
Abstract
The SARS-CoV-2 pandemic and its unprecedented global societal and economic disruptive impact has marked the third zoonotic introduction of a highly pathogenic coronavirus into the human population. Although the previous coronavirus SARS-CoV and MERS-CoV epidemics raised awareness of the need for clinically available therapeutic or preventive interventions, to date, no treatments with proven efficacy are available. The development of effective intervention strategies relies on the knowledge of molecular and cellular mechanisms of coronavirus infections, which highlights the significance of studying virus–host interactions at the molecular level to identify targets for antiviral intervention and to elucidate critical viral and host determinants that are decisive for the development of severe disease. In this Review, we summarize the first discoveries that shape our current understanding of SARS-CoV-2 infection throughout the intracellular viral life cycle and relate that to our knowledge of coronavirus biology. The elucidation of similarities and differences between SARS-CoV-2 and other coronaviruses will support future preparedness and strategies to combat coronavirus infections.
Similar content being viewed by others
Introduction
Coronaviruses (CoVs) are a highly diverse family of enveloped positive-sense single-stranded RNA viruses. They infect humans, other mammals and avian species, including livestock and companion animals, and are therefore not only a challenge for public health but also a veterinary and economic concern. Within the order of Nidovirales and the suborder of Coronavirineae lies the family Coronaviridae. The latter is further specified into the subfamily of Orthocoronavirinae, which consists of four genera: alphacoronavirus, betacoronavirus, gammacoronavirus and deltacoronavirus. Whereas alphacoronaviruses and betacoronaviruses exclusively infect mammalian species, gammacoronaviruses and deltacoronaviruses have a wider host range that includes avian species. Human and animal coronavirus infections mainly result in respiratory and enteric diseases1,2.
Human coronaviruses, such as HCoV-229E and HCoV-OC43, have long been known to circulate in the population and they, together with the more recently identified HCoV-NL63 and HCoV-HKU1, cause seasonal and usually mild respiratory tract infections associated with symptoms of the ‘common cold’. In strong contrast, severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV-2, which have emerged in the human population over the past 20 years, are highly pathogenic. By infecting bronchial epithelial cells, pneumocytes and upper respiratory tract cells in humans, SARS-CoV, MERS-CoV and SARS-CoV-2 infections can develop into severe, life-threatening respiratory pathologies and lung injuries for which no specific prophylactic or therapeutic treatment has been approved to date.
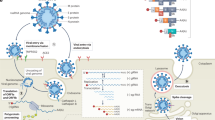
The initial steps of coronavirus infection involve the specific binding of the coronavirus spike (S) protein to the cellular entry receptors, which have been identified for several coronaviruses and include human aminopeptidase N (APN; HCoV-229E), angiotensin-converting enzyme 2 (ACE2; HCoV-NL63, SARS-CoV and SARS-CoV-2) and dipeptidyl peptidase 4 (DPP4; MERS-CoV). The expression and tissue distribution of entry receptors consequently influence viral tropism and pathogenicity. During the intracellular life cycle (Fig. 1), coronaviruses express and replicate their genomic RNA to produce full-length copies that are incorporated into newly produced viral particles. Coronaviruses possess remarkably large RNA genomes flanked by 5′ and 3′ untranslated regions that contain cis-acting secondary RNA structures essential for RNA synthesis. At the 5′ end, the genomic RNA features two large open reading frames (ORFs; ORF1a and ORF1b) that occupy two-thirds of the capped and polyadenylated genome. ORF1a and ORF1b encode 15–16 non-structural proteins (nsp), of which 15 compose the viral replication and transcription complex (RTC) that includes, amongst others, RNA-processing and RNA-modifying enzymes and an RNA proofreading function necessary for maintaining the integrity of the >30 kb coronavirus genome3. ORFs that encode structural proteins and interspersed ORFs that encode accessory proteins are transcribed from the 3′ one-third of the genome to form a nested set of subgenomic mRNAs (sg mRNAs). Coronavirus accessory proteins are highly variable sets of virus-specific proteins that display limited conservation even within individual species but they are principally thought to contribute to modulating host responses to infection and are determinants of viral pathogenicity4,5. Nevertheless, the molecular functions of many accessory proteins remain largely unknown owing to the lack of homologies to accessory proteins of other coronaviruses or to other known proteins6.

a | The coronavirus virion consists of structural proteins, namely spike (S), envelope (E), membrane (M), nucleocapsid (N) and, for some betacoronaviruses, haemagglutinin-esterase (not shown). The positive-sense, single-stranded RNA genome (+ssRNA) is encapsidated by N, whereas M and E ensure its incorporation in the viral particle during the assembly process. S trimers protrude from the host-derived viral envelope and provide specificity for cellular entry receptors. b | Coronavirus particles bind to cellular attachment factors and specific S interactions with the cellular receptors (such as angiotensin-converting enzyme 2 (ACE2)), together with host factors (such as the cell surface serine protease TMPRSS2), promote viral uptake and fusion at the cellular or endosomal membrane. Following entry, the release and uncoating of the incoming genomic RNA subject it to the immediate translation of two large open reading frames, ORF1a and ORF1b. The resulting polyproteins pp1a and pp1ab are co-translationally and post-translationally processed into the individual non-structural proteins (nsps) that form the viral replication and transcription complex. Concordant with the expression of nsps, the biogenesis of viral replication organelles consisting of characteristic perinuclear double-membrane vesicles (DMVs), convoluted membranes (CMs) and small open double-membrane spherules (DMSs) create a protective microenvironment for viral genomic RNA replication and transcription of subgenomic mRNAs (sg mRNAs) comprising the characteristic nested set of coronavirus mRNAs. Translated structural proteins translocate into endoplasmic reticulum (ER) membranes and transit through the ER-to-Golgi intermediate compartment (ERGIC), where interaction with N-encapsidated, newly produced genomic RNA results in budding into the lumen of secretory vesicular compartments. Finally, virions are secreted from the infected cell by exocytosis. Key steps inhibited by compounds that are currently being validated and which represent attractive antiviral targets are highlighted in red. An, 3′ polyA sequence; cap, 5′ cap structure; dsRNA, double-stranded RNA; L, leader sequence; RdRP, RNA-dependent RNA polymerase.
Despite the previous public health emergencies caused by the SARS-CoV and MERS-CoV outbreaks and the impact of the ongoing SARS-CoV-2 pandemic on society and human health, intervention strategies to combat coronavirus infections are only in their early stages and await proof of clinical efficacy. Their development intimately relies on the deepened understanding of basic mechanisms of coronavirus gene functions as well as of the molecular interactions with host factors. Since the discovery of the first coronavirus (avian infectious bronchitis virus) in the 1930s7 and the discovery of the first human coronaviruses (HCoV-229E and HCoV-OC43) in the 1960s8,9, the coronavirus research field has made substantial progress in understanding the basic principles of coronavirus replication and pathogenesis (Box 1). This advancement was accelerated after the emergence of SARS-CoV in 2002 and MERS-CoV in 2012 and has broadened our view on coronaviruses as zoonotic pathogens that can severely affect human health. Moreover, the unprecedented speed and technical progress of coronavirus research that has become evident in a few months after the appearance of SARS-CoV-2 at the end of 2019 has led to a rapidly growing understanding of this newly emerging pathogen and of its associated disease, COVID-19. In this Review, we discuss key aspects of coronavirus biology and their implications for SARS-CoV-2 infections as well as the treatment and prevention strategies.

Entry of coronaviruses
Coronavirus S proteins are homotrimeric class I fusion glycoproteins that are divided into two functionally distinct parts (S1 and S2) (Fig. 2). The surface-exposed S1 contains the receptor-binding domain (RBD) that specifically engages the host cell receptor, thereby determining virus cell tropism and pathogenicity. The transmembrane S2 domain contains heptad repeat regions and the fusion peptide, which mediate the fusion of viral and cellular membranes upon extensive conformational rearrangements10,11,12. Shortly after the 2002–2003 SARS-CoV outbreak, ACE2 was identified as the functional receptor that enables infection by SARS-CoV13. The high genomic and structural homology between the S proteins of SARS-CoV and SARS-CoV-2 (76% amino acid identity) supported the identification of ACE2 as the cell-surface receptor for SARS-CoV-2 (refs12,14,15,16). Remarkably, essential SARS-CoV contact residues that interact with ACE2 were highly conserved in SARS-CoV-2 as well as in members of the species Severe acute respiratory syndrome-related coronavirus that use ACE2 or have similar amino acid side chain properties14,15,17,18,19. These data were corroborated by the atomic resolution of the interface between the SARS-CoV-2 S protein and ACE2 (refs16,19,20,21). By contrast, the bat Severe acute respiratory syndrome-related coronavirus RaTG13 S sequence (93.1% nucleotide identity to SARS-CoV-2) shows conservation of only one out of six amino acids directly involved in ACE2 binding, even though, based on the entire genomic sequence, RaTG13 is the closest relative of SARS-CoV-2 known to date (96.2%)14 (Box 2).

a | Schematic illustration of coronavirus spike, indicating domain 1 and domain 2. The receptor-binding motif (RBM) is located on S1 and the fusion peptide (FP), heptad repeat 1 (HR1), HR2 and the transmembrane (TM) domains are located on S2. The cleavage sites are indicated. The colour code designates conserved spike regions surrounding the angiotensin-converting enzyme 2 (ACE2)-binding domain among severe acute respiratory syndrome-related coronaviruses (SARSr-CoVs) and high amino acid sequence variations within the site of receptor interaction. b | Amino acid alignment of human SARS-CoV-2 (Wuhan-Hu-1) and SARS-CoV (Frankfurt-1), bat (RaTG13, RmYN02, CoVZC45 and CoVZXC21) and pangolin (MP789, P1E) SARSr-CoVs. The spike gene sequence alignment was performed using MUSCLE and using the default settings and codon alignment, then translated into amino acids using MEGA7, version 7.0.26. The alignment was coloured according to percentage amino acid similarity with a Blosum 62 score matrix. The colour code designates conserved spike regions surrounding the ACE2-binding domain among SARSr-CoVs and high amino acid sequence variations within the site of receptor interaction. The insertion of a polybasic cleavage site (PRRAR, amino acids 681 to 685) in Wuhan-Hu-1 is indicated, and similar insertions are depicted in bat SARSr-CoV RmYN02. c | Within the spike sequence, the ACE2 receptor-binding motif (amino acids 437 to 509, black line) is depicted. The spike contact residues for ACE2 interaction are marked with asterisks.
These data suggest that, much like during the evolution of SARS-CoV, frequent recombination events between severe acute respiratory syndrome-related coronaviruses that coexist in bats probably favoured the emergence of SARS-CoV-2 (ref.22). Indeed, predicted recombination breakpoints divide the S gene into three parts. The middle part of the S protein (amino acids 1,030–1,651, encompassing the RBD) is most similar to SARS-CoV and bat severe acute respiratory syndrome-related coronaviruses WIV1 and RsSHC014, all of which use human ACE2 as a cellular entry receptor23. However, the amino-terminal and carboxy-terminal parts of the SARS-CoV-2 S protein (amino acids 1–1,029 and 1,651–3,804, respectively) are more closely related to severe acute respiratory syndrome-related coronaviruses ZC45 and ZXC21. These observations highlight the importance of recombination as a general mechanism contributing to coronavirus diversity and might therefore drive the emergence of future pathogenic human coronaviruses from bat reservoirs. This emphasizes the need for surveillance to determine the breadth of diversity of severe acute respiratory syndrome-related coronaviruses, to evaluate how frequently recombination events take place in the field and to understand which virus variants have the potential to infect humans. Increased surveillance is thus instrumental to improve our preparedness for future outbreaks of severe acute respiratory syndrome-related coronaviruses.
Besides receptor binding, the proteolytic cleavage of coronavirus S proteins by host cell-derived proteases is essential to permit fusion24,25. SARS-CoV has been shown to use the cell-surface serine protease TMPRSS2 for priming and entry, although the endosomal cysteine proteases cathepsin B (CatB) and CatL can also assist in this process24,25,26,27,28. Concordantly, the simultaneous inhibition of TMPRSS2, CatB and CatL efficiently prevents SARS-CoV entry into in vitro cell cultures29. TMPRSS2 is expressed in the human respiratory tract and thus strongly contributes to both SARS-CoV spread and pathogenesis. Notably, SARS-CoV-2 entry relies mainly on TMPRSS2 rather than on CatB and CatL, as inhibition of TMPRSS2 was sufficient to prevent SARS-CoV-2 entry in lung cell lines and primary lung cells15,30. These data support the evaluation of the TMPRSS2 inhibitors camostat mesylate and nafamostat mesylate in clinical trials, since in vitro studies have demonstrated their potent antiviral activity against emerging coronaviruses, including SARS-CoV-2 (refs29,31,32).
Given these similarities in receptor usage and cleavage requirements, it is surprising that SARS-CoV and SARS-CoV-2 display marked differences in virus replication efficiency and spread. SARS-CoV primarily targets pneumocytes and lung macrophages in lower respiratory tract tissues, where ACE2 is predominantly expressed, consistent with the lower respiratory tract disease resulting from SARS-CoV infection and the limited viral spread33,34,35. By contrast, SARS-CoV-2 replicates abundantly in upper respiratory epithelia, where ACE2 is also expressed, and is efficiently transmitted36,37,38.
Different host cell tropism, replication kinetics and transmission of SARS-CoV and SARS-CoV-2 might be determined by S protein–ACE2 binding affinities. For example, it has been reported that the S protein and ACE2 binding affinity is correlated with disease severity in SARS-CoV infections18. The affinity of the SARS-CoV-2 RBD to ACE2 has been shown to be similar16,19 or stronger20,30 than that of the SARS-CoV RBD. However, the binding affinity of the entire SARS-CoV-2 S protein to ACE2 seems to be equal or lower than that of SARS-CoV, suggestive of a less exposed RBD16,28,30. In addition to ACE2, attachment and entry factors, such as cellular glycans and integrins or neuropilin 1, may also have an impact on the observed phenotypic differences of SARS-CoV and SARS-CoV-2 (refs39,40,41,42,43).
A peculiar feature of the SARS-CoV-2 S protein is the acquisition of a polybasic cleavage site (PRRAR) at the S1–S2 boundary, which permits efficient cleavage by the prototype proprotein convertase furin. Cleavage results in enhanced infection and has been proposed to be a key event in SARS-CoV-2 evolution as efficient S protein cleavage is required for successful infection and is a main determinant in overcoming species barriers10,11,12,15,16,28,30,44,45,46. This pre-processing of the SARS-CoV-2 S protein by furin may contribute to the expanded cell tropism and zoonotic potential and might increase transmissibility16,46. Importantly, such cleavage sites have not been identified in other members of the Sarbecovirus genus46. However, there are multiple instances of furin-like cleavage site acquisitions that occurred independently during coronavirus evolution and similar cleavage sites are present in other human coronaviruses such as HCoV-HKU1 (ref.47), HCoV-OC43 (ref.48) and MERS-CoV49. Recently, an independent insertion of amino acids (PAA) at the same region of the S protein has been identified in the bat coronavirus RmYN02 (ref.50). Such independent insertion events highlight the zoonotic potential of bat severe acute respiratory syndrome-related coronaviruses and may increase the possibility of future outbreaks.
The importance of coronavirus S protein-mediated receptor binding and temporally coordinated conformational rearrangements that result in membrane fusion make this process a prime target of innate and adaptive antiviral responses. Notably, a screen involving several hundred interferon-stimulated genes identified lymphocyte antigen 6 family member E (Ly6E) as a potent inhibitor of coronavirus fusion51. Ly6E-mediated inhibition of coronavirus entry was demonstrated for various coronaviruses, including SARS-CoV-2, and seems to have pivotal importance in protecting the haematopoietic immune cell compartment in a mouse model of coronavirus infection. Moreover, the exposure of S protein on the surface of the virion results in the induction of specific neutralizing humoral immune responses52. Coronavirus S proteins are heavily glycosylated, which promotes immune evasion by shielding epitopes from neutralizing antibodies16,53,54. Nevertheless, sera from patients with SARS and COVID-19 can neutralize SARS-CoV and SARS-CoV-2, respectively15,28. Several specific or cross-reactive antibodies that bind the SARS-CoV-2 S protein have been recently reported and their administration to infected patients could potentially provide immediate protection55,56,57,58. Human monoclonal antibodies from previous hybridoma collections from SARS-CoV S protein-immunized transgenic mice55 or from the memory B cell repertoire of convalescent patients with SARS and COVID-19 have been shown to either directly interfere with RBD–ACE2 interaction55,57,58,59 or to destabilize intermediate pre-fusion conformations upon binding different epitopes55,56. Taken together, the exploitation of a combination of multiple neutralizing antibodies that do not compete for overlapping epitopes may not only result in synergistic improvements but also impede the appearance of escape mutations.

Viral gene expression and RNA synthesis
Genome translation
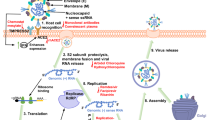
The release of the coronavirus genome into the host cell cytoplasm upon entry marks the onset of a complex programme of viral gene expression, which is highly regulated in space and time. The translation of ORF1a and ORF1b from the genomic RNA produces two polyproteins, pp1a and pp1ab, respectively. The latter results from a programmed –1 ribosomal frameshift at the short overlap of ORF1a and ORF1b4. Ribosome profiling revealed that the efficiency of the frameshift between ORF1a and ORF1b lies between 45% and 70% in the case of SARS-CoV-2 (ref.60), similar to that measured for mouse hepatitis virus (MHV)61. This determines the stoichiometry between pp1a and pp1ab, with pp1a being approximately 1.4–2.2 times more expressed than pp1ab60. Sixteen non-structural proteins are co-translationally and post-translationally released from pp1a (nsp1–11) and pp1ab (nsp1–10, nsp12–16) upon proteolytic cleavage by two cysteine proteases that are located within nsp3 (papain-like protease; PLpro) and nsp5 (chymotrypsin-like protease) (Fig. 3). The protease residing in nsp5 is frequently referred to as 3C-like protease (3CLpro), because of its similarities to the picornaviral 3C protease, or as main protease (Mpro), because it is responsible for proteolytic processing of the majority of polyprotein cleavage sites. Proteolytic release of nsp1 is known to occur rapidly62, which enables nsp1 to target the host cell translation machinery63,64,65. Nsp2–16 compose the viral RTC and are targeted to defined subcellular locations where interactions with host cell factors determine the course of the replication cycle66,67,68. Nsp2–11 are believed to provide the necessary supporting functions to accommodate the viral RTC, such as modulating intracellular membranes, host immune evasion and providing cofactors for replication, whereas nsp12–16 contain the core enzymatic functions involved in RNA synthesis, RNA proofreading and RNA modification4,67. RNA synthesis is performed by the nsp12 RNA-dependent RNA polymerase (RdRP) and its two cofactors nsp7 and nsp8, the latter with proposed primase or 3′-terminal adenylyltransferase activity4,67,69,70. Notably, nsp14 provides a 3′–5′ exonuclease activity that assists RNA synthesis with a unique RNA proofreading function71. The coronavirus capping machinery, which is not yet fully elucidated, is composed of nsp10, which functions as a cofactor, nsp13, which provides the RNA 5′-triphosphatase activity, and nsp14 and nsp16, which perform the functions of N7-methyltransferase and 2′-O-methyltransferase, respectively67,72,73,74. Notably, one key enzyme typically involved in the formation of the 5′ cap structure, the guanylyltransferase, has not yet been identified in coronaviruses.

Coronavirus polyprotein processing and domains of non-structural proteins (nsp) are illustrated for severe acute respiratory syndrome-related coronaviruses. Proteolytic cleavage of the polyproteins pp1a and pp1ab is facilitated by viral proteases residing in nsp3 (PLpro) and nsp5 (Mpro). PLpro proteolytically releases nsp1, nsp2, nsp3 and the amino terminus of nsp4 from the polyproteins pp1a and pp1ab (indicated by the blue arrows). Mpro proteolytically releases nsp5–16 and the carboxy terminus of nsp4 from the polyproteins pp1a and pp1ab (indicated by the red arrows)176. Conserved domains and known functions are schematically depicted for nsp1–16 (refs4,66,67,177). DMV, double-membrane vesicle; DPUP, Domain Preceding Ubl2 and PLpro; EndoU, endoribonuclease; ExoN, exoribonuclease; HEL, helicase; Mac I–III, macrodomains 1–3; Mpro, main protease; NiRAN, nidovirus RdRP-associated nucleotidyltransferase; NMT, guanosine N7-methyltransferase; OMT, ribose 2′-O-methyltransferase; PLpro, papain-like protease; Pr, primase or 3′-terminal adenylyl-transferase; RdRP, RNA-dependent RNA polymerase; TM, transmembrane domains; Ubl, ubiquitin-like domain; Y, Y and CoV-Y domain; ZBD, zinc-binding domain.
The establishment of the viral RTC is crucial for virus replication and thus a promising target for antivirals against SARS-CoV-2. One such target is Mpro, which resides in nsp5. Mpro releases the majority of nsps from the polyproteins and is essential for the viral life cycle. Furthermore, as Mpro is very sequence specific, compounds that structurally mimic those cleavage sites can specifically target the viral protease with little or no impact on host cellular proteases75,76,77. Based on structural analysis of the protein, multiple research groups have successfully developed lead compounds that block Mpro function in cell culture assays, thus providing frameworks that could aid in rapid drug discovery75,77.
RNA synthesis
Viral genomic replication is initiated by the synthesis of full-length negative-sense genomic copies, which function as templates for the generation of new positive-sense genomic RNA. These newly synthesized genomes are used for translation to generate more nsps and RTCs or are packaged into new virions. A hallmark of coronaviruses and most members of the order Nidovirales is the discontinuous viral transcription process, first proposed by Sawicki and Sawicki78, that produces a set of nested 3′ and 5′ co-terminal subgenomic RNAs (sgRNAs)78,79 (Fig. 4). During negative-strand RNA synthesis, the RTC interrupts transcription following the encounter of transcription regulatory sequences (TRSs) that are located upstream to most ORFs in the 3′ one-third of the viral genome. At these TRS elements, also called TRS ‘body’, the synthesis of the negative-strand RNA stops and is re-initiated at the TRS adjacent to a leader sequence (TRS-L) located at about 70 nucleotides from the 5′ end of the genome78,79,80,81,82. This discontinuous step of coronavirus RNA synthesis involves the interaction between complementary TRSs of the nascent negative strand RNA (negative-sense TRS body) and the positive strand genomic RNA (positive-sense TRS-L). Upon re-initiation of RNA synthesis at the TRS-L region, a negative strand copy of the leader sequence is added to the nascent RNA to complete the synthesis of negative-strand sgRNAs. The discontinuous step of negative strand RNA synthesis results in the production of a set of negative-strand sgRNAs that are then used as templates to synthesize a characteristic nested set of positive-sense sg mRNAs that are translated into structural and accessory proteins. Although the coronavirus sg mRNAs are structurally polycistronic, it is assumed that they are functionally monocistronic and that only the first ORF at the 5′ end, which is absent in the next smaller sgRNA, is translated from each sgRNA78,81.

Schematic depiction of coronaviral RNA synthesis. Full-length positive-sense genomic RNA is used as a template to produce both full-length negative-sense copies for genome replication and subgenomic negative-sense RNAs (–sgRNA) to produce the subgenomic mRNAs (sg mRNA). The negative strand RNA synthesis involving a template switch from a body transcription regulatory sequences (TRS-B) to the leader TRS (TRS-L) is illustrated to produce one sg mRNA. This process can take place at any TRS-B and will collectively result in the production of the characteristic nested set of coronaviral mRNAs.
The TRS elements for SARS-CoV-2 have already been determined by RNA sequencing analyses of viral RNAs80,83. Like for SARS-CoV, the consensus TRS core of SARS-CoV-2 is 5′-ACGAAC-3′ and eight sg mRNAs have been shown to be produced in SARS-CoV-2-infected cells (sg mRNAs 2–9). In addition to canonical sgRNAs, recent reports also determined the existence of numerous non-canonical RNA products of discontinuous transcription, including fusions of the 5′ leader sequence to unexpected 3′ sites, TRS-L independent long-distance fusions, and local fusions resulting in small deletions mainly in the structural and accessory genes60,80. However, it remains to be determined whether all of these non-canonical sgRNAs truly arise by discontinuous transcription or whether they represent RNAs that result from recombination. Nevertheless, similar findings were previously reported for other coronaviruses, including MHV61 and HCoV-229E81, which indicates an enhanced coding potential for coronaviruses80. Overall, these unexpected fusion events may drive coronavirus evolution through variant generation, and novel ORFs could encode additional accessory proteins that are involved in either viral replication or modulation of the host immune response60,80.
The RdRP residing in nsp12 is the centrepiece of the coronavirus RTC and has been suggested as a promising drug target as it is a crucial enzyme in the virus life cycle both for replication of the viral genome but also for transcription of sgRNAs. The structure of the SARS-CoV-2 RdRP nsp12 and its cofactors nsp7 and nsp8 has been elucidated and shows a high degree of conservation to the SARS-CoV structure69,84,85. The amino acid sequence of the SARS-CoV and SARS-CoV-2 RdRPs show a >95% similarity with most changes located in the nidovirus RdRP-associated nucleotidyltransferase domain, which, despite being a genetic marker of Nidovirales, has yet to be functionally elucidated69. The structural similarities of the RdRP active site, including conserved key amino acid residues, with other positive-sense RNA viruses suggest the possibility to repurpose known drugs that are effective against other RNA viruses69. One of the most promising candidates is the phosphoramidate remdesivir (RDV), which, in its triphosphate form, acts as a substrate for viral RdRPs and competes with ATP86. RDV has shown potential as an antiviral agent against a broad range of RNA viruses, including Filoviridae (for example, Ebola virus), Paramyxoviridae (for example, Nipah virus) and Pneumoviridae (for example, respiratory syncytial virus) as well as other coronaviruses, including SARS-CoV and MERS-CoV86,87. The RdRP of SARS-CoV-2 selectively incorporates RDV over ATP, which subsequently results in a delayed-chain termination86,88. In contrast to classic nucleoside analogues that lead to immediate termination of the synthesis reaction after incorporation, the RdRP continues for three nucleotides after RDV has been incorporated before chain termination. Nucleotide analogues like RDV may have limited efficacy owing to the proofreading function of the exonuclease domain contained in nsp14 (ExoN)89. The corrective function that is exerted by ExoN is not only responsible for maintaining the stability of the coronavirus genome but also enables the excision of erroneous mutagenic nucleotides71,89. The mode of action observed for RDV might be an explanation for its increased efficiency over other nucleoside analogues as the delayed-chain termination could lead to improved evasion from the proofreading function of nsp14. The current model suggests steric hindrance as a likely reason for termination, disturbing the positioning of the RNA and thus hampering the translocation to the next position86,88. RDV was shown to reduce virus replication of SARS-CoV-2 in vitro90 and was demonstrated to restrict clinical symptoms of SARS-CoV-2 in rhesus macaques upon early pre-symptomatic treatment91. However, a recent randomized, double-blind, placebo-controlled clinical trial in humans with severe COVID-19 showed limited clinical efficacy of RDV treatment92 and further studies will be necessary. Another promising candidate is the purine analogue favipiravir (FPV), which has been shown to effectively target multiple RNA viruses93. Although the mechanism of action is not yet completely understood, a recent study of the in vitro mechanism of FPV suggested a combination of chain termination, slowed RNA synthesis and lethal mutagenesis as the mode of action against SARS-CoV-2, which indicates that FPV might be used to effectively restrict viral replication93. Indeed, results of an experimental pilot study showed that using FPV as treatment against COVID-19 led to increased recovery and faster viral clearance times in treated patients compared to control treatments94. Clinical studies with both RDV and FPV are currently ongoing and will establish whether these compounds are effective antivirals to treat coronavirus infections93.
Expression of structural and accessory proteins
The ORFs encoding the structural proteins (that is, S protein, envelope (E) protein, membrane (M) protein and nucleocapsid (N) protein) are located in the 3′ one-third of coronavirus genomes. Interspersed between these ORFs are the ORFs encoding for so-called accessory proteins. The structural proteins of SARS-CoV-2 have not yet been assessed in terms of their role in virus assembly and budding. In general, coronavirus structural proteins assemble and assist in the budding of new virions at the endoplasmic reticulum (ER)-to-Golgi compartment that are suggested to exit the infected cell by exocytosis95,96,97. However, recent evidence shows that betacoronaviruses, including MHV and SARS-CoV-2, rather egress infected cells via the lysosomal trafficking pathway98. During this process, viral interference with lysosomal acidification, lysosomal enzyme activity and antigen presentation was demonstrated.
At least five ORFs encoding accessory genes have been reported for SARS-CoV-2: ORF3a, ORF6, ORF7a, ORF7b and ORF8 (GenBank entry NC_045512.2) as well as potentially ORF3b99 and ORF9b100, the latter of which is probably expressed as a result of leaky scanning of the sgRNA of the nucleocapsid protein80,99,101. In addition, ORF10 has been postulated to be located downstream of the N gene. However, not all of these ORFs have been experimentally verified yet and the exact number of accessory genes of SARS-CoV-2 is still debated80,102. For example, in the case of ORF10, recent sequencing data questioned whether ORF10 is actually expressed, as the corresponding sgRNA could only be detected once in the entire dataset80. Furthermore, using proteomics approaches, the ORF10 protein has not been found in infected cells100,102, whereas ribosome profiling data suggested that ORF10 may be translated60.
The accessory genes display a high variability among coronavirus groups and usually show no sequence similarity with other viral and cellular proteins. Although they are not required for virus replication in cell culture4,5, they are, to some extent, conserved within the respective virus species and are suspected to have important roles in the natural host. Indeed, in the case of SARS-CoV, it was shown that at least ORF3b, ORF6 and ORF9b function as interferon antagonists6,102,103,104. There are some notable differences between the accessory genes of SARS-CoV-2 and SARS-CoV, with the latter having a total of eight described accessory genes (ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8a, ORF8b and ORF9b). In SARS-CoV-2, ORF3b contains a premature stop codon and is thus substantially shorter than the SARS-CoV variant. Although there are indications that ORF3b could exhibit its interferon antagonistic function also in a truncated form99, it has not yet been found to be expressed at the protein level in virus-infected cells100,102. SARS-CoV-2 ORF8 shows an especially low homology to SARS-CoV ORF8. The coding sequence of SARS-CoV ORF8 went through a gradual deletion over the course of the SARS-CoV epidemic. Whereas the early isolates from human patients contained a full-length ORF8, a deletion of 29 nucleotides was observed in all SARS-CoV strains during the middle-to-late stages. This deletion caused the split of one full-length ORF8 into two truncated gene products, ORF8a and ORF8b. Furthermore, less frequent deletion events were also observed, including an 82-nucleotide deletion and a 415-nucleotide deletion, which led to a complete loss of ORF8 (refs105,106), suggesting a possible benefit of SARS-CoV ORF8 deletions in vivo. Notably, however, reconstitution of SARS-CoV ORF8 by reverse genetics was associated with slightly increased fitness in cell culture106. Recently, SARS-CoV-2 ORF8 was reported to bind to major histocompatibility complex and mediate its degradation in cell culture107. This indicates that SARS-CoV-2 ORF8 might mediate a form of immune evasion, which is not the case for the split SARS-CoV ORF8a or ORF8b107. Interestingly, a mutant SARS-CoV-2 strain found in Singapore displayed a deletion of 382 nucleotides in the region of ORF8, indeed spanning most of the ORF and the adjacent the TRS. This may indicate a tendency towards host adaption and decreased pathogenicity108 or, alternatively, that the ORF8 protein is dispensable in humans, whereas it is required in the natural host.
Replication compartments
Primary interactions between nsps and host cell factors during the early coronavirus replication cycle initiate the biogenesis of replication organelles66,109,110. Although mechanisms underlying replication organelle formation are not fully understood, the concerted role of the membrane-spanning nsp3, nsp4 and nsp6 has been implicated in diverting host endomembranes into replication organelles111,112,113. Detailed electron microscopy investigations have described the phenotypic appearance and extent of membrane modifications induced by coronaviruses to accommodate viral replication. Coronavirus infection, like many other positive-sense RNA viruses, manifests in the generation of ER-derived and interconnected perinuclear double-membrane structures such as double-membrane vesicles (DMVs), convoluted membranes and the recently discovered double-membrane spherules112,114,115,116. Interestingly, these structures are highly dynamic and develop during the viral life cycle114,117. Even though replicase subunits — notably SARS-CoV nsp3, nsp5 and nsp8 — have been shown to be anchored on convoluted membranes, to date, the specific location of viral RNA synthesis remains the most intriguing unanswered question114,117. Double-stranded RNA (dsRNA), commonly considered as viral replication intermediates, segregates into the DMV interior97,114,118. Consistently, viral RNA synthesis was shown to occur within DMVs by using metabolic labelling of newly synthesized viral RNA in the context of SARS-CoV, MERS-CoV and infectious bronchitis virus infections116. Although, until recently, no openings towards the cytosol have been observed97,114, molecular pores involving nsp3 were demonstrated to span DMVs in MHV-infected cells118. These newly identified structures, which were also observed in SARS-CoV-2-infected cells, provide a connection between the dsRNA-containing DMV interior and the cytosol, thereby hypothetically rendering newly synthesized viral RNAs available for translation and encapsidation into nascent virions118. They also provide new opportunities to experimentally address the origin, fate and trafficking routes of viral RNAs contained in DMVs.
Replication organelles are a conserved and characteristic feature of coronavirus replication and, consistent with suggested roles of rewired intracellular membranes in the context of other positive-sense RNA virus infections, they provide a propitious niche with adequate concentrations of macromolecules necessary for RNA synthesis while preventing the exposure of viral replication intermediates to cytosolic innate immune sensors95,119. The functional dissection of coronavirus replication organelles has proven challenging as their contributions to viral fitness and pathogenesis are indistinguishable from functions provided by enzymes of the RTC, which are anchored on the membranes of the replication organelle120,121,122. Nevertheless, recent studies revealed the overall composition of the coronavirus RTC, with nsp2–nsp16 and the nucleocapsid protein comprising the viral components68,123. Moreover, several genetic and proteomic screening approaches aimed at deciphering essential coronavirus–host interactions and the RTC microenvironment identified supportive roles of the ER and the early secretory system as well as related vesicular trafficking pathways for efficient replication68,124,125,126 and provided a comprehensive list of cellular proteins that are in close proximity to the coronaviral RTC68,127,128,129. Collectively, these studies, in combination with advanced electron microscopy, provide ground for future studies to dissect the microarchitecture of the coronaviral RTC in relation to remodelled ER-derived membranes and to functionally link those structures to processes taking place in close proximity to the RTC such as translation, replication and transcription of viral RNA.
Virus–host interactions and host response
A successful intracellular coronavirus life cycle invariably relies on critical molecular interactions with host proteins that are repurposed to support the requirements of the virus. This includes host factors required for virus entry (such as the entry receptor and host cell proteases), factors required for viral RNA synthesis and virus assembly (such as ER and Golgi components and associated vesicular trafficking pathways) and factors required for the translation of viral mRNAs (such as critical translational initiation factors)68,124,125,126,127,128,129.
A first systematic expression study of SARS-CoV-2 proteins and subsequent affinity purification followed by mass spectrometry identified more than 300 potential coronavirus–host protein interactions. Although outside the context of a SARS-CoV-2 infection, interactors of individually overexpressed SARS-CoV-2 proteins uncovered several cellular processes reminiscent of those of other coronaviruses that are likely to also be involved in the SARS-CoV-2 life cycle130. Importantly, 69 compounds, either FDA approved or at different stages of clinical development, that target putative SARS-CoV-2 protein interactors were foregrounded, a subset of which efficiently prevented SARS-CoV-2 replication in vitro. These systematic screening approaches of large compound libraries that target host proteins provide means of rapidly identifying antiviral (repurposed) drugs and accelerated clinical availability131. However, a detailed functional characterization of conserved host pathways that promote coronavirus replication will guide the development of efficacious targeted therapeutics against coronavirus infections.
In addition, coronaviruses efficiently evade innate immune responses. Virus–host interactions in this context are multifaceted and include strategies to hide viral pathogen-associated molecular patterns, such as replication intermediates (dsRNA), that may be sensed by cytosolic pattern recognition receptors132,133. DMVs have been proposed to shield dsRNA and sites of viral RNA synthesis; however, experimental proof supporting this idea has not yet been obtained. The coronaviral RTC also contributes to innate immune evasion through several nsp-encoded functions. These include PLpro-mediated deubiquitylation activity134,135, de-ADP-ribosylation by nsp3-encoded macro domains136, RNA-modifying enzymatic activities such as 5′-cap N7-methylation and 2′-O-methylation (nsp14 and nsp16, respectively)74,137,138, and exonuclease139 and endoribonuclease140,141 activities (nsp14 and nsp15, respectively). Although these mechanisms have been elucidated in considerable detail for several prototype coronaviruses, data for SARS-CoV-2 are not yet available.
Besides the well-conserved functions residing in the nsps that comprise the RTC, additional mechanisms to counteract innate immune responses are known for coronaviruses. For example, nsp1 is rapidly proteolytically released from pp1a and pp1ab and affects cellular translation in the cytoplasm to favour viral mRNAs over cellular mRNA, and thereby also decreases the expression of type I and III interferons and of other host proteins of the innate immune response. Indeed, a first structural and functional analysis of SARS-CoV-2 nsp1 showed binding of nsp1 to ribosomes and nsp1-mediated impairment of translation64. Furthermore, several coronavirus accessory proteins are known to affect innate immune responses, most prominently MHV NS2 and MERS-CoV ORF4b proteins, that have 2′,5′-phosphodiesterase activity to antagonize the OAS–RNase L pathway142. Although this activity is not predicted for any accessory protein of SARS-CoV or SARS-CoV-2, the ORF3b, ORF6 and N proteins of SARS-CoV have been shown to interfere at multiple levels of the cellular interferon signalling pathway, thereby efficiently inhibiting innate immune responses103. Interestingly, an initial report recently suggested a similar role of SARS-CoV-2 ORF3b as an effective interferon antagonist99. Although this property remains to be demonstrated in the context of viral infection, these results suggest that SARS-CoV-2 shares some preserved accessory protein activities with SARS-CoV that interfere with antiviral host responses.
Coronavirus biology and COVID-19
Our knowledge on SARS-CoV-2 replication, gene function and host interactions is accumulating at unprecedented speed and it will be important to link those findings to the disease induced by SARS-CoV-2 infection, COVID-19. Thus, there is a need to establish experimental systems, such as representative animal models to study the transmission and pathogenicity of SARS-CoV-2, primary airway epithelial cultures and organoids to study SARS-CoV-2 replication and host responses to infection in relevant cell types, and reverse genetics systems to study the specific gene functions of SARS-CoV-2 (Table 1). These tools will be instrumental to understanding how the molecular biology of SARS-CoV-2 affects the development of COVID-19.
As we currently understand, SARS and COVID-19 are a consequence of virus-encoded functions and delayed interferon responses and, in severe cases, they are associated with dysregulated immune responses and immunopathologies143,144. Indeed, rapid and uncontrolled viral replication of SARS-CoV has been demonstrated to evade the host innate immune activation during its initial steps. As a consequence, the increase in aberrant pro-inflammatory responses and immune cell infiltration in the lungs provoke tissue damage and contribute to the clinical manifestation of SARS145.
Consistently, host responses, such as cytokine expression, that are known to drive inflammation and immunopathologies have been assessed in studies that revealed that SARS-CoV-2 considerably affects the transcriptional landscape of infected cells by inducing inflammatory cytokine and chemokine signatures38,146,147. Although interferon responses have been shown to potently impair SARS-CoV-2 replication, only moderate induction of type I interferon, type II interferon and interferon-stimulated genes was reported38,147.
Together, these effects may translate into strong and dysregulated pro-inflammatory responses, while cells display low innate antiviral defence activation as revealed by single-cell transcriptomic studies of nasopharyngeal and bronchial patient samples38,146,148,149. In severe COVID-19 cases, as opposed to mild cases, aberrant recruitment of inflammatory macrophages and infiltration of T lymphocytes, including cytotoxic T cells, as well as of neutrophils have been measured in the lung146,149. The accumulating evidence of dysregulated pro-inflammatory responses during SARS-CoV-2 infections has led to the use of immune modulators to inhibit hyperactivated pathogenic immune responses143,144,150,151.
Conclusions
In contrast to the SARS-CoV epidemic of almost 20 years ago, improved technologies, such as transcriptomics, proteomics, single-cell RNA sequencing, global single-cell profiling of patient samples, advanced primary 3D cell cultures and rapid reverse genetics, have been valuable tools to understand and tackle SARS-CoV-2 infections. Furthermore, several existing animal models initially established for SARS-CoV are applicable to study SARS-CoV-2 and will help to identify the critical viral and host factors that impact on COVID-19. We need to understand why SARS-CoV-2, in contrast to SARS-CoV, is replicating so efficiently in the upper respiratory tract and which viral and host determinants are decisive on whether COVID-19 patients will develop mild or severe disease152,153,154. Finally, we need to put the first encouraging studies on SARS-CoV-2 into the context of coronavirus biology to develop efficacious strategies to treat COVID-19 and to develop urgently needed vaccines.
References
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020). Most recent update of the coronavirus taxonomy by the International Committee on Taxonomy of Viruses after the emergence of SARS-CoV-2. Coined the virus name SARS-CoV-2.
Corman, V. M., Muth, D., Niemeyer, D. & Drosten, C. Hosts and sources of endemic human coronaviruses. Adv. Virus Res. 100, 163–188 (2018).
Gorbalenya, A. E., Enjuanes, L., Ziebuhr, J. & Snijder, E. J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 117, 17–37 (2006).
Perlman, S. & Netland, J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7, 439–450 (2009).
Masters, P. S. The molecular biology of coronaviruses. Adv. Virus Res. 65, 193–292 (2006).
Liu, D. X., Fung, T. S., Chong, K. K. L., Shukla, A. & Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 109, 97–109 (2014).
Schalk, A. & Hawn, M. C. An apparently new respiratory disease of baby chicks. J. Am. Vet. Med. Assoc. 78, 413–423 (1931).
Hamre, D. & Procknow, J. J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. Med. 121, 190–193 (1966).
McIntosh, K., Dees, J. H., Becker, W. B., Kapikian, A. Z. & Chanock, R. M. Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc. Natl Acad. Sci. USA 57, 933–940 (1967).
Tortorici, M. A. & Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 105, 93–116 (2019).
Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 3, 237–261 (2016).
Letko, M., Marzi, A. & Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 5, 562–569 (2020). First functional assessment of the interaction of the SARS-CoV-2 Spike protein receptor binding domain with the cellular receptor ACE2.
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020). Functional assessment of SARS-CoV-2 entry into host cells highlighting the importance of the ACE2 receptor and the cellular protease TMPRSS2 as entry factors.
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292 (2020). Recommended resource describing key aspects of SARS-CoV-2 Spike features.
Li, F., Li, W., Farzan, M. & Harrison, S. C. Structural biology: structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309, 1864–1868 (2005).
Li, W. et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 24, 1634–1643 (2005).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature https://doi.org/10.1038/s41586-020-2180-5 (2020).
Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature https://doi.org/10.1038/s41586-020-2179-y (2020).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Boni, M. F. et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. https://doi.org/10.1038/s41564-020-0771-4 (2020).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature https://doi.org/10.1038/s41586-020-2008-3 (2020).
Gierer, S. et al. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J. Virol. 87, 5502–5511 (2013).
Matsuyama, S. et al. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 84, 12658–12664 (2010).
Simmons, G. et al. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl Acad. Sci. USA 102, 11876–11881 (2005).
Belouzard, S., Chu, V. C. & Whittaker, G. R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl Acad. Sci. USA 106, 5871–5876 (2009).
Ou, X. et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 11, 1620 (2020).
Kawase, M., Shirato, K., van der Hoek, L., Taguchi, F. & Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 86, 6537–6545 (2012).
Shang, J. et al. Cell entry mechanisms of SARS-CoV-2. Proc. Natl Acad. Sci. USA 117, 11727–11734 (2020).
Yamamoto, M. et al. Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome Coronavirus s protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob. Agents Chemother. 60, 6532–6539 (2016).
Hoffmann, M. et al. Nafamostat mesylate blocks activation of SARS-CoV-2: New treatment option for COVID-19. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.00754-20 (2020).
Hamming, I. et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203, 631–637 (2004).
Shieh, W. J. et al. Immunohistochemical, in situ hybridization, and ultrastructural localization of SARS-associated coronavirus in lung of a fatal case of severe acute respiratory syndrome in Taiwan. Hum. Pathol. 36, 303–309 (2005).
Leung, G. M. et al. The epidemiology of severe acute respiratory syndrome in the 2003 Hong Kong epidemic: an analysis of all 1755 patients. Ann. Intern. Med. 141, 662–673 (2004).
Wölfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature https://doi.org/10.1038/s41586-020-2196-x (2020). Demonstrates replication of SARS-CoV-2 in the upper respiratory tract and provides detailed analysis of virus production and shedding over the course of COVID-19.
Ziegler, C. G. K. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035 (2020).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell https://doi.org/10.1016/j.cell.2020.04.026 (2020).
Sigrist, C. J., Bridge, A. & Le Mercier, P. A potential role for integrins in host cell entry by SARS-CoV-2. Antiviral Res. https://doi.org/10.1016/j.antiviral.2020.104759 (2020).
Li, W. et al. Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc. Natl Acad. Sci. USA 114, E8508–E8517 (2017).
de Haan, C. A. M. et al. Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J. Virol. 79, 14451–14456 (2005).
Cantuti-Castelvetri, L. et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and provides a possible pathway into the central nervous system. Preprint at bioRxiv https://doi.org/10.1101/2020.06.07.137802 (2020).
Daly, J. L. et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Preprint at bioRxiv https://doi.org/10.1101/2020.06.05.134114 (2020).
Menachery, V. D. et al. Trypsin treatment unlocks barrier for zoonotic bat coronavirus infection. J. Virol. 94, e01774–19 (2020). Highlights the requirements for cross-species transmission and the zoonotic potential of coronaviruses.
Coutard, B. et al. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 176, 104742 (2020).
Hoffmann, M., Kleine-Weber, K.-W. & Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 78, 779–784.e5 (2020).
Chan, C. M. et al. Spike protein, S, of human coronavirus HKU1: role in viral life cycle and application in antibody detection. Exp. Biol. Med. 233, 1527–1536 (2008).
Le Coupanec, A. et al. Cleavage of a neuroinvasive human respiratory virus spike glycoprotein by proprotein convertases modulates neurovirulence and virus spread within the central nervous system. PLoS Pathog. 11, e1005261 (2015).
Mille, J. K., Whittaker, G. R., Millet, J. K. & Whittaker, G. R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl Acad. Sci. USA 111, 15214–15219 (2014).
Zhou, H. et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. https://doi.org/10.1016/j.cub.2020.05.023 (2020).
Pfaender, S. et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol. https://doi.org/10.1038/s41564-020-0769-y (2020).
Buchholz, U. J. et al. Contributions of the structural proteins of severe respiratory syndrome coronavirus to protective immunity. Proc. Natl Acad. Sci. USA 101, 9804–9809 (2004).
Walls, A. C. et al. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat. Struct. Mol. Biol. 23, 899–905 (2016).
Watanabe, Y., Allen, J. D., Wrapp, D., McLellan, J. S. & Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science https://doi.org/10.1126/science.abb9983 (2020).
Pinto, D. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020).
Wang, C. et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 11, 2251 (2020).
Cao, Y. et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182, 73–84 (2020).
Wrapp, D. et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 181, 1436–1441 (2020).
Wu, Y. et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 368, 1274–1278 (2020).
Finkel, Y. et al. The coding capacity of SARS-CoV-2. Preprint at bioRxiv https://doi.org/10.1101/2020.05.07.082909 (2020).
Irigoyen, N. et al. High-resolution analysis of coronavirus gene expression by RNA Sequencing and ribosome profiling. PLoS Pathog. 12, e1005473 (2016). First ribosomal profiling analysis on cells infected by a coronavirus, providing a comprehensive overview on coronavirus gene expression.
Denison, M. R. & Perlman, S. Translation and processing of mouse hepatitis virus virion RNA in a cell-free system. J. Virol. 60, 12–18 (1986).
Kamitani, W. et al. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl Acad. Sci. USA 103, 12885–12890 (2006).
Thoms, M. et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science https://doi.org/10.1126/science.abc8665 (2020).
Schubert, K. et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. https://doi.org/10.1038/s41594-020-0511-8 (2020). Together with Thoms, M. et al., this article demonstrates binding of SARS-CoV-2 nsp1 to the 40S ribosomal subunit mRNA entry channel that leads to translation inhibition.
Sims, A. C., Ostermann, J. & Denison, M. R. Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes. J. Virol. 74, 5647–5654 (2000).
Snijder, E. J., Decroly, E. & Ziebuhr, J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv. Virus Res. 96, 59–126 (2016).
V’kovski, P. et al. Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity-labeling. eLife 8, e42037 (2019).
Gao, Y. et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 368, 779–782 (2020). Elucidates the molecular structure of the RNA-dependent RNA polymerase of SARS-CoV-2.
Tvarogová, J. et al. Identification and characterization of a human coronavirus 229E Nonstructural protein 8-associated RNA 3′-terminal adenylyltransferase activity. J. Virol. https://doi.org/10.1128/JVI.00291-19 (2019).
Eckerle, L. D., Lu, X., Sperry, S. M., Choi, L. & Denison, M. R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 81, 12135–12144 (2007). Experimental evidence for the involvement of the nsp14 exonuclease activity in coronavirus genome integrity, which led, in subsequent studies, to the concept of an RNA proofreading function during coronavirus RNA synthesis.
Ivanov, K. A. & Ziebuhr, J. Human coronavirus 229E nonstructural protein 13: characterization of duplex-unwinding, nucleoside triphosphatase, and RNA 5′-triphosphatase activities. J. Virol. 78, 7833–7838 (2004).
Chen, Y. et al. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 7, e1002294 (2011).
Chen, Y. et al. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl Acad. Sci. USA 106, 3484–3489 (2009). Discovery of N7 methyltransferase in SARS-CoV nsp14 as part of its RNA capping machinery.
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 368, 409–412 (2020).
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300, 1763–1767 (2003).
Jin, Z. et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 582, 289–293 (2020).
Sawicki, S. G. & Sawicki, D. L. Coronaviruses use discontinuous extension for synthesis of subgenome-length negative strands. Adv. Exp. Med. Biol. 380, 499–506 (1995). Description of the discontinuous RNA synthesis mechanism used to produce coronaviral subgenomic RNAs. Established the existence and functional importance of viral subgenomic negative-sense RNAs.
Sola, I., Almazán, F., Zúñiga, S. & Enjuanes, L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu. Rev. Virol. 2, 265–288 (2015).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921 (2020).
Viehweger, A. et al. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 29, 1545–1554 (2019).
Di, H., McIntyre, A. A. & Brinton, M. A. New insights about the regulation of Nidovirus subgenomic mRNA synthesis. Virology 517, 38–43 (2018).
Thao, T. T. N. et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature https://doi.org/10.1038/s41586-020-2294-9 (2020).
Yin, W. et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368, 1499–1504 (2020).
Hillen, H. S. et al. Structure of replicating SARS-CoV-2 polymerase. Nature 584, 154–156 (2020).
Gordon, C. J. et al. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 295, 6785–6797 (2020). Describes insights of the mechanism of action of remdesivir such as the delayed chain termination that results from incorporation of remdesivir into the SARS-CoV-2 nascent RNA by the RNA-dependent RNA polymerase.
Sheahan, T. P. et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl Med. 9, eaal3653 (2017).
Wang, Q. et al. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell 182, 417–428 (2020).
Ferron, F. et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl Acad. Sci. USA 115, E162–E171 (2017).
Wang, M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 30,, 269–271 (2020).
Williamson, B. N. et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Preprint at bioRxiv https://doi.org/10.1101/2020.04.15.043166 (2020).
Wang, Y. et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 395, 1569–1578 (2020).
Shannon, A. et al. Favipiravir strikes the SARS-CoV-2 at its Achilles heel, the RNA polymerase. Preprint at bioRxiv https://doi.org/10.1101/2020.05.15.098731 (2020).
Cai, Q. et al. Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering https://doi.org/10.1016/j.eng.2020.03.007 (2020).
Stertz, S. et al. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 361, 304–315 (2007).
de Haan, C. A. & Rottier, P. J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 64, 165–230 (2005).
Klein, S. et al. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Preprint at bioRxiv https://doi.org/10.1101/2020.06.23.167064 (2020). Detailed ultrastructural investigation of the SARS-CoV-2 replication structures and assembly process, notably detecting viral (presumably double-stranded) RNA inside DMVs.
Ghosh, S. et al. β-Coronaviruses use lysosomal organelles for cellular egress. Preprint at bioRxiv https://doi.org/10.1101/2020.07.25.192310 (2020).
Konno, Y. et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is further increased by a naturally occurring elongation variant. Preprint at bioRxiv https://doi.org/10.1101/2020.05.11.088179 (2020).
Bojkova, D. et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583, 469–230 (2020).
Xu, K. et al. Severe acute respiratory syndrome coronavirus accessory protein 9b is a virion-associated protein. Virology 388, 279–285 (2009).
Davidson, A. D. et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 12, 68 (2020).
Kopecky-Bromberg, S. A., Martínez-Sobrido, L., Frieman, M., Baric, R. A. & Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 81, 548–557 (2007).
Shi, C.-S. et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 193, 3080–3089 (2014).
Chinese SARS Molecular Epidemiology Consortium. Molecular evolution of the SARS coronavirus, during the course of the SARS epidemic in China. Science 303, 1666–1669 (2004).
Muth, D. et al. Attenuation of replication by a 29 nucleotide deletion in SARS-coronavirus acquired during the early stages of human-to-human transmission. Sci. Rep. 8, 15177 (2018).
Zhang, Y. et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through potently downregulating MHC-I. Preprint at bioRxiv https://doi.org/10.1101/2020.05.24.111823 (2020).
Su, Y. C. F. et al. Discovery and genomic characterization of a 382-nucleotide deletion in ORF7B and orf8 during the early evolution of SARS-CoV-2. MBio 11, e01610–20 (2020).
Brockway, S. M., Lu, X. T., Peters, T. R., Dermody, T. S. & Denison, M. R. Intracellular localization and protein interactions of the gene 1 protein p28 during mouse hepatitis virus replication. J. Virol. 78, 11551–11562 (2004).
Gosert, R., Kanjanahaluethai, A., Egger, D., Bienz, K. & Baker, S. C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 76, 3697–3708 (2002). First evidence that coronaviruses induce double-membrane structures in the cytoplasm of infected cells.
Angelini, M. M., Akhlaghpour, M., Neuman, B. W. & Buchmeier, M. J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. MBio 4, e00524–13 (2013).
Oudshoorn, D. et al. Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. MBio https://doi.org/10.1128/mBio.01658-17 (2017).
Lundin, A. et al. Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the middle East respiratory syndrome virus. PLoS Pathog. 10, e1004166 (2014).
Knoops, K. et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6, e226 (2008). Landmark study revealing the extent of SARS-CoV membranous replication compartments by electron tomography and the localization of viral nsps and dsRNA in infected cells.
Maier, H. J. et al. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. MBio 4, e00801-13 (2013).
Snijder, E. J. et al. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 18, e3000715 (2020).
Ulasli, M., Verheije, M. H., de Haan, C. A. & Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell Microbiol. 12, 844–861 (2010).
Wolff, G. et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 369, 1395–1398 (2020). Identification of a pore connecting the interior of the coronavirus DMVs to the cytosol, thereby providing a plausible concept for how viral RNAs can exit DMVs.
Overby, A. K., Popov, V. L., Niedrig, M. & Weber, F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J. Virol. 84, 8470–8483 (2010).
Al-Mulla, H. M. et al. Competitive fitness in coronaviruses is not correlated with size or number of double-membrane vesicles under reduced-temperature growth conditions. MBio 5, e01107-13 (2014).
Maier, H. J. et al. Extensive coronavirus-induced membrane rearrangements are not a determinant of pathogenicity. Sci. Rep. 6, 27126 (2016).
Oudshoorn, D. et al. Antiviral innate immune response interferes with the formation of replication-associated membrane structures induced by a positive-strand RNA virus. MBio https://doi.org/10.1128/mBio.01991-16 (2016).
Athmer, J. et al. In situ tagged nsp15 reveals interactions with coronavirus replication/transcription complex-associated proteins. MBio https://doi.org/10.1128/MBIO.02320-16 (2017).
Knoops, K. et al. Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus RNA synthesis and virus-induced remodeling of endoplasmic reticulum membranes. J. Virol. 84, 833–846 (2010).
Zhang, L., Zhang, Z. P., Zhang, X. E., Lin, F. S. & Ge, F. Quantitative proteomics analysis reveals BAG3 as a potential target to suppress severe acute respiratory syndrome coronavirus replication. J. Virol. 84, 6050–6059 (2010).
de Wilde, A. H. et al. A kinome-wide small interfering RNA screen identifies proviral and antiviral host factors in severe acute respiratory syndrome coronavirus replication, including double-stranded RNA-activated protein kinase and early secretory pathway proteins. J. Virol. 89, 8318–8333 (2015).
Reggiori, F. et al. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 7, 500–508 (2010).
Wong, H. H. et al. Genome-wide screen reveals valosin-containing protein requirement for coronavirus exit from endosomes. J. Virol. 89, 11116–11128 (2015).
Schneider, M. et al. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J. Virol. 86, 10112–10122 (2012).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020).
Riva, L. et al. A large-scale drug repositioning survey for SARS-CoV-2 antivirals. Preprint at bioRxiv https://doi.org/10.1101/2020.04.16.044016 (2020).
Li, J., Liu, Y. & Zhang, X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J. Virol. 84, 6472–6482 (2010).
Cervantes-Barragan, L. et al. Control of coronavirus infection through plasmacytoid dendritic-cell- derived type I interferon. Blood 109, 1131–1137 (2007).
Zhang, W. et al. Potent and selective inhibition of pathogenic viruses by engineered ubiquitin variants. PLoS Pathog. 13, e1006372 (2017).
Clementz, M. A. et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 84, 4619–4629 (2010).
Grunewald, M. E. et al. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog. 15, e1007756 (2019).
Zust, R. et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12, 137–143 (2011).
Daffis, S. et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468, 452–456 (2010). Together with Zust, R. et al., this article defines a role for viral cap-1 2′-O-methylation in evading host innate immune responses.
Minskaia, E. et al. Discovery of an RNA virus 3′->5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl Acad. Sci. USA 103, 5108–5113 (2006).
Kindler, E. et al. Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog. 13, e1006195 (2017).
Deng, X. et al. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl Acad. Sci. USA 114, E4251–E4260 (2017).
Canton, J. et al. MERS-CoV 4b protein interferes with the NF-kappaB-dependent innate immune response during infection. PLoS Pathog. 14, e1006838 (2018).
Tay, M. Z., Poh, C. M., Rénia, L., MacAry, P. A. & Ng, L. F. P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 20, 363–337 (2020).
Vabret, N. et al. Immunology of COVID-19: current state of the science. Immunity 52, 910–941 (2020).
Channappanavar, R. et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19, 181–193 (2016). Study revealing the importance of a swift and strong type I interferon response to prevent lethal SARS-CoV infection in a mouse model.
Chua, R. L. et al. Cross-talk between the airway epithelium and activated immune cells defines severity in COVID-19. Preprint at medRxiv https://doi.org/10.1101/2020.04.29.20084327 (2020).
V’kovski, P. et al. Disparate temperature-dependent virus - host dynamics for SARS-CoV-2 and SARS-CoV in the human respiratory epithelium. Preprint at bioRxiv https://doi.org/10.1101/2020.04.27.062315 (2020).
Giamarellos-Bourboulis, E. J. et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 27, 992–1000 (2020).
Liao, M. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844 (2020).
Zhong, J., Tang, J., Ye, C. & Dong, L. The immunology of COVID-19: is immune modulation an option for treatment? Lancet Rheumatol. 2, e428–e436 (2020).
Li, H. et al. Impact of corticosteroid therapy on outcomes of persons with SARS-CoV-2, SARS-CoV, or MERS-CoV infection: a systematic review and meta-analysis. Leukemia 34, 1503–1511 (2020).
Zhang, X. et al. Viral and host factors related to the clinical outcome of COVID-19. Nature https://doi.org/10.1038/s41586-020-2355-0 (2020).
Bost, P. et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell https://doi.org/10.1016/j.cell.2020.05.006 (2020).
Wen, W. et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 6, 31 (2020).
Shan, C. et al. Infection with novel coronavirus (SARS-CoV-2) causes pneumonia in the rhesus macaques. Cell Res. 30, 670–677 (2020).
Munster, V. J. et al. Respiratory disease in rhesus macaques inoculated with SARS-CoV-2. Nature https://doi.org/10.1038/s41586-020-2324-7 (2020).
McAuliffe, J. et al. Replication of SARS coronavirus administered into the respiratory tract of African Green, rhesus and cynomolgus monkeys. Virology https://doi.org/10.1016/j.virol.2004.09.030 (2004).
Woolsey, C. B. et al. Establishment of an African green monkey model for COVID-19. Preprint at bioRxiv https://doi.org/10.1101/2020.05.17.100289 (2020).
Dinnon, K. H. et al. A mouse-adapted SARS-CoV-2 model for the evaluation of COVID-19 medical countermeasures. Preprint at bioRxiv https://doi.org/10.1101/2020.05.06.081497 (2020).
Bao, L. et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 583, 830–833 (2020).
Netland, J., Meyerholz, D. K., Moore, S., Cassell, M. & Perlman, S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. https://doi.org/10.1128/jvi.00737-08 (2008).
Shi, J. et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS–coronavirus 2. Science 368, 1016–1020 (2020).
Kim, Y. Y. Il et al. Infection and rapid transmission of SARS-CoV-2 in ferrets. Cell Host Microbe https://doi.org/10.1016/j.chom.2020.03.023 (2020).
Park, S. J. et al. Antiviral efficacies of FDA-approved drugs against SARS-COV-2 infection in ferrets. MBio https://doi.org/10.1128/mBio.01114-20 (2020).
Martina, B. E. E. et al. SARS virus infection of cats and ferrets. Nature https://doi.org/10.1038/425915a (2003).
Sia, S. F. et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature https://doi.org/10.1038/s41586-020-2342-5 (2020).
Chan, J. F. W. et al. Simulation of the clinical and pathological manifestations of Coronavirus Disease 2019 (COVID-19) in golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa325 (2020).
Zhou, J. et al. Infection of bat and human intestinal organoids by SARS-CoV-2. Nat. Med. https://doi.org/10.1038/s41591-020-0912-6 (2020).
Monteil, V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell https://doi.org/10.1016/j.cell.2020.04.004 (2020).
Allison, S. J. SARS-CoV-2 infection of kidney organoids prevented with soluble human ACE2. Nat. Rev. Nephrol. https://doi.org/10.1038/s41581-020-0291-8 (2020).
Hui, K. P. Y. et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: an analysis in ex-vivo and in-vitro cultures. Lancet Respir. Med. https://doi.org/10.1016/S2213-2600(20)30193-4 (2020).
Hou, Y. J. et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell https://doi.org/10.1016/j.cell.2020.05.042 (2020).
Xie, X. et al. An infectious cDNA clone of SARS-CoV-2. Cell Host Microbe 27, 841–848 (2020).
Day, C. W. et al. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology https://doi.org/10.1016/j.virol.2009.09.023 (2009).
Roberts, A. et al. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. https://doi.org/10.1371/journal.ppat.0030005 (2007).
Thiel, V. et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 84, 2305–2315 (2003).
Lei, J., Kusov, Y. & Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 149, 58–74 (2018).
Boursnell, M. E. G. et al. Completion of the sequence of the genome of the coronavirus avian infectious bronchitis virus. J. Gen. Virol. 68, 57–77 (1987).
Koetzner, C. A., Parker, M. M., Ricard, C. S., Sturman, L. S. & Masters, P. S. Repair and mutagenesis of the genome of a deletion mutant of the coronavirus mouse hepatitis virus by targeted RNA recombination. J. Virol. 66, 1841–1848 (1992).
Van Der Most, R. G., Heijnen, L., Spaan, W. J. M. & De Groot, R. J. Homologous RNA recombination allows efficient introduction of site-specific mutations into the genome of coronavirus mhv-a59 via synthetic co-replicating rnas. Nucleic Acids Res. 20, 3375–3381 (1992).
Thiel, V., Herold, J., Schelle, B. & Siddell, S. G. Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus The GenBank accession number of the sequence reported in this paper is AF304460. J. Gen. Virol. 82, 1273–1281 (2001).
Almazán, F. et al. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc. Natl Acad. Sci. USA 97, 5516–5521 (2000).
Zhong, N. S. et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 362, 1353–1358 (2003).
Ksiazek, T. G. et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966 (2003).
van der Hoek, L. et al. Identification of a new human coronavirus. Nat. Med. 10, 368–373 (2004).
Woo, P. C. et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 79, 884–895 (2005).
Garbino, J. et al. A prospective hospital-based study of the clinical impact of non-severe acute respiratory syndrome (non-SARS)-related human coronavirus infection. Clin. Infect. Dis. 43, 1009–1015 (2006).
Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D. & Fouchier, R. A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820 (2012).
Memish, Z. A. et al. Middle East respiratory syndrome coronavirus in Bats, Saudi Arabia. Emerg. Infect. Dis. 19, 1819–1823 (2013).
Haagmans, B. L. et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet Infect. Dis. 14, 140–145 (2014).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Böhmer, M. M. et al. Investigation of a COVID-19 outbreak in Germany resulting from a single travel-associated primary case: a case series. Lancet Infect. Dis. 20, 920–928 (2020).
Xu, Z. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8, 420–422 (2020).
Guan, W. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020).
Zhang, Y. Z. & Holmes, E. C. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell https://doi.org/10.1016/j.cell.2020.03.035 (2020).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science 302, 276–278 (2003).
Lam, T. T. Y. et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature https://doi.org/10.1038/s41586-020-2169-0 (2020).
Ge, X. Y. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature https://doi.org/10.1038/nature12711 (2013).
Cui, J., Li, F. & Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192 (2019).
Acknowledgements
Work in the authors laboratory was supported by the Swiss National Science Foundation (SNF grants #173085, 196644, 182880) and the Federal Ministry of Education and Research, Germany (BMBF; grant RAPID, #01KI1723A).
Author information
Authors and Affiliations
Contributions
P.V., A.K., S.S., H.S. and V.T. researched data for the article and substantially contributed to the discussion of content. P.V., A.K., S.S. and VT wrote the article and reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Microbiology thanks M. Kikkert, who co-reviewed with V. van Vliet, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
COVID-19 Dashboard: https://coronavirus.jhu.edu/map.html
Glossary
- Nested set
-
Refers to the nested set of coronavirus 5′-coterminal and 3′-coterminal RNAs. Most nidoviruses (Nidus, nest, latin) share this mechanism of transcription and are grouped into the order Nidovirales.
- Zoonotic pathogens
-
Animal pathogens that can infect and replicate in humans.
- Recombination breakpoints
-
Distinct sites in the viral genome that are associated with a high frequency of exchange of genetic material between related viruses during co-infection of the same host cell.
- Integrins
-
Proteins that bind carbohydrate moieties found on proteins of the extracellular matrix or on cell-surface glycoproteins.
- Hybridoma
-
Clonal cells resulting from the fusion of B lymphoblasts and lymphoid myeloma cells. Hybridoma cells are used for the production of monoclonal antibodies.
- Escape mutations
-
Nucleotide changes that enable evasion from a selective pressure. Frequently used to describe changes in the viral genome that impair the efficiency of antibodies or antiviral compounds.
- Transcription regulatory sequences
-
(TRSs). Direct the leader-body junction during synthesis of coronavirus subgenomic RNAs.
- Accessory genes
-
Sets of coronavirus genes that encode proteins that are neither the non-structural proteins 1–16 (encoded in ORF1a/b and composing the replication and transcription complex) nor the canonical coronavirus structural proteins S, E, M and N. Usually dispensable in cell culture.
- Major histocompatibility complex
-
Major histocompatibility complex (MHC) molecules are cell-surface proteins that present a repertoire of proteins currently being expressed in the cell or proteins that have been uptaken by the cell in the form of MHC-bound peptides (called epitopes). T cells can recognize the MHC–peptide complexes and are stimulated to exert their immune functions upon recognition of MHC-bound pathogen-derived peptides.
- Double-membrane vesicles
-
(DMVs). DMVs are characteristic membranous structures induced by coronaviruses and many other positive-sense RNA viruses. They anchor the replication and transcription complex and support viral replication and RNA synthesis.
- OAS–RNase L pathway
-
There are several cellular 2′,5′-oligoadenylate synthetases that produce 2′,5′-oligoadenylates upon stimulation by double-stranded RNA. 2′,5′-oligoadenylates can stimulate the cellular ribonuclease RNase L that degrades cellular and viral RNAs as part of the antiviral host defence.
Rights and permissions
About this article

Cite this article
V’kovski, P., Kratzel, A., Steiner, S. et al. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol 19, 155–170 (2021). https://doi.org/10.1038/s41579-020-00468-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-020-00468-6
This article is cited by
-
Development of nanoparticles incorporated with quercetin and ACE2-membrane as a novel therapy for COVID-19
Journal of Nanobiotechnology (2024)
-
Altered expression of serum lncRNA CASC2 and miRNA-21-5p in COVID-19 patients
Human Genomics (2024)
-
nCoV-19 therapeutics using cucurbitacin I structural derivatives: an in silico approach
Future Journal of Pharmaceutical Sciences (2024)
-
Proteomic analysis of SARS-CoV-2 particles unveils a key role of G3BP proteins in viral assembly
Nature Communications (2024)
-
Synergistic inhibition effects of andrographolide and baicalin on coronavirus mechanisms by downregulation of ACE2 protein level
Scientific Reports (2024)
