
Abstract
Protein kinases play a major role in cellular activation processes, including signal transduction by diverse immunoreceptors. Given their roles in cell growth and death and in the production of inflammatory mediators, targeting kinases has proven to be an effective treatment strategy, initially as anticancer therapies, but shortly thereafter in immune-mediated diseases. Herein, we provide an overview of the status of small molecule inhibitors specifically generated to target protein kinases relevant to immune cell function, with an emphasis on those approved for the treatment of immune-mediated diseases. The development of inhibitors of Janus kinases that target cytokine receptor signalling has been a particularly active area, with Janus kinase inhibitors being approved for the treatment of multiple autoimmune and allergic diseases as well as COVID-19. In addition, TEC family kinase inhibitors (including Bruton’s tyrosine kinase inhibitors) targeting antigen receptor signalling have been approved for haematological malignancies and graft versus host disease. This experience provides multiple important lessons regarding the importance (or not) of selectivity and the limits to which genetic information informs efficacy and safety. Many new agents are being generated, along with new approaches for targeting kinases.
Similar content being viewed by others

Introduction
The extraordinary advances in the basic science of immunology have provided therapeutic approaches that have revolutionized outcomes in patients with inflammatory and immune-mediated diseases1,2. These include numerous successful therapeutic monoclonal antibodies and engineered recombinant cytokine receptors. Advances in deciphering the biochemical pathways of immune cell signalling3,4 similarly led to the generation of small molecule therapies that complement biologics. In this Review, we focus on protein kinase inhibitors and their use in immune-mediated and inflammatory disorders. Although this field has been reviewed numerous times5,6, it moves extraordinarily quickly, with over 60 small molecule protein kinase inhibitors now approved and hundreds more in development6. We provide a brief history of targeted kinase inhibitors, which first emerged for the treatment of cancer. We then discuss how purposefully targeting signal transduction pathways used by major classes of immunoreceptors has led to the effective treatment of numerous immune-mediated disorders. We focus on Janus kinase (JAK) inhibitors as they comprise one of the most active and successful areas, but we also review the successful targeting of TEC family kinases and other emerging protein kinase drug targets. We compare the ways in which kinase inhibitors may be similar yet distinct from biologics targeting cells and cytokines, and we consider future approaches and opportunities in this field.
Overview of protein kinases
Reversible protein phosphorylation by kinases and phosphatases is a fundamental cellular regulatory mechanism, important for controlling protein activity during key cellular processes such as cell cycle, cell growth, differentiation, movement, metabolism and apoptosis7,8. Phosphorylation by protein kinases converts signals from outside the cell to downstream readouts within the cell by facilitating protein interactions and translocation and by altering protein conformations. These changes lead to modification of downstream enzymes, specific gene transcription and protein degradation8. The actions of protein kinases are reversed by protein phosphatases.
The human genome contains more than 518 protein kinases, comprising 1.7% of human genes, as well as an additional 20 lipid kinases9. Protein kinases, also known as phosphotransferases, catalyse the transfer of the γ-phosphate from a purine nucleotide triphosphate (that is, ATP and GTP) to the hydroxyl groups of their protein substrates by generating phosphate monoesters using protein alcohol groups (on serine and threonine residues) and/or protein phenolic groups (on tyrosine residues) as phosphate acceptors. Thus, protein kinases can be classified by the amino acid substrate preference: serine–threonine kinases, tyrosine kinases and dual kinases (which phosphorylate serine, threonine or tyrosine residues). Protein tyrosine kinases make up around 10% of the total kinase family and are often involved in proximal receptor signalling. Almost all protein kinases have catalytic domains that belong to a single eukaryotic protein kinase superfamily.
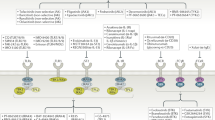
T cells, B cells and innate immune cells express different classes of cytokine receptors and multichain immune recognition receptors (including T cell receptors (TCRs), B cell receptors (BCRs), Fc receptors (FcRs) for IgG, natural killer (NK) cell receptors and C-type lectin receptors) that use phosphorylation to trigger the first steps of activation or are linked to kinases via adaptor molecules (Fig. 1). The main kinases involved in immune cell signalling include: receptor tyrosine kinases (RTKs), receptor serine kinases, non-RTKs such as the JAKs and the SRC, SYK and TEC family of tyrosine kinases, as well as a larger group of downstream serine–threonine kinases7,10,11 (Fig. 1). RTKs and receptor serine kinases have intrinsic phosphotransferase activity induced by ligand binding, whereas many immune receptors including antigen receptors and cytokine receptors lack this intrinsic enzyme activity and recruit cytoplasmic tyrosine kinases. For example, JAKs are noncovalently associated with cytokine receptors and phosphorylate both the receptor and signal transducers and activators of transcription (STATs) to induce gene expression. Engagement of antigen receptors induces rapid activation of SRC family kinases, leading to the recruitment and activation of SYK in B cells and ZAP70 in T cells, which phosphorylate downstream adaptor molecules that recruit TEC family kinases and phospholipase Cγ. Activated phospholipase Cγ cleaves membrane-bound phosphatidylinositol 4,5-bisphosphate into inositol trisphosphate, which induces Ca2+ mobilization and activation of calcineurin and diacylglycerol, which activates protein kinase C, RAS and mitogen-activated protein kinase pathways. Together, with signals from co-stimulatory molecules, these cascades activate nuclear factor of activated T cells, nuclear factor-κB and AP-1 transcription factors, along with phosphoinositide 3-kinase, mTOR and AKT pathways. It is notable that some intermediates in the signalling cascades of different receptors are shared and others are distinct (Fig. 1).

Key immune receptors expressed by T cells, B cells and innate immune cells include different classes of cytokine receptors and multichain immune recognition receptors. Only a subset of downstream pathways are shown, which are relevant to the inhibitors discussed in this Review. The prominent kinases involved in immune receptor signalling include: receptor tyrosine kinases, receptor serine kinases, non-receptor tyrosine kinases such as the Janus kinases (JAKs) and the SRC (such as LYN), SYK and TEC (such as BTK) families of tyrosine kinases, as well as the larger group of downstream serine–threonine kinases. These are represented here with ligands, receptor classes, kinases and key downstream signalling cascades. BCR, B cell receptor; BMP, bone morphogenetic protein; BTK, Bruton’s tyrosine kinase; CSF, colony-stimulating factor; DAG, diacylglycerol; GPCR, G-protein-coupled receptor; IKKα, inhibitor of nuclear factor-κB kinase subunit-α; IKKβ, inhibitor of nuclear factor-κB kinase subunit-β; IKKγ, inhibitor of nuclear factor-κB kinase subunit-γ; IRAK, IL-1 receptor-associated kinase; JNK, JUN N-terminal kinase; MAPK, mitogen-activated protein kinase; MEKK, mitogen-activated protein kinase kinase; MIRR, multichain immune recognition receptor; mTOR, mammalian target of rapamycin; MYD88, myeloid differentiation primary response 88; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor-κB; PAMP, pathogen-associated molecular pattern; PI3K, phosphoinositide 3-kinase; PKC, protein kinase C; PLCγ, phospholipase Cγ; RIP, receptor-interacting serine–threonine protein kinase; ROCK, RHO-associated protein kinase; STAT, signal transducer and activator of transcription; SYK, spleen tyrosine kinase; TCR, T cell receptor; TGFβ, transforming growth factor-β; TLR, Toll-like receptor; TNF, tumour necrosis factor; TNFR, tumour necrosis factor receptor; TRADD, tumour necrosis factor receptor type 1-associated death domain; TRAF, TNFR-associated factor.
Some kinases have selective expression in immune cells, but many kinases are broadly expressed; although the former would represent logical targets to develop new therapies for autoimmune and inflammatory diseases, inhibitors of kinases with broad expression also turn out to be safe and effective drugs6,10.
Targeting kinases: in the beginning
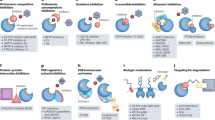
Many oncogenes and their cellular counterparts are kinases; thus, interfering with pathological phosphorylation in cancer seemed a logical treatment strategy12. However, given how ubiquitous protein kinases are for critical cellular functions and the conservation of the ATP-binding region across kinase classes, there was also reasonable skepticism about whether specificity could be attained with kinase inhibitors. We now know that despite structural similarities of protein tyrosine kinases in their ATP-bound active state, structural differences in the inactive conformation and gatekeeper residues in kinase domains allow for the development of selective protein kinase inhibitors6,10. The first inhibitors of protein phosphorylation were not purposefully designed to do so (Box 1). In addition, there are opportunities beyond targeting kinase domains, including allosteric inhibitors and targeted protein degradation (discussed subsequently).
ABL kinases
Selective targeting of a kinase was first accomplished in the early 2000s with the approval of imatinib for the treatment of chronic myeloid leukaemia (CML) (Fig. 2), a major advance that moved away from broadly cytotoxic agents to more targeted therapy for molecular abnormalities of cancer13,14. Imatinib selectively targets the fusion protein BCR–ABL tyrosine kinase generated by a chromosomal translocation associated with most cases of CML15,16. Imatinib was developed from the lead compound 2-phenylaminopyrimidine and modified by the introduction of methyl and pyridyl groups to confer enhanced selectivity and inhibitory activity against ABL kinases. The addition of an N-methyl-piperazine enhanced the aqueous solubility and oral bioavailability of imatinib17.

The timeline lists the year of approval and indication, starting with the approval of cyclosporine for allograft transplantation (in 1989) to the approvals of JAK inhibitors (JAKinibs) for immune-mediated diseases to date. AA, alopecia areata; AD, atopic dermatitis; BCR, B cell receptor; BTK, Bruton’s tyrosine kinase; CLL, chronic lymphocytic leukaemia; CML, chronic myeloid leukaemia; EGFR, epidermal growth factor receptor; GVHD, graft versus host disease; ITP, immune thrombocytopenia; JAK, Janus kinase; JIA, juvenile idiopathic arthritis; PsA, psoriatic arthritis; RA, rheumatoid arthritis; ROCK; RHO-associated kinase; SYK, spleen tyrosine kinase; TGFβ, transforming growth factor-β; VEGFR, vascular endothelial growth factor receptor.
The successful use of imatinib is associated with a reduction in the proportion of BCR–ABL cells in the bone marrow. In many cases, the response to treatment lasts for decades18,19,20. However, resistance to imatinib can develop and is typically associated with the acquisition of mutations of the ABL kinase domain that alter binding of imatinib21. Second-generation ABL kinase inhibitors (nilotinib and dasatinib) were introduced to address imatinib resistance22,23 (Fig. 2). Notably, dasatinib has less specificity for ABL compared with imatinib and has activity against SRC family kinases but is conversely more tolerant of mutations in the ABL kinase domain. Third-generation inhibitors (ponatinib and bosutinib) were then developed to overcome the ABL T315I mutation, which confers resistance to previous generations of ABL kinase inhibitors24.
In the immune system, imatinib has effects on lymphocytes, mast cells and macrophages, inhibiting signal transduction pathways that lead to the secretion of pro-inflammatory cytokines25. In addition to its role in several haematological malignancies, late-phase clinical studies of imatinib have been initiated for the treatment of COVID-19, pulmonary hypertension and pain with sickle cell anaemia19. In these cases, imatinib appears to reverse capillary leak. In COVID-19, hypoxaemic respiratory failure owing to capillary leak and alveolar oedema is a major complication; experimental and early clinical data suggest that imatinib helps to reverse this process. In vitro models suggest that imatinib limits arginine-mediated endothelial barrier dysfunction by enhancing RAC1 activity and enforcing adhesion of endothelial cells to the extracellular matrix26. In sickle cell disease, imatinib appears to protect the integrity of the erythrocyte membrane (NCT03997903).
Receptor tyrosine kinases
Although targeting BCR–ABL kinase activity is an effective therapy for CML with minimal side effects, we now know that imatinib targets kinases beyond BCR–ABL, including RTKs, and has utility beyond this setting. In haematopoietic cells, imatinib targets the stem cell factor RTK (KIT) and platelet-derived growth factor receptor-α (PDGFRα), which are key oncogenic drivers in most gastrointestinal stromal cell tumours27. Imatinib has revolutionized the treatment of this disorder and is now the first-line therapy, despite never having undergone prospective clinical trials. Mutations in KIT and PDGFRα are also seen in several rare myeloproliferative diseases such as systemic mastocytosis, hypereosinophilic syndrome and/or chronic eosinophilic leukaemia28. In a meta-analysis of published case reports, imatinib was shown to be the most widely used therapy for these conditions after corticosteroids29.
Another PDGFR inhibitor is nintedanib, which also inhibits about 20 other kinases, including fibroblast growth factor receptors, vascular endothelial growth factor receptors30, colony-stimulating factor 1 receptor (CSF1R) and the SRC family kinase LCK31. Nintedanib is approved for the treatment of idiopathic pulmonary fibrosis32; it inhibits the release of multiple cytokines by human peripheral blood mononuclear cells or T cells. Nintedanib appears to decrease uncontrolled proliferation of lung fibroblasts and differentiation of fibroblasts into myofibroblasts, which deposit extracellular matrix into the interstitial spaces in these diseases33. Data indicate that nintedanib prevents pro-fibrotic macrophage polarization and expression of the macrophage polarization marker CCL18, which has been associated with disease progression34.
CSF1R is expressed in monocytes, tissue-resident macrophages, dendritic cells and osteoclasts and is activated by macrophage CSF1 and IL-34. Mutations affecting CSF1R are seen in 10% of cases of histiocytosis, a rare group of clonal, often localized, myeloproliferative disorders that include Erdheim–Chester disease and Langerhans cell histiocytosis subtypes35. The presence of these mutations leads to robust cytokine-independent cell growth in vitro. CSF1R signalling has also been implicated in rheumatoid arthritis (RA), amyotrophic lateral sclerosis and graft versus host disease (GVHD). In RA, CSF1R is highly expressed by proliferating fibroblast-like synoviocytes, suggesting a role in proliferation of these cell types36. In amyotrophic lateral sclerosis, CSF1R has a role in the invasion of macrophages into peripheral nerves37. In GVHD, CSF1R-expressing macrophages from donors infiltrate the skin, leading to cutaneous inflammation38. The CSF1R antagonist antibody axatilimab has completed small phase I/II clinical trials for the treatment of chronic GVHD that are resistant to multiple lines of therapy and has shown some promise39. By contrast, the small molecule CSF1R inhibitor edicotinib was not efficacious in RA40.
Structurally similar to RTKs are receptor serine–threonine kinases41, which are discussed in Box 2.
JAK family kinase inhibitors
Although targeting a single cytokine or cytokine receptor has revolutionized therapy for immune-mediated diseases, this approach is not successful in all patients and often does not result in durable remission42. Therefore, it is not surprising that targeting signalling pathways shared by multiple cytokine receptors with small molecules became an attractive and extremely successful approach. As such, we focus on studies of JAKs as key pharmaceutical targets for immune-mediated and inflammatory diseases.
JAKs (comprising JAK1, JAK2, JAK3 and TYK2) associate with receptors for type I and II cytokines that lack intrinsic RTK activity, inducing downstream cell signalling for multiple cell functions43 (Fig. 3). Of the JAK family members, JAK1 is activated by the largest number of type I and II cytokines, including type I interferons (IFNs), IFNγ, IL-2, IL-4, IL-6, IL-13 and thymic stromal lymphopoietin; JAK2 is activated by erythropoietin, thrombopoietin, growth hormone, IL-3, IL-5, IL-12, IL-23, CSF1 and CSF2 (Fig. 3). Moreover, most cytokine receptors require the use of more than one JAK. The essential function of JAKs has been established by identifying genetic variants in patients and by the generation of knockout mice44,45,46,47 (Table 1). Specifically, gene targeting of Jak1 and Jak2 in mice is lethal;48 JAK1 and JAK2 are essential for signalling downstream of many cytokines that impact processes in multiple tissues and cells including haematopoiesis, host defence, immunoregulation, body growth and metabolism. JAK1 loss-of-function mutations cause primary immunodeficiency in humans associated with atypical mycobacterial infection, whereas JAK1 gain-of-function mutations are associated with systemic immune dysregulation and hypereosinophilic syndrome47. Somatic JAK2 gain-of-function mutations are associated with three common myeloproliferative neoplasms: primary polycythemia vera, essential thrombocythemia and primary myelofibrosis, consistent with the role of the JAK2-associated haematopoietic cytokines erythropoietin, thrombopoietin and CSFs45. Loss-of-function mutations of TYK2 in humans and mice are also associated with immunodeficiency characterized by increased susceptibility to bacterial, viral and fungal infections46, reflecting its importance for signalling by IFNs, IL-10 family cytokines, IL-12, IL-23 and IL-27. JAK3 loss-of-function mutations result in severe combined immunodeficiency, owing to the disruption of signalling by receptors containing the common γ-chain (for γc cytokines IL-2, IL-4, IL-7, IL-9 and IL-21)44. These phenotypes highlight the relatively limited functions of TYK2 and JAK3 versus the broad actions JAK1 and JAK2; the spectrum of cytokines impacted is relevant for the mechanism of action, efficacy and adverse events of JAK inhibitors (JAKinibs).

The receptor class, specific Janus kinases (JAKs) and associated cytokines are grouped by functional and immunological effects and disease associations. JAK inhibitors (JAKinibs) are listed from the less-selective first-generation JAKinibs and panJAKinibs (at the top) to the more selective JAKinibs (at the bottom). CSF, colony-stimulating factor; EPO, erythropoietin; GH, growth hormone; gp130, glycoprotein 130; IFN, interferon; ILC, innate lymphoid cell; OSM, oncostatin M; PRL, prolactin; SCID, severe combined immunodeficiency; TH1 cell, T helper 1 cell; TH17 cell, T helper 17 cell; TPO, thrombopoietin; Treg cell, regulatory T cell; TSLP, thymic stromal lymphopoietin.
Mechanisms of action of JAKinibs in immune-mediated disease
First-generation JAKinibs block both JAK1 and JAK2. Consequently, in GVHD, JAKinibs can attenuate allogeneic donor T cell activation by blocking the action of IFNs in promoting antigen presentation as well as the action of many effector cytokines49,50. JAKinibs impact diseases mediated by type 1 (IFNγ-driven), type 2 (IL-4-driven, IL-5-driven or IL-13-driven) or type 3 (IL-17-driven or IL-22-driven) immune responses (Fig. 3), with effects on both innate and adaptive immune cells, as well as on non-immune cells, stromal cells and neurons, as targeted cytokine receptors are expressed by virtually all cells. Both type 1 and type 2 immune responses are implicated in the pathological mechanisms of alopecia areata (AA)51,52; although the biologic dupilumab (anti-IL-4Rα, which blocks both IL-4 and IL-13) can be efficacious in some patients, it is not approved for AA53,54,55. Thus, the broader actions of JAKinibs may be advantageous for this autoimmune disorder. In skin obtained from mice and humans with alopecia, transcriptomic profiling revealed expression of IFN response signature genes and several γc cytokines including IL-2 and IL-15. Use of JAKinibs eliminates the IFN signature and development of alopecia in a mouse model, as well as induces near-complete hair regrowth in patients treated with the JAKinib ruxolitinib and multiple other JAKinibs56. In vitiligo, skin-resident memory T cells demonstrate skewed JAK1-dependent and JAK2-dependent type 1 and type 2 cytokine profiles that stimulate melanocyte and keratinocyte inflammatory responses, which can be inhibited by ruxolitinib57. Consistent with the expression of cytokine receptors on neurons, JAKinibs can also reduce pain and itch associated with inflammatory skin diseases58. In these examples, JAKinibs have the advantage over biologics that target a single mediator by blocking more than one of the canonical responses and potentially different endotypes of the disease.
Cytokines do not act in isolation but induce the expression of other cytokines; for example, IL-6 induces the JAK-independent pro-inflammatory cytokines IL-1, IL-17 and tumour necrosis factor (TNF). Cells pretreated with JAKinibs and then activated by lipopolysaccharide and other Toll-like receptor ligands that induce IL-6 produce less IL-1 and TNF59. The IL-1R antagonist anakinra is efficacious in various autoinflammatory diseases, such as haemophagocytic lymphohistiocytosis, pointing to a prominent role of IL-1. Thus, JAKinibs may be efficacious in these diseases via this indirect mechanism59,60. The capacity of first-generation JAKinibs to inhibit a wide variety of cytokines may be an important aspect of their efficacy (as illustrated by their efficacy in COVID-19; discussed subsequently), but this broader effect can make it difficult to link their efficacy with a single mechanism61.
First-generation approved JAKinibs
Ruxolitinib was the first JAKinib to be approved by the FDA for the treatment of myeloproliferative diseases: primary polycythemia vera, essential thrombocythemia and primary myelofibrosis (Table 2 and Fig. 2). Activating mutations of JAK2 are found in 50% (primary myelofibrosis) to 90% (primary polycythemia vera) of cases62,63,64 (Table 1); thus, ruxolitinib was originally designed as a JAK2 inhibitor. However, it was subsequently found to also inhibit JAK1. This dual targeting was initially seen as a limitation of the drug for these JAK2-mediated myeloproliferative diseases. However, patients with primary myelofibrosis have systemic symptoms, including night sweats and loss of appetite, that may be JAK1-dependent rather than JAK2-dependent65,66,67. Furthermore, ruxolitinib has now been licensed for the treatment of inflammatory diseases; its success in this setting is likely related to its ability to inhibit JAK1. Ruxolitinib is also now approved for the treatment of acute and chronic GVHD49,65,68. Topical ruxolitinib is used as a treatment for atopic dermatitis (AD) and vitiligo, potentially avoiding side effects associated with systemic use of this inhibitor69 (Table 2 and Fig. 2).
Tofacitinib was the first FDA-approved JAKinib for an inflammatory disease70,71. It was originally intended to be a JAK3 inhibitor. Studies in mouse models and patients with severe combined immunodeficiency owing to γc cytokine and JAK3 deficiencies predicted that a specific JAK3 inhibitor would be a potent immunosuppressant, leading to profound T and NK cell lymphopenia and consequently an increased risk of infections72. Given the important role of IL-2 in maintaining FOXP3+ regulatory T cells, it was conceivable that loss of IL-2-induced signalling through the use of JAK3 inhibitors would exacerbate autoimmune disease73. However, tofacitinib is immunosuppressive without causing lymphopenia74, which may be related to its ability to inhibit JAK1 in addition to JAK3. Tofacitinib is now approved for the treatment of RA, psoriatic arthritis (PsA), ankylosing spondyloarthritis, ulcerative colitis and polyarticular course juvenile idiopathic arthritis70,71,75,76,77,78 (Table 2 and Figs. 2 and 3). Tofacitinib has shown promise as a therapy for dermatomyositis79; in particular, studies have demonstrated improved survival in patients with anti-MDA5 dermatomyositis with life-threatening interstitial lung disease80,81. Finally, children with gain-of-function STAT1 mutations or loss-of-function mutations in the STAT1 inhibitor SOCS1 have enhanced type 1 immune responses at the cost of loss of type 3 immunity, resulting in chronic mucocutaneous candidiasis and splenomegaly. Tofacitinib blocks IFN signalling by inhibiting STAT1 activation and has been effective in treating these two conditions82,83. For a drug conceived from research on patients with primary immunodeficiency, it is gratifying to see its use in treating patients with inborn errors of immunity.
Baricitinib, a JAK1 and JAK2 inhibitor, is approved for the treatment of RA, AD, AA and COVID-19 (Table 2 and Figs. 2 and 3). However, two phase III studies of baricitinib in systemic lupus erythematosus (SLE) (NCT03616912 and NCT03616964) showed discordant results for the primary outcome measure84. There are ongoing studies using first-generation JAKinibs, including topical forms, in multiple other immune conditions (Table 3).
Use of JAKinibs in COVID-19
At the start of the COVID-19 pandemic, it was noted that much of the lung pathology seen in critically ill patients developed in the weeks following clearance of virus, suggesting a role for cytokine-mediated tissue damage. Although it may seem counterintuitive to use an immunosuppressive drug in a viral infection, it was discovered that the corticosteroid dexamethasone reduced mortality in patients with COVID-19 requiring oxygen for ventilatory support85. Similarly, the potential use of JAKinibs in cytokine-mediated immunopathology was quickly appreciated on the basis of the proven efficacy in a preclinical model of sepsis and their immunosuppressive effects59. Besides its anti-inflammatory activity, baricitinib was reported to have antiviral activity by inhibiting numb-associated kinases and adaptor protein kinases that are required for viral entry and by decreasing IFN-mediated expression of the SARS-CoV-2 receptor, angiotensin-converting enzyme 2 (refs. 86,87,88). On this basis, baricitinib was investigated for the treatment of COVID-19 (NCT04640168) and subsequently approved for use in patients hospitalized with COVID-19 requiring oxygen for ventilatory support89,90. At present, more patients have been treated with baricitinib for COVID-19 than all other indications combined. Other JAKinibs have shown efficacy in COVID-19 in smaller studies. For instance, in patients hospitalized with COVID-19-associated pneumonia (NCT04469914), tofacitinib treatment decreased risk of death or respiratory failure compared with placebo91. This efficacy in COVID-19, as well as some positive outcomes reported with haemophagocytic lymphohistiocytosis60, suggests the potential use of JAKinibs in other scenarios associated with cytokine storm, such as septic shock. However, more research is needed to address the timing and context of JAKinib use in these clinical settings, including potential generation of parenteral JAKinibs.
Side effects of first-generation JAKinibs
Given the broad impact of JAKinibs on cytokine signalling and the phenotypes associated with genetic deletion of JAKs (Table 1), increased rates of infection were expected in individuals treated with JAKinibs. Indeed, increased infections, including serious and opportunistic infections, are seen in patients treated with JAKinibs. A retrospective observational study reviewing the World Health Organization database of adverse events associated with first-generation JAKinibs (ruxolitinib, tofacitinib and baricitinib) found an increased association with viral (herpes and influenza viruses), fungal and mycobacterial infections; varicella zoster virus infection and reactivation were significantly greater in patients treated with first-generation JAKinibs than in patients treated with conventional disease-modifying antirheumatic drugs (DMARDs)92. The risk of infection is increased when JAKinibs are used at high doses or in combination with immunosuppressive drugs, as illustrated in early transplantation trials93. Increased risk of viral infections may be due to inhibition of IFN signalling and type 1 immune responses and reduced numbers of NK cells94.
Because first-generation JAKinibs target JAK2, which is activated downstream of receptors important in blood cell development such as the erythropoietin receptor, anaemia and other cytopenias can occur; typically, these are not limiting adverse events. Venous thromboembolism and hyperlipidaemia have also been observed with the use of JAKinibs, the latter is also associated with biologics that block IL-6 (refs. 95,96). The mechanisms and cytokines underlying increased risk of venous thrombosis and pulmonary embolism remain unclear.
In an open-label safety trial comparing tofacitinib with TNF inhibitors for the treatment of active RA with at least one cardiovascular risk factor, opportunistic infections, major adverse cardiovascular events, malignancy, venous thrombosis and all-cause death were greater in the tofacitinib group after median 4-year follow-up97. On the basis of these findings, the FDA added a warning for tofacitinib and other JAKinibs, highlighting these increased risks in the context of RA98. However, 95% confidence intervals for these hazard ratios extend below one, indicating that further study is needed. The lack of an untreated control group in this study further complicates interpretation of the study, making it unclear whether these differences are simply due to the relative efficacy of TNF inhibitors compared with JAKinibs in suppressing inflammation, or whether JAKinibs were associated with risk beyond that associated with disease.
A recent follow-up study found that the risk for malignancy associated with tofacitinib was highest among those with a history of atherosclerotic cardiovascular disease or significant cardiovascular risk, indicating some shared risk factors99. Inflammatory diseases such as RA are known to be associated with increased cardiovascular disease and cancer; controversy regarding these relative risks remains. Additional studies of long-term safety of tofacitinib showed increased risk for opportunistic infection, major adverse cardiovascular events and venous thrombosis, consistent with previous studies. These studies note that the increased cancer risk was stable over time100. In a post hoc analysis of patients with RA on tofacitinib with about 10 years of follow-up and dose changes at investigator discretion, safety data were similar between doses for multiple adverse effects, including varicella zoster viral infection, serious infections, deep vein thromboses and pulmonary embolism101. The European Alliance of Associations for Rheumatology Task Force for RA treatment noted that, after failure of a conventional DMARD, a JAKinib may be considered after taking relevant risk factors into account. This recommendation was based on the absence of evidence for greater risk of tofacitinib versus TNF inhibitors in patients without risk factors. Notably, it cannot be excluded that other DMARDs would have similar risks if subjected to an outcome-based randomized controlled trial102.
In a small study assessing the impact of tofacitinib on vascular complications in SLE, surrogate vascular end points suggested that tofacitinib might reduce risk. Tofacitinib use was associated with reduced formation of neutrophil extracellular traps, which promote vascular disease in lupus103. Although the evidence is modest, ruxolitinib is associated with a reduced incidence of thrombosis in patients with myeloproliferative disease, a patient group with high incidence of thromboembolic disease104. This could be a property of ruxolitinib or more likely because JAK inhibition blocks the pro-thrombotic consequences of myeloproliferation.
Second-generation JAKinibs
JAKinibs have evolved in multiple ways since the first generation, with efforts to improve selectivity and pharmacokinetics such as prolonged half-life105. Several relatively selective JAK1 inhibitors have been generated (Fig. 3): upadacitinib is approved for RA, PsA, ankylosing spondyloarthritis, ulcerative colitis and AD; abrocitinib is approved for AD106 and filgotinib is approved in the European Union and Japan for RA (Table 2). Upadacitinib was reported to have greater selectivity for JAK1 versus JAK2 when compared with first-generation JAKinibs107, although not all assays support this selectivity. Upadacitinib use is still associated with anaemia, which may reflect a residual blockade of JAK2, as well as an increased risk of venous thrombosis. Abrocitinib can be associated with hyperlipidaemia, major adverse cardiovascular events, venous thromboembolism, varicella zoster virus infection and decreased platelet counts108. Abrocitinib can also be associated with cytopenias that may be related to JAK1 inhibition; the IL-6 family cytokines oncostatin M and IL-11 are both drivers of haematopoiesis and depend on JAK1 for signalling. Oncostatin M-deficient mice have impaired haematopoiesis109, and use of oncostatin M-blocking antibodies is associated with reduced blood cell counts in clinical trials110. There is still uncertainty regarding the degree of selectivity of these supposed JAK1 inhibitors and whether they still negatively impact haematological, clotting and cardiovascular risk. More data and investigation are required to definitively know whether there are significant differences between newer and older agents.
Compared with JAK1 and JAK2, TYK2 is used by a narrower spectrum of cytokine receptors, and thus TYK2 inhibitors would be expected to be beneficial in diseases mediated by type 1 IFNs, IL-12 and IL-23 (Fig. 3), consistent with reduced risk of autoimmunity in humans with TYK2 loss-of-function variants111 (Table 1). Selective TYK2 inhibitors include ropsacitinib (PF-06826647, an ATP competitor)112, deucravacitinib, TAK-279, VTX958 and ESK-001113; the latter four agents bind to the pseudokinase domain of TYK2, which is a novel approach that confers greater selectivity than approaches that rely on binding the kinase domain114. Deucravacitinib has recently been approved for the treatment of plaque psoriasis115 (Table 2). Phase II clinical trial studies show efficacy in PsA116 and SLE117. However, deucravacitinib was not efficacious in a phase II trial in inflammatory bowel disease, perhaps owing to inhibition of the anti-inflammatory cytokines IL-10, IL-22 and IL-27. Although examination of larger patient cohorts will be needed, so far anaemias and hyperlipidaemia do not appear to be associated with deucravacitinib use. Infections, including varicella zoster virus infections, are associated with deucravacitinib use, but reportedly, inhibition of TYK2 preserves IFNλ signalling, which may provide residual antiviral activity118. Brepocitinib (PF-06700841) is a JAK1 and TYK2 inhibitor that has proved to be successful in phase III clinical trials for the treatment of severe AA119 and is currently being investigated in a phase II clinical trial for scarring alopecia (NCT05076006). A new TYK2 inhibitor, KL130008, is being studied in a phase II clinical trial for AA (NCT05496426).
JAK3 has the most restricted expression among the JAKs, and targeting JAK3 inhibits a limited number of cytokines (Fig. 3). In this respect, a JAK3-selective inhibitor could avoid haematological and cardiovascular complications. Decernotinib is reported be a selective JAK3 inhibitor that showed efficacy in RA, but its use is limited by multiple drug interactions. Decernotinib is metabolized by aldehyde oxidase to a metabolite that, in turn, inhibits CYP3A4, which is key for the inactivation of many common drugs120,121. An irreversible covalent JAK3-selective inhibitor (Z583) has been developed and showed efficacy in a collagen-induced arthritis mouse model, with potential for other inflammatory diseases122. Ritlecitinib is a JAK3 and TEC kinase inhibitor that has been studied for the treatment of RA and AA119,123,124. The blocking of two distinct kinase families may provide broader actions for efficacy and different, potentially less severe, side effects owing to inhibition of other JAK family members, although this remains to be determined. Other agents with broader kinase specificity have been developed including fedratinib and pacritinib, which are competitive inhibitors of JAK2 and the RTK FLT3 that are approved for the treatment of primary myelofibrosis125.
In addition to systemic use, there are multiple JAKinibs available for topical use in inflammatory skin conditions, limiting side effects associated with systemic use. Topical use of delgocitinib and ruxolitinib is approved for the treatment of AD and ruxolitinib for vitiligo69,126,127 (Table 2). Topical application of JAKinibs is also being tested in phase I–III studies for the treatment of chronic hand dermatitis (NCT05233410, 05219864 and 05293717) and GVHD (NCT03954236). Along with topical use of JAKinibs, inhaled agents are being developed. LAS194046 and AZD0449, both inhaled JAKinibs, were shown to decrease allergic lung inflammation in rats128,129. AZD0449 (a JAK1 inhibitor) has successfully completed phase I clinical trials (NCT03766399) and others are ongoing (NCT04769869). Nezulcitinib (TD-0903) successfully completed phase II clinical trials for severe COVID-19 (ref. 130). As with inhaled corticosteroids, inhaled JAKinibs could have improved safety profiles. Non-absorbable gut-selective JAKinibs have also been developed131, including OST-122, which is in phase II clinical trials for the treatment of ulcerative colitis (NCT04353791).
Another therapeutic strategy for JAKinibs involves combining them with other drugs. In patients with RA, the combination of methotrexate and tofacitinib proved beneficial to those previously refractory to methotrexate132. In severe COVID-19, baricitinib has been combined with direct-acting antivirals such as remdesivir; the combination of JAK2 inhibitors such as fedratinib with antiviral or anticytokine therapy has also been used and proposed133. Other combination therapy approaches will require further study, including potential combined use with biologics, bearing in mind that this could increase adverse events.
In summary, JAKinibs are firmly established as a therapeutic option for various conditions. The adverse events associated with JAKinibs are largely expected given the range of cytokines they block, although additional investigation into cardiovascular risk and clotting abnormalities is needed. With respect to efficacy and safety of JAKinibs, it is worth reflecting on the extent to which genetic phenotypes do or do not predict JAKinib safety. The lethal phenotypes of germline deletion of Jak2 and Jak1 were initially interpreted by some as a major limitation. Although blocking JAK2 may have downsides, first-generation JAKinibs are effective and appear relatively safe, particularly in younger populations without cardiovascular risk, contrasting the knockout phenotype.
At the same time, the degree of selectivity of agents and precisely how they act at cellular, molecular and pathological levels to mediate their therapeutic effect remain unclear. Developing a mechanistic understanding is simpler for biological agents that target single mediators than for JAKinibs, which target numerous pathways and can have different tissue-specific responses depending on the JAKinib used123. Targeting the JAK pseudokinase domain, such as is the case for deucravacitinib, may lead to improved selectivity. Advances in understanding structural details of cytokine receptors and JAKs along with improved molecular modelling may help to distinguish which inhibitor might be best for a certain inflammatory disease134. Furthermore, improved high-throughput platforms for biomarkers and improved specificity of JAKinibs should provide mechanistic insights; machine learning algorithms based on molecular fingerprints are being used to identify new targets135.
SYK family kinase inhibitors
SYK is a non-receptor tyrosine kinase essential for proximal FcR and BCR signalling that functions similar to its homologue protein tyrosine kinase ZAP70 in TCR signalling. The SYK inhibitor fostamatinib is currently approved for the treatment of immune thrombocytopenia136 (Table 1). In this autoimmune disease, autoreactive IgG antibodies target and destroy platelets through SYK-dependent FcγR-mediated phagocytosis by macrophages137. SYK family inhibitors have been proposed for the treatment of refractory chronic urticaria, an IgE-mediated allergic reaction in which cross-linking of the high-affinity FcR for IgE on mast cells leads to production of histamine, leukotrienes, prostaglandins and cytokines. The SYK inhibitor GSK-2646264 has completed phase I clinical trials for the treatment of refractory chronic urticaria (NCT02424799) and cutaneous lupus (NCT02927457). An SYK inhibitor has also been successfully used in a mouse model of a primary immunodeficiency with multi-organ inflammation caused by a dominant gain-of-function SYK mutation138.
Gusacitinib (ASN-002), a JAK and SYK inhibitor that inhibits FcR and BCR signalling, as well as cytokine signalling, has shown efficacy in AD139,140 (Table 3). In the glucose-6-phosphate isomerase arthritis model, the combination of tofacitinib and a SYK inhibitor was superior to either drug individually141.
TEC family kinase inhibitors
The TEC family of non-receptor tyrosine kinases includes Bruton’s tyrosine kinase (BTK), TEC protein tyrosine kinase, bone marrow kinase on chromosome X, IL-2-inducible T cell kinase (ITK) and tyrosine protein kinase TXK (also known as RLK). BTK was the first kinase associated with a human primary immunodeficiency, X-linked agammaglobulinaemia, which is characterized by defective B cell development and impaired antibody production142 (Table 1). Indeed, BTK has an essential role in signalling from the BCR, activating phospholipase Cγ-mediated generation of the secondary messengers inositol trisphosphate and diacylglycerol and downstream gene transcription143 (Fig. 1). In T cells, this activity is shared between ITK, TEC and TXK, with ITK playing the main role. Besides its well-known role in BCR signalling, BTK participates in signalling downstream of chemokine receptors, Toll-like receptors, the NLRP3 inflammasome144 and multiple FcRs in innate immune cells145.
Consistent with the critical requirement for BTK in B cells, BTK inhibitors are effective in treating several mature B cell neoplastic diseases, including chronic lymphocytic leukaemia, lymphoplasmacytic lymphoma (also known as Waldenström macroglobulinaemia) and mantle-cell lymphoma146 (Table 2). However, the requirement for BTK in antibody production has also led to interest in BTK inhibitors for the treatment of antibody-mediated autoimmune diseases147. Ibrutinib, a covalent irreversible inhibitor binding residue Cys481 in the BTK kinase domain, was the first BTK inhibitor to be developed in 2007 (ref. 148). Ibrutinib inhibits other kinases that have equivalent cysteine residues in their ATP-binding cleft, including ITK, TEC, some SRC family kinases and epidermal growth factor receptor. Side effects of ibrutinib include bleeding, atrial fibrillation, rashes and hypertension, which have been attributed in part to effects on TEC, epidermal growth factor receptor and other kinases. Newer generations of BTK covalent inhibitors have been developed, including the FDA-approved drugs acalabrutinib and zanubrutinib, which have greater specificity and fewer side effects. All these drugs have been used for treating malignancies, but several have potential for the treatment of autoimmune diseases including multiple sclerosis, Sjögren disease and refractory chronic urticaria (Table 3). However, resistance to ibrutinib and other covalent binders can occur, often not only owing to mutations affecting Cys481 but also owing to other mutations in BTK, as well as mutations affecting its downstream target, phospholipase Cγ. More recently, non-covalent and reversible inhibitors have been developed that interact with multiple other residues in the ATP-binding site, providing potential protection against resistance and greater selectivity for BTK148. Approximately 22 BTK inhibitors are in development, with at least 13 in clinical trials for immune-mediated diseases149,150.
BTK inhibitors have also been tested for their ability to interfere with the ‘cytokine storm’ seen in patients with severe COVID-19 (ref. 151). It has been proposed that BTK inhibition may suppress the excessive inflammation and pro-inflammatory cytokine production caused by innate immune responses to SARS-CoV-2 and provide protection against severe lung injury152. Patients with X-linked agammaglobulinaemia who were infected with SARS-CoV-2 were originally reported to manifest relatively mild disease courses possibly owing to decreased innate immune cell activation in the context of relatively intact T cell-mediated immunity153. However, more recent data indicate that some patients with X-linked agammaglobulinaemia fail to clear SARS-CoV-2, requiring rehospitalization154. These observations highlight both the importance of humoral immunity for protection against SARS-CoV-2 and the potential pitfalls of using immunosuppression during viral illness.
ITK is the TEC family kinase that is most highly expressed in T cells, where it functions to regulate the magnitude of TCR signalling and T cell differentiation143. The BTK inhibitor ibrutinib is also a potent inhibitor of ITK and has subsequently been investigated for the treatment of several inflammatory conditions involving T cells; it was the first drug to be licensed for the treatment of GVHD155. Indeed, ITK-deficient mice are resistant to GVHD156, suggesting that ITK is a critical target in this treatment. ITK-deficient mice are also resistant to airway inflammation, and multiple ITK inhibitors have been developed with the goal of treating asthma. However, although administration of a selective ITK inhibitor or ITK deficiency prevents development of allergic asthma in murine models, administration of an ITK inhibitor after induction of asthma worsened outcomes157. That observation was attributed to effects on T cell reactivation-induced cell death, although other mechanisms may contribute, including increased responsiveness to IL-2 (ref. 158). ITK deficiency in mice or treatment with ITK inhibitors also prevents development of experimental autoimmune encephalomyelitis, a mouse model for multiple sclerosis159, although it has not yet been evaluated in humans for this purpose. JTE051 is an oral ITK inhibitor that completed a phase II clinical trial for RA, but showed no significant improvement in the ACR20 response rate at week 12 (ref. 90). However, ritlecitinib, a JAK3 and TEC inhibitor, as noted earlier, has shown efficacy in treating RA, AA and vitiligo in clinical trials119,123,124,160.
Conclusions and future predictions
Over the past 20 years, great progress has been made in advancing protein kinase inhibitors in the clinic, starting with cancer but now substantially impacting autoimmune, allergic and even infectious diseases including COVID-19. The success of imatinib highlights the strategy of purposefully targeting kinases, both as anticancer drugs and inhibitors of the immune system. Yet, this dual utility is not universally applicable; mTOR inhibitors have had modest success as anticancer drugs compared with their efficacy in allogeneic transplantation and immune modulation (Box 1). By contrast, inhibitors of components of mitogen-activated protein kinase signalling cascades including BRAF have had success as chemotherapies but generated little interest as inhibitors of immune function, although efforts in this area and others are ongoing (Box 2).
Beyond therapeutic efficacy, targeted therapies, as well as biologics, can elucidate immunopathogenic mechanisms and disease endotypes; indeed, truly selective agents can give clues to underlying pathogenesis. Unlike the specificity of biologics, small molecule kinase inhibitors, especially competitive ATP antagonists, have relative degrees of selectivity. In addition, many kinases mediate signalling by diverse ligands and even different families of ligands. This is true of first-generation JAKinibs — even when efficacious, it can be difficult to assign this to specific cytokines. Nevertheless, detailed assessment of biomarkers and gene expression may reveal clues to disease pathogenesis and mechanism of action.
Many kinase inhibitors have demonstrated efficacy in immune-mediated disease, but equally, if not more, important is safety. Delivering the drug directly to the target organ or cell is a longstanding solution for limiting systemic adverse events. For example, several JAKinibs are available topically to treat inflammatory skin conditions or as inhaled agents to treat asthma. This is analogous to the success of topical and inhaled steroids that are now mainstays of therapy in both conditions. Similarly, parenteral injection of steroids is widely used, but many approved kinase inhibitors have not been generated as parenteral formulations.
In principle, the ideal protein kinase inhibitor is selective, with minimal off-target side effects. However, it has become clear that some kinase inhibitors interact not only with other kinases but also with G-protein-coupled receptors and bromodomain-containing proteins. For instance, the JAK2 inhibitor fedratinib also targets bromodomain-containing protein 4, which could add to its efficacy but also lead to adverse events. Alternative strategies for designing kinase inhibitors have potential to maximize selectivity beyond competitive ATP agonists. Strategies that are already being used are generation of irreversible kinase inhibitors and allosteric inhibitors. Many human protein kinases have cysteine residues in, or proximal to, the ATP-binding site; examples of inhibitors that target these residues include covalent BTK inhibitors and the JAK3 and TEC inhibitor ritlecitinib. In addition, as the catalytic domain of protein kinases is highly conserved, cysteine residues distal from the ATP-binding site may also be targeted. Only a small proportion of protein kinases have a catalytically inactive kinase domain; targeting the JAK pseudokinase domain was the strategy used to develop deucravacitinib. More recently, a covalent, allosteric JAK1 inhibitor, VVD-118313, that targets the pseudokinase domain has also been reported161. Interestingly, endogenous compounds including metabolites can bind outside catalytic domains. For instance, the Krebs cycle-derived metabolite itaconate inhibits IL-4 signalling and binds to multiple JAK1 cysteines162. Furthermore, residues other than cysteines such as lysine, tyrosine, histidine and methionine can be used to generate irreversible inhibitors.
Deucravacitinib is also notable in that it is deuterated. Because deuterium–carbon bonds are stronger than hydrogen–carbon bonds, this modification can improve pharmacodynamics, with the potential for increasing potency and selectivity. Ideally, these options for rational design improve efficacy and safety. The macrolide antibiotics sirolimus and tacrolimus inhibit discrete signalling pathways by indirect mechanisms with a high degree of specificity even when they bind to the same protein (Box 1). This suggests that there are other novel ways of devising specific signalling inhibitors yet to be discovered. Ideally, agents with improved selectivity will offer even greater opportunities to elucidate immunopathological mechanisms.
Moreover, other alternative strategies in targeting kinases may evolve in the future. Another strategy for targeting kinases is targeted protein degradation, which uses small molecules (degraders) to recruit E3 ubiquitin ligases and to promote proteolysis163,164. This pathway is now known to be important for thalidomide and related compounds, which target cereblon that binds to the E3 ubiquitin ligase cullin-RING ligase 4 and expands its repertoire of target proteins for degradation165,166. Although one utility of targeted protein degradation is expanding the spectrum of ‘druggable’ molecules, screens have also been used to accelerate the generation of specific kinase degraders167. Targeted protein degradation using small molecule degraders to promote proteolysis may be another promising strategy. KT-474 is an IRAK4 degrader being tested in a phase I clinical study in healthy volunteers and in patients with AD and hidradenitis suppurativa168 (NCT04772885).
Finally, depending on the disease and pathway inhibited, it may be better to partially suppress rather than completely inhibit kinase function to improve safety and tolerability, particularly in the case of JAK and TEC family inhibitors that affect the development of multiple haematopoietic cell lineages. This strategy may be particularly relevant to inflammatory and autoimmune diseases in which dampened responses may lead to acceptable clinical outcomes versus in cancers in which full suppression may be needed. It remains to be seen whether protein inhibitors with short half-lives that may partially suppress pathways could be used in conjunction with longer acting immunosuppressants for chronic inflammation. In short, advances are coming quickly but in many respects this is just the beginning; ideally, our ability to assess the action of immune-related kinase inhibitors and their contribution to immunopathology in real time will improve our ability to use these drugs effectively — and more importantly, safely.
Change history
27 November 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41577-023-00976-5
References
O’Shea, J. J., Kanno, Y. & Chan, A. C. In search of magic bullets: the golden age of immunotherapeutics. Cell 157, 227–240 (2014).
McInnes, I. B. & Gravallese, E. M. Immune-mediated inflammatory disease therapeutics: past, present and future. Nat. Rev. Immunol. 21, 680–686 (2021).
Courtney, A. H., Lo, W. L. & Weiss, A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem. Sci. 43, 108–123 (2018).
Ross, S. H. & Cantrell, D. A. Signaling and function of interleukin-2 in T lymphocytes. Annu. Rev. Immunol. 36, 411–433 (2018).
Zarrin, A. A., Bao, K., Lupardus, P. & Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 20, 39–63 (2021).
Attwood, M. M., Fabbro, D., Sokolov, A. V., Knapp, S. & Schioth, H. B. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat. Rev. Drug Discov. 20, 839–861 (2021).
Ardito, F., Giuliani, M., Perrone, D., Troiano, G. & Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 40, 271–280 (2017).
Deribe, Y. L., Pawson, T. & Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672 (2010).
Duong-Ly, K. C. & Peterson, J. R. The human kinome and kinase inhibition. Curr. Protoc. Pharmacol. https://doi.org/10.1002/0471141755.ph0209s60 (2013).
Arter, C., Trask, L., Ward, S., Yeoh, S. & Bayliss, R. Structural features of the protein kinase domain and targeted binding by small molecule inhibitors. J. Biol. Chem. 298, 102247 (2022).
Goodridge, H. S. & Harnett, M. M. Introduction to immune cell signalling. Parasitology 130, S3–S9 (2005).
Zhang, Z., Bu, L., Luo, J. & Guo, J. Targeting protein kinases benefits cancer immunotherapy. Biochim. Biophys. Acta Rev. Cancer 1877, 188738 (2022).
Druker, B. J. et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 355, 2408–2417 (2006).
O’Brien, S. G. et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 348, 994–1004 (2003). This is one of the first examples of the use of a selective kinase inhibitor for myeloproliferative disease.
Ren, R. Mechanisms of BCR–ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 5, 172–183 (2005).
Zimmermann, J., Buchdunger, E., Mett, H., Meyer, T. & Lydon, N. B. Potent and selective inhibitors of the Abl-kinase: phenylamino-pyrimidine (PAP) derivatives. Bioorg. Med. Chem. Lett. 7, 187–192 (1997).
Ayala-Aguilera, C. C. et al. Small molecule kinase inhibitor drugs (1995–2021): medical indication, pharmacology, and synthesis. J. Med. Chem. 65, 1047–1131 (2022).
Manning, G., Whyte, D. B., Martinez, R., Hunter, T. & Sudarsanam, S. The protein kinase complement of the human genome. Science 298, 1912–1934 (2002).
Sacha, T. Imatinib in chronic myeloid leukemia: an overview. Mediterr. J. Hematol. Infect. Dis. 6, e2014007 (2014).
Stanley, E. R. & Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 6, a021857 (2014).
Shelley, A. J. Biosystematics and distribution of simuliid vectors of human onchocerciasis in South America. Mem. Inst. Oswaldo Cruz 83, 399–403 (1988).
Kantarjian, H. M. et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood 110, 3540–3546 (2007).
Ali, S. et al. Dasatinib may overcome the negative prognostic impact of KIR2DS1 in newly diagnosed patients with chronic myeloid leukemia. Blood 120, 697–698 (2012). This paper describes the need for second-generation ABL kinase inhibitors.
Cortes, J. E. et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 369, 1783–1796 (2013).
Azizi, G. & Mirshafiey, A. Imatinib mesylate: an innovation in treatment of autoimmune diseases. Recent Pat. Inflamm. Allergy Drug Discov. 7, 259–267 (2013).
Aman, J. et al. Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation 126, 2728–2738 (2012).
Taymaz-Nikerel, H., Eraslan, S. & Kirdar, B. Insights into the mechanism of anticancer drug imatinib revealed through multi-omic analyses in yeast. OMICS 24, 667–678 (2020).
Tefferi, A. Molecular drug targets in myeloproliferative neoplasms: mutant ABL1, JAK2, MPL, KIT, PDGFRA, PDGFRB and FGFR1. J. Cell Mol. Med. 13, 215–237 (2009). This paper highlights critical drug therapy targets in myeloproliferative disease.
Requena, G. et al. Clinical profile and treatment in hypereosinophilic syndrome variants: a pragmatic review. J. Allergy Clin. Immunol. Pract. 10, 2125–2134 (2022).
Hilberg, F. et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 68, 4774–4782 (2008).
Wollin, L. et al. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 54, 1900161 (2019).
Flaherty, K. R. et al. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 381, 1718–1727 (2019).
Wollin, L. et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 45, 1434–1445 (2015).
Prasse, A. et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 179, 717–723 (2009).
Durham, B. H. et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat. Med. 25, 1839–1842 (2019).
Hu, X. et al. Imatinib inhibits CSF1R that stimulates proliferation of rheumatoid arthritis fibroblast-like synoviocytes. Clin. Exp. Immunol. 195, 237–250 (2019).
Martinez-Muriana, A. et al. CSF1R blockade slows the progression of amyotrophic lateral sclerosis by reducing microgliosis and invasion of macrophages into peripheral nerves. Sci. Rep. 6, 25663 (2016).
Alexander, K. A. et al. CSF-1-dependant donor-derived macrophages mediate chronic graft-versus-host disease. J. Clin. Invest. 124, 4266–4280 (2014).
Lee, S. J. et al. Safety, tolerability, and efficacy of axatilimab, a CSF-1R humanized antibody, for chronic graft-versus-host disease after 2 or more lines of systemic treatment. Blood 138, 263–263 (2021).
Genovese, M. C. et al. Results from a phase IIA parallel group study of JNJ-40346527, an oral CSF-1R inhibitor, in patients with active rheumatoid arthritis despite disease-modifying antirheumatic drug therapy. J. Rheumatol. 42, 1752–1760 (2015).
Moses, H. L., Roberts, A. B. & Derynck, R. The discovery and early days of TGF-beta: a historical perspective. Cold Spring Harb. Perspect. Biol. 8, a021865 (2016).
Karlsson, J. A. et al. Treatment response to a second or third TNF-inhibitor in RA: results from the South Swedish Arthritis Treatment Group Register. Rheumatology 47, 507–513 (2008).
Gadina, M. et al. Janus kinases to jakinibs: from basic insights to clinical practice. Rheumatology 58, i4–i16 (2019).
Pesu, M. et al. Jak3, severe combined immunodeficiency, and a new class of immunosuppressive drugs. Immunol. Rev. 203, 127–142 (2005). This Review describes the critical role of JAK3 signalling in immune cells and defines a form of severe combined immunodeficiency that is driven by a variant in JAK3 signalling. This sparked a collaboration between industry and academia to use the inhibitor tofacitinib for immune-mediated disorders.
Tefferi, A. A refined diagnostic algorithm for polycythemia vera that incorporates mutation screening for JAK2(V617F). Curr. Hematol. Malig. Rep. 1, 81–86 (2006).
Ghoreschi, K., Laurence, A. & O’Shea, J. J. Janus kinases in immune cell signaling. Immunol. Rev. 228, 273–287 (2009).
Witalisz-Siepracka, A. et al. Loss of JAK1 drives innate immune deficiency. Front. Immunol. 9, 3108 (2018).
Aringer, M. et al. Janus kinases and their role in growth and disease. Life Sci. 64, 2173–2186 (1999).
Zeiser, R. et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N. Engl. J. Med. 382, 1800–1810 (2020).
Jagasia, M. et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label phase 2 trial. Blood 135, 1739–1749 (2020). This is one of the first clinical trials of a JAKinib for the treatment of GVHD.
Waskiel-Burnat, A. et al. The role of serum Th1, Th2, and Th17 cytokines in patients with alopecia areata: clinical implications. Cells 10, 3397 (2021).
Petukhova, L. et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature 466, 113–117 (2010).
Guttman-Yassky, E. et al. Phase 2a randomized clinical trial of dupilumab (anti-IL-4Ralpha) for alopecia areata patients. Allergy 77, 897–906 (2022).
McKenzie, P. L. & Castelo-Soccio, L. Dupilumab therapy for alopecia areata in pediatric patients with concomitant atopic dermatitis. J. Am. Acad. Dermatol. 84, 1691–1694 (2021).
Hendricks, A. J., Lio, P. A. & Shi, V. Y. Dupilumab and alopecia: causative or therapeutic? Dermatology 235, 306–307 (2019).
Xing, L. et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat. Med. 20, 1043–1049 (2014). This is a seminal paper showing that AA is immune-mediated and that JAK inhibitors can reverse the disease. This led to the first human trials of JAKinibs for AA.
Martins, C. et al. Vitiligo skin T cells are prone to produce type 1 and type 2 cytokines to induce melanocyte dysfunction and epidermal inflammatory response through Jak signaling. J. Invest. Dermatol. 142, 1194–1205.e7 (2022).
Kim, B. S. The translational revolution of itch. Neuron 110, 2209–2214 (2022).
Ghoreschi, K. et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 186, 4234–4243 (2011).
Keenan, C., Nichols, K. E. & Albeituni, S. Use of the JAK inhibitor ruxolitinib in the treatment of hemophagocytic lymphohistiocytosis. Front. Immunol. 12, 614704 (2021).
Schwartz, D. M. et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 16, 843–862 (2017).
Jones, A. V. et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 106, 2162–2168 (2005). This paper identifies that JAK2 gain-of-function mutations are important for the pathogenesis of chronic myeloproliferative disease and prompts the use of JAK inhibition as a therapeutic approach.
Kralovics, R. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 (2005).
Steensma, D. P. et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both ‘atypical’ myeloproliferative disorders and myelodysplastic syndromes. Blood 106, 1207–1209 (2005).
Verstovsek, S. et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 363, 1117–1127 (2010).
Harrison, C. et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 366, 787–798 (2012).
Verstovsek, S. et al. The clinical benefit of ruxolitinib across patient subgroups: analysis of a placebo-controlled, phase III study in patients with myelofibrosis. Br. J. Haematol. 161, 508–516 (2013).
Quintas-Cardama, A. et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 115, 3109–3117 (2010).
Papp, K. et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: results from 2 phase 3, randomized, double-blind studies. J. Am. Acad. Dermatol. 85, 863–872 (2021).
Burmester, G. R. et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet 381, 451–460 (2013).
Rakieh, C. & Conaghan, P. G. Tofacitinib for treatment of rheumatoid arthritis. Adv. Ther. 30, 713–726 (2013).
Robinette, M. L. et al. Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol. 11, 50–60 (2018).
Ballesteros-Tato, A. Beyond regulatory T cells: the potential role for IL-2 to deplete T-follicular helper cells and treat autoimmune diseases. Immunotherapy 6, 1207–1220 (2014).
van Vollenhoven, R. et al. Evaluation of the short-, mid-, and long-term effects of tofacitinib on lymphocytes in patients with rheumatoid arthritis. Arthritis Rheumatol. 71, 685–695 (2019).
Mease, P. et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N. Engl. J. Med. 377, 1537–1550 (2017).
Sandborn, W. J. et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 376, 1723–1736 (2017).
Deodhar, A. et al. Tofacitinib for the treatment of ankylosing spondylitis: a phase III, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 80, 1004–1013 (2021).
Ruperto, N. et al. Tofacitinib in juvenile idiopathic arthritis: a double-blind, placebo-controlled, withdrawal phase 3 randomised trial. Lancet 398, 1984–1996 (2021).
Paudyal, A. et al. JAK-inhibitors for dermatomyositis: a concise literature review. Dermatol. Ther. 34, e14939 (2021).
Chen, Z., Wang, X. & Ye, S. Tofacitinib in amyopathic dermatomyositis-associated interstitial lung disease. N. Engl. J. Med. 381, 291–293 (2019).
Fan, L. et al. A retrospective analysis of outcome in melanoma differentiation-associated gene 5-related interstitial lung disease treated with tofacitinib or tacrolimus. J. Rheumatol. 49, 1356–1364 (2022).
Chaimowitz, N. S., Ebenezer, S. J., Hanson, I. C., Anderson, M. & Forbes, L. R. STAT1 gain of function, type 1 diabetes, and reversal with JAK inhibition. N. Engl. J. Med. 383, 1494–1496 (2020).
Michniacki, T. F. et al. SOCS1 haploinsufficiency presenting as severe enthesitis, bone marrow hypocellularity, and refractory thrombocytopenia in a pediatric patient with subsequent response to JAK inhibition. J. Clin. Immunol. 42, 1766–1777 (2022).
Morand, E. F. et al. Efficacy and safety of baricitinib in patients with systemic lupus erythematosus: results from two randomised, double-blind, placebo-controlled, parallel-group, phase 3 studies. Ann. Rheum. Dis. 81, 237 (2022).
The RECOVERY Collaborative Group. Dexamethasone in hospitalized patients with Covid-19. N. Engl. J. Med. 384, 693–704 (2021).
Stebbing, J. et al. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 20, 400–402 (2020).
Stebbing, J. et al. JAK inhibition reduces SARS-CoV-2 liver infectivity and modulates inflammatory responses to reduce morbidity and mortality. Sci. Adv. 7, eabe4724 (2021).
Zhang, X., Zhang, Y., Qiao, W., Zhang, J. & Qi, Z. Baricitinib, a drug with potential effect to prevent SARS-CoV-2 from entering target cells and control cytokine storm induced by COVID-19. Int. Immunopharmacol. 86, 106749 (2020).
King, B. et al. Two phase 3 trials of baricitinib for alopecia areata. N. Engl. J. Med. 386, 1687–1699 (2022).
Blaess, J. et al. Immunosuppressive agents for rheumatoid arthritis: a systematic review of clinical trials and their current development stage. Ther. Adv. Musculoskelet. Dis. 12, 1759720X20959971 (2020).
Guimaraes, P. O. et al. Tofacitinib in patients hospitalized with Covid-19 pneumonia. N. Engl. J. Med. 385, 406–415 (2021).
Hoisnard, L. et al. Adverse events associated with JAK inhibitors in 126,815 reports from the WHO pharmacovigilance database. Sci. Rep. 12, 7140 (2022).
Vincenti, F. et al. Randomized phase 2b trial of tofacitinib (CP-690,550) in de novo kidney transplant patients: efficacy, renal function and safety at 1 year. Am. J. Transpl. 12, 2446–2456 (2012).
Sunzini, F., McInnes, I. & Siebert, S. JAK inhibitors and infections risk: focus on herpes zoster. Ther. Adv. Musculoskelet. Dis. 12, 1759720X20936059 (2020).
Yates, M. et al. Venous thromboembolism risk with JAK inhibitors: a meta-analysis. Arthritis Rheumatol. 73, 779–788 (2021). In this meta-analysis, thromboembolic risk factors for use of JAKinibs in patients with rheumatoid arthritis are identified. This led to warnings from multiple safety agencies.
Rose-John, S. Interleukin-6 family cytokines. Cold Spring Harb. Perspect. Biol. 10, a028415 (2018).
Ytterberg, S. R. et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N. Engl. J. Med. 386, 316–326 (2022).
U.S. Food & Drug Administration. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death (2021).
Curtis, J. R. et al. Malignancy risk with tofacitinib versus TNF inhibitors in rheumatoid arthritis: results from the open-label, randomised controlled ORAL surveillance trial. Ann. Rheum. Dis. 82, 331–343 (2023).
Cohen, S. B. et al. Long-term safety of tofacitinib up to 9.5 years: a comprehensive integrated analysis of the rheumatoid arthritis clinical development programme. RMD Open 6, e001395 (2020).
Mueller, R. B. et al. Effect of dose adjustments on the efficacy and safety of tofacitinib in patients with rheumatoid arthritis: a post hoc analysis of an open-label, long-term extension study (ORAL Sequel). Clin. Rheumatol. 41, 1045–1055 (2022).
Smolen, J. S. et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann. Rheum. Dis. 82, 3–18 (2023).
Hasni, S. A. et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat. Commun. 12, 3391 (2021).
Abe, M. Replication of ColE1 plasmid deoxyribonucleic acid in a thermosensitive dnaA mutant of Escherichia coli. J. Bacteriol. 141, 1024–1030 (1980).
Li, N. et al. Randomized, double-blinded, placebo-controlled phase I study of the pharmacokinetics, pharmacodynamics, and safety of KL130008, a novel oral JAK inhibitor, in healthy subjects. Eur. J. Pharm. Sci. 176, 106257 (2022).
Silverberg, J. I. et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 156, 863–873 (2020).
Parmentier, J. M. et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2, 23 (2018).
Simpson, E. L. et al. Integrated safety analysis of abrocitinib for the treatment of moderate-to-severe atopic dermatitis from the phase II and phase III clinical trial program. Am. J. Clin. Dermatol. 22, 693–707 (2021).
Tanaka, M. et al. Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood 102, 3154–3162 (2003).
Reid, J. et al. In vivo affinity and target engagement in skin and blood in a first-time-in-human study of an anti-oncostatin M monoclonal antibody. Br. J. Clin. Pharmacol. 84, 2280–2291 (2018).
Diogo, D. et al. TYK2 protein-coding variants protect against rheumatoid arthritis and autoimmunity, with no evidence of major pleiotropic effects on non-autoimmune complex traits. PLoS ONE 10, e0122271 (2015).
Gerstenberger, B. S. et al. Discovery of tyrosine kinase 2 (TYK2) inhibitor (PF-06826647) for the treatment of autoimmune diseases. J. Med. Chem. 63, 13561–13577 (2020).
Loo, W. J. et al. Clinical implications of targeting the JAK–STAT pathway in psoriatic disease: emphasis on the TYK2 pathway. J. Cutan. Med. Surg. 27 (suppl. 1), 3S–24S (2023).
Wrobleski, S. T. et al. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J. Med. Chem. 62, 8973–8995 (2019). This paper describes the use of allosteric inhibitors and selective inhibition of TYK2 to reduce off-target effects.
Chimalakonda, A. et al. Selectivity profile of the tyrosine kinase 2 inhibitor deucravacitinib compared with Janus kinase 1/2/3 inhibitors. Dermatol. Ther. 11, 1763–1776 (2021).
Mease, P. J. et al. Efficacy and safety of selective TYK2 inhibitor, deucravacitinib, in a phase II trial in psoriatic arthritis. Ann. Rheum. Dis. 81, 815–822 (2022).
Morand, E. et al. Deucravacitinib, a tyrosine kinase 2 inhibitor, in systemic lupus erythematosus: a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 75, 242–252 (2023).
Schnepf, D. et al. Selective Janus kinase inhibition preserves interferon-lambda-mediated antiviral responses. Sci. Immunol. 6, eabd5318 (2021).
King, B. et al. A phase 2a randomized, placebo-controlled study to evaluate the efficacy and safety of the oral Janus kinase inhibitors ritlecitinib and brepocitinib in alopecia areata: 24-week results. J. Am. Acad. Dermatol. 85, 379–387 (2021).
Genovese, M. C., van Vollenhoven, R. F., Pacheco-Tena, C., Zhang, Y. & Kinnman, N. VX-509 (Decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol. 68, 46–55 (2016).
Zetterberg, C. et al. VX-509 (Decernotinib)-mediated CYP3A time-dependent inhibition: an aldehyde oxidase metabolite as a perpetrator of drug–drug interactions. Drug Metab. Dispos. 44, 1286–1295 (2016).
Chen, C. et al. A highly selective JAK3 inhibitor is developed for treating rheumatoid arthritis by suppressing gammac cytokine-related JAK-STAT signal. Sci. Adv. 8, eabo4363 (2022).
Guttman-Yassky, E. et al. Ritlecitinib and brepocitinib demonstrate significant improvement in scalp alopecia areata biomarkers. J. Allergy Clin. Immunol. 149, 1318–1328 (2022).
Robinson, M. F. et al. Efficacy and safety of PF-06651600 (Ritlecitinib), a novel JAK3/TEC inhibitor, in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 72, 1621–1631 (2020).
Talpaz, M. & Kiladjian, J. J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 35, 1–17 (2021).
Rosmarin, D. et al. Ruxolitinib cream for treatment of vitiligo: a randomised, controlled, phase 2 trial. Lancet 396, 110–120 (2020).
Nakagawa, H. et al. Delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: a phase 3, randomized, double-blind, vehicle-controlled study and an open-label, long-term extension study. J. Am. Acad. Dermatol. 82, 823–831 (2020). A report of a phase III clinical trial showing efficacy of topical JAK inhibitors for the treatment of atopic dermatitis.
Milara, J. et al. The pan-JAK inhibitor LAS194046 reduces neutrophil activation from severe asthma and COPD patients in vitro. Sci. Rep. 12, 5132 (2022).
Nilsson, M. et al. Characterization of selective and potent JAK1 inhibitors intended for the inhaled treatment of asthma. Drug Des. Devel Ther. 16, 2901–2917 (2022).
Singh, D. et al. A phase 2 multiple ascending dose study of the inhaled pan-JAK inhibitor nezulcitinib (TD-0903) in severe COVID-19. Eur. Respir. J. 58, 2100673 (2021).
Alexander, M., Luo, Y., Raimondi, G., O’Shea, J. J. & Gadina, M. Jakinibs of all trades: inhibiting cytokine signaling in immune-mediated pathologies. Pharmaceuticals 15, 48 (2021).
van der Heijde, D. et al. Tofacitinib in combination with methotrexate in patients with rheumatoid arthritis: clinical efficacy, radiographic, and safety outcomes from a twenty-four-month, phase III study. Arthritis Rheumatol. 71, 878–891 (2019).
Seif, F., Pornour, M. & Mansouri, D. Combination of JAKinibs with methotrexate or anti-cytokine biologics in patients with severe COVID-19. Int. Arch. Allergy Immunol. 181, 648–649 (2020).
Sk, M. F., Jonniya, N. A., Roy, R. & Kar, P. Unraveling the molecular mechanism of recognition of selected next-generation antirheumatoid arthritis inhibitors by Janus kinase 1. ACS Omega 7, 6195–6209 (2022).
Yang, M. et al. Machine learning models based on molecular fingerprints and an extreme gradient boosting method lead to the discovery of JAK2 inhibitors. J. Chem. Inf. Model. 59, 5002–5012 (2019). This paper shows the use of machine learning to identify new JAK inhibitors. This method also led to the use of JAKinibs for the treatment of COVID-19-related illness.
Paik, J. Fostamatinib: a review in chronic immune thrombocytopenia. Drugs 81, 935–943 (2021).
Provan, D. & Semple, J. W. Recent advances in the mechanisms and treatment of immune thrombocytopenia. eBioMedicine 76, 103820 (2022).
Wang, L. et al. Gain-of-function variants in SYK cause immune dysregulation and systemic inflammation in humans and mice. Nat. Genet. 53, 500–510 (2021).
Pavel, A. B. et al. Oral Janus kinase/SYK inhibition (ASN002) suppresses inflammation and improves epidermal barrier markers in patients with atopic dermatitis. J. Allergy Clin. Immunol. 144, 1011–1024 (2019).
Bissonnette, R. et al. The oral Janus kinase/spleen tyrosine kinase inhibitor ASN002 demonstrates efficacy and improves associated systemic inflammation in patients with moderate-to-severe atopic dermatitis: results from a randomized double-blind placebo-controlled study. Br. J. Dermatol. 181, 733–742 (2019).
Llop-Guevara, A. et al. Simultaneous inhibition of JAK and SYK kinases ameliorates chronic and destructive arthritis in mice. Arthritis Res. Ther. 17, 356 (2015).
Lindvall, J. M. et al. Bruton’s tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol. Rev. 203, 200–215 (2005).
Berg, L. J., Finkelstein, L. D., Lucas, J. A. & Schwartzberg, P. L. Tec family kinases in T lymphocyte development and function. Annu. Rev. Immunol. 23, 549–600 (2005).
Pal Singh, S., Dammeijer, F. & Hendriks, R. W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 17, 57 (2018).
Weber, A. N. R. et al. Bruton’s tyrosine kinase: an emerging key player in innate immunity. Front. Immunol. 8, 1454 (2017).
Burger, J. A. & Wiestner, A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat. Rev. Cancer 18, 148–167 (2018).
Ringheim, G. E., Wampole, M. & Oberoi, K. Bruton’s tyrosine kinase (BTK) inhibitors and autoimmune diseases: making sense of BTK inhibitor specificity profiles and recent clinical trial successes and failures. Front. Immunol. 12, 662223 (2021).
Zain, R. & Vihinen, M. Structure–function relationships of covalent and non-covalent BTK inhibitors. Front. Immunol. 12, 694853 (2021).
Perutelli, F., Montalbano, M. C., Boccellato, E., Coscia, M. & Vitale, C. Beyond ibrutinib: novel BTK inhibitors for the treatment of chronic lymphocytic leukemia. Curr. Opin. Oncol. 34, 757–767 (2022).
Smith, C. I. E., Brown, J. R. & Zain, R. Editorial: new insights on Bruton’s tyrosine kinase inhibitors. Front. Immunol. 12, 804735 (2021).
Rezaei, M., Barati, S., Babamahmoodi, A., Dastan, F. & Marjani, M. The possible role of Bruton tyrosine kinase inhibitors in the treatment of COVID-19: a review. Curr. Ther. Res. Clin. Exp. 96, 100658 (2022).
Stack, M. et al. BTK inhibitors for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): a systematic review. Clin. Immunol. 230, 108816 (2021).
Hovey, J. G., Tolbert, D. & Howell, D. Burton’s agammaglobulinemia and COVID-19. Cureus 12, e11701 (2020).
Drzymalla, E. et al. COVID-19-related health outcomes in people with primary immunodeficiency: a systematic review. Clin. Immunol. 243, 109097 (2022).
Jaglowski, S. M. & Blazar, B. R. How ibrutinib, a B-cell malignancy drug, became an FDA-approved second-line therapy for steroid-resistant chronic GVHD. Blood Adv. 2, 2012–2019 (2018).
Mammadli, M. et al. Targeting interleukin-2-inducible T-cell kinase (ITK) differentiates GVL and GVHD in allo-HSCT. Front. Immunol. 11, 593863 (2020).
Sun, Y. et al. Inhibition of the kinase ITK in a mouse model of asthma reduces cell death and fails to inhibit the inflammatory response. Sci. Signal. 8, ra122 (2015).
Gomez-Rodriguez, J. et al. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J. Exp. Med. 211, 529–543 (2014).
Lechner, K. S., Neurath, M. F. & Weigmann, B. Role of the IL-2 inducible tyrosine kinase ITK and its inhibitors in disease pathogenesis. J. Mol. Med. 98, 1385–1395 (2020).
Ezzedine, K. et al. Efficacy and safety of oral ritlecitinib for the treatment of active nonsegmental vitiligo: a randomized phase 2b clinical trial. J. Am. Acad. Dermatol. 88, 395–403 (2023).
Kavanagh, M. E. et al. Selective inhibitors of JAK1 targeting an isoform-restricted allosteric cysteine. Nat. Chem. Biol. 18, 1388–1398 (2022).
Runtsch, M. C. et al. Itaconate and itaconate derivatives target JAK1 to suppress alternative activation of macrophages. Cell Metab. 34, 487–501.e8 (2022).
Burslem, G. M. & Crews, C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 181, 102–114 (2020).
Mullard, A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 20, 247–250 (2021).
Ito, T. et al. Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010).
Jan, M., Sperling, A. S. & Ebert, B. L. Cancer therapies based on targeted protein degradation — lessons learned with lenalidomide. Nat. Rev. Clin. Oncol. 18, 401–417 (2021).
Donovan, K. A. et al. Mapping the degradable kinome provides a resource for expedited degrader development. Cell 183, 1714–1731.e10 (2020).
Mullard, A. IRAK4 degrader to take on innate immunity. Nat. Biotechnol. 38, 1221–1223 (2020).
O’Riordan, C. E. et al. X-linked immunodeficient mice with no functional Bruton’s tyrosine kinase are protected from sepsis-induced multiple organ failure. Front. Immunol. 11, 581758 (2020).
Streubel, B., Vinatzer, U., Willheim, M., Raderer, M. & Chott, A. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia 20, 313–318 (2006).
Flores, C., Fouquet, G., Moura, I. C., Maciel, T. T. & Hermine, O. Lessons to learn from low-dose cyclosporin-A: a new approach for unexpected clinical applications. Front. Immunol. 10, 588 (2019).
Calne, R. Y. et al. Cyclosporin A initially as the only immunosuppressant in 34 recipients of cadaveric organs: 32 kidneys, 2 pancreases, and 2 livers. Lancet 2, 1033–1036 (1979).
Scalea, J. R., Levi, S. T., Ally, W. & Brayman, K. L. Tacrolimus for the prevention and treatment of rejection of solid organ transplants. Expert. Rev. Clin. Immunol. 12, 333–342 (2016).
Nakahara, T., Morimoto, H., Murakami, N. & Furue, M. Mechanistic insights into topical tacrolimus for the treatment of atopic dermatitis. Pediatr. Allergy Immunol. 29, 233–238 (2018).
Park, Y. J., Yoo, S. A., Kim, M. & Kim, W. U. The role of calcium-calcineurin-NFAT signaling pathway in health and autoimmune diseases. Front. Immunol. 11, 195 (2020).
Hogan, P. G., Chen, L., Nardone, J. & Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232 (2003).
Griffith, J. P. et al. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12–FK506 complex. Cell 82, 507–522 (1995).
Livi, G. P. Halcyon days of TOR: reflections on the multiple independent discovery of the yeast and mammalian TOR proteins. Gene 692, 145–155 (2019).
Di Maira, T., Little, E. C. & Berenguer, M. Immunosuppression in liver transplant. Best. Pract. Res. Clin. Gastroenterol. 46-47, 101681 (2020).
Lo, Y. C., Lee, C. F. & Powell, J. D. Insight into the role of mTOR and metabolism in T cells reveals new potential approaches to preventing graft rejection. Curr. Opin. Organ. Transpl. 19, 363–371 (2014).
Palsson-McDermott, E. M. & O’Neill, L. A. J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 30, 300–314 (2020).
Schena, F. P. et al. Conversion from calcineurin inhibitors to sirolimus maintenance therapy in renal allograft recipients: 24-month efficacy and safety results from the CONVERT trial. Transplantation 87, 233–242 (2009).
Braun, W. Transplantation: sirolimus plus calcineurin inhibitors in transplantation. Nat. Rev. Nephrol. 5, 252–254 (2009).
Dansirikul, C., Duffull, S. B., Morris, R. G. & Tett, S. E. Relationships between sirolimus dosing, concentration and outcomes in renal transplant recipients. Br. J. Clin. Pharmacol. 60, 560–565 (2005).
Murakami, M. et al. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell Biol. 24, 6710–6718 (2004).
Batlle, E. & Massague, J. Transforming growth factor-beta signaling in immunity and cancer. Immunity 50, 924–940 (2019).
Hatton, R. D. TGF-beta in Th17 cell development: the truth is out there. Immunity 34, 288–290 (2011).
Zhang, S. The role of transforming growth factor beta in T helper 17 differentiation. Immunology 155, 24–35 (2018).
Massague, J. TGFbeta in cancer. Cell 134, 215–230 (2008).
Kim, B. G., Malek, E., Choi, S. H., Ignatz-Hoover, J. J. & Driscoll, J. J. Novel therapies emerging in oncology to target the TGF-beta pathway. J. Hematol. Oncol. 14, 55 (2021).
Nakayama, S. et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 82, 210–217 (2008).
Bi, Y. et al. A disease progression model of longitudinal lung function decline in idiopathic pulmonary fibrosis patients. J. Pharmacokinet. Pharmacodyn. 48, 55–67 (2021).
Wang, Z., Wesche, H., Stevens, T., Walker, N. & Yeh, W. C. IRAK-4 inhibitors for inflammation. Curr. Top. Med. Chem. 9, 724–737 (2009).
Gobin, K. et al. IRAK4 deficiency in a patient with recurrent pneumococcal infections: case report and review of the literature. Front. Pediatr. 5, 83 (2017).
von Bernuth, H., Picard, C., Puel, A. & Casanova, J. L. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur. J. Immunol. 42, 3126–3135 (2012).
Petrova, T. et al. Prevention and partial reversion of the lupus phenotype in ABIN1[D485N] mice by an IRAK4 inhibitor. Lupus Sci. Med. 8, e000573 (2021).
Winkler, A. et al. The interleukin-1 receptor-associated kinase 4 inhibitor PF-06650833 blocks inflammation in preclinical models of rheumatic disease and in humans enrolled in a randomized clinical trial. Arthritis Rheumatol. 73, 2206–2218 (2021).
Arthur, J. S. C. & Ley, S. C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692 (2013).
Rincón, M., Flavell, R. A. & Davis, R. A. The JNK and P38 MAP kinase signaling pathways in T cell-mediated immune responses. Free. Radic. Biol. Med. 28, 1328–1337 (2000).
Yokota, T. & Wang, Y. p38 MAP kinases in the heart. Gene 575, 369–376 (2016).
Chapman, P. B. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516 (2011).
Hauschild, A. et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380, 358–365 (2012).
Flaherty, K. T. et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 367, 107–114 (2012).
Grob, J. J. et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. Lancet Oncol. 16, 1389–1398 (2015).
Lee, J. C. et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746 (1994).
Gaur, R. et al. CC-99677, a novel, oral, selective covalent MK2 inhibitor, sustainably reduces pro-inflammatory cytokine production. Arthritis Res. Ther. 24, 199 (2022).
El Masri, R. & Delon, J. RHO GTPases: from new partners to complex immune syndromes. Nat. Rev. Immunol. 21, 499–513 (2021).
Pernis, A. B., Ricker, E., Weng, C. H., Rozo, C. & Yi, W. Rho kinases in autoimmune diseases. Annu. Rev. Med. 67, 355–374 (2016).
Flynn, R. et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 127, 2144–2154 (2016).
Louis, C., Burns, C. & Wicks, I. TANK-binding kinase 1-dependent responses in health and autoimmunity. Front. Immunol. 9, 434 (2018).
Thomson, D. W. & Bergamini, G. Recent progress in small molecule TBK1 inhibitors: a patent review (2015–2020). Expert Opin. Ther. Pat. 31, 785–794 (2021).
Scarneo, S. et al. Development and efficacy of an orally bioavailable selective TAK1 inhibitor for the treatment of inflammatory arthritis. ACS Chem. Biol. 17, 536–544 (2022).
Darling, N. J. & Cohen, P. Nuts and bolts of the salt-inducible kinases (SIKs). Biochem. J. 478, 1377–1397 (2021).
Lu, R. Q. et al. SGK1, a critical regulator of immune modulation and fibrosis and a potential therapeutic target in chronic graft-versus-host disease. Front. Immunol. 13, 822303 (2022).
Khor, B. et al. The kinase DYRK1A reciprocally regulates the differentiation of Th17 and regulatory T cells. eLife 4, e05920 (2015).
Liu, T. et al. DYRK1A inhibitors for disease therapy: current status and perspectives. Eur. J. Med. Chem. 229, 114062 (2022).
Acknowledgements
This work was supported by the US NIH Intramural Research Program, National Institute of Arthritis and Musculoskeletal and Skin Diseases and National Institute of Allergy and Infectious Diseases. Owing to how fast this field is moving, we apologize for any approvals and clinical trials we may have missed.
Author information
Authors and Affiliations
Contributions
L.C.-S. and H.K. contributed equally. All authors contributed to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The NIH holds a patent entitled ‘Janus family kinases and identification of immune modulators’ US7070972B1. J.J.O’S. receives income from the US Government for royalties. All other authors have no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks L. Berg, I. McInnes, A. Najm and A. Zarrin for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- ACR20 response
-
American College of Rheumatology response to intervention defined as at least 20% improvement in both the tender joint count and swollen joint count and at least 20% improvement in 3–5 other core set measures (such as pain and physical function).
- Alopecia areata
-
(AA). An autoimmune disease of the hair follicle characterized by non-scarring hair loss. Cytotoxic CD8+NKG2D+ T cells and interferon-γ have an important role in its development.
- Amyotrophic lateral sclerosis
-
A severe neurodegenerative disease defined by loss of upper and lower motor neurons with associated accumulation of protein aggregates in cells. There are alterations in T cells, monocytes, complement and cytokines in the peripheral blood of patients with this disease.
- Ankylosing spondyloarthritis
-
A form of arthritis that causes inflammation in the joints and ligaments of the spine and overtime causes the bones to fuse. Males carrying the MHC class I allele HLA-B27 have an increased risk of developing the disease.
- Atopic dermatitis
-
(AD). A chronic pruritic skin condition that is characterized by inflammation, redness and irritation of the skin. It has been associated with increased production of T helper 2 cytokines.
- Chronic eosinophilic leukaemia
-
A myeloproliferative neoplasm of the blood with clonal overproduction of eosinophils in the bone marrow.
- Chronic mucocutaneous candidiasis
-
Hereditary immunodeficiency syndromes associated with chronic non-invasive Candida infection of the skin, nails and mucous membranes owing to dysfunctional T cells.
- Chronic myeloid leukaemia
-
(CML). Indolent cancer with clonal increases in myeloblasts in the bone marrow and blood. It is often marked by a chromosome change called the Philadelphia chromosome, giving rise to the fusion protein BCR–ABL.
- Dermatomyositis
-
An inflammatory disease that leads to chronic muscle inflammation (myositis), muscle weakness and skin rash.
- Graft versus host disease
-
An immune response mounted against the recipient of an allograft by immunocompetent donor T cells that are derived from the graft. Typically, it is seen in the context of allogeneic bone marrow transplantation.
- Haemophagocytic lymphohistiocytosis
-
A severe, life-threatening, systemic inflammatory syndrome characterized by uncontrolled proliferation of activated lymphocytes and macrophages and cytokine storm. Primary forms are caused by genetic variants commonly affecting cytotoxic lymphocyte function. Secondary forms can occur following viral infections, such as Epstein–Barr virus, or cancers, such as leukaemia.
- Hidradenitis suppurativa
-
A painful, chronic relapsing inflammatory skin condition characterized by follicular occlusion, scarring and sinus tracts in apocrine-bearing areas of the skin. Aberrant activation of innate immunity and microbiome dysbiosis contribute to its pathogenesis.
- Hypereosinophilic syndrome
-
A group of blood disorders characterized by sustained overproduction of eosinophils that enter various tissues leading to damage to these organs.
- Hyperlipidaemia
-
A condition in which there are too many lipids such as cholesterol and triglycerides in the blood. Over time, these lipids can become deposited in arteries and increase the risk of blockages.
- Idiopathic pulmonary fibrosis
-
A chronic inflammatory disease of the lungs characterized by scarring, leading to a progressive and irreversible decline in lung function.
- Immune thrombocytopenia
-
A blood disorder of low platelet counts owing to autoimmune activity against platelet antigens, leading to easy bruising and bleeding.
- Juvenile idiopathic arthritis
-
Type of chronic rheumatic disease in children characterized by progressive joint destruction and sometimes systemic inflammation. A complex interaction between lymphocytes, monocytes, macrophages and neutrophils triggers the disease.
- Major adverse cardiovascular events
-
A composite end point classically defined to include myocardial infarction, stroke and cardiovascular death.
- mTOR
-
A conserved protein kinase that regulates cellular metabolism, autophagy, protein translation, cell growth and survival in response to environmental cues. The immunosuppressive drug rapamycin inhibits mTOR complex 1 by binding FKBP12 and is used in allograft transplantation.
- Myeloproliferative diseases
-
Blood cancers caused by changes in bone marrow stem cells. The most common types are chronic myelogenous leukaemia, polycythemia vera, chronic idiopathic myelofibrosis, essential thrombocytopenia and chronic eosinophilic leukaemia.
- Psoriatic arthritis
-
(PsA). A form of arthritis that affects some individuals with psoriasis leading to inflammation of the joints and entheses (sites where tendons and ligaments attach to bone).
- Refractory chronic urticaria
-
Defined by the presence of evanescent wheals, angioedema or both for longer than 6 weeks that is not controlled by higher dose non-sedating H1 antihistamines in combination with other standard therapies.
- Scarring alopecia
-
A group of hair disorders associated with inflammation leading to destruction of the hair follicle and hair loss.
- Septic shock
-
A life-threatening condition in which dangerously low blood pressures occur secondary to an immune reaction to a systemic infection.
- Systemic lupus erythematosus
-
(SLE). A chronic immune disease and most common form of lupus in which there is a breakdown of self-tolerance with activation of autoreactive T and B cells. Widespread inflammation leads to tissue damage and can affect the joints, skin, brain, kidneys and blood vessels.
- Systemic mastocytosis
-
A blood disorder with increased mast cells in the blood, which leads to the release of vasoactive cell mediators. This leads to various symptoms including anaphylaxis, flushing, gastrointestinal and neuropsychiatric complaints.
- Systemic sclerosis
-
A chronic disease characterized by diffuse fibrosis and vascular abnormalities in the skin, joints and internal organs.
- Ulcerative colitis
-
Chronic inflammation of the bowels in which abnormal reactions of both the innate and adaptive immune system cause inflammation and ulcers on the inner lining of the large intestine. Antibodies to resident microbiota highlight influence of B cells, and impact of cytokines leads to sustained inflammation.
- Venous thromboembolism
-
A condition in which a blood clot (thrombus) form in a deep vein, known as venous thrombosis. It can also be associated with or without pulmonary embolism, whereby a thrombus breaks off (embolizes) and flows to the lungs to lodge there.
- Vitiligo
-
A chronic inflammatory condition of the skin in which pigment-producing melanocytes are targeted leading to patches of visible depigmentation. The key immune cells include T helper 1 cells, cytotoxic T cells, regulatory and memory T cells as well as dendritic cells and natural killer cells. Key mediators such as interferon-γ and IL-15 are also implicated in its pathogenesis.
- X-linked agammaglobulinaemia
-
An inherited immune disease caused by an inability to produce B cells or inability of B cells to make immunoglobulins.
Rights and permissions
About this article

Cite this article
Castelo-Soccio, L., Kim, H., Gadina, M. et al. Protein kinases: drug targets for immunological disorders. Nat Rev Immunol 23, 787–806 (2023). https://doi.org/10.1038/s41577-023-00877-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-023-00877-7
This article is cited by
-
JAK-STAT signaling in inflammation and stress-related diseases: implications for therapeutic interventions
Molecular Biomedicine (2023)
-
Structural basis of a redox-dependent conformational switch that regulates the stress kinase p38α
Nature Communications (2023)
