and movement.
Rights and permissions
About this article

Cite this article
Canovas, B., Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol 22, 346–366 (2021). https://doi.org/10.1038/s41580-020-00322-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41580-020-00322-w
This article is cited by
-
Unconventional activation of PRKDC by TNF-α: deciphering its crucial role in Th1-mediated inflammation beyond DNA repair as part of the DNA-PK complex
Journal of Inflammation (2024)
-
Chaga mushroom extract suppresses oral cancer cell growth via inhibition of energy metabolism
Scientific Reports (2024)
-
Dysregulation of miRNA–mRNA expression in fetal growth restriction in a caloric restricted mouse model
Scientific Reports (2024)
-
TGF-β signaling in health, disease, and therapeutics
Signal Transduction and Targeted Therapy (2024)
-
Plant-derived bioactive compounds as key players in the modulation of immune-related conditions
Phytochemistry Reviews (2024)
