
Abstract
Cryptococcus spp., in particular Cryptococcus neoformans and Cryptococcus gattii, have an enormous impact on human health worldwide. The global burden of cryptococcal meningitis is almost a quarter of a million cases and 181,000 deaths annually, with mortality rates of 100% if infections remain untreated. Despite these alarming statistics, treatment options for cryptococcosis remain limited, with only three major classes of drugs approved for clinical use. Exacerbating the public health burden is the fact that the only new class of antifungal drugs developed in decades, the echinocandins, displays negligible antifungal activity against Cryptococcus spp., and the efficacy of the remaining therapeutics is hampered by host toxicity and pathogen resistance. Here, we describe the current arsenal of antifungal agents and the treatment strategies employed to manage cryptococcal disease. We further elaborate on the recent advances in our understanding of the intrinsic and adaptive resistance mechanisms that are utilized by Cryptococcus spp. to evade therapeutic treatments. Finally, we review potential therapeutic strategies, including combination therapy, the targeting of virulence traits, impairing stress response pathways and modulating host immunity, to effectively treat infections caused by Cryptococcus spp. Overall, understanding of the mechanisms that regulate anti-cryptococcal drug resistance, coupled with advances in genomics technologies and high-throughput screening methodologies, will catalyse innovation and accelerate antifungal drug discovery.
Similar content being viewed by others
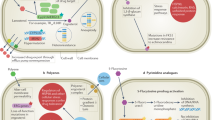

Introduction
Fungal pathogens infect more than a billion people worldwide, with invasive fungal infections having a higher mortality rate and causing more annual deaths than tuberculosis or malaria1. Despite the alarming impact of these infectious agents on human health, current antifungal drug treatments for invasive fungal infections are limited to polyenes, azoles and echinocandins2 (Fig. 1; Table 1). The polyenes target and deplete the essential membrane lipid ergosterol from the plasma membrane, whereas the azoles directly block ergosterol biosynthesis by inhibiting the function of lanosterol 14α-demethylase. The echinocandins disrupt fungal cell wall integrity by inhibiting production of the key cell wall component (1,3)-β-d-glucan2. Finally, the pyrimidine analogue flucytosine (also known as 5-fluorocytosine) functions as an antimetabolite that ultimately blocks DNA synthesis; however, frequent resistance development precludes use as a monotherapy. Unfortunately, widespread antifungal use has fuelled the rapid emergence of drug resistance to all three classes of antifungal agents3. Among the predominant fungi impacting human health, the basidiomycete Cryptococcus neoformans species complex is of particular concern (Box 1). C. neoformans and Cryptoccocus gattii are the major species responsible for life-threatening cryptococcal meningitis; immunocompromised individuals are most vulnerable, but there are also reports of cryptococcal infections in immunocompetent hosts4,5. An estimated 223,100 cases of cryptococcal meningitis occur globally each year, leading to 181,100 deaths6. The majority of deaths are reported from resource-limited countries owing to a lack of access to drugs and the high cost of effective treatments, signifying the urgency in developing affordable therapeutics against these deadly fungal pathogens7.

Cryptococcus spp. achieve resistance to therapeutics by several different mechanisms. Current antifungal agents and their cellular targets are depicted. The polyenes directly target ergosterol whereas the azoles target the ergosterol biosynthetic enzyme Erg11. The pyrimidine analogues block DNA and RNA synthesis. The echinocandins target the glucan synthase Fks1, which is crucial for cell wall synthesis and integrity, although Cryptococcus spp. display inherent resistance to this class of antifungal molecules (dotted line). Within the nucleus, genetic plasticity is generated by aneuploidy, formation of hypermutator strains and transposon movement, which can result in rapid and often transient adaptation to antifungal assault. These adaptations can lead to resistance by overexpression or alteration of the drug target, inactivation of proteins required for drug target engagement or increased expression of efflux pumps. Formation of the cell capsule and thickening and alterations to the cell wall (including melanin production) can also increase tolerance to antifungal treatment. Finally, Cryptococcus can form antifungal-resistant titan cells, which are defined by a large cell size (>10 µm), high ploidy (>4C), a thick cell wall and a highly crosslinked capsule.
In this Review, we focus on current treatment standards to combat cryptococcal disease, as well as the challenges and advances in addressing this global health threat. Specifically, we describe the intrinsic and adaptive mechanisms employed by Cryptococcus spp. that drive antifungal resistance and immune evasion, and we discuss progress in the development of novel antifungal drugs and treatments.
Treatments for cryptococcal infections
Treatment options for invasive cryptococcal infection are limited, given that Cryptococcus spp. are intrinsically resistant to echinocandins and that they employ an arsenal of defences that enable resistance to azoles2,8. As such, the polyene amphotericin B has been the primary treatment for cryptococcal infection for more than half a century, despite the fact that it causes substantial toxicity and that its availability is limited by economic and logistical constraints2,4. Liposome bilayer-coated amphotericin B (LAmB) has been developed to reduce toxicity while retaining antifungal activity, mitigating the dosage limitation of conventional amphotericin B deoxycholate (DAmB)9. These lipid formulations show great promise, such that a phase II trial demonstrated that a single high dose of LAmB is of equivalent efficacy to a 7-day course of DAmB10. A new form of encochleated oral amphotericin B (CAmB), where amphotericin B is encapsulated in a lipid-containing crystal nanoparticle, is effectively delivered to the central nervous system and displays antifungal efficacy in a mouse model11. CAmB is now being tested in clinical trials, and the results will be of particular importance as this formulation would mitigate the costly requirement for intravenous delivery of other forms of the drug12. An alternate approach to mitigate toxicity and improve efficacy involves combination treatment. Specifically, a clinical trial with patients who are HIV-positive and suffering from cryptococcal meningitis showed that the combination of amphotericin B with the fluorinated pyrimidine analogue flucytosine, followed by the triazole fluconazole, improved survival rates compared with amphotericin B alone and minimized toxic effects13.
The success of a phase III trial testing the combination regimen of amphotericin B and flucytosine prompted the WHO to update its guidelines for the treatment of cryptococcal disease in patients infected with HIV14,15. Specifically, the treatment includes an initial induction phase consisting of 1 week of amphotericin B and flucytosine, followed by 1 week of fluconazole at a high dose, a consolidation phase of 8 weeks of fluconazole at an intermediate dose, and then a maintenance phase of fluconazole at a low dose until immune reconstitution15. These and other studies reinforce the importance of attaining early fungicidal activity to effectively clear Cryptococcus spp. from the cerebrospinal fluid and improve patient outcome16. The guidelines also recommend cryptococcal antigen screening among all individuals living with AIDS and fluconazole prophylaxis when antigen screening is not available15. The phase III trial reported 10-week mortality of 24%, which is a considerable improvement compared with the current ~70% mortality rate in many low- and middle-income countries in Africa6,14. In low- and middle-income countries, flucytosine is not available owing to market failure and lack of in-country registration, whereas access to amphotericin B and its liposomal form is limited due to the high cost and lack of safe intravenous administration7. Scarcity of these two drugs has resulted in the widespread use of the much less effective fluconazole monotherapy, which explains the dramatically high mortality rate in these low- and middle-income African countries4,7.
Antifungal resistance mechanisms
Unlike other systemic fungal infections, cryptococcal infections are extremely challenging to treat owing to the unique characteristics of Cryptococcus spp. For example, Cryptococcus spp. display extraordinary genomic plasticity and physiological adaptability, which allows resilience to antifungal assault (Fig. 1). These traits can be inherited, and are often transient, such that they are lost when the stress is removed17,18. Treatment options are potentially further undermined by environmental exposure to cellular stressors. Laboratory studies have shown that selection for resistance to environmental agrochemicals, primarily through the upregulation of the azole target gene ERG11 and drug efflux pumps, can lead to cross-resistance to clinically deployed antifungals19,20. Thus, a better understanding of how resistance evolves is needed if more effective therapeutic strategies are to be developed.
Genomic plasticity
The ability of Cryptococcus to alter its genomic architecture in the face of antifungal stress primarily occurs through a phenomenon referred to as heteroresistance. This phenomenon was first described in 1999, when researchers observed that a subset of Cryptococcus cells were able to grow at azole concentrations well above the inhibitory concentration in vitro21. Heteroresistance has major clinical implications, as the proportion of resistant cells increases over the course of therapy, making this mechanism a major contributor to relapse during azole maintenance therapy22,23. Heteroresistance primarily occurs through the formation of aneuploid cells, both in vitro and in vivo17,22,24 (Fig. 1), most commonly involving disomy of chromosome 1, which contains ERG11 and the predominant efflux pump gene AFR1 (ref.17). Disomy of chromosome 4 is the second most common aneuploidy conferring azole heteroresistance, which is largely attributed to three genes: SEY1, which encodes a GTPase, and GLO3 and GCS2, which encode ADP-ribosylation factor proteins25. Aneuploidy has been reported for other chromosomes, including chromosomes 6, 10, 11 and 14, although the specific genetic elements responsible for the selective advantage remain unclear17,22,25,26. Finally, although the mechanism of aneuploidy formation in Cryptococcus spp. remains enigmatic, research suggests that either mis-segregation of chromosomes or compromised nuclear division leads to the formation of aneuploid cells under azole stress27,28,29.
In contrast to the large-scale genome alterations underpinning heteroresistance, a subset of Cryptococcus isolates have been identified as hypermutator strains30,31, which contain mutations in core genes in the DNA mismatch repair pathway, most notably MSH2 (refs30,32,33) (Fig. 1). However, hypermutator phenotypes can also result from mutations in other genes, such as MSL1 and PMS1 (ref.30). These mutations result in an ~200-fold higher mutation rate than in standard laboratory strains30 and preferentially affect genes containing homopolymer runs33. In addition, a mutation in the proofreading domain of POL3 (encoding the catalytic subunit of DNA polymerase δ) was also shown to confer the hypermutator phenotype31. Whereas evolutionary theory predicts that hypermutator strains will be eliminated from populations34, evidence suggests that they persist in other pathogenic fungi35,36 and are not detrimental to survival in vivo30. Of concern is that hypermutator isolates derived from both the clinic and the laboratory lead to the rapid emergence of resistance to drugs such as rapamycin, FK506 (ref.33), flucytosine32, fluconazole and even amphotericin B30. Recent work exploring the mutations that confer resistance to flucytosine in C. gattii established that hypermutator strains have a 15% higher rate of resistance to this prodrug (the active drug is fluorouracil) when compared with isolates that do not possess a hypermutator mutation. This enhanced resistance was caused by loss-of-function mutations in the genes encoding the purine-cytosine permease Fcy2 and the uracil phosphoribosyltransferase Fur1, which import and process the prodrug, respectively, as well as a mutation in the gene encoding Uxs1, which produces UDP-xylose, a precursor in capsule biosynthesis32.
A final mechanism through which genomic plasticity can enable resistance is the movement of transposable elements (Fig. 1). Transposons have been implicated in antibiotic resistance in bacteria37 and a 2020 report linked transposons to drug resistance in C. neoformans38. Resistance was mediated by inactivation of the genes encoding the targets of 5-fluoroorotic acid, Ura3 and Ura5, by the movement of the DNA transposon T1 and the retrotransposon TCN12 within the fungal genome38. Furthermore, these transposable elements also mediated resistance to rapamycin, FK506 and flucytosine38. Finally, transposon-driven mutagenesis was enhanced in multiple strains at 37 °C compared with that at 30 °C, suggesting that the body temperature of a mammalian host may amplify this phenomenon38.
Morphological alterations
Cryptococcus spp. have a unique cell wall structure and composition, which is composed not only of glucans, chitin, chitosan, mannoproteins and GPI-anchored proteins (which are also present in the cell walls of other fungi) but also of an exopolysaccharide capsule and the pigment melanin (Fig. 1). Homeostasis of this crucial, dynamic cellular component is vital for cell integrity and survival39. Changes in the Cryptococcus capsule and cell wall contribute to resistance and virulence. For example, C. neoformans cells with thicker capsules exhibit amphotericin B resistance and enhanced virulence40,41, and the thicker cell walls of older C. neoformans cells have been implicated in their increased antifungal resistance42. Finally, the ability of fungi to undergo dramatic morphological transitions is exemplified by the formation of Cryptococcus titan cells, which exhibit numerous alterations that likely contribute to drug resistance43 (Box 2).
Intrinsic resistance to echinocandins
The inherent resistance of Cryptococcus spp. to echinocandins is a key limitation in the treatment of cryptococcosis8. This resistance is paradoxical, as β-1,3-glucan synthase, the enzymatic target of the echinocandins, is essential in Cryptococcus and has been demonstrated to be potently inhibited by echinocandins biochemically44,45. A genetic screen of more than 10,000 C. neoformans mutants provided insight into this inherent resistance46. This screen identified CDC50 (encoding the β-subunit of a lipid flippase enzyme) as integral to echinocandin resistance and further demonstrated its involvement in maintaining lipid asymmetry of the phospholipid membrane46. In addition, caspofungin treatment of a cdc50Δ mutant resulted in heightened intracellular calcium and hyperactivation of calcineurin-dependent stress responses47. A forward genetic screen to identify suppressor mutations that restore caspofungin resistance in a cdc50Δ background revealed that inactivation of the calcium channel protein Crm1 reduced intracellular calcium, normalized calcineurin signalling and reinstated caspofungin resistance47. Cdc50 directly interacts with Crm1 and negatively regulates its expression47. Therefore, it was postulated that echinocandin resistance in Cryptococcus is mediated by calcineurin signalling (via Crm1), which is overstimulated as a result of CDC50 deletion, thereby triggering cell death47. These data highlight a pivotal role of stress responses in mediating echinocandin resistance, which is exemplified by the identification of 14 caspofungin-sensitive C. neoformans mutants that are defective in the activation of stress responses, including calcineurin-dependent responses48.
Potential therapeutic targets
Multiple approaches have been used to advance our understanding of Cryptococcus biology and to identify novel essential genes, virulence factors and stress-response regulators that might serve as novel therapeutic targets to combat cryptococcal disease (Box 3).
Essential genes
Elucidating fungal-specific essential genes that can serve as therapeutic targets has been the focus of research for many years. A study assessing more than 2,500 insertional haploid C. neoformans mutants identified 32 genes for which the sporulated haploid mutant was inviable in laboratory conditions49. These genes, together with previously established essential genes, define a shortlist of genes that are conserved across fungi but are not present in humans, including TRR1, FOL1, MGM101, FAS1, FAS2, HOM3, THR1 and IPC1 (ref.49). Comparative genomics identified the thioredoxin reductase Trr1 as a valuable fungal-specific drug target50. Furthermore, depletion of either FAS1 or FAS2, which encode the two subunits of the essential fatty acid synthase, reduces Cryptococcus spp. viability in vitro and in vivo51. Shifting the definition of essentiality to include those genes that are required for growth in vivo (that is, infection), an assessment of transcription factors and kinases that are necessary for establishing meningitis in mice identified Pdr802, Hob1 and Sre1 as factors that are crucial for Cryptococcus adhesion to and crossing of the blood–brain barrier and survival in the brain parenchyma52,53. In particular, Hob1 is a master regulator of brain infectivity by controlling the expression of factors that are known to have a key role in this process53. Furthermore, core virulence factors were identified, including the transcription factor Gat201, which is implicated in inhibiting macrophage phagocytosis, and the kinase Cdc7, which is involved in melanization and drug resistance52,53,54.
Virulence factors
Targeting virulence factors, which are not required for the growth of the pathogen but, instead, are crucial for causing disease, opens up a plethora of new targets for drug development. Primary virulence factors in Cryptococcus spp. include the polysaccharide capsule, melanin production and cell wall integrity, thermotolerance and secreted extracellular enzymes. The Cryptococcus polysaccharide capsule modulates immune responses, enhances pathogenicity and confers protection against oxidative stress55,56. Therefore, the machinery responsible for capsule biosynthesis and structure, such as the xylosyltransferase Cxt1 (ref.57) and the lactonohydrolase Lhc1 (ref.41), represent ideal anti-virulence targets. Deletion of the gene encoding either of these enzymes impairs capsule linkage and reduces the fungal burden in infections in mice41,57. Furthermore, Lhc1 modulates immune responses, blocking phagocytosis of C. neoformans by macrophage-like cell lines in vitro41. Given the importance of the capsule, mutant screens have focused on identifying acapsular strains. Deletion of CAP10, CAP60 and PXB1 not only blocks capsule production but also leads to attenuated virulence52,58. Finally, the macrolides clarithromycin and azithromycin, as well as thiazole derivatives, disrupt the integrity and reduce the thickness of the capsule, resulting in enhanced phagocytosis of Cryptococcus by murine macrophages59,60, thus demonstrating the therapeutic potential of using small molecules to impair Cryptococcus spp. capsule formation.
The fungal cell wall is a dynamic structure that is important not only for cell integrity but also for modulating the host immune response39. Thus, many researchers have investigated the function of cell wall-related genes, finding that deletion of these genes has pleiotropic effects, including reduced virulence. This includes genes involved in the synthesis of β-1,6-glucan (such as KRE5, KRE6 and SKN1)61, the protective pigment melanin (such as the laccase gene LAC1)62, chitin in the cell wall (including the chitin synthase gene CHS3 and the regulator protein gene CSR2)63 and chitosan (exemplified by the chitin deacetylase genes CDA1, CDA2 and CDA3)64,65. In particular, melanin is a major virulence factor that protects fungal cells from oxidative damage, antifungal assault and high temperature, and also modulates host immune responses39. Melanin is produced by laccases, which are upregulated in C. neoformans clinical and environmental isolates66, and laccase-deficient mutants display attenuated pulmonary dissemination in vivo67. A screen of compounds that specifically inhibit melanization found a new role for the pyrimidine analogue flucytosine in inhibiting pigmentation in a DNA polymerase β-dependent manner68.
During infection, extracellular enzymes (such as lipases, proteases and DNases) are secreted by Cryptococcus cells to damage host tissue, promote virulence and interfere with the immune response58. For example, phospholipase B1 (encoded by PLB1) facilitates Cryptococcus adherence to and invasion of the host cell, contributes to fungal cell wall integrity and degrades host cell membranes, thereby providing nutrients that support fungal survival69,70. PLB1 mutants display attenuated virulence in mice owing to impaired titan cell formation, proliferation and survival within macrophages71. Finally, the secreted peptide Qsp1 is an important virulence determinant that acts as an autoregulatory signalling molecule and is transported into the fungal cell through Opt1 to initiate a diverse transcriptional response, including the production of proteases and activation of cell wall integrity72,73.
Comparative genomic studies have also provided new evolutionary insights into the genes involved in virulence. A genome-wide association study of C. neoformans isolates found sequence differences between clinical and environmental isolates in genes involved in virulence and stress responses, including CCK2 (encoding a casein kinase)74. Additionally, comparative analysis of a series of isolates collected over the course of an infection revealed that later-stage isolates possessed enhanced thermotolerance, capsule production and stress adaptation compared with earlier-stage isolates75. Overall, these studies highlight how much remains to be learned about the genes required for Cryptococcus virulence in vivo.
Stress responses
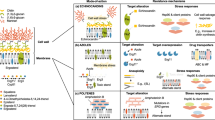
Stress responses in fungal pathogens are instrumental in enabling these organisms to survive diverse environmental insults. Important advances have been made in targeting fungal stress response proteins using structure-guided design of compounds. For example, the protein phosphatase calcineurin is essential for diverse drug-resistance and virulence phenotypes in pathogenic fungi76. However, as current calcineurin inhibitors such as FK506 are immunosuppressive (through inhibition of host calcineurin), fungal-selective calcineurin inhibitors are needed76. Fortunately, the crystal structure of the two subunits of calcineurin in a complex with the inhibitor FK506 and the FK506-binding protein FKBP12 from four prominent fungal pathogens, including C. neoformans, has been solved. These structures identified a specific residue that is conserved in the fungal FKBP12 but not the human FKBP12 and is crucial for FK506-mediated inhibition of calcineurin77, enabling the design of APX879, an FK506 analogue with an acetohydrazine substitution of the C22 carbonyl, which has 70-fold lower immunosuppressive effects than FK506 while retaining antifungal activity77 (Fig 2; Table 1). Other fungal-selective calcineurin inhibitors have been obtained by screening FK506 analogues to identify compounds that synergize with fluconazole to clear fungal infections in mice78.

The monoclonal antibody 18B7 binds to glucuronoxylomannan (GXM), a key capsular polysaccharide that modulates anti-cryptococcal immune responses129. Glucuronoxylomannogalactan (GXMGal) is another major capsular polysaccharide in Cryptococcus spp. Benzothioureas (BTUs) affect the integrity of the cell wall by inhibiting the late post-Golgi secretory pathway95. APX001 (also known as fosmanogepix) targets Gwt1, an inositol acyltransferase required for glycosylphosphatidylinositol (GPI)-anchor biosynthesis, and thereby blocks the cell wall localization of GPI-anchored mannoproteins97. Clofazimine is a broad-spectrum antifungal compound that induces cell membrane stress, potentiating the activity with clinically used antifungals115. The hydrazycin N′-(3-bromo-4-hydroxybenzylidene)-2-methylbenzohydrazide (BHBM) and its derivative 3-bromo-N′-(3-bromo-4-hydroxybenzylidene)benzohydrazide (B0) target the synthesis of the sphingolipid glucosylceramide (GlcCer)100. APX879 and Compound 112 (CMPD#112) are stress response inhibitors that target the phosphatase calcineurin and the chaperone protein Hsp90, respectively77,82. The natural product ibomycin perturbs the multivesicular body (MVB) pathway, and thereby disrupts membrane function90. The extent to which these compounds have been investigated varies: the mechanism of action has been predicted using an indirect assay (red), the proximal target has been verified using a direct assay (orange) or the molecule has been tested for safety and/or efficacy in clinical trials (green). The chemical structure of the highlighted molecules is depicted. ER, endoplasmic reticulum.
Structure-guided design has also been performed to optimize fungal-selective inhibitors of Hsp90. Hsp90 is an essential molecular chaperone that stabilizes a myriad of cellular proteins. Hsp90 inhibition has been shown to impair C. neoformans virulence traits, such as capsule assembly, and increase susceptibility to antifungal agents79,80. An inhibitor of Hsp90 with 25-fold greater selectivity for Candida albicans Hsp90 than for human HSP90 (ref.81) and aminopyrazole-substituted resorcylate amides with more than 30-fold greater selectivity for C. neoformans Hsp90 than for human HSP90 (ref.82) have been identified (Fig. 2; Table 1). Although promising, further study is needed to improve the whole-cell activity of these molecules while maintaining fungal target specificity82.
Additional studies have explored the potential of inhibiting TOR signalling for antifungal drug development, as TOR is a highly conserved master regulator of various cellular processes, including ribosome biogenesis, metabolic pathways and stress responses83. The C. neoformans homologue of mTOR kinase, Tor1, is essential for Cryptococcus stress response signalling, virulence84,85 and fluconazole tolerance (that is, the ability of a drug-susceptible organism to grow in the presence of an antifungal drug at concentrations above the minimum inhibitory concentration)86. However, evidence exists for divergence in regulation of TOR signalling in C. neoformans and other fungi85, which would have important implications for the development of a broad-spectrum antifungal therapeutic.
Promising chemical matter
The development of new antifungal molecules remains a considerable challenge, as most compound libraries have been designed to maximize their ‘drug-like’ properties with respect to mammalian targets and physiology, whereas antifungal drugs require unique physicochemical properties, owing in part to the need for these molecules to transverse the fungal cell wall. Dark chemical matter, which are molecules that show no biological activity when tested in numerous screening programmes, offer a potentially exciting source of novel antifungal therapeutics. Systematic analysis of these apparently inactive compounds revealed that they are not biologically inert, and even identified a potent inhibitor of C. neoformans growth in vitro87. Furthermore, because of the unique neurological features of cryptococcal disease, antifungal molecules are needed that have good central nervous system penetration, and they should be able to be administered orally to ensure widespread access within developing countries with limited resources. This realization has driven antifungal screening efforts towards the use of non-traditional compound libraries. For example, the Malaria Box and the Pathogen Box of Medicines for Malaria Venture, which are collections of diverse molecules that are active against malaria and neglected tropical diseases, have now been screened for molecules with activity against fungal pathogens, including C. neoformans88,89. Furthermore, screening a diverse collection of natural product-producing actinomycete strains led to identification of the glycosylated macrolactone ibomycin as an inhibitor of C. neoformans growth, owing to its ability to permeate the fungal cell wall and affect membrane function by disrupting endosomal trafficking90 (Fig. 2; Table 1). In addition to employing non-traditional compound libraries for the discovery of novel antifungals, alternative high-throughput screening platforms that specifically identify fungicidal molecules have been developed91,92. These platforms, coupled with genetic and proteomic methods to unveil the targets of compounds93,94, have been instrumental in identifying novel drug targets and efficacious compounds.
The fungal cell wall is an essential and complex organelle that facilitates the interaction of the fungus with its environment and maintains cellular integrity. This structure is absent from mammalian cells, and therefore great efforts have been made to develop antifungal agents that target this unique structure directly or that impair its biosynthesis. A high-throughput screen of >300,000 compounds from the US National Institutes of Health (NIH) Molecular Libraries Program was undertaken to identify agents with fungicidal activity against C. neoformans, where secondary assays prioritized compounds that interfered with fungal cell wall integrity91. Through this endeavour, N-substituted benzothioureas (BTUs) were highlighted as a scaffold that is capable of inhibiting activation of the cell wall integrity pathway in Cryptococcus spp.91 (Fig. 2; Table 1). Specifically, BTUs were implicated as direct inhibitors of the Sec4-class small GTPase Sav1, leading to impaired function of the late post-Golgi secretory pathway and highlighting a unique way to compromise cell wall integrity and inhibit fungal cell growth95. Gwt1, an important virulence factor and the enzyme responsible for catalysing an early step in glycosylphosphatidylinositol (GPI)-anchor biosynthesis96, has also emerged as a promising antifungal target. The Gwt1 inhibitor APX001 has demonstrated in vitro and in vivo efficacy against Cryptococcus spp.97 (Fig. 2; Table 1). APX001, alone or in combination with fluconazole, significantly decreased the fungal burden in both the lungs and the brain in a mouse model of cryptococcal infection98. Clinical trials with APX001 for the treatment of cryptococcosis are starting imminently, and phase II trials for the treatment of invasive infections caused by Aspergillus spp. and Candida spp. are currently underway (NCT02957929, NCT02956499 and NCT04240886), highlighting the potential broad-spectrum utility of this drug candidate. Finally, biosynthesis of sphingolipids is emerging as an attractive target given the function of these lipids in mediating signal transduction, cell regulation and virulence in fungal pathogens99. A screen of synthetic small molecules that specifically target the synthesis of the fungal sphingolipid glucosylceramide (GluCer) identified two compounds — N′-(3-bromo-4-hydroxybenzylidene)-2-methylbenzohydrazide (BHBM) and its derivative 3-bromo-N′-(3-bromo-4-hydroxybenzylidene)benzohydrazide (B0) — as highly effective antifungal agents in multiple mouse models of cryptococcosis100 (Fig. 2; Table 1).
In addition to the development of drugs with novel targets, antifungal molecules with improved efficacy and fungal selectivity have been developed to take advantage of established targets, such as Erg11 (also known as Cyp51). The tetrazole VT-1598 is a rationally designed Erg11 inhibitor that has increased fungal selectivity compared with other approved Erg11 inhibitors, including fluconazole101 (Table 1). Similar to other clinically approved azoles, VT-1598 displays broad-spectrum activity against diverse fungi, including a strong therapeutic potential in a mouse model of cryptococcal meningitis101.
Repurposing old drugs
To expedite the development of novel antifungal agents, compounds developed for other therapeutic means can be repurposed if they are discovered to also possess antifungal properties. In this context, previously completed preclinical studies provide crucial information regarding the therapeutic potential of these molecules. For example, benzimidazoles are a class of anthelmintic compounds that have been in clinical use for decades102. Fenbendazole displays potent in vitro activity against C. neoformans and C. gattii, as well as in vivo activity in a mouse model of cryptococcosis103. The efficacy of fenbendazole was attributed to its growth-inhibitory properties, its inhibition of Cryptococcus virulence determinants and its inhibition of fungal proliferation inside macrophages103. Another example is the macrolides, commonly used antibiotics that target protein synthesis, which reduce capsule formation in C. gattii, impairing this key virulence trait and resulting in enhanced phagocytosis and inflammatory cytokine production by macrophages in culture59.
Drug repurposing can be further advanced by selective optimization of approved drugs to enhance the potency or selectivity of desired ‘off-target’ activities. For example, the antimalarial compound mefloquine has limited activity against C. albicans and C. neoformans104 but mefloquine derivatives have been identified that have improved in vitro antifungal activity and that impede C. neoformans capsule formation and melanization104. Unfortunately, these derivatives also displayed a marginal increase in their in vitro toxicity against human cells compared with mefloquine104.
Perhaps the most notable example of compounds being repurposed to combat cryptococcosis involves the drugs sertraline and tamoxifen, as both of these agents advanced to clinical trials (Table 1). Originally developed as an antidepressant, sertraline acts synergistically with fluconazole in a mouse model of systemic cryptococcosis, reducing the fungal burden105. Similarly, tamoxifen, an oestrogen receptor modulator that is used to treat and prevent breast cancer and osteoporosis, also displayed potent anti-cryptococcal activity in vitro106 and in a mouse model of disseminated cryptococcosis107. Unfortunately, despite promising results in phase I/II clinical trials, sertraline treatment alone or in combination with amphotericin B and fluconazole did not improve patient outcomes in a phase III trial with individuals being treated for cryptococcal meningits108. A phase II clinical trial to assess the efficacy and safety of tamoxifen (300 mg daily) as an adjunct therapy to the standard therapy of amphotericin B and fluconazole for the treatment of cryptococcal meningitis (NCT03112031) has completed109; however, this trial found no therapeutic benefit of this combination compared with the standard therapy alone110.
Screening of clinically approved drugs not only identified promising lead compounds but also revealed trends in the classes of molecules that are active against Cryptococcus spp. For example, a screen with the Prestwick Chemical Library of 1,120 off-patent drugs and molecules identified 31 molecules with fungilytic activity111. Of these 31 molecules, the largest class of inhibitors identified were antipsychotic agents, with general antiseptic agents following close behind111. Subsequent studies to identify compounds that augment amphotericin B and fluconazole activity against Cryptococcus spp. also identified numerous antipsychotic drugs, including sertraline, trifluoperazine and thioridazine112,113. Analysis of the chemical structures of drugs with anti-cryptococcal activity revealed a general structure comprising a hydrophobic or lipophilic moiety linked to a cationic amine substituent111. The amphipathic nature of this motif permits the crossing of biological membranes, a property that is important for traversing the blood–brain barrier and accessing cryptococcal cells that reside within the lysosome or phagolysosome of host cells114.
Drug combination therapy
Synergistic combination therapies are an increasingly important strategy to extend the efficacy of currently used drugs2. Combination therapy has the potential to confer enhanced efficacy and fungal selectivity while reducing vulnerability to the evolution of resistance2. This is illustrated by inhibitors of Hsp90, calcineurin and other stress response regulators, which show promise in combination with azoles for inhibiting Cryptococcus spp. growth76,79,80. To identify synergistic pairs on a larger scale, researchers employed high-throughput screening methods coupled with chemical–genetic profiling112,115,116,117,118. A study assessing the combination of known antifungals with ~3,600 bioactive compounds identified clofazimine as a broad-spectrum adjuvant therapy that is capable of acting synergistically with both caspofungin and fluconazole against C. neoformans, among other fungal species115 (Fig. 2; Table 1).
Chemical–genetic mapping of synergistic compound pairs has been explored in Saccharomyces cerevisiae, but pathogen-specific analyses have revealed that many compounds display species-specific chemical–genetic interactions118. A high-throughput approach has been developed to predict synergistic and antagonistic drug pairs using at least one known synergistic drug pair and chemical–genetic data sets118,119. This approach, termed the overlap2 method, has been applied to both fungal and bacterial pathogens. For example, the overlap2 method was applied to a compound library enriched for FDA-approved compounds against C. neoformans116,117,118,119 and successfully predicted that the anticholinergic drug dicyclomine HCl with fluconazole would serve as a potent anti-cryptococcal combination therapy. Indeed, the combination of these drugs displayed enhanced efficacy in a mouse model of cryptococcal meningitis119. Several β-lactam antibiotics were identified as antagonistic with fluconazole in C. neoformans, an important consideration given that susceptible patient populations often have comorbidities119. Overall, the continued pursuit of drug combinations that display efficacy against Cryptococcus spp. will be imperative for the development of effective treatment options that are recalcitrant to the development of resistance.
Immunotherapy and vaccine development
It has become increasingly evident that interactions between Cryptococcus spp. and host immune responses dictate disease progression4, demanding a greater understanding of pathogen–host immune interactions. Studies in animal models have emphasized the importance of CD4+ T cell-mediated immunity (particularly T helper 1 cells) in protection against cryptococcosis120,121. This immune response is mediated by the production of cytokines, including interleukin-2 (IL-2), IL-12, tumour necrosis factor (TNF) and interferon-γ (IFNγ), which in turn recruit phagocytes that help to clear the infection120,122. A strong IFNγ response is indicative of a good prognosis in patients with cryptococcosis123,124. Two phase II clinical trials investigated the safety and efficacy of administering exogenous, recombinant IFNγ as a cryptococcal therapy and found that addition of adjuvant IFNγ to the standard of care enhanced fungal clearance from the cerebrospinal fluid with no reported increase in adverse effects124,125.
In addition to cell-mediated immunity, pioneering work established that a monoclonal antibody specific for the C. neoformans polysaccharide capsule was protective against cryptococcal infection in mice126,127. Given this, antibodies against conserved structures on the cryptococcal cell have been targeted as potential therapeutic strategies128,129. A well-characterized example is 18B7, a mouse-derived monoclonal antibody directed against glucuronoxylomannan, the primary component of the C. neoformans polysaccharide capsule129 (Fig. 2; Table 1). 18B7 is capable of modulating C. neoformans gene expression and metabolism, resulting in increased susceptibility to antifungal drugs130. Furthermore, 18B7 behaves as a catalytic antibody that is capable of hydrolysis of the C. neoformans capsule131. With promising anti-cryptococcal effects in rodent models, 18B7 was evaluated in a phase I/II trial in patients with AIDS-related cryptococcal meningitis132. Administration of 18B7 was safe and transiently reduced serum antigen levels at high doses but exhibited poor cerebrospinal fluid penetration132. Further research is needed to devise strategies to target anti-cryptococcal antibodies to clinically relevant sites within the body.
A major challenge for vaccine development is that the vaccine needs to be effective in immunocompromised individuals, particularly those deficient in CD4+ T cells. Interestingly, in a mouse model of cryptococcosis, immunization with a C. neoformans strain that was genetically engineered to produce IFNγ (H99γ) induced protective immunity in mice depleted of CD4+ T cells133,134. Furthermore, immunocompetent mice immunized with H99γ that were subsequently depleted of both CD4+ T cells and CD8+ T cells were protected upon challenge with wild-type C. neoformans133. Similar protective effects were seen with an avirulent but live mutant strain of C. neoformans lacking sterylglucosidase (sglΔ)134,135. Heat-killed mutant strains have also been reported to induce strong T helper 1 cell responses, resulting in full protection against C. neoformans challenge65,136. For example, immunization with a heat-killed C. neoformans mutant for the F-box protein Fbp1 (HK-fbp1) resulted in protection against Cryptococcus infection in mice depleted of CD4+ T cells137. Finally, there is evidence suggesting that a successful vaccine may require an appropriate adjuvant and antigen delivery systems to induce sufficient protective immunity. Glucan particles generated from acapsular mutant strains of C. neoformans and C. gattii show great promise as a delivery system and adjuvant for cryptococcal vaccines138. Specifically, mice vaccinated with glucan particles containing cryptococcal antigens mounted antigen-specific T cell responses and sustained protection against cryptococcosis138. An important consideration in moving forward with these immunomodulatory therapies is the potential for the development of immune reconstitution inflammatory syndrome, a hyperactive inflammatory response that occurs during immune restoration in select individuals, particularly patients with AIDS who are undergoing antiretroviral therapy139. Given that co-infections with C. neoformans and other pathogens represent a significant risk factor for the development of immune reconstitution inflammatory syndrome, this adverse reaction needs to be carefully monitored when advancing novel immunotherapies.
Conclusions
With the continued threat that Cryptococcus infections pose to public health, the need to strengthen the antifungal drug development pipeline has never been greater. Fortunately, enhanced interest among the scientific community to investigate and develop new antifungal strategies, including the targeting of essential genes or cellular functions, impeding virulence factors, inhibiting stress-response signalling, employing compound combinations and developing immunomodulatory therapies, offers great hope for the future. These motivations, coupled with advances in genomics technologies, the application of structure-guided drug design and the expansion of screening efforts to include structurally diverse compound libraries, will undoubtedly result in key discoveries to help the hundreds of thousands of individuals suffering from Cryptococcus infections worldwide.
References
Brown, G. D. et al. Hidden killers: human fungal infections. Sci. Transl Med. 4, 165rv113 (2012). This seminal review highlights the profound impact that fungal pathogens have on human health.
Lee, Y., Puumala, E., Robbins, N. & Cowen, L. E. Antifungal drug resistance: molecular mechanisms in Candida albicans and beyond. Chem. Rev. https://doi.org/10.1021/acs.chemrev.0c00199 (2020).
Fisher, M. C., Hawkins, N. J., Sanglard, D. & Gurr, S. J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 360, 739–742 (2018). This important review highlights the impact that drug-resistant fungal species have on human health.
Perfect, J. R. et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 50, 291–322 (2010).
Kwon-Chung, K. J. et al. Cryptococcus neoformans and Cryptococcus gattii, the etiologic agents of cryptococcosis. Cold Spring Harb. Perspect. Med. 4, a019760 (2014).
Rajasingham, R. et al. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect. Dis. 17, 873–881 (2017).
Loyse, A. et al. Leave no one behind: response to new evidence and guidelines for the management of cryptococcal meningitis in low-income and middle-income countries. Lancet Infect. Dis. 19, e143–e147 (2019).
Denning, D. W. Echinocandin antifungal drugs. Lancet 362, 1142–1151 (2003).
Stone, N. R., Bicanic, T., Salim, R. & Hope, W. Liposomal amphotericin B (AmBisome®): a review of the pharmacokinetics, pharmacodynamics, clinical experience and future directions. Drugs 76, 485–500 (2016).
Jarvis, J. N. et al. Short-course high-dose liposomal amphotericin B for human immunodeficiency virus-associated cryptococcal meningitis: a phase 2 randomized controlled trial. Clin. Infect. Dis. 68, 393–401 (2019).
Lu, R. et al. Efficacy of oral encochleated amphotericin B in a mouse model of cryptococcal meningoencephalitis. mBio 10, e00724-19 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04031833 (2019).
Day, J. N. et al. Combination antifungal therapy for cryptococcal meningitis. N. Engl. J. Med. 368, 1291–1302 (2013). This important clinical trial shows that the combination of amphotericin B and flucytosine improves survival of patients with cryptococcal meningitis.
Molloy, S. F. et al. Antifungal combinations for treatment of cryptococcal meningitis in Africa. N. Engl. J. Med. 378, 1004–1017 (2018). This seminal study provides guidelines adopted by the WHO for combatting cryptococcal infections.
World Health Organization. Guidelines for the diagnosis, prevention and management of cryptococcal disease in HIV-infected adults, adolescents and children (WHO, 2018). These guidelines provide the current most effective treatment regimen recommendations for the control of cryptococcal meningitis in HIV-infected patients.
Williamson, P. R. et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis and therapy. Nat. Rev. Neurol. 13, 13–24 (2017).
Sionov, E., Lee, H., Chang, Y. C. & Kwon-Chung, K. J. Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog. 6, e1000848 (2010).
Sionov, E., Chang, Y. C., Garraffo, H. M. & Kwon-Chung, K. J. Heteroresistance to fluconazole in Cryptococcus neoformans is intrinsic and associated with virulence. Antimicrob. Agents Chemother. 53, 2804–2815 (2009).
Bastos, R. W. et al. From the environment to the host: how non-azole agrochemical exposure affects the antifungal susceptibility and virulence of Cryptococcus gattii. Sci. Total Environ. 681, 516–523 (2019).
Bastos, R. W. et al. Environmental triazole induces cross-resistance to clinical drugs and affects morphophysiology and virulence of Cryptococcus gattii and C. neoformans. Antimicrob. Agents Chemother. 62, e01179-17 (2018).
Mondon, P. et al. Heteroresistance to fluconazole and voriconazole in Cryptococcus neoformans. Antimicrob. Agents Chemother. 43, 1856–1861 (1999).
Stone, N. R. et al. Dynamic ploidy changes drive fluconazole resistance in human cryptococcal meningitis. J. Clin. Invest. 129, 999–1014 (2019). This paper presents the first clinical samples to demonstrate fluconazole heteroresistance due to aneuploidy, achieved through increased efflux levels.
Rhodes, J. et al. A population genomics approach to assessing the genetic basis of within-host microevolution underlying recurrent cryptococcal meningitis infection. G3 7, 1165–1176 (2017).
Sionov, E., Chang, Y. C. & Kwon-Chung, K. J. Azole heteroresistance in Cryptococcus neoformans: emergence of resistant clones with chromosomal disomy in the mouse brain during fluconazole treatment. Antimicrob. Agents Chemother. 57, 5127–5130 (2013).
Ngamskulrungroj, P. et al. Characterization of the chromosome 4 genes that affect fluconazole-induced disomy formation in Cryptococcus neoformans. PLoS ONE 7, e33022 (2012).
Gerstein, A. C. et al. Polyploid titan cells produce haploid and aneuploid progeny to promote stress adaptation. mBio 6, e01340-15 (2015).
Altamirano, S. et al. Fluconazole-induced ploidy change in Cryptococcus neoformans results from the uncoupling of cell growth and nuclear division. mSphere 2, e00205–e00217 (2017).
Chang, Y. C., Khanal Lamichhane, A. & Kwon-Chung, K. J. Cryptococcus neoformans, unlike Candida albicans, forms aneuploid clones directly from uninucleated cells under fluconazole stress. mBio 9, e01290-18 (2018).
Okagaki, L. H. et al. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 6, e1000953 (2010).
Boyce, K. J. et al. Mismatch repair of DNA replication errors contributes to microevolution in the pathogenic fungus Cryptococcus neoformans. mBio 8, e00595-17 (2017). This report is one of the first of a hypermutator strain in clinical isolates, which has a fitness advantage owing to mutations in mismatch repair genes.
Boyce, K. J., Cao, C., Xue, C. & Idnurm, A. A spontaneous mutation in DNA polymerase POL3 during in vitro passaging causes a hypermutator phenotype in Cryptococcus species. DNA Repair 86, 102751 (2020).
Billmyre, R. B., Applen Clancey, S., Li, L. X., Doering, T. L. & Heitman, J. 5-Fluorocytosine resistance is associated with hypermutation and alterations in capsule biosynthesis in Cryptococcus. Nat. Commun. 11, 127 (2020).
Billmyre, R. B., Clancey, S. A. & Heitman, J. Natural mismatch repair mutations mediate phenotypic diversity and drug resistance in Cryptococcus deuterogattii. eLife 6, e28802 (2017).
Tenaillon, O., Le Nagard, H., Godelle, B. & Taddei, F. Mutators and sex in bacteria: conflict between adaptive strategies. Proc. Natl Acad. Sci. USA 97, 10465–10470 (2000).
Healey, K. R. et al. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat. Commun. 7, 11128 (2016).
Steenwyk, J. L. et al. Extensive loss of cell-cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. PLoS Biol. 17, e3000255 (2019).
Babakhani, S. & Oloomi, M. Transposons: the agents of antibiotic resistance in bacteria. J. Basic Microbiol. 58, 905–917 (2018).
Gusa, A. et al. Transposon mobilization in the human fungal pathogen Cryptococcus is mutagenic during infection and promotes drug resistance in vitro. Proc. Natl Acad. Sci. USA 117, 9973–9980 (2020). This paper is the first description of transposon-mediated antifungal resistance in Cryptococcus spp.
Garcia-Rubio, R., de Oliveira, H. C., Rivera, J. & Trevijano-Contador, N. The fungal cell wall: Candida, Cryptococcus, and Aspergillus species. Front. Microbiol. 10, 2993 (2019).
Cordoba, S., Afeltra, J. & Vitale, R. G. Evaluation of the in vitro activity of amphotericin B by time-kill curve methodology against large and small capsulate C. neoformans isolates. Diagn. Microbiol. Infect. Dis. 71, 260–262 (2011).
Park, Y. D. et al. A role for LHC1 in higher order structure and complement binding of the Cryptococcus neoformans capsule. PLoS Pathog. 10, e1004037 (2014).
Bouklas, T. et al. Old Cryptococcus neoformans cells contribute to virulence in chronic cryptococcosis. mBio 4, e00455-13 (2013).
Zafar, H., Altamirano, S., Ballou, E. R. & Nielsen, K. A titanic drug resistance threat in Cryptococcus neoformans. Curr. Opin. Microbiol. 52, 158–164 (2019). This seminal review summarizes the current understanding of the mechanisms underlying antifungal resistance of the Cryptococcus titan cell.
Thompson, J. R. et al. A glucan synthase FKS1 homolog in Cryptococcus neoformans is single copy and encodes an essential function. J. Bacteriol. 181, 444–453 (1999).
Maligie, M. A. & Selitrennikoff, C. P. Cryptococcus neoformans resistance to echinocandins: (1,3)β-glucan synthase activity is sensitive to echinocandins. Antimicrob. Agents Chemother. 49, 2851–2856 (2005).
Huang, W. et al. Lipid flippase subunit Cdc50 mediates drug resistance and virulence in Cryptococcus neoformans. mBio 7, e00478-16 (2016). This paper provides foundational genetic insights into the intrinsic echinocandin resistance of Cryptococcus.
Cao, C., Wang, Y., Husain, S., Soteropoulos, P. & Xue, C. A mechanosensitive channel governs lipid flippase-mediated echinocandin resistance in Cryptococcus neoformans. mBio 10, e01952-19 (2019).
Pianalto, K. M., Billmyre, R. B., Telzrow, C. L. & Alspaugh, J. A. Roles for stress response and cell wall biosynthesis pathways in caspofungin tolerance in Cryptococcus neoformans. Genetics 213, 213–227 (2019).
Ianiri, G. & Idnurm, A. Essential gene discovery in the basidiomycete Cryptococcus neoformans for antifungal drug target prioritization. mBio 6, 02334-14 (2015).
Abadio, A. K. et al. Comparative genomics allowed the identification of drug targets against human fungal pathogens. BMC Genomics 12, 75 (2011).
Chayakulkeeree, M., Rude, T. H., Toffaletti, D. L. & Perfect, J. R. Fatty acid synthesis is essential for survival of Cryptococcus neoformans and a potential fungicidal target. Antimicrob. Agents Chemother. 51, 3537–3545 (2007).
Liu, O. W. et al. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 135, 174–188 (2008).
Lee, K. T. et al. Fungal kinases and transcription factors regulating brain infection in Cryptococcus neoformans. Nat. Commun. 11, 1521 (2020). This study presents an extensive analysis of Cryptococcus kinases and transcription factors that are required for each major stage of infection, and represents a major resource for future work.
Lee, K. T. et al. Systematic functional analysis of kinases in the fungal pathogen Cryptococcus neoformans. Nat. Commun. 7, 12766 (2016).
Zaragoza, O. Basic principles of the virulence of Cryptococcus. Virulence 10, 490–501 (2019).
McFadden, D., Zaragoza, O. & Casadevall, A. The capsular dynamics of Cryptococcus neoformans. Trends Microbiol. 14, 497–505 (2006).
Klutts, J. S. & Doering, T. L. Cryptococcal xylosyltransferase 1 (Cxt1p) from Cryptococcus neoformans plays a direct role in the synthesis of capsule polysaccharides. J. Biol. Chem. 283, 14327–14334 (2008).
Almeida, F., Wolf, J. M. & Casadevall, A. Virulence-associated enzymes of Cryptococcus neoformans. Eukaryot. Cell 14, 1173–1185 (2015).
Nakamura, S. et al. Macrolides inhibit capsule formation of highly virulent Cryptococcus gattii and promote innate immune susceptibility. Antimicrob. Agents Chemother. 63, e02364-18 (2019).
de Sa, N. P. et al. Thiazole derivatives act on virulence factors of Cryptococcus spp. Med. Mycol. 57, 84–91 (2019).
Gilbert, N. M. et al. KRE genes are required for β-1,6-glucan synthesis, maintenance of capsule architecture and cell wall protein anchoring in Cryptococcus neoformans. Mol. Microbiol. 76, 517–534 (2010).
Zhu, X., Gibbons, J., Garcia-Rivera, J., Casadevall, A. & Williamson, P. R. Laccase of Cryptococcus neoformans is a cell wall-associated virulence factor. Infect. Immun. 69, 5589–5596 (2001).
Banks, I. R. et al. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot. Cell 4, 1902–1912 (2005).
Upadhya, R. et al. Cryptococcus neoformans Cda1 and its chitin deacetylase activity are required for fungal pathogenesis. mBio 9, e02087-18 (2018).
Upadhya, R. et al. Induction of protective immunity to cryptococcal infection in mice by a heat-killed, chitosan-deficient strain of Cryptococcus neoformans. mBio 7, e00547-16 (2016).
Lahiri, S. et al. In vitro expression of vital virulent genes of clinical and environmental isolates of Cryptococcus neoformans/gattii in endothelial cells of human blood–brain barrier. J. Mycol. Med. 29, 239–244 (2019).
Noverr, M. C., Williamson, P. R., Fajardo, R. S. & Huffnagle, G. B. CNLAC1 is required for extrapulmonary dissemination of Cryptococcus neoformans but not pulmonary persistence. Infect. Immun. 72, 1693–1699 (2004).
Zimbres, A. C. G. et al. Pharmacological inhibition of pigmentation in Cryptococcus. FEMS Yeast Res. 19, foy119 (2019).
Wright, L. C. et al. Cryptococcal lipid metabolism: phospholipase B1 is implicated in transcellular metabolism of macrophage-derived lipids. Eukaryot. Cell 6, 37–47 (2007).
Siafakas, A. R. et al. Cell wall-linked cryptococcal phospholipase B1 is a source of secreted enzyme and a determinant of cell wall integrity. J. Biol. Chem. 282, 37508–37514 (2007).
Evans, R. J. et al. Cryptococcal phospholipase B1 is required for intracellular proliferation and control of titan cell morphology during macrophage infection. Infect. Immun. 83, 1296–1304 (2015).
Lee, H., Chang, Y. C., Nardone, G. & Kwon-Chung, K. J. TUP1 disruption in Cryptococcus neoformans uncovers a peptide-mediated density-dependent growth phenomenon that mimics quorum sensing. Mol. Microbiol. 64, 591–601 (2007).
Homer, C. M. et al. Intracellular action of a secreted peptide required for fungal virulence. Cell Host Microbe 19, 849–864 (2016).
Desjardins, C. A. et al. Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans. Genome Res. 27, 1207–1219 (2017).
Chen, Y. et al. Microevolution of serial clinical isolates of Cryptococcus neoformans var. grubii and C. gattii. mBio 8, e00166-17 (2017).
Juvvadi, P. R., Lamoth, F. & Steinbach, W. J. Calcineurin as a multifunctional regulator: unraveling novel functions in fungal stress responses, hyphal growth, drug resistance, and pathogenesis. Fungal Biol. Rev. 28, 56–69 (2014).
Juvvadi, P. R. et al. Harnessing calcineurin–FK506–FKBP12 crystal structures from invasive fungal pathogens to develop antifungal agents. Nat. Commun. 10, 4275 (2019). This paper presents an excellent example of successful structure-guided drug design to develop fungal-specific calcineurin inhibitors.
Lee, Y. et al. In vitro and in vivo asssessment of FK506 analogs as novel antifungal drug candidates. Antimicrob. Agents Chemother. 62, e01627-18 (2018).
Chatterjee, S. & Tatu, U. Heat shock protein 90 localizes to the surface and augments virulence factors of Cryptococcus neoformans. PLoS Negl. Trop. Dis. 11, e0005836 (2017).
Cordeiro, R. A. et al. Inhibition of heat-shock protein 90 enhances the susceptibility to antifungals and reduces the virulence of Cryptococcus neoformans/Cryptococcus gattii species complex. Microbiology 162, 309–317 (2016).
Whitesell, L. et al. Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus. Nat. Commun. 10, 402 (2019).
Huang, D. S. et al. Design and synthesis of fungal-selective resorcylate aminopyrazole Hsp90 inhibitors. J. Med. Chem. 63, 2139–2180 (2020).
Bahn, Y. S. & Jung, K. W. Stress signaling pathways for the pathogenicity of Cryptococcus. Eukaryot. Cell 12, 1564–1577 (2013).
Lee, H., Khanal Lamichhane, A., Garraffo, H. M., Kwon-Chung, K. J. & Chang, Y. C. Involvement of PDK1, PKC and TOR signalling pathways in basal fluconazole tolerance in Cryptococcus neoformans. Mol. Microbiol. 84, 130–146 (2012).
So, Y. S., Lee, D. G., Idnurm, A., Ianiri, G. & Bahn, Y. S. The TOR pathway pays pleiotropic roles in growth and stress responses of the fungal pathogen Cryptococcus neoformans. Genetics 212, 1241–1258 (2019).
Berman, J. & Krysan, D. J. Drug resistance and tolerance in fungi. Nat. Rev. Microbiol. 18, 319–331 (2020).
Wassermann, A. M. et al. Dark chemical matter as a promising starting point for drug lead discovery. Nat. Chem. Biol. 11, 958–966 (2015). This paper reaffirms that dark chemical matter compounds can be attractive lead compounds and demonstrates the potential for anti-cryptococcal drug discovery.
Jung, E. H., Meyers, D. J., Bosch, J. & Casadevall, A. Novel antifungal compounds discovered in medicines for Malaria Venture’s Malaria Box. mSphere 3, e00537-17 (2018).
Mayer, F. L. & Kronstad, J. W. Discovery of a novel antifungal agent in the Pathogen Box. mSphere 2, e00120-17 (2017).
Robbins, N. et al. Discovery of ibomycin, a complex macrolactone that exerts antifungal activity by impeding endocytic trafficking and membrane function. Cell Chem. Biol. 23, 1383–1394 (2016).
Hartland, K. et al. High-throughput screen in Cryptococcus neoformans identifies a novel molecular scaffold that inhibits cell wall integrity pathway signaling. ACS Infect. Dis. 2, 93–102 (2016).
Rabjohns, J. L. A. et al. A high-throughput screening assay for fungicidal compounds against Cryptococcus neoformans. J. Biomol. Screen. 19, 270–277 (2014).
Park, Y. D. et al. Identification of multiple cryptococcal fungicidal drug targets by combined gene dosing and drug affinity responsive target stability screening. mBio 7, e01073-16 (2016).
Xue, A., Robbins, N. & Cowen, L. E. Advances in fungal chemical genomics for the discovery of new antifungal agents. Ann. NY Acad. Sci. https://doi.org/10.1111/nyas.14484 (2021).
Beattie, S. R. et al. Benzothiourea derivatives target the secretory pathway of the human fungal pathogen Cryptococcus neoformans. ACS Infect. Dis. 6, 529–539 (2020).
Zhao, Q. et al. GWT1 encoding an inositol acyltransferase homolog is required for laccase repression and stress resistance in the basidiomycete Cryptococcus neoformans. FEMS Yeast Res. 15, fov086 (2015).
Shaw, K. J. et al. In vitro and in vivo evaluation of APX001A/APX001 and other Gwt1 inhibitors against Cryptococcus. Antimicrob. Agents Chemother. 62, e00523-18 (2018).
Rittershaus, P. C. et al. Glucosylceramide synthase is an essential regulator of pathogenicity of Cryptococcus neoformans. J. Clin. Invest. 116, 1651–1659 (2006).
Singh, A. et al. Methylation of glycosylated sphingolipid modulates membrane lipid topography and pathogenicity of Cryptococcus neoformans. Cell Microbiol. 14, 500–516 (2012).
Mor, V. et al. Identification of a new class of antifungals targeting the synthesis of fungal sphingolipids. mBio 6, e00647 (2015). This work identifies a new class of antifungal agents that target sphingolipids in several fungal pathogens and that display fungal-selective activity and synergism with currently used therapies.
Garvey, E. P., Sharp, A. D., Warn, P. A., Yates, C. M. & Schotzinger, R. J. The novel fungal CYP51 inhibitor VT-1598 is efficacious alone and in combination with liposomal amphotericin B in a murine model of cryptococcal meningitis. J. Antimicrob. Chemother. 73, 2815–2822 (2018).
McKellar, Q. A. & Scott, E. W. The benzimidazole anthelmintic agents — a review. J. Vet. Pharmacol. Ther. 13, 223–247 (1990).
de Oliveira, H. C. et al. Fenbendazole controls in vitro growth, virulence potential, and animal infection in the Cryptococcus model. Antimicrob. Agents Chemother. 64, e00286-20 (2020).
Montoya, M. C., Beattie, S., Alden, K. M. & Krysan, D. J. Derivatives of the antimalarial drug mefloquine are broad-spectrum antifungal molecules with activity against drug-resistant clinical isolates. Antimicrob. Agents Chemother. 64, e02331-19 (2020).
Zhai, B., Wu, C., Wang, L., Sachs, M. S. & Lin, X. The antidepressant sertraline provides a promising therapeutic option for neurotropic cryptococcal infections. Antimicrob. Agents Chemother. 56, 3758–3766 (2012).
Dolan, K. et al. Antifungal activity of tamoxifen: in vitro and in vivo activities and mechanistic characterization. Antimicrob. Agents Chemother. 53, 3337–3346 (2009).
Butts, A. et al. Estrogen receptor antagonists are anti-cryptococcal agents that directly bind EF hand proteins and synergize with fluconazole in vivo. mBio 5, e00765-13 (2014).
Rhein, J. et al. Efficacy of adjunctive sertraline for the treatment of HIV-associated cryptococcal meningitis: an open-label dose-ranging study. Lancet Infect. Dis. 16, 809–818 (2016).
Ngan, N. T. T. et al. A randomized open label trial of tamoxifen combined with amphotericin B and fluconazole for cryptococcal meningitis. Wellcome Open Res. 4, 8 (2019).
Day, J. Best options in cryptococcosis. 29th ECCMID https://www.escmid.org/escmid_publications/escmid_elibrary/material/?mid=70711 (2019).
Butts, A. et al. A repurposing approach identifies off-patent drugs with fungicidal cryptococcal activity, a common structural chemotype, and pharmacological properties relevant to the treatment of cryptococcosis. Eukaryot. Cell 12, 278–287 (2013).
Spitzer, M. et al. Cross-species discovery of syncretic drug combinations that potentiate the antifungal fluconazole. Mol. Syst. Biol. 7, 499 (2011).
Rossi, S. A. et al. Identification of off-patent drugs that show synergism with amphotericin B or that present antifungal action against Cryptococcus neoformans and Candida spp. Antimicrob. Agents Chemother. 64, e01921-19 (2020).
Banks, W. A. Characteristics of compounds that cross the blood–brain barrier. BMC Neurol. 9, S3 (2009).
Robbins, N. et al. An antifungal combination matrix identifies a rich pool of adjuvant molecules that enhance drug activity against diverse fungal pathogens. Cell Rep. 13, 1481–1492 (2015).
Wambaugh, M. A. & Brown, J. C. S. High-throughput identification of synergistic drug combinations by the overlap2 method. J. Vis. Exp. 135, 57241 (2018).
Wambaugh, M. A., Shakya, V. P. S., Lewis, A. J., Mulvey, M. A. & Brown, J. C. S. High-throughput identification and rational design of synergistic small-molecule pairs for combating and bypassing antibiotic resistance. PLoS Biol. 15, e2001644 (2017).
Brown, J. C. S. et al. Unraveling the biology of a fungal meningitis pathogen using chemical genetics. Cell 159, 1168–1187 (2014). This work establishes a chemical–genetic platform providing an approach to explore antifungal activities of novel compounds and predict synergistic antifungal combinations that are effective against Cryptococcus spp.
Wambaugh, M. A. et al. Synergistic and antagonistic drug interactions in the treatment of systemic fungal infections. eLife 9, e54160 (2020).
Wozniak, K. L. et al. Insights into the mechanisms of protective immunity against Cryptococcus neoformans infection using a mouse model of pulmonary cryptococcosis. PLoS ONE 4, e6854 (2009).
Hole, C. R. & Wormley, F. L. Jr Vaccine and immunotherapeutic approaches for the prevention of cryptococcosis: lessons learned from animal models. Front. Microbiol. 3, 291 (2012).
Caballero Van Dyke, M. C. & Wormley, F. L. Jr. A call to arms: quest for a cryptococcal vaccine. Trends Microbiol. 26, 436–446 (2018).
Jarvis, J. N. et al. Determinants of mortality in a combined cohort of 501 patients with HIV-associated cryptococcal meningitis: implications for improving outcomes. Clin. Infect. Dis. 58, 736–745 (2014).
Jarvis, J. N. et al. Adjunctive interferon-γ immunotherapy for the treatment of HIV-associated cryptococcal meningitis: a randomized controlled trial. AIDS 26, 1105–1113 (2012).
Pappas, P. G. et al. Recombinant interferon-γ1b as adjunctive therapy for AIDS-related acute cryptococcal meningitis. J. Infect. Dis. 189, 2185–2191 (2004).
Dromer, F., Charreire, J., Contrepois, A., Carbon, C. & Yeni, P. Protection of mice against experimental cryptococcosis by anti-Cryptococcus neoformans monoclonal antibody. Infect. Immun. 55, 749–752 (1987).
Dromer, F., Salamero, J., Contrepois, A., Carbon, C. & Yeni, P. Production, characterization, and antibody specificity of a mouse monoclonal antibody reactive with Cryptococcus neoformans capsular polysaccharide. Infect. Immun. 55, 742–748 (1987). This report is the first of a monoclonal antibody that provides protection against experimental cryptococcosis, challenging the long-standing dogma that antibodies are unimportant in anti-cryptococcal immunity.
Rachini, A. et al. An anti-β-glucan monoclonal antibody inhibits growth and capsule formation of Cryptococcus neoformans in vitro and exerts therapeutic, anticryptococcal activity in vivo. Infect. Immun. 75, 5085–5094 (2007).
Casadevall, A. et al. Characterization of a murine monoclonal antibody to Cryptococcus neoformans polysaccharide that is a candidate for human therapeutic studies. Antimicrob. Agents Chemother. 42, 1437–1446 (1998).
McClelland, E. E., Nicola, A. M., Prados-Rosales, R. & Casadevall, A. Ab binding alters gene expression in Cryptococcus neoformans and directly modulates fungal metabolism. J. Clin. Invest. 120, 1355–1361 (2010).
Bowen, A., Wear, M. P., Cordero, R. J., Oscarson, S. & Casadevall, A. A monoclonal antibody to Cryptococcus neoformans glucuronoxylomannan manifests hydrolytic activity for both peptides and polysaccharides. J. Biol. Chem. 292, 417–434 (2017).
Larsen, R. A. et al. Phase I evaluation of the safety and pharmacokinetics of murine-derived anticryptococcal antibody 18B7 in subjects with treated cryptococcal meningitis. Antimicrob. Agents Chemother. 49, 952–958 (2005).
Wozniak, K. L., Young, M. L. & Wormley, F. L. Jr Protective immunity against experimental pulmonary cryptococcosis in T cell-depleted mice. Clin. Vaccine Immunol. 18, 717–723 (2011).
Wormley, F. L. Jr., Perfect, J. R., Steele, C. & Cox, G. M. Protection against cryptococcosis by using a murine γ interferon-producing Cryptococcus neoformans strain. Infect. Immun. 75, 1453–1462 (2007).
Rella, A. et al. Role of sterylglucosidase 1 (Sgl1) on the pathogenicity of Cryptococcus neoformans: potential applications for vaccine development. Front. Microbiol. 6, 836 (2015).
Zhai, B. et al. Development of protective inflammation and cell-mediated immunity against Cryptococcus neoformans after exposure to hyphal mutants. mBio 6, e01433-15 (2015).
Wang, Y., Wang, K., Masso-Silva, J. A., Rivera, A. & Xue, C. A heat-killed Cryptococcus mutant atrain induces host protection against multiple invasive mycoses in a murine vaccine model. mBio 10, e02145-19 (2019).
Specht, C. A. et al. Vaccination withrecombinant Cryptococcus proteins in glucan particles protects mice against cryptococcosis in a manner dependent upon mouse atrain and cryptococcal species. mBio 8, e01872-17 (2017).
Barber, D. L., Andrade, B. B., Sereti, I. & Sher, A. Immune reconstitution inflammatory syndrome: the trouble with immunity when you had none. Nat. Rev. Microbiol. 10, 150–156 (2012).
Ikeda, R., Shinoda, T., Fukazawa, Y. & Kaufman, L. Antigenic characterization of Cryptococcus neoformans serotypes and its application to serotyping of clinical isolates. J. Clin. Microbiol. 16, 22–29 (1982).
Franzot, S. P., Salkin, I. F. & Casadevall, A. Cryptococcus neoformans var. grubii: separate varietal status for Cryptococcus neoformans serotype A isolates. J. Clin. Microbiol. 37, 838–840 (1999).
Meyer, W. et al. Molecular typing of IberoAmerican Cryptococcus neoformans isolates. Emerg. Infect. Dis. 9, 189–195 (2003).
Hagen, F. et al. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet. Biol. 78, 16–48 (2015).
Kwon-Chung, K. J. et al. The case for adopting the “species complex” nomenclature for the etiologic agents of cryptococcosis. mSphere 2, e00357-16 (2017).
Hagen, F. et al. Importance of resolving fungal nomenclature: the case of multiple pathogenic species in the Cryptococcus genus. mSphere 2, e00238-17 (2017).
Cruickshank, J. G., Cavill, R. & Jelbert, M. Cryptococcus neoformans of unusual morphology. Appl. Microbiol. 25, 309–312 (1973).
Zaragoza, O. et al. Fungal cell gigantism during mammalian infection. PLoS Pathog. 6, e1000945 (2010).
Zaragoza, O. & Nielsen, K. Titan cells in Cryptococcus neoformans: cells with a giant impact. Curr. Opin. Microbiol. 16, 409–413 (2013).
Dambuza, I. M. et al. The Cryptococcus neoformans titan cell is an inducible and regulated morphotype underlying pathogenesis. PLoS Pathog. 14, e1006978 (2018).
Okagaki, L. H. et al. Cryptococcal titan cell formation is regulated by G-protein signaling in response to multiple stimuli. Eukaryot. Cell 10, 1306–1316 (2011).
Mukaremera, L. et al. Titan cell production in Cryptococcus neoformans reshapes the cell wall and capsule composition during infection. Cell Surf. 1, 15–24 (2018).
Wiesner, D. L. et al. Chitin recognition via chitotriosidase promotes pathologic type-2 helper T cell responses to cryptococcal infection. PLoS Pathog. 11, e1004701 (2015).
Toffaletti, D. L., Rude, T. H., Johnston, S. A., Durack, D. T. & Perfect, J. R. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J. Bacteriol. 175, 1405–1411 (1993).
Janbon, G. et al. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLoS Genet. 10, e1004261 (2014).
Goranov, A. I. & Madhani, H. D. Functional profiling of human fungal pathogen genomes. Cold Spring Harb. Perspect. Med. 5, a019596 (2014).
Jung, K. W. et al. Systematic functional profiling of transcription factor networks in Cryptococcus neoformans. Nat. Commun. 6, 6757 (2015).
Fan, Y. & Lin, X. Multiple applications of a transient CRISPR–Cas9 coupled with electroporation (TRACE) system in the Cryptococcus neoformans species complex. Genetics 208, 1357–1372 (2018).
Idnurm, A., Reedy, J. L., Nussbaum, J. C. & Heitman, J. Cryptococcus neoformans virulence gene discovery through insertional mutagenesis. Eukaryot. Cell 3, 420–429 (2004).
Esher, S. K., Granek, J. A. & Alspaugh, J. A. Rapid mapping of insertional mutations to probe cell wall regulation in Cryptococcus neoformans. Fungal Genet. Biol. 82, 9–21 (2015).
Ory, J. J., Griffith, C. L. & Doering, T. L. An efficiently regulated promoter system for Cryptococcus neoformans utilizing the CTR4 promoter. Yeast 21, 919–926 (2004).
Ruff, J. A., Lodge, J. K. & Baker, L. G. Three galactose inducible promoters for use in C. neoformans var. grubii. Fungal Genet. Biol. 46, 9–16 (2009).
Acknowledgements
The authors thank all members of the Cowen laboratory for helpful discussions. L.E.C. is supported by the Canadian Institutes of Health Research Foundation Grant (FDN-154288), National Institutes of Health (NIH) National Institute of Allergy and Infectious Diseases (NIAID) R01 Grants (1R01AI127375-01, 1R01AI120958-01A1) and an NIH NIAID R21 Grant (1R21AI141080-01), is a Canada Research Chair (Tier 1) in Microbial Genomics & Infectious Disease and is co-director of the CIFAR Fungal Kingdom: Threats & Opportunities programme.
Author information
Authors and Affiliations
Contributions
K.R.I., N.M.R. and C.F. researched data for the article; K.R.I., N.M.R., C.F., N.R. and L.E.C. made substantial contributions to discussion of the content; K.R.I. and N.M.R wrote the article; and C.F., N.R. and L.E.C. reviewed/edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
L.E.C. is a co-founder and shareholder in Bright Angel Therapeutics, a platform company for development of novel antifungal therapeutics, and is a consultant for Boragen, a small-molecule development company focused on leveraging the unique chemical properties of boron chemistry for crop protection and animal health. K.R.I., N.M.R., C.F. and N.R. declare no competing interests.
Additional information
Peer review information
Nature Reviews Microbiology thanks K. Nielsen (who co-reviewed with K. Jackson), T. Bicanic and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Iyer, K.R., Revie, N.M., Fu, C. et al. Treatment strategies for cryptococcal infection: challenges, advances and future outlook. Nat Rev Microbiol 19, 454–466 (2021). https://doi.org/10.1038/s41579-021-00511-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-021-00511-0
This article is cited by
-
Deciphering antifungal and antibiofilm mechanisms of isobavachalcone against Cryptococcus neoformans through RNA-seq and functional analyses
Microbial Cell Factories (2024)
-
Brain glucose induces tolerance of Cryptococcus neoformans to amphotericin B during meningitis
Nature Microbiology (2024)
-
A dual-targeting antifungal is effective against multidrug-resistant human fungal pathogens
Nature Microbiology (2024)
-
Clinical treatment of cryptococcal meningitis: an evidence-based review on the emerging clinical data
Journal of Neurology (2024)
-
Immunoinformatic-guided designing and evaluating protein and mRNA-based vaccines against Cryptococcus neoformans for immunocompromised patients
Journal of Genetic Engineering and Biotechnology (2023)
