
Abstract
Protein phosphatases act as key regulators of multiple important cellular processes and are attractive therapeutic targets for various diseases. Although extensive effort has been dedicated to phosphatase-targeted drug discovery, early expeditions for competitive phosphatase inhibitors were plagued by druggability issues, leading to the stigmatization of phosphatases as difficult targets. Despite challenges, persistent efforts have led to the identification of several drug-like, non-competitive modulators of some of these enzymes — including SH2 domain-containing protein tyrosine phosphatase 2, protein tyrosine phosphatase 1B, vascular endothelial protein tyrosine phosphatase and protein phosphatase 1 — reigniting interest in therapeutic targeting of phosphatases. Here, we discuss recent progress in phosphatase drug discovery, with emphasis on the development of selective modulators that exhibit biological activity. The roles and regulation of protein phosphatases in immune cells and their potential as powerful targets for immuno-oncology and autoimmunity indications are assessed.
Similar content being viewed by others

Introduction
Phosphorylation of intracellular proteins is a major post-translational modification that regulates nearly every biological process1. Protein phosphorylation is reversible and dynamic, controlled by the opposing activities of protein kinases, which catalyse protein phosphorylation, and protein phosphatases, which remove the phosphate. Of all the post-translational modifications that govern protein function, phosphorylation is one of the most prevalent and thus has been one of the most-studied mechanisms regulating cell signal transduction. Phosphorylation is used by the cell to regulate protein function by altering protein folding, localization, interactions, stability and activity. Perturbations in the regulation of protein phosphorylation caused by anomalous activities of kinases or phosphatases can have major impacts on cellular processes, including survival, growth, migration, differentiation and energy metabolism. Consequently, dysregulated protein phosphorylation is implicated in numerous human diseases, including cancer, diabetes and neurological disorders2,3.
Phosphatases are widely expressed in the immune system and as such act as key regulators of signalling in multiple types of immune cell. For example, in T cells, phosphatases control signalling at multiple nodes, including early events downstream of T cell receptor (TCR) engagement, signalling through inhibitory receptors such as PD1 and pathways that control the differentiation and functions of T helper cells (TH cells) and regulatory T cells (Treg cells)4,5. Phosphatases regulate various aspects of myeloid cell function, such as antigen presentation and phagocytosis, inhibitory ‘don’t eat me’ signalling, and differentiation and function of myeloid-derived suppressor cells (MDSCs)6,7. Given their expression profiles and key roles in immune cell signalling, this enzyme superfamily holds potential as drug targets to treat autoimmune disease and for cancer immunotherapy; in fact, recent seminal papers suggest that modulating the activity of key phosphatases can lead to tumour growth control in vivo8,9,10,11,12,13,14.
The first drug to target a phosphatase to be FDA approved was the calcineurin inhibitor cyclosporine15. Since the initial approval of cyclosporine in 1983 for immunosuppression following organ transplantation, additional cyclosporine formulations and the calcineurin inhibitors tacrolimus/FK506, pimecrolimus and voclosporin have been approved for the prevention of organ transplant rejection and as immunosuppressants in rheumatoid arthritis (RA), lupus nephritis, psoriasis, atopic dermatitis, keratoconjunctivitis sicca and more15,16. Another drug, fingolimod/FTY720, indirectly activates protein phosphatase 2A (PP2A) by binding to its negative regulator su(var)3-9, enhancer of zeste, trithorax (SET) and was FDA approved for the treatment of multiple sclerosis (MS) in 2010 (refs. 15,16). However, further introduction of phosphatase-modulating agents into the clinic was severely hampered by historic difficulties in targeting these enzymes17.
Efforts to drug protein tyrosine phosphatases (PTPs), such as PTP1B and SH2 domain-containing PTP 2 (SHP2), that were focused on orthosteric targeting were confounded by the highly conserved and charged PTP active site, which attracts potent inhibitors with limited therapeutic potential owing to lack of selectivity, cellular permeability and/or bioavailability17. Protein serine/threonine phosphatases (PSPs), on the other hand, form multimeric complexes with diverse combinations and require detailed biological knowledge to determine which complex to target18. In both cases, a lack of structural information on full-length PTPs and PSP holoenzymes has limited the options for developing drugs that target these proteins.
More recently, however, phosphatases have been selectively targeted pre-clinically with orally bioavailable therapies, and novel phosphatase targets have emerged because of new biological insights8,9,10,11,12,13,14. As a result, a major resurgence in interest in these enzymes has occurred. Although most of the recent compounds are allosteric inhibitors19,20,21,22,23,24,25,26,27, other novel modalities are increasingly common (Fig. 1). Additionally, despite previous challenges, competitive targeting of the phosphatase active site has not been completely abandoned, especially in cases where highly potent agents can be obtained or the active site contains structurally unique features28,29,30,31.

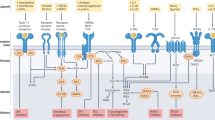
a,b, Orthosteric inhibitors bind to the phosphatase active site. a, Competitive inhibitors compete with substrate for binding to the phosphatase. b, Uncompetitive inhibitors bind to a phosphatase–substrate complex, preventing completion of catalysis. c, Oxidizing protein tyrosine phosphatase (PTP) inhibitors lead to oxidation of the PTP catalytic Cys. d, Irreversible inhibitors render the phosphatase inactive by covalently modifying the active site. e, Allosteric inhibitors induce or stabilize a catalytically unfavourable conformation of the phosphatase. f, Protein–protein interaction inhibitors disrupt or prevent complex formation between a phosphatase and its binding partner. g, Protein serine/threonine phosphatases (PSPs) can be inhibited by targeting specific regulatory subunits. h, PSPs can be activated by molecules that stabilize specific holoenzyme complexes. i, Phosphatases can be targeted by decoy biologics (targeting the extracellular region of receptor PTPs (RPTPs) or an intracellular region) or through antibodies. j, Proteolysis-targeting chimera (PROTAC) molecules target the phosphatase for degradation by bringing it into close proximity with an E3 ubiquitin ligase. CDC25, cell division cycle 25; EYA2, eyes absent 2; LMPTP, low-molecular-weight PTP; mAb, monoclonal antibody; PRL, phosphatase of regenerating liver; SET, su(var)3-9, enhancer of zeste, trithorax; SHP2, SH2 domain-containing PTP 2; STEP, striatum-enriched PTP; TC-PTP, T cell PTP; VE-PTP, vascular endothelial PTP; WIP1, wild-type p53-induced phosphatase 1.
Owing to new emerging biology on the roles of phosphatases in autoimmunity and their potential as tumour immunotherapy targets, coupled with new strategies to modulate these enzymes, this Review focuses on the roles of phosphatases in immune-mediated diseases. Examples of novel strategies to tackle these tough yet increasingly tractable targets are provided, emphasizing methods that identify selective modulators with biological activity.
Protein phosphatase families
The human protein phosphatome consists of 189 genes32, and protein phosphatases can be classified on the basis of catalytic mechanism into the PTP, PSP and haloacid dehalogenase (HAD) phosphatase superfamilies33,34,35,36 (Table 1). Multiple enzymes from all superfamilies are being investigated as candidate drug targets for immune-mediated disorders and other diseases. The distinction between PTPs, PSPs and HADs is based on catalytic mechanism; it should be noted that some phosphatases in the PTP or PSP superfamily dephosphorylate phospho-Ser/Thr or phospho-Tyr substrate residues, respectively, and some HAD phosphatases dephosphorylate substrates on phospho-Ser/Thr or phospho-Tyr residues. Furthermore, other phosphatases, such as phosphatase and tensin homologue (PTEN) and RNA guanylyltransferase and 5′-phosphatase (RNGTT), can dephosphorylate non-protein substrates, such as lipids or nucleic acids, respectively.
The PTP superfamily
PTPs are single-unit enzymes, many of which contain domains in addition to the catalytic domain, determining regulatory mechanisms, subcellular localization and substrate specificity33. PTPs consist of Cys-based and His-based families classified by the chief catalytic residue involved in nucleophilic substrate attack during catalysis.
The majority of PTPs are Cys-based and characterized by a conserved CX5R motif, in which the catalytic Cys acts as a nucleophile during catalysis, and the Arg assists in substrate binding33. Cys-based PTPs can be further subdivided into three classes. Class 1 is the largest and includes classical phospho-Tyr-specific receptor PTPs (RPTPs), non-receptor PTPs and dual-specificity PTPs (DSPs). DSPs can dephosphorylate phospho-Tyr and phospho-Ser/Thr residues as well as lipids and are subclassified as classic MAP kinase phosphatases (MKPs) or atypical DSPs. Class 2 Cys-based PTPs show homology to some bacterial arsenate reductases and consist of low-molecular-weight PTP (LMPTP or LMW-PTP) and suppressor of Sua7-2 (SSu72). Cys-based class 3 contains three cell division cycle 25 (CDC25) PTPs, which contain a rhodanese phosphatase domain.
His-based PTPs consist of the two ubiquitin-associated and Src homology 3 domain-containing (UBASH3) phosphatases, which are part of the histidine phosphatase (HP) superfamily33,37. These enzymes contain a catalytic RHG motif and use His as the nucleophile.
The PSP superfamily
PSPs consist of phosphoprotein phosphatase (PPP) and metal-dependent protein phosphatase (PPM) families34,35. Both PPPs and PPMs require divalent metal cations for catalysis.
There are seven PPP enzymes in humans: PP1, PP2A, PP2B (calcineurin), PP4, PP5, PP6 and PP7. Several members of the PPP family are multimeric enzymes34,35. PP1 forms dimeric holoenzymes consisting of a catalytic and a regulatory subunit, and PP2A forms mostly trimeric holoenzymes with a scaffolding, a catalytic and a regulatory subunit. This multimeric nature allows for extensive diversity among holoenzymes in post-translational regulation, subcellular localization, substrate specificity and ultimately, in their biological roles in signalling.
PPMs contain Mg2+/Mn2+ in the active site and consist primarily of the single-unit PP2C enzymes (most PPMs) and heterodimeric pyruvate dehydrogenase phosphatases (PDPs)38.
The HAD superfamily
HAD phosphatases use an Asp residue as a catalytic nucleophile and Mg2+ as a cofactor and contain a DxDx(V/T) active-site signature motif36,38. The four eyes absent (EYA) phosphatases contain a C-terminal EYA domain with phospho-Tyr phosphatase activity and also contain an N-terminal domain with phospho-Thr phosphatase activity, although whether this is intrinsic or mediated through interaction with PP2A remains to be clarified39. Other HAD phosphatases — FIIF-associating component of RNA polymerase II CTD phosphatase (FCP) and small CTD phosphatase (SCP) — dephosphorylate phospho-Ser residues of the CTD of RNA polymerase II38.
Phosphatase targets in tumour immunotherapy and autoimmune disorders
Phosphatases are targeted using numerous different modalities, and many of these agents have been or are being developed as immune modifiers (Tables 2–4). Several protein phosphatases in immune cells have emerged in the past few years as candidate therapeutic targets for autoimmunity and tumour immunotherapy indications. As examples discussed below, among the most validated targets are T cell PTP (TC-PTP) and SHP2, with small-molecule inhibitors currently in clinical trials for tumour immunotherapy indications. Activating TC-PTP and SHP1 may also provide novel strategies for combating autoimmune diseases. Biological data suggest that inhibition of PP2A could be a strategy in autoimmunity and for tumour immunotherapy in combination with checkpoint inhibition. Evidence is also building for phosphatase of regenerating liver 3 (PRL3) as a target for an antibody-dependent cell-mediated cytotoxicity approach against tumours (Box 1).
The roles of phosphatases in cancer cell signalling is a mature field, and several excellent reviews on drugging cancer phosphatases are available18,40,41,42. Recent studies also point to stromal and glial cell-expressed phosphatases — especially receptor PTPs and subunits of PP1 and PP2A — as potential key players and targets for autoimmune diseases and cancer43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58.
In the following sections, we primarily discuss the relevant biology of protein phosphatases for which modulating agents are available and demonstrate efficacy in vivo as tumour immunotherapeutics or treatments for autoimmunity. For a comprehensive list of phosphatases holding potential as targets for autoimmunity or tumour immunotherapy — including those for which target validation is in a nascent stage — the reader is referred to Boxes 2 and 3, respectively.
TC-PTP: biology and therapeutic implications
TC-PTP is a ubiquitous non-receptor PTP encoded by PTPN2. Alternative splicing generates a major form, TC45 (45 kDa), as well as TC48 (48 kDa). TC45 is targeted to the nucleus through a bipartite nuclear localization signal and shuttles to the cytoplasm in response to cellular stimuli such as insulin, epidermal growth factor, tumour necrosis factor (TNF) and interferon-γ (IFNγ), where it can access substrates in the cytoplasm or at the plasma membrane. TC48 contains a hydrophobic C terminus that localizes it to the endoplasmic reticulum, where it has more restricted access to substrates59. TC-PTP inhibits signalling downstream of numerous pro-inflammatory cytokines, including IL-2, IL-6, IL-15 and IFNγ, by dephosphorylating and inhibiting JAKs and STATs. TC-PTP also inhibits TCR signalling by dephosphorylating Src family kinase (SFK) activation motifs and dephosphorylates growth factor receptors such as epidermal, platelet-derived and vascular endothelial growth factor receptors (EGFR, PDGFR and VEGFR)60. TC45 enzymatic activity is regulated through auto-inhibition by its disordered C-terminal region61,62. Upon adhesion to collagen, the cytoplasmic tail of α1 integrin binds to and activates TC45 by displacing its auto-inhibitory region62,63. The discovery of this naturally occurring allosteric regulation mechanism suggests potential for allosteric control of TC-PTP activity by modulating placement of its disordered C-terminal region.
TC-PTP has reported roles in autoimmune disease, cancer and cancer immunosurveillance and is a highly attractive target for immune-mediated diseases8,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80. TC-PTP is an immunological rheostat, modulation of which tips the immune system towards suppression of autoimmunity and inflammation or activation of antitumour responses. As discussed below, TC-PTP activation for autoimmunity would likely attenuate disease severity through multiple mechanisms owing to its autoimmune disease-promoting role in various cell types, while inhibiting TC-PTP could be useful for all aspects of T cell-mediated immunotherapy, including boosting chimeric antigen receptor (CAR) T cell function. Data suggest that inhibition of TC-PTP in tumour cells would offer the added benefit of rendering tumours more susceptible to immune cell infiltration and attack by cytotoxic T cells8,77.
TC-PTP in autoimmune disorders
Multiple lines of evidence suggest TC-PTP as a candidate target for activation to treat autoimmunity. Certain alleles of PTPN2 are risk factors for autoimmune diseases including type 1 diabetes mellitus, RA, Crohn’s disease and ulcerative colitis (reviewed elsewhere81). Autoimmune-associated single-nucleotide polymorphism (SNP) rs1893217 decreases levels of PTPN2 mRNA by 40% in T cells82, and SNP rs2542151 reduces TC-PTP protein in colonic lamina propria fibroblasts of patients with Crohn’s disease83, suggesting that TC-PTP loss is a risk factor for autoimmunity.
TC-PTP deletion enhances autoimmunity in mice
Mice with a genetic deletion of Ptpn2 die within 3–4 weeks of birth from inflammatory disease accompanied by lymphocytic organ infiltration84, and inducible haematopoietic TC-PTP deletion in adult mice causes systemic inflammation and autoimmunity64. Evidence from several mouse models supports a protective role for TC-PTP in autoimmune diabetes, colitis and inflammatory arthritis.
T cell-specific TC-PTP deletion accelerates diabetes onset and increases incidence of salivary gland lymphocyte infiltration — a characteristic of Sjögren syndrome — and colitis65. In the OT-I–OVA mouse system, TC-PTP-deficient OT-I T cell transfer into recipients bearing ovalbumin (OVA) on pancreatic β-cells results in β-cell destruction and diabetes66.
Global heterozygous TC-PTP deletion67, or deletion in T cells68, macrophages69,70 or intestinal epithelial cells (IECs)71 enhances experimental colitis67,68,69,70,71,72. Mice that lack TC-PTP in T cells show enhanced numbers of IFNγ+CD4+ T cells, liver inflammatory infiltration and autoantibodies68. Adoptive transfer of TC-PTP-deficient naive CD4+ T cells leads to greater expansion of IFNγ+ and IFNγ+IL-17+ cells, suggesting that TC-PTP inhibits the expansion of pathogenic T cell subsets during intestinal inflammation68. In macrophages, TC-PTP knockout (KO) exacerbates colitis through an IL-1β-mediated mechanism69 and enhances colon epithelial permeability in mice through an IL-6-dependent mechanism69,70. IEC-specific TC-PTP KO mice also exhibit exacerbated experimental colitis71.
The evidence that TC-PTP deletion in any of these cell types exacerbates disease suggests that they collectively mediate the colitis-protective role of this phosphatase. This is further supported by the enhanced colitis that occurs upon global heterozygous TC-PTP deletion67 and suggests that systemic TC-PTP activation may provide an effective colitis-treating strategy without the need for specific cell-targeting approaches.
TC-PTP deficiency similarly exacerbates inflammatory arthritis in mice. Heterozygous TC-PTP deletion worsens disease severity in the SKG model of inflammatory arthritis, by enhancing IL-6-induced STAT3 phosphorylation in Treg cells, which encourages their conversion into pathogenic IL-17-producing ‘exTreg’ cells73. Heterozygous TC-PTP deletion in T cells or Treg cells also enhances arthritis73. Global or inducible Treg cell-specific heterozygous TC-PTP deletion in SKG mice similarly enhances colitis-induced arthritis, which is accompanied by exTreg accumulation in the arthritic joint74. The role of TC-PTP in suppressing both intestinal and joint inflammation might explain the known connection between these pathologies in patients with RA, perhaps through Treg cell migration from the colon to the joints75. Although the action of TC-PTP in arthritis is mediated through Treg cells, global heterozygous TC-PTP deletion leads to a similar phenotype, suggesting again that specific cell-targeting approaches would not be needed for TC-PTP activation in RA.
TC-PTP activation ameliorates autoimmunity in mice
Proof-of-principle evidence of TC-PTP activation as a treatment for autoimmunity was demonstrated using the dietary polyamine spermidine. This compound was identified as a TC-PTP activator from high-throughput screening of commercial small-molecule libraries for full-length TC45 agonists85. Spermidine does not bind to the TC-PTP catalytic domain86; instead it competes with the α1 integrin cytoplasmic tail for TC45 binding, suggesting that it may relieve auto-inhibition by the TC45 C terminus85. Oral spermidine administration during dextran sodium sulfate (DSS) colitis induction protects mice from weight loss and colonic inflammation and damage76. Although spermidine is a polypharmacological molecule unlikely to serve as a clinical candidate, these findings provide a key demonstration that targeting TC-PTP regions outside of the catalytic domain can provide a means to activate the phosphatase using a small molecule and further suggest that TC-PTP activation may serve as a strategy for treating autoimmune diseases for which TC-PTP has a protective role, such as colitis and RA.
TC-PTP in tumour immunotherapy
Whereas TC-PTP activation could be beneficial in autoimmune conditions, inhibiting this phosphatase could enhance tumour immunotherapies by both sensitizing tumours to cytotoxic T cell killing and enhancing antitumour responses of T cells. In an in vivo genetic screen using CRISPR–Cas9 genome editing in transplanted B16 melanoma tumours in mice, TC-PTP deletion increased sensitivity of tumours to an immunotherapy that comprises an anti-PD1 antibody and a tumour cell vaccine (GVAX)79. TC-PTP-deficient B16 tumours display increased levels of surface antigen-loaded major histocompatibility complex class I (MHC-I) and more infiltrating immune cells, including CD8+ T cells expressing granzyme B, IFNγ or TNF. TC-PTP also emerged as a target for sensitizing tumour cells to death from CD8+ T cells or IFNγ80. TC-PTP knockdown likely sensitizes melanoma cell lines to IFNγ by augmenting STAT1, STAT3 and STAT5 phosphorylation and MHC-I, MHC-II and PDL1 expression. In the anti-PDL1-resistant YUMM1.1 mouse melanoma model, stable TC-PTP knockdown reduces tumour growth and renders tumours sensitive to inhibition by an anti-PDL1 antibody; this sensitivity depends on the presence of T cells87.
TC-PTP deletion enhances antitumour immunity in mice
TC-PTP-deficient CD8+ T cells display enhanced antitumour immunity compared with wild-type cells when transferred into multiple tumour xenograft models77. These cells exhibit a type of ‘terminal’ exhaustion still capable of killing tumour cells owing to retention of cytolytic capabilities77. Mice that lack TC-PTP in haematopoietic cells completely clear MC38 tumours and B16 tumours and are more responsive to GVAX–anti-PD1 immunotherapy77. TC-PTP in CD8+ T cells may be particularly important, as depletion of CD8+ T cells, but not CD4+ T cells, abolishes the capacity of TC-PTP-deficient T cells to control MC38 tumour growth and render tumours responsive to anti-PD1 immunotherapy78.
TC-PTP deletion in T cells also protects aged Tp53+/− mice against tumour development and suppresses growth of implanted mammary carcinomas, increasing numbers of tumour-activated CD4+ and CD8+ effector/memory cells8. Tumour Treg cells are also increased, although this is not sufficient to overcome the antitumour effects of Ptpn2-deficient effector/memory T cells. TC-PTP inhibition likely enhances Treg cell destabilization and conversion to a non-immunosuppressive phenotype, although this has not yet been tested in a tumour setting8. Adoptively transferred TC-PTP-deficient T cells block tumour growth in mice and show decreased levels of classic exhaustion markers PD1 and LAG3, and increased levels of CD44. Although terminal exhaustion was not assessed in this study, these and the findings discussed above suggest that lack of TC-PTP in CD8+ T cells enhances their cytolytic function.
TC-PTP deletion or inhibition enhances CAR T cell function
TC-PTP deficiency or inhibition enhances CAR T cell immunotherapy, promoting the generation, antigen-specific activation and cytotoxicity of CD8+ HER2 CAR T cells ex vivo and repressing HER2+ E0771 mammary tumour growth and CD8+ T cell infiltration8. In mice that receive TC-PTP-deficient CAR T cells, tumours are controlled in the absence of autoimmunity and overt morbidity up to 70 days after transfer.
Experiments with the highly selective (+)-methoxyacetic acid-based TC-PTP inhibitor compound 8 demonstrate boosted CAR T cell function upon TC-PTP inhibition8. Compound 8 increases antigen-specific cytotoxic potential of murine CD8+ HER2 CAR T cells to that of TC-PTP-deficient CAR T cells, but does not affect TC-PTP-deficient CAR T cells8. Treatment of human CAR T cells that target the Lewis Y (LY) antigen — which is overexpressed in many cancers — with compound 8 enhances IFNγ and TNF expression in response to CAR crosslinking or exposure to LY-expressing ovarian carcinoma cells8.
TC-PTP inhibitors for tumour immunotherapy
In support of TC-PTP as an immunotherapy target, two orally bioavailable TC-PTP inhibitors developed by Calico and AbbVie, ABBV-CLS-579 and ABBV-CLS-484 (patent publication WO/2019/246513), are currently undergoing phase I clinical trials for locally advanced or metastatic tumours (NCT04777994 and NCT04417465).
However, owing to high conservation between TC-PTP and PTP1B, identification of TC-PTP-selective molecules with favourable pharmacological profiles has historically been challenging. Structural studies indicate that these two enzymes are regulated allosterically through their C termini by different mechanisms62,88 — suggesting potential for selective targeting by approaching regions outside the catalytic domain. T cell PTP1B deficiency or pharmacological PTP1B inhibition has recently been reported to inhibit growth of mammary, melanoma and colorectal tumours, suggesting that, similarly to TC-PTP, PTP1B deficiency can enhance T cell-mediated antitumour responses89. Furthermore, the dual PTP1B–TC-PTP inhibitor developed by Merck Frosst Canada, called 1B/TC90, enhances monocyte-derived dendritic cell (moDC) maturation. Administration of moDCs treated with 1B/TC to tumour-bearing mice suppressed tumour growth, and treatment of moDCs derived from patients with pancreatic cancer with IB/TC enhances IFNγ+ co-cultured autologous T cells91. Therefore, although selective targeting is usually desired in drug discovery efforts, in the case of TC-PTP for tumour immunotherapy, a stringent requirement for selectivity over PTP1B may not be necessary, and, in fact, limited selectivity may even boost therapeutic efficacy.
SHP2: biology and therapeutic implications
SHP2 is encoded by the PTPN11 gene, is ubiquitously expressed and, like its homologue SHP1, comprises tandem SH2 domains (N-SH2 and C-SH2), a PTP domain and a disordered C-terminal tail that contains Tyr phosphorylation sites. SHP2 is regulated by an intramolecular auto-inhibited conformation in which residues of N-SH2 insert into the PTP domain catalytic cleft92,93 (Fig. 2a). Binding of SHP2 SH2 domains to phospho-Tyr residues of interacting proteins shifts the enzyme to an open, active conformation94. Additionally, Tyr phosphorylation in the tail provides docking sites for interacting proteins or its C-SH2, which also enhance SHP2 activation94. SHP2 can also exist in a partially open state in which the N-SH2 allows access to the active site95.

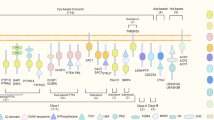
a, SH2 domain-containing PTP 2 (SHP2) is regulated by an auto-inhibited intramolecular mechanism. In the open conformation, the catalytic domain of SHP2 remains accessible for interaction with substrate. In the closed or inactive conformation, the N-SH2 domain blocks access to the catalytic site, rendering SHP2 inactive. SHP1 undergoes a similar regulation mechanism. b,c, Models for the PD1–SHP2 interaction. b, Two-step activation model. Before PD1 engagement on T cells, SHP2 resides in the auto-inhibited closed conformation. PDL1 binding recruits SHP2 to the phosphorylated immunoreceptor tyrosine-based switch motif (ITSM). Phosphorylation of the immunoreceptor tyrosine-based inhibitory motif (ITIM) unfolds SHP2 into its active conformation. c, Dimerization model. SHP2 induces PD1 dimerization through N-SH2 and C-SH2 binding to the phospho-ITSM on two distinct PD1 molecules. TCR, T cell receptor.
SHP2 promotes RAS–RAF–MAPK signalling downstream of receptor tyrosine kinases (RTKs)40,96. Proposed mechanisms include Tyr dephosphorylation of negative regulators of this pathway — such as RAS–GAP, RAS–GAP binding sites of RTKs, CSK binding sites of paxillin and PAG/CBP, and the Grb2/SOS binding site of Sprouty — and direct dephosphorylation of RAS, inhibiting its binding to RAF40,96. SHP2 also promotes other signalling pathways including PI3K–AKT, JAK–STAT and nuclear factor-κB (NF-κB)40,96. PTPN11 is considered an oncogene, as gain-of-function mutations lead to neoplasms and Noonan syndrome97. These mutations activate SHP2 by destabilizing interactions between the PTP and N-SH2 domains, promoting the open conformation of SHP2.
SHP2 has long been considered a drug target for cancer (reviewed elsewhere18,40,98). Below, we focus on the potential for SHP2 in autoimmunity and its recent emergence as a tumour immunotherapy target.
SHP2 inhibition in autoimmune disorders
So far, a small number of studies have suggested a role for SHP2 in autoimmunity and potential for SHP2 as an autoimmunity target99,100,101.
PTPN11, which encodes SHP2, is located within a linkage disequilibrium block that is associated with RA99. Inducible heterozygous SHP2 deletion in myeloid cells reduces inflammatory arthritis in mice, suggesting an arthritis-promoting action of SHP2 in myeloid cells100. Furthermore, treatment with the bidentate SHP2 inhibitor 11a-1, derived from a precursor that interacts with the SHP2 active site and nearby β5−β6 loop, attenuates inflammatory arthritis in mice100,101. Additionally, SHP2 activity is increased in peripheral blood mononuclear cells (PBMCs) and spleen isolates from systemic lupus erythematosus (SLE)-susceptible MRL-lpr mice, and 11a-1 treatment reduces disease in these mice101.
These findings suggest that further exploration of the role of SHP2 in RA, SLE and potentially other autoimmune diseases is warranted.
SHP2 in tumour immunotherapy
SHP2 inhibition has been considered a strategy for enhancing tumour immunity owing to the proposed roles of SHP2 as an effector of inhibitory PD1 signalling in T cells102 and colony-stimulating factor 1 receptor (CSF1R) signalling in myeloid cells103; the latter can promote tumour growth by reprogramming tumour-associated macrophages (TAMs) to an immunosuppressive M2 state104.
SHP2 in T cell PD1 signalling
SHP2 is considered a positive regulator of T cell activation by dephosphorylating inhibitory sites of TCR signalling mediators and activating sites of negative regulators105. However, SHP2 is also considered a key mediator of PD1 inhibitory receptor signalling in T cells following docking to the phospho-Tyr-containing immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) in the PD1 cytoplasmic region. As PD1 expression is induced by T cell activation, a possible explanation for this dual role in T cell signalling is that SHP2 initially promotes T cell activation upon TCR ligand engagement and later dampens signalling by complexing with PD1. Multiple studies have described a PD1–SHP2 interaction106,107,108, yet there is no consensus model for the precise nature of the complex or the functional necessity of SHP2 in PD1 signalling.
SHP2 emerged as the most abundant PD1 interactor in studies of cell-free reconstitution systems mimicking the T cell plasma membrane and MS-based quantitative proteomics of pervanadate-treated T cells106,107. Mutation of the phosphorylation site in either the ITIM (Tyr223Phe) or ITSM (Tyr248Phe) reduced the PD1–SHP2 interaction and strongly reduced the phosphatase activity of SHP2, while double mutation of both sites abolishes the interaction106,108. In Jurkat cells overexpressing PD1, mutation of either Tyr nearly equally inhibited the block in IL-2 or IFNγ production upon Jurkat engagement with programmed cell death 1 ligand 2 (ref. 108).
These results support a two-step activation model (Fig. 2b) in which before PD1 engagement on T cells, SHP2 resides in the auto-inhibited conformation. PD1 ligand binding recruits SHP2 to the phosphorylated ITSM; however, ITIM phosphorylation is needed to unfold SHP2 into its active conformation and propagate inhibitory PD1 signalling108. An alternative dimerization model (Fig. 2c) proposes SHP2 inducing PD1 dimerization through N-SH2 and C-SH2 binding to phospho-ITSM-Tyr248 on two PD1 molecules109. The dimerization model is supported by data collected in Jurkat–PD1 and biophysical analyses demonstrating that SHP2 can bind to two PD1 molecules solely through the pITSM, and that mutation of ITSM-Tyr248 or either SHP2 SH2 domain, but not mutation of ITIM-Tyr223, disrupts the PD1–SHP2 interaction109,110. Additionally, SHP2 induces PD1–PD1 interaction that is blocked by mutation of ITSM-Tyr248 or either SHP2 SH2 domain, but not by ITIM-Tyr223 (ref. 109). The dimerization model is further corroborated by NMR spectroscopy, which reveals that whereas SHP2 C-SH2 only binds to pITSM in a pITIM–pITSM peptide, SHP2 N-SH2 binds to either phospho-motif111, and pITSM has a higher affinity for either SHP2 SH2 domain than pITIM110.
Despite convincing data regarding PD1–SHP2 complex formation, some studies show PD1 signalling can inhibit T cell activation in the absence of SHP2 (refs. 107,112). There may be cellular contexts in which SHP1 compensates for loss of SHP2 in maintaining PD1 signalling107, or where PD1 can inhibit T cell activation in the absence of both phosphatases112.
SHP2 deletion in mouse models of tumour immunity
In mouse xenograft models, T cell-specific SHP2 deletion yields varying results. In a metastatic melanoma model, T cell SHP2 deficiency has no effect on survival and late-stage tumour size, and metastasis is increased113. Tumour-bearing mice carrying SHP2 deficiency in T cells display higher serum IL-6 levels and increased MDSCs. MDSC accumulation and tumour growth in these mice are inhibited by IL-6 blockade. In the MC38 colon adenocarcinoma model, tumour growth and tumour-infiltrating lymphocytes (TILs) are unaffected by lack of SHP2 in T cells114. Anti-PD1 antibody treatment improves tumour control irrespective of mouse genotype, suggesting that PD1 inhibitory function occurs in the absence of SHP2. On the other hand, another report shows T cell-specific SHP2 deficiency enhancing control of MC38 tumour growth, with tumours displaying increased activated CD8+ T cells9.
Myeloid-specific SHP2 deletion inhibits B16 melanoma growth and increases tumour levels of chemoattractant CXCL9, IFNγ-induced CXCL9 production by macrophages and CD8+ T cell tumour infiltration115. Accordingly, CXCL9 or IFNγ neutralization promotes tumour growth in myeloid SHP2-deficient mice.
SHP2 inhibitors in tumour immunotherapy
As discussed below, several orally bioavailable allosteric SHP2 inhibitors have been developed and some are undergoing clinical trials for cancers. These compounds act as ‘molecular glue’, stabilizing the auto-inhibited state of SHP2 by binding the pocket created by the closed conformation of the enzyme20,21 and are highly selective over other phosphatases and kinases. SHP099 (refs. 20,21) was developed following a screening for full-length SHP2 inhibition in the presence of bis-phosphorylated IRS1 peptide to open the enzyme. Pyrazine derivative TNO155 lacks the hERG2 binding and phototoxicity problems of SHP099 (ref. 22) and is being tested in phase I and I/II clinical trials (NCT03114319, NCT04000529, NCT04330664, NCT04294160, NCT04292119, NCT04185883 and NCT04699188).
SHP099 administration to mice has shown promising results in tumour models. Oral or intraperitoneal SHP099 administration to mice bearing CT26 or MC38 colon carcinomas or 4T1 breast tumours decreases tumour growth9,10,116. The antitumour effects of SHP099 are likely due to immune cells, as, first, MC38 and CT26 cells carry PTPN11 variants insensitive to SHP099; second, SHP2 KO in 4T1 cells slows tumour growth, but less than SHP099 administration; and third, SHP099 efficacy is impaired in immunocompromised mice9,10. Combining SHP099 with anti-PD1 inhibits MC38 tumour growth more than either monotherapy alone9. Oral TNO155 administration inhibits growth of MC38 tumours as a monotherapy and CT26 tumours in combination with anti-PD1 (ref. 10).
In combination with anti-PD1, SHP099 or TNO155 treatment tends to enhance activation and tumour infiltration of CD8+ T cells9,10 yet reduce overall CD45+ immune cell infiltration and alter TAM composition10. SHP2 KO in 4T1 cells promotes CD8 T cell tumour infiltration and activation similarly to SHP099, but does not affect TAMs, suggesting that the effects on CD8+ TILs are due to SHP2 loss or inhibition in the tumour, whereas the effects on TAMs are due to SHP2 inhibition in myeloid or other non-tumoural cells10. Together with the above-described findings using myeloid-specific SHP2 deletion115, these data suggest that CD8+ T cell tumour infiltration can be influenced by SHP2 loss or inhibition in the tumour or myeloid cells, depending on the model used.
In addition, oral SHP099 administration combined with radiation plus anti-PDL1 enhances control of anti-PD1-resistant 344SQ non-small-cell lung carcinoma (NSCLC) and PANC-02 pancreatic tumours at local and abscopal sites, improves survival compared with radiation plus anti-PDL1, and reduces 344SQ lung metastases117. SHP099 does not inhibit 344SQ cell viability in culture, and depletion of CD8- or F4/80-expressing cells reduces the beneficial effect of triple therapy on survival and lung metastasis, confirming that it is largely mediated by antitumour immunity.
SHP2 inhibition is also being considered in combination with KRAS-G12C inhibitors for enhancing immunity against tumours carrying the KRASG12C mutation. Indeed, SHP099 increases GDP occupancy of KRAS, enabling enhanced efficacy of G12C inhibitors that target GDP-bound KRAS-G12C11. SHP099 in combination with the KRAS-G12C inhibitor ARS1620 (ARS) regresses tumours in a mouse pancreatic ductal adenocarcinoma (PDAC) model and increases survival following treatment termination. SHP099–ARS therapy in combination with anti-PD1 regresses tumours more than SHP099–ARS or either single agent plus anti-PD1. SHP099 also enhances ARS antitumour efficacy against KRASG12C-carrying patient-derived pancreatic cancer and NSCLC models. Tumours of SHP099–ARS combination-treated mice show increased T cells and decreased tumour CD11b+ myeloid cells.
The allosteric SHP2 inhibitor RMC-4550 was developed following evaluation of Revolution Medicines’ collection of methyl-pyrazine compounds23. Derivative RMC-4630 is undergoing phase I and I/II clinical trials (NCT03634982, NCT04916236, NCT03989115, NCT04185883 and NCT04418661), with preliminary results indicating disease control in five of seven patients with KRASG12C NSCLC102.
Oral RMC-4550 administration slows tumour growth in mouse colon carcinoma, breast carcinoma and B cell lymphoma models, with no effect on colon tumours in RAG2 KO mice or after CD8+ T cell depletion, suggesting a T cell-mediated action103. RMC-4550 inhibition of CT26 tumours is additive with anti-PD1, anti-CTLA4 or anti-CSF1R antibodies, and RMC-4550 in combination with anti-PD1 increases time to EMT6 endpoint tumour burden. RMC-4550 treatment also alters tumour immune cell composition and increases MHC-I and PDL1 expression on tumour cells. RMC-4550 has no effect on T cell proliferation or cytokine release but inhibits growth of CSF1-differentiated bone marrow-derived cells and primary human monocytes and blocks anti-proliferative effects of MDSCs on CD8+ T cells.
Considering that SHP099 or TNO155 and RMC-4550 inhibit SHP2 through a similar allosteric mechanism103, it remains to be determined why the effects of RMC-4550 on TAM composition in colorectal cancer tumours103 are observed for SHP099 or TNO155 only when administered in combination with anti-PD1 (ref. 10). Moreover, in both breast cancer tumours and colorectal cancer tumours, SHP099 administration leads to reduced tumour infiltration of CD45+ cells9,10, while reduced immune cell infiltration was not reported for RMC-4550 (ref. 103). Possible explanations for these differences in effects on the tumour microenvironment (TME) may be variations in experimental models used or selectivity profiles of these compounds. Regardless, taken together, they demonstrate that SHP2 inhibition skews the TME towards an enhanced cytotoxic T cell and reduced suppressor myeloid cell phenotype.
Another SHP2 inhibitor IACS-13909 was identified through collaboration between Navire Pharma, Inc. and University of Texas MD Anderson Cancer Center by structure-based design24. Derivative BBP-398 (IACS-15509) is undergoing phase I clinical trials as monotherapy and — in partnership with Bristol Meyers Squibb — as combination with PD1 and KRAS-G12C inhibitors (NCT04528836, NCT05375084 and NCT05480865). Jacobio developed SHP2 inhibitors JAB-3068 and JAB-3312, which are undergoing phase I/II clinical trials as monotherapy and — in partnership with AbbVie — as combination with PD1 or MEK inhibitors (NCT03518554, NCT03565003, NCT04721223, NCT04045496, NCT04121286 and NCT04720976). Another inhibitor, RLY-1971, developed by Relay Therapeutics is undergoing phase I trials as monotherapy and — in partnership with Genentech — in combination with a KRAS-G12C inhibitor (NCT04252339 and NCT05487235).
The prominent cell types that mediate the antitumour efficacy of SHP2 inhibitors have not yet been completely clarified. Although there is strong evidence for a role of SHP2 in PD1 signalling, tumour immunotherapy studies in mice have yielded conflicting results. Myeloid cells may play a major part in the tumour-promoting actions of SHP2, and the effects of SHP2 inhibition on T cells may in part be secondary to the effects on myeloid cells. The above-described reports of SHP2 inhibition using SHP2-deleted or inhibitor-insensitive tumour cells suggest that tumour cell SHP2 contributes to suppression of T cells during cancer. Further biological studies using conditionally deleted mice and/or SHP2-modified cells with varying tumour models would help to clarify this.
SHP1: biology and therapeutic implications
SHP1 is a non-receptor PTP encoded by PTPN6 and is expressed in haematopoietic cells and, under a different promoter, in epithelial cells primarily as a distinctive isoform differing in the first few N-terminal amino acids of the protein. Haematopoietic SHP1 is primarily cytosolic whereas epithelial SHP1 localizes to the nucleus, suggesting that these forms of SHP1 may act on different substrates118.
SHP1 is composed of tandem N-terminal SH2 domains (N-SH2 and C-SH2), a PTP catalytic domain and a C-terminal region with Tyr phosphorylation sites118,119. Like SHP2, SHP1 is regulated by auto-inhibition in which N-SH2 blocks access of substrate to the active site. Interaction between N-SH2 and phospho-Tyr peptides relieves this mechanism and increases SHP1 enzymatic activity120.
SHP1 is a negative regulator of immune cell activation, as it controls signalling from multiple immune cell surface receptors, including the TCR118,121. SHP1 targets in T cells include TCRζ, LCK, FYN, zeta chain-associated protein of 70 kDa (ZAP70), SH2 domain-containing leukocyte protein of 76 kDa (SLP76), phosphoinositide 3-kinases (PI3Ks) and VAV. SHP1 also regulates signalling through cytokine receptors by dephosphorylating JAKs and STATs. Owing to its potent role in immune cell signalling, SHP1 is considered a target for autoimmunity and tumour immunotherapy.
SHP1 in autoimmune disorders
Several lines of evidence support activation of SHP1 in autoimmune diseases. Peripheral T cells from patients with RA show delayed recruitment of SHP1 to the TCR–APC synapse122, and PBMCs and peripheral monocyte-derived macrophages from patients with MS have reduced SHP1 expression123,124. Treatment of PBMCs of patients with MS with IFNβ — a treatment for MS — induces SHP1 activity; SHP1 knockdown in PBMCs abolishes the anti-inflammatory effects of IFNβ on these cells, suggesting that SHP1 may contribute to the therapeutic effects of this cytokine125.
SHP1 deletion causes autoimmunity in mice
In various mouse models, SHP1 deficiency leads to autoimmune manifestations. Mice homozygous for motheaten (me) or viable motheaten (mev) mutations exhibit SHP1 mutant alleles (referred to here as ‘motheaten’), which cause aberrant SHP1 splicing, and develop severe autoimmune and immunodeficiency syndromes121. Motheaten mice exhibit increased peripheral blood monocytes and neutrophils, autoantibodies, glomerulonephritis, immunodeficiency, increased Treg cells, severe tissue inflammation and damage, early death from interstitial pneumonitis and more severe disease in the experimental autoimmune encephalomyelitis (EAE) model121,126.
T cell-specific SHP1 deletion enhances TCR-induced proliferation of CD8+ T cells127, renders CD4+ T cells resistant to Treg cell-mediated suppression128 and promotes accumulation of memory T cells129. Deletion of SHP1 in B cells, dendritic cells and neutrophils leads to various manifestations of autoimmunity130,131,132,133,134.
SHP1 activators in autoimmune disease
Given the clear role of SHP1 as an immunological regulator, interest has arisen in activating SHP1 as a potential therapy for autoimmune indications, especially as transgenic SHP1 overexpression protects mice from inflammatory arthritis in the cartilage proteoglycan-induced arthritis (PGIA) model without noticeable adverse effects135. The FDA-approved multi-kinase inhibitors sorafenib and regorafenib (approved for some cancers), nintedanib (approved for pulmonary fibrosis) and dovitinib (under consideration for renal cell carcinoma) activate SHP1 (refs. 136,137,138,–139). Deletion of the SHP1 N-SH2 domain or D61A point mutation (this residue stabilizes the closed conformation) abolishes their activation of SHP1, suggesting that these drugs free the protein from its auto-inhibited state136,137,138,139. In the PGIA model, oral regorafenib administration beginning when disease is not yet evident significantly reduces arthritis incidence and severity but leads to weight loss and increased mortality135. Later administration upon initial signs of arthritis significantly decreases arthritis severity without significant weight loss.
SHP1 in tumour immunotherapy
Evidence is building that inhibiting SHP1 may provide a strategy for enhancing adoptive T cell immunotherapy. SHP1 phosphatase activity is increased in TILs with low lytic activity, and SHP1 activity is required for the tumour cell-induced non-lytic phenotype140. In line with this observation, several studies show that SHP1 deficiency enhances adoptive T cell immunotherapy141,142. As discussed below, the efficacy of adoptively transferred SHP1-deficient T cells varies as a monotherapy among different tumour models; however, SHP1 inhibition is likely to be efficacious in combination with PD1/PDL1 inhibition.
Although the biological role of SHP1 in tumour immunotherapy — including the prominent cell type mediating its action — is not completely clear, global inducible SHP1 deletion and chemical inhibition are efficacious against growth of some tumours in mice. However, as we discuss below, SHP1 inhibition might be deleterious in the case of certain cancers, with direct SHP1 inhibition in the tumour cells opposing the immunotherapeutic action of SHP1 inhibition in the immune system. Additionally, SHP1 inhibition is likely to have significant toxic side-effects. Taken together, for immunotherapy, SHP1 may be a target best suited to adoptive cell transfer therapies.
SHP1 deletion enhances antitumour immunity in mice
Tumour-specific SHP1-deficient CD8+ T cells show enhanced in vitro and in vivo expansion in response to tumour antigen compared with SHP2-replete CD8+ T cells141,142. Upon adoptive transfer, tumour-specific SHP1-deficient CD8+ T cells improve therapeutic outcome in mice with disseminated leukaemia141 and prevent tumour metastasis in mice carrying B16-F10 melanoma tumours142. SHP1 knockdown in adoptively transferred OT-I T cells does not affect OVA peptide-expressing B16-F10 tumour growth; however, in combination with a checkpoint blockade immunotherapy cocktail, SHP1 knockdown impairs growth of high-affinity antigen-expressing tumours and low-affinity antigen-expressing tumours that are non-responsive to checkpoint blockade alone142. SHP1 knockdown does not increase the number of OT-I cells found in tumours, but enhances endogenous CD8+ T cell infiltration and CXCR3 and GzmB expression in tumours expressing low-affinity antigens, suggesting that SHP1 knockdown boosts immune activation in the TME142. Inhibiting SHP1 in combination with checkpoint blockade therapy is thus likely useful against tumours expressing low-affinity ligands. Tumour antigens are often self-peptides recognized by T cells with low affinity, eliciting suboptimal immune responses and limiting effectiveness of checkpoint inhibitor therapy143. SHP1 inhibition may be especially helpful in the case of tumours expressing low-affinity antigens with low or no responsiveness to checkpoint blockade.
Mice with global, inducible SHP1 deletion develop features reminiscent of motheaten mice, including splenomegaly and lung inflammation144. Immune-rich E0771 breast adenocarcinoma and MC38 colon adenocarcinoma tumours, but not poorly immunogenic B16-F10 melanoma tumours, grow poorly in mice with an inducible SHP1 deletion144. E0771 tumours from SHP1-deficient mice contain higher percentages of activated, antigen-experienced CD4+ and CD8+ T cells and increased effector T cell to Treg cell ratio. MC38 tumours from SHP1-deficient mice also show increased numbers of CD8+ T cells. The increased control of tumour growth is unlikely to be due solely to T cells, as T cell-specific SHP1 deletion does not affect MC38 tumour growth. SHP1-deficient human and mouse macrophages display increased phagocytosis, presumably through loss of phagocytosis-inhibiting SIRPα signals; thus, macrophages are a likely candidate cell type for mediation of the antitumour action of SHP1 loss at least in part. However, this has not yet been demonstrated using conditional SHP1 deletion, as tumour studies have been precluded in dendritic cell and neutrophil SHP1-deficient strains owing to intense immune activation.
SHP1 inhibitors enhance antitumour immunity in mice
SHP1 inhibitors have been developed and investigated as potential antitumour immunotherapies. In cells, the SHP1 inhibitor TPI-1 — identified by screening a library against recombinant SHP1 catalytic domain145 — shows tenfold selectivity over SHP2, little activity on MKP1, and increases phosphorylation of numerous SHP1 targets but not SHP2 and CD45 targets. TPI-1 increases the number and percentage of IFNγ+ cells in cultured mouse splenocytes and human peripheral blood cells, and in mouse spleens in vivo. TPI-1 does not inhibit the growth of B16 cells in culture, yet slows tumour growth in xenografted mice after oral or subcutaneous administration, although not in athymic nude mice. A TPI-1 analogue, TPI-1a4, also reduces the growth of xenografted melanoma and colon cancer cell lines.
SHP1 binds to the leukocyte-associated immunoglobulin-like receptor 1 (LAIR1) and mediates LAIR1-promoted T cell exhaustion upon LAIR1 binding to collagen146. Collagen levels are increased in PD1/PDL1 blockade-resistant 344SQ lung tumours in xenografted mice, and TPI-1 administration reduces growth of these tumours, decreasing exhausted and increasing activated CD8+ TIL populations. TPI-1 combination with anti-PD1 inhibits lung metastasis and increases primary and metastatic CD8+ TILs.
Inhibition of SHP1 could have the added benefit of enhancing natural killer (NK) cell antitumour activity147. TME hypoxia reduces NK cell cytotoxicity towards tumour cells; however, SHP1 knockdown or inhibition with TPI-1 partially restores hypoxic NK cell cytotoxicity.
SHP1 activation for direct treatment of tumour cells
It is noteworthy that SHP1 is expressed in some cancers, and increased levels are found in ovarian and some high-grade breast tumours119. Consistent with its negative regulatory role in cell proliferation and migration and invasion, higher SHP1 mRNA levels are associated with a better survival outcome in many carcinomas119; thus, SHP1 is viewed as a tumour suppressor in the context of certain carcinomas, and SHP1 activation has been suggested for treatment via direct action on the tumour cells. The sorafenib analogues SC-43 and SC-40 show increased potency compared with sorafenib148. Oral SC-43 or SC-40 administration in subcutaneous hepatocellular carcinoma tumour-bearing mice increases SHP1 phosphatase activity in the tumour and decreases tumour size.
PP2A: biology and therapeutic implications
PP2A is a ubiquitously expressed heterotrimeric enzyme that comprises a scaffolding subunit PP2A-A (encoded by PPP2R1A or PPP2R1B), a catalytic subunit PP2A-C (PPP2CA or PPP2CB) and a regulatory subunit PP2A-B (one of multiple subunits categorized into four subfamilies, B55, B56, PR70/72 and striatin (STRN)). Holoenzymes consist of A/B/C subunit heterotrimers, except for STRN-including STRN-interacting phosphatase and kinase (STRIPAK) complexes, which contain additional core units35,149,150. The multisubunit nature of PP2A allows for extensive diversity among interactors and substrates. PP2A is inhibited by Tyr307 phosphorylation and Leu309 demethylation in the PP2A-C C-terminal tail35,149. PP2A is also inhibited by interaction with oncogenic proteins such as cancerous inhibitor of PP2A (CIP2A), SET and cAMP-regulated phosphoprotein 19 (ARPP19)35,149. CIP2A and SET selectively inhibit B56-containing PP2A complexes, and ARPP19 inhibits B55-containing PP2A complexes35,149. Whereas PP2A subunits B55 and B56 display tumour-suppressive roles, oncogenic roles have been reported for STRN3 and STRN4 PP2A subunits, suggesting that the B subunit is a crucial determinant of the role of the heterotrimer in cellular function and cancer35,149.
PP2A regulates numerous cellular processes, including growth, cell cycle, mitosis, differentiation and apoptosis35,149. PP2A is considered a tumour suppressor through inhibition of oncogenic regulators such as MYC, ERK, protein kinase B (AKT) and B cell lymphoma 2 (BCL-2)35,149. PP2A inhibition can occur in cancer owing to genetic mutations, phosphorylation and/or demethylation of the PP2A-C C terminus, or upregulation of endogenous PP2A regulators35,149. The non-phosphorylated form of the FDA-approved drug fingolimod (also known as FTY720) activates PP2A by displacing SET from the PP2A catalytic domain and displays antitumour activity18. Recently reported non-phosphorylatable analogues CC11 (ref. 151) and CM-1231 (ref. 152) also activate PP2A by displacing SET and inhibit leukaemic cell growth. PP2A-activating small-molecule derivatives of perphenazine have been developed, such as ‘small-molecule activators of PP2A’ (SMAPs)153,154,155. These compounds stabilize specific PP2A heterotrimers and display antitumour action in vivo153,155, although doubts have been raised regarding the specificity of their action in vivo156. However, PP2A also has roles in DNA damage repair; thus, PP2A inhibition is being pursued as a strategy to induce synthetic lethality, sensitizing cancer cells to DNA-damaging radiation and chemotherapeutics18,157.
PP2A also has regulatory roles in the immune system; most reports on this topic have focused on T cells. Knockdown of the PP2A catalytic domain enhances T cell IL-2 production, suggesting that PP2A negatively regulates T cell activation158. Concordantly, loss of CIP2A, the negative regulator of PP2A, leads to inhibitory effects on T cells159. CD4+ T cells from CIP2A KO mice show reduced CD69 expression following TCR stimulation, and after immunization with Listeria monocytogenes, CIP2A KO mice display reduced frequency of splenic CD4+ and CD8+ T cells and reduced ability to control growth of the bacterium159.
PP2A also has regulatory roles in CD4+ T cell subsets. PP2A promotes Treg cell suppressor function by enhancing Treg cell IL-2 receptor expression and through inhibition of mTOR complex 1 (mTORC1) signalling160,161,162. Inducible loss of PP2A catalytic activity in Treg cells reduces their suppressive capacity160.
Several studies suggest that PP2A promotes TH17 cell differentiation and IL-17 production. T cell transgenic overexpression of PP2A in mice promotes CD4+ T cell IL-17 production by facilitating chromatin accessibility at the Il17 locus163,164. PP2A-C-deficient CD4+ T cells show aberrant RORγt regulation, reduced IL-17A transcription and reduced TH17 cell differentiation in vitro, and loss of PP2A-C in mice reduces TH17 cells165. Treatment of T cells with PP2A/PP5 inhibitor cantharidin inhibits TH17 cell differentiation in vitro but not in PP2A-deficient T cells165. In accordance, the PP2A negative regulator CIP2A inhibits TH17 cell differentiation and IL-17 production166. CIP2A expression is reduced in human TH17 cells compared with undifferentiated CD4+ T cells, and CIP2A deficiency enhances differentiation of human and mouse TH17 cells and enhances human CD4+ T cell STAT3-Tyr705 phosphorylation. However, this same study showed that in in vitro differentiated human TH17 cells, PP2A inhibition by PP2A-A knockdown (preventing formation of PP2A complexes) or treatment with okadaic acid increases IL-17 production, and PP2A activation with SET-targeting fingolimod/FTY720 inhibits IL-17 production166, suggesting that PP2A regulation of TH17 cell function may be complex and finely tuned by differing roles of distinct PP2A-B subunit-containing complexes.
PP2A also acts as a promoter of B cell function. Loss of functional PP2A in B cells through PPP2R1A deficiency impairs B cell class switch recombination, plasmablast differentiation and immunoglobulin production in vitro, and in vivo leads to decreased germinal centre formation, plasmablast differentiation and serum immunoglobulin levels, and poor response to immunization167.
PP2A inhibition is being explored for sensitization of cells to cancer treatments, immunotherapy and for the treatment of autoimmune disease, and PP2A-activating strategies are sought to enhance the tumour suppressor functions of PP2A. When designing strategies to chemically target PP2A, its multimeric nature makes modulator development more difficult; however, it also provides opportunities for development of selective agents. Selectivity could potentially be conferred by targeting specific heterotrimers or skewing the balance between different heterotrimers by stabilizing or disrupting particular complexes. Deeper characterization of the biological roles of individual heterotrimers in immuno-oncology and autoimmunity will help to clarify the validation of PP2A as a target for specific immune indications and help to develop efficacious modulating strategies.
Below, we focus on the potential of PP2A as an immunological target for autoimmune disease and tumour immunotherapy. Excellent reviews18,157 are available on the non-immunological aspects of drugging PP2A for cancer.
PP2A in autoimmune disorders
Studies suggest that PP2A inhibition could be beneficial in SLE through actions on B cells and T cells or against TH17 cell-driven diseases such as MS or psoriatic arthritis. In humans, polymorphisms in the PPP2CA gene associate with SLE168 and PP2A-C expression is increased in T cells158,169 and B cells167 from patients with SLE . PP2A-C knockdown restores defective IL-2 production by T cells from patients with SLE158. Additionally, PP2A regulatory B55α subunit (PPP2R2A) expression is increased in T cells from patients with SLE170.
PP2A overexpression enhances autoimmunity in mice
PP2A overexpression in mice leads to autoimmune manifestations through actions on T cells and B cells. Transgenic mice carrying T cell PP2A-C overexpression produce excessive IL-17 and develop glomerulonephritis abrogated by IL-17 neutralization163. B cells from lupus-prone mice and activated B cells show increased PP2A-A and PP2A-C expression and increased PP2A catalytic activity167. Transgenic overexpression of PP2A-B regulatory subunit G5PR (PPP2R3C) in lymphoid cells leads to IgM and IgG anti-double-stranded DNA (dsDNA) autoantibodies and autoimmunity in aged female mice171.
PP2A deletion attenuates autoimmunity in mice
Loss of PP2A in T cells or B cells in mice attenuates autoimmunity. Deletion of PP2A-C (PPP2CA) in T cells or PP2A inhibition with cantharidin protects against EAE165. These mice show reduced TH17 but not TH1 or Treg cells, and CD4+ T cells from these mice show reduced IL-17A expression and impaired ability to differentiate into TH17 cells165. Additionally, T cell B55α subunit (PPP2R2A) deficiency protects against EAE and reduces TH17 cell differentiation in vitro and in vivo, but also reduces TH1 cell numbers170.
The autoimmune-protective effects of PP2A loss in T cells are unlikely to be mediated through Treg cells, as PP2A promotes Treg cell suppressor function160,161,162. Inducible Treg cell-specific loss of PP2A catalytic activity reduces Treg cell suppressive function and causes spontaneous multi-organ autoimmunity160. The success of PP2A inhibition in autoimmunity may depend in part on the predominance of Treg versus non-Treg immune cell subpopulations in the context of particular autoimmune diseases.
In the B cell compartment, mice that lack functional B cell PP2A through PPP2R1A deficiency develop attenuated lupus-like manifestations following disease induction, including reduced anti-nucleus IgG titres and decreased kidney IgG deposition. These mice show impairment of germinal centre formation and plasmablast–plasma cell differentiation and reduced serum IgM, IgG, IgG1 and IgA levels167.
PP2A in tumour immunotherapy
PP2A is emerging as a target for enhancing antitumour responses of T cells. Silencing of PPP2R2D (which encodes the PP2A regulatory subunit) in OT-I T cells or in TRP1 CD4+ T cells causes their accumulation in B16-OVA tumours, inhibits apoptosis and enhances their proliferation and production of IFNγ, IL-2 and granulocyte–macrophage colony-stimulating factor (GM-CSF)172. PPP2R2D silencing in either CD4+ or CD8+ cells enhances their antitumour activity when adoptively transferred into tumour-bearing mice. Furthermore, PP2A phosphatase activity mediates suppression of T cells caused by elevation of extracellular potassium concentrations ([K+]e), which can occur when necrotic tumours release potassium into the TME extracellular fluid173. PP2A knockdown, overexpression of dominant negative PP2A or PP2A inhibition with okadaic acid restores CD8+ T cell IFNγ production in the presence of enhanced [K+]e173.
PP2A inhibition enhances antitumour immunity in mice
Recent studies demonstrate that the PP2A/PP5 inhibitor LB-100 (also known as LB-1) enhances effectiveness of checkpoint inhibitor therapy in mice, further suggesting PP2A inhibition as a potential tumour immunotherapy strategy12,13,174. LB-100 is a derivative of norcantharidin, a demethylated analogue of a natural compound, cantharidin, which was isolated from beetles and used traditionally in China and ancient Europe for medicinal purposes175,176. LB-100 was identified as a tumour radio- and chemo-sensitizing agent, and completed a phase I clinical trial for treatment of solid tumours in combination with docetaxel (NCT01837667). The compound is also undergoing a phase II trial in recurrent glioblastoma (NCT03027388), a phase I/II trial in myelodysplastic syndromes (NCT03886662) and a phase Ib trial in combination with three standard chemotherapeutic agents in extensive-stage small-cell lung cancer (NCT04560972).
LB-100 administration in combination with an anti-PD1 blocking antibody to CT26 colorectal carcinoma tumour-bearing mice slows tumour growth and extends survival, in contrast to single-agent therapy12. Combination therapy enhances CD8+ T cell activation and tumour infiltration. Tumour rejection does not occur in combination-treated immunocompromised NSG mice or CD8+ T cell-depleted mice, confirming the importance of T cells in its therapeutic action. As combination-treated, completely regressed mice do not develop re-implanted CT26 tumours, this suggests secondary antigen-specific tumour memory12. Combination therapy with anti-PD1 is also beneficial against growth of B16 melanoma12 and striatum-implanted GL261 glioblastoma13 tumours; ablation of CD4+ or CD8+ T cells abrogates glioblastoma rejection in combination therapy-treated mice13. In co-culture experiments, LB-100 enhances IFNγ production by CD8+ T cells, leading to upregulation of PDL1 expression on co-cultured glioblastoma cells, providing a possible explanation of why combination with PD1 blockade is needed for LB-100 immunotherapeutic efficacy in vivo.
At present, it is unclear whether inhibiting PP2A for immunotherapy would require combination with checkpoint inhibitor therapy. The studies with LB-100 suggest that its immunotherapeutic functions require combination with anti-PD1; however, whether this is owing to features of LB-100 or the mechanism of action of PP2A is not yet elucidated.
PTPN22: biology and therapeutic implications
PTPN22 is a non-receptor PTP expressed in haematopoietic cells. PTPN22 contains an N-terminal PTP domain, an inter-domain and a CTD that includes four proline‐rich sequence motifs, the first of which binds to the SH3 domain of C‐terminal Src family kinase (CSK)177.
PTPN22 inhibits TCR signalling by dephosphorylating early signalling mediators such as SFKs, ZAP70, TCRζ, Vav and valosin-containing protein178. Other regulatory roles for PTPN22 have emerged, including inhibiting Treg cell immunosuppressive functions, B cell IL-6R signalling, myeloid cell NOD2 signalling, dendritic cell dectin 1 signalling and IFNα receptor signalling, promoting type 1 interferon response downstream pattern recognition receptors in myeloid cells, inflammasome activation by dephosphorylating myeloid cell NLR family pyrin domain-containing 3 (NLRP3), neutrophil effector functions and mast cell IgE receptor signalling.
The structure of only the catalytic domain is known; thus, although PTPN22 contains extensive regions outside the catalytic domain, current lack of structural knowledge regarding these regions makes it difficult to design an approach to target them.
The roles of PTPN22 in immune cell signalling, including what is understood about the mechanisms that regulate its functions, have been extensively reviewed elsewhere. The interested reader is referred to several excellent reviews on this topic178,179,180,181,182,183,184.
PTPN22 in autoimmune disorders
The PTPN22C1858T SNP — which encodes a missense substitution of Arg for Trp at position 620 (R620W), disrupting the interaction with CSK178,185 — is a major shared autoimmunity locus and the most significant risk factor for RA outside the MHC locus in individuals of European descent186. PTPN22C1858T is also associated with type 1 diabetes mellitus, SLE, anti-neutrophil cytoplasmic antibody-associated vasculitis, psoriatic arthritis and other autoimmune diseases. Although its pathogenic mechanism of action is still the subject of much investigation, carriers of this variant have altered T and B cell functions and higher frequencies of autoreactive B cell receptor repertoires187,188,189,190. It is currently unknown whether a PTPN22 inhibitor would treat autoimmune disease in carriers of the variant. Interestingly, carriers of another PTPN22 variant with reduced phosphatase activity, PTPN22G788A, encoding Arg263Glu in the catalytic domain, are at reduced risk of developing RA191 and SLE192, suggesting that reduced PTPN22 catalytic activity is indeed protective against some autoimmune diseases.
Unlike deletion of some of the other phosphatases mentioned in this Review, mice that lack PTPN22 appear to be healthy in normal conditions. These mice exhibit accumulation of effector memory T cells in aged animals, increased CD4+ T cell help to B cells, expanded follicular helper T cells and germinal centres, and expanded Treg cells with enhanced suppressive and adhesive functions193,194,195. PTPN22-deficient mice display reduced severity of TH17-dependent autoimmune arthritis196, EAE194 and anaphylaxis197, yet develop more severe DSS-induced colitis198,199,200 and increased diabetes frequency on the non-obese diabetic background201. In lupus models, PTPN22-deficient mice display accelerated IFNα-induced lupus-like disease202 and increased autoantibody production in females in the BXSB model of SLE203.
Although several PTPN22 inhibitors have been reported, few are suitable for in vivo use; thus, studies of PTPN22 inhibition in autoimmunity models are lacking. The PTPN22 inhibitor LTV-1 (ref. 204), which appears to interact with the phosphate-binding loop of the active site of PTPN22 and a nearby hydrophobic pocket204, does reduce the frequency of autoreactive B cells in a mouse model of impaired B cell tolerance190. LTV-1 exhibits a mostly competitive mechanism of action and high selectivity for PTPN22 over SHP1, CD45 and PTP-PEST, but only threefold selectivity over PTP1B and TC-PTP.
However, for autoimmunity, PTPN22 target validation remains tenuous. There is extensive genetic evidence associating PTPN22 with disease pathogenesis, yet there is currently a lack of substantial preclinical evidence for PTPN22 inhibition attenuating established autoimmunity.
PTPN22 in tumour immunotherapy
Owing to its inhibitory role in early TCR signalling and T cell activation, PTPN22 has been considered a target for augmenting T cell-mediated tumour immunity, especially after findings that adoptively transferred PTPN22-deficient OT-I T cells are more effective at controlling growth of OVA peptide-expressing xenografted lymphoma and ovarian carcinoma tumours205,206. Transfer of PTPN22-deficient OT-I T cells also augments efficacy of a transforming growth factor-β (TGFβ)-blocking antibody205. However, a more recent study found that PTPN22 deficiency does not affect the efficiency of adoptively transferred OT-I T cells against OVA peptide-expressing AT3 mammary or MC38 tumours and importantly does not improve the efficacy of human or mouse CAR T cells against solid tumours in vivo, suggesting that PTPN22 is an unlikely target for adoptive T cell therapies207.
Despite this, global PTPN22 deletion augments the growth-inhibitory effects of anti-PDL1 on MC38 and CT26 tumours and enhances effects of anti-PDL1 on MC38 tumour CD8+ T cell infiltration and CD8+ T cell to Treg cell ratio14,208. PTPN22-deficient mice suppress MC38 tumour growth even in the absence of checkpoint inhibitor administration, with increased CD4+ and CD8+ T cell, macrophage and NK cell tumour infiltration14. PTPN22-deficient mice also inhibit AT3-OVA murine mammary tumours207 and reject Hepa1-6.x1 mouse hepatoma and E.G7-OVA thymic lymphoma tumours14,208. The remission of Hepa1-6.x1 mouse tumours is completely reversed by anti-CD4 depleting antibody and nearly blocked by an anti-CD8 antibody — indicating a major role of T cells — and partially reversed by an anti-IFNAR antibody208.
In humans, the autoimmune-associated PTPN22-R620W variant confers protection from non-melanoma skin cancer208, melanoma and gastrointestinal and central nervous system cancers14, and confers enhanced response to checkpoint inhibitor immunotherapy14,208. Mice carrying homozygous R619W knock-in alteration (homologous to the human autoimmune-associated R620W alteration) display enhanced control of xenografted Hepa1-6.x1 tumour growth compared with wild-type mice, although are less effective at controlling tumour growth than mice that lack PTPN22 (refs. 208,209). PTPN22-R619W-mutant mice also show significantly greater control of xenografted immunogenic B16-OVA tumours — but not mice that lack adaptive immunity — and immunogenic MC38 tumours, with enhanced tumour immune cell infiltration in both models209. B16-OVA tumours of PTPN22-R619W-mutant mice display enhanced expression of markers of T cell and myeloid cell activation and enhanced infiltration of type 1 conventional dendritic cells209. However, no significant differences were observed between PTPN22-R619W-mutant and wild-type mice in the control of poorly immunogenic LLC and B16-F10 tumour growth209.
PTPN22 inhibition in tumour immunotherapy
As proof of concept of PTPN22 inhibition, catalytically inactive PTPN22-C227S knock-in mice are equally effective at inhibiting Hepa1-6.x1 tumour growth as PTPN22-deficient mice, suggesting that the tumour-promoting action of PRPN22 is dependent on its catalytic activity208. Moreover, treatment of mice carrying MC38 or CT26 tumours with the newly developed PTPN22 inhibitor L-1 significantly reduces tumour growth14 — and more so in combination with anti-PDL1 — and enhances tumour macrophages and CD8+ and CD4+ T cells14. L-1, a quinolone derivative, was identified by screening a small library of drug-like molecules for inhibition of PTPN22 (ref. 14). L-1 contains the quinolone derivative core with an l-alanine linker and biphenyl carboxylic group and inhibits PTPN22 competitively and shows sevenfold to tenfold selectivity over other PTPs. Importantly, L-1 treatment has no effect on MC38 tumour growth in PTPN22-deficient mice, suggesting that its therapeutic effects are mediated through host-expressed PTPN22. Partial depletion of tumour F4/80+ macrophages in L-1-treated mice diminished the efficacy of L-1, suggesting that its antitumour effects are mediated at least in part through macrophages.
Concluding remarks and future perspectives
The phosphatase field is undergoing explosive growth in the development of phosphatase-modulating agents. These advances coupled with increasing insights into biological functions of phosphatases indicate that enzymes once deemed undruggable may in fact be tractable targets for various human diseases.
Among the most impactful series of events stimulating the explosion in efforts to drug phosphatases have been the discovery of allosteric phosphatase modulators, the discovery of additional phosphatase modulators with unique mechanisms of action and the advancement of phosphatase inhibitors to clinical trials.
The discovery of allosteric phosphatase modulators coupled with structural data revealing their mechanisms of action has been instrumental in establishing that, although these enzymes lack a shared allosteric mechanism as found in kinases, allosteric targeting is still possible enzyme by enzyme. Key developments in this area were the discovery of trodusquemine as an allosteric PTP1B inhibitor, demonstrating that the phosphatase activity of PTP1B can be selectively modulated by targeting a non-catalytic disordered region and the importance of alternative structural methods such as NMR when co-crystallization is not possible25; the discovery of the allosteric WIP1 inhibitor GSK2830371, along with the use of photoaffinity labelling mass spectrometry to localize its binding to a unique ‘flap’ subdomain that confers selective inhibition of WIP1 (ref. 27); and the discovery of the allosteric ‘molecular glue’ SHP2 inhibitor SHP099 and co-crystallization indicating that the inhibitor holds the enzyme in a naturally occurring catalytically unfavourable conformation used within the cell as an intramolecular regulation mechanism20,21.
In addition to allosterics, the discovery of other phosphatase modulators with unique mechanisms of action has shown that alternative targeting strategies are possible taking the enzyme target into account. The oxidation-stabilizing PTP1B inhibitor chelerythrine210 takes advantage of an intracellular physiological mechanism to control phosphatase activity, and the competitive ‘induced fit’ LMPTP inhibitor compound 28 (ref. 31) and the uncompetitive ‘molecular cork’ LMPTP inhibitor compounds 23 (ref. 29) and 6g30 demonstrate that orthosteric targeting is a viable approach for phosphatases with active sites that contain structurally unique features.
The advancement of phosphatase inhibitors to clinical trials has tremendously boosted global interest in re-examining enzymes within this extended family and even traditional drugging approaches that were deemed unlikely to work until recently. Key examples include clinical trials with the competitive picomolar VE-PTP inhibitor AKB-9778, also demonstrating that competitive orthosteric drugging is a viable option if sufficient potency can be obtained; the allosteric SHP2 inhibitor TNO155; and the PP2A/PP5 inhibitor LB-100.
Among current targets for immune-mediated diseases, TC-PTP and SHP2 have the most compelling evidence for a tumour immunotherapy indication. Substantial preclinical validation data from multiple immuno-oncology angles has been collected for TC-PTP using genetically modified mouse models, and its biological mechanisms are well understood. To date, few TC-PTP inhibitors are available, but recent advancement of the Calico/AbbVie compounds to phase I trials further demonstrates tractability for TC-PTP in this indication. Although the mechanism of action of SHP2 in immuno-oncology is not yet completely elucidated, convincing preclinical data with multiple SHP2 inhibitors with known modes of action have been collected. Owing to the long-standing status of SHP2 as a cancer target, multiple inhibitors from various sources have been developed, and substantial knowledge regarding the structure and regulation of this enzyme is available.
Although substantial progress has been made in the validation of phosphatase targets for immuno-oncology, validation of these enzymes as targets for autoimmune diseases has lagged behind. Compelling data using genetically modified mouse models are building a case for PP2A inhibition as a potential therapeutic strategy for SLE and TH17-mediated autoimmune diseases and for TC-PTP activation as a potential therapy for RA and/or inflammatory bowel disease. However, at present, further validation studies using selective targeting agents in autoimmune models are still needed. Interesting preliminary studies suggest that SHP1 activation and SHP2 inhibition could be beneficial against RA and potentially SLE (for SHP2 inhibition); however, these early findings need to be further substantiated. Although PTPN22 has long been of interest for autoimmunity owing to its causal role in autoimmune disease pathogenesis, the question of whether inhibition of PTPN22 would be beneficial against a specific autoimmune disease still remains. To address this, there is a need for high-quality tool compounds and specific cell-targeting mice to study the biological role of PTPN22 in specific disease contexts.
Despite advances in the development of modulators of different types of phosphatases, increased molecular understanding of the target of interest and the development of cell-based assays to confirm intracellular target engagement and desired biological effects are essential. As these superfamilies of enzymes are not currently amenable to a platform-based approach, molecular knowledge of the individual enzyme in question is essential for phosphatases, as the strategies to target them at this point need to be tailored to each particular enzyme. Given past problems that have plagued phosphatase drug discovery, cell-based assays are also crucial for early identification of promising scaffolds. For orthosteric targeting, a key additional barrier that remains is achieving selectivity. For allosteric modulator development, there is a further need for appropriate high-throughput screening assays to identify such modulators and for alternative structural methods to characterize their modalities when co-crystallization is not an option. Furthermore, a critical barrier may be that some targeted allosteric pockets cannot be bound by small molecules with high affinity.
Depending on the indication, a ubiquitous or wide expression profile may be a barrier for some phosphatase targets. However, for certain indications such as immunotherapy, a wide tissue expression profile coupled with convergent mechanisms of action among different cell types could prove to be an asset (for example, TC-PTP and SHP2). For autoimmune indications, a restricted expression profile, such as to immune cells and/or local stromal cells, may reduce the risks of on-target toxicities (for example, SHP1 and PTPN22).
Over the next decade, we anticipate that several phosphatase-targeted drugs will attain FDA approval as tumour immunotherapeutics. Successful drugging of phosphatases for immuno-oncology coupled with further preclinical validation studies in autoimmune models will likely lead to commencement of clinical trials for phosphatase-targeting agents in autoimmune disorders as well. We also envision intensified interest in using and developing new methods to characterize the structure, catalytic dynamics and biological interactions of phosphatases to more deeply understand how to approach this intriguing and untapped family of targets.
References
Hunter, T. The genesis of tyrosine phosphorylation. Cold Spring Harb. Perspect. Biol. 6, a020644 (2014).
Tonks, N. K. Protein tyrosine phosphatases–from housekeeping enzymes to master regulators of signal transduction. FEBS J. 280, 346–378 (2013).
Roskoski, R. Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 100, 1–23 (2015).
Castro-Sanchez, P., Teagle, A. R., Prade, S. & Zamoyska, R. Modulation of TCR signaling by tyrosine phosphatases: from autoimmunity to immunotherapy. Front. Cell Dev. Biol. 8, 608747 (2020).
Pike, K. A. & Tremblay, M. L. Protein tyrosine phosphatases: regulators of CD4 T cells in inflammatory bowel disease. Front. Immunol. 9, 2504 (2018).
Zhang, W. et al. Advances in anti-tumor treatments targeting the CD47/SIRPα axis. Front. Immunol. 11, 18 (2020).
Spalinger, M. R., Schwarzfischer, M. & Scharl, M. The role of protein tyrosine phosphatases in inflammasome activation. Int. J. Mol. Sci. 21, 5481 (2020).
Wiede, F. et al. PTPN2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J. 39, e103637 (2020). This publication demonstrates that loss of TC-PTP in T cells enhances multiple angles of T cell antitumour immunity and that inhibition of TC-PTP enhances CAR T cell function.
Zhao, M. et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm. Sin. B 9, 304–315 (2019). This publication demonstrates that allosteric inhibition of SHP2 in mice enhances antitumour immunity alone and in combination with anti-PD1.
Wang, Y. et al. SHP2 blockade enhances anti-tumor immunity via tumor cell intrinsic and extrinsic mechanisms. Sci. Rep. 11, 1399 (2021).
Fedele, C. et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 218, e20201414 (2021).
Ho, W. S. et al. Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat. Commun. 9, 2126 (2018). This publication describes antitumour efficacy of PP2A/PP5 inhibitor LB-100 in combination with anti-PD1 in mice through a T cell-mediated action.
Maggio, D. et al. Inhibition of protein phosphatase-2A with LB-100 enhances antitumor immunity against glioblastoma. J. Neurooncol. 148, 231–244 (2020). This report describes efficacy of PP2A/PP5 inhibitor LB-100 at controlling glioblastoma growth in combination with anti-PD1 in mice through a T cell-mediated action.
Ho, W. J. et al. Systemic inhibition of PTPN22 augments anticancer immunity. J. Clin. Invest. 131, e146950 (2021). This report demonstrates that inhibition of PTPN22 reduces growth of xenografted colorectal cancer tumours in mice, alone and in combination with anti-PDL1.
Safarini, O. A., Keshavamurthy, C. & Patel, P. Calcineurin inhibitors. StatPearls [online] https://www.ncbi.nlm.nih.gov/books/NBK558995/ (updated 24 Nov 2022).
Ponticelli, C., Reggiani, F. & Moroni, G. Old and new calcineurin inhibitors in lupus nephritis. J. Clin. Med. 10, 4832 (2021).
Stanford, S. M. & Bottini, N. Targeting tyrosine phosphatases: time to end the stigma. Trends Pharmacol. Sci. 38, 524–540 (2017).
Vainonen, J. P., Momeny, M. & Westermarck, J. Druggable cancer phosphatases. Sci. Transl Med. 13, eabe2967 (2021).
Krishnan, N., Konidaris, K. F., Gasser, G. & Tonks, N. K. A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J. Biol. Chem. 293, 1517–1525 (2018).
Chen, Y. N. et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152 (2016). This publication describes discovery of the first allosteric SHP2 inhibitor SHP099, which acts as a molecular glue to hold the enzyme in an auto-inhibited conformation.
Garcia Fortanet, J. et al. Allosteric Inhibition of SHP2: identification of a potent, selective, and orally efficacious phosphatase inhibitor. J. Med. Chem. 59, 7773–7782 (2016).
LaMarche, M. J. et al. Identification of TNO155, an allosteric SHP2 inhibitor for the treatment of cancer. J. Med. Chem. 63, 13578–13594 (2020). Report of the TNO155 allosteric SHP2 inhibitor, a SHP099 derivative with improved features.
Nichols, R. J. et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol. 20, 1064–1073 (2018).
Sun, Y. et al. Allosteric SHP2 inhibitor, IACS-13909, overcomes EGFR-dependent and EGFR-independent resistance mechanisms toward osimertinib. Cancer Res. 80, 4840–4853 (2020).
Gannam, Z. T. K. et al. An allosteric site on MKP5 reveals a strategy for small-molecule inhibition. Sci. Signal. 13, eaba3043 (2020).
Anantharajan, J. et al. Structural and functional analyses of an allosteric EYA2 phosphatase inhibitor that has on-target effects in human lung cancer cells. Mol. Cancer Ther. 18, 1484–1496 (2019).
Gilmartin, A. G. et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 10, 181–187 (2014).
Hussain, R. M., Neiweem, A. E., Kansara, V., Harris, A. & Ciulla, T. A. Tie-2/angiopoietin pathway modulation as a therapeutic strategy for retinal disease. Expert Opin. Investig. Drugs 28, 861–869 (2019).
Stanford, S. M. et al. Diabetes reversal by inhibition of the low-molecular-weight tyrosine phosphatase. Nat. Chem. Biol. 13, 624–632 (2017).
Stanford, S. M. et al. Discovery of orally bioavailable purine-based inhibitors of the low-molecular-weight protein tyrosine phosphatase. J. Med. Chem. 64, 5645–5653 (2021).
He, R. et al. Inhibition of low molecular weight protein tyrosine phosphatase by an induced-fit mechanism. J. Med. Chem. 59, 9094–9106 (2016).
Chen, M. J., Dixon, J. E. & Manning, G. Genomics and evolution of protein phosphatases. Sci. Signal. 10, eaag1796 (2017).
Alonso, A. & Pulido, R. The extended human PTPome: a growing tyrosine phosphatase family. FEBS J. 283, 1404–1429 (2016).
Brautigan, D. L. & Shenolikar, S. Protein serine/threonine phosphatases: keys to unlocking regulators and substrates. Annu. Rev. Biochem. 87, 921–964 (2018).
Fowle, H., Zhao, Z. & Grana, X. PP2A holoenzymes, substrate specificity driving cellular functions and deregulation in cancer. Adv. Cancer Res. 144, 55–93 (2019).
Seifried, A., Schultz, J. & Gohla, A. Human HAD phosphatases: structure, mechanism, and roles in health and disease. FEBS J. 280, 549–571 (2013).
Tsygankov, A. Y. TULA proteins as signaling regulators. Cell. Signal. 65, 109424 (2020).
Shi, Y. Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 (2009).
Roychoudhury, K. & Hegde, R. S. The eyes absent proteins: unusual HAD family tyrosine phosphatases. Int. J. Mol. Sci. 22, 3925 (2021).
Frankson, R. et al. Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. 77, 5701–5705 (2017).
Lazo, J. S., McQueeney, K. E., Burnett, J. C., Wipf, P. & Sharlow, E. R. Small molecule targeting of PTPs in cancer. Int. J. Biochem. Cell Biol. 96, 171–181 (2018).
Elson, A. Stepping out of the shadows: oncogenic and tumor-promoting protein tyrosine phosphatases. Int. J. Biochem. Cell Biol. 96, 135–147 (2018).
Ohtake, Y. et al. Protein tyrosine phosphatase sigma regulates autoimmune encephalomyelitis development. Brain Behav. Immun. 65, 111–124 (2017).
Luo, F. et al. Modulation of proteoglycan receptor PTPsigma enhances MMP-2 activity to promote recovery from multiple sclerosis. Nat. Commun. 9, 4126 (2018).
Pendleton, J. C. et al. Chondroitin sulfate proteoglycans inhibit oligodendrocyte myelination through PTPsigma. Exp. Neurol. 247, 113–121 (2013).
Niknam, P., Raoufy, M. R., Fathollahi, Y. & Javan, M. Modulating proteoglycan receptor PTPsigma using intracellular sigma peptide improves remyelination and functional recovery in mice with demyelinated optic chiasm. Mol. Cell. Neurosci. 99, 103391 (2019).
Aschner, Y. et al. Protein tyrosine phosphatase α mediates profibrotic signaling in lung fibroblasts through TGF-β responsiveness. Am. J. Pathol. 184, 1489–1502 (2014).
Aschner, Y. et al. Protein tyrosine phosphatase-α amplifies transforming growth factor-β-dependent profibrotic signaling in lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 319, L294–L311 (2020).
Stanford, S. M. et al. Receptor protein tyrosine phosphatase α-mediated enhancement of rheumatoid synovial fibroblast signaling and promotion of arthritis in mice. Arthritis Rheumatol. 68, 359–369 (2016).
Sacchetti, C. et al. PTP4A1 promotes TGFβ signaling and fibrosis in systemic sclerosis. Nat. Commun. 8, 1060 (2017).
Clavarino, G. et al. Unfolded protein response gene GADD34 is overexpressed in rheumatoid arthritis and related to the presence of circulating anti-citrullinated protein antibodies. Autoimmunity 49, 172–178 (2016).
Plazy, C. et al. Letter to the Editor: Protein phosphatase 1 subunit Ppp1r15a/GADD34 is overexpressed in systemic lupus erythematosus and related to the expression of type I interferon response genes. Autoimmun. Rev. 18, 211–213 (2019).
Lin, W. et al. Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-γ. Am. J. Pathol. 173, 1508–1517 (2008).
Chen, Y., Kunjamma, R. B., Weiner, M., Chan, J. R. & Popko, B. Prolonging the integrated stress response enhances CNS remyelination in an inflammatory environment. eLife 10, e65469 (2021).
Chen, Y. et al. Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 142, 344–361 (2019).
Doody, K. M. et al. Targeting phosphatase-dependent proteoglycan switch for rheumatoid arthritis therapy. Sci. Transl Med. 7, 288ra276 (2015).
Svensson, M. N. D. et al. Synoviocyte-targeted therapy synergizes with TNF inhibition in arthritis reversal. Sci. Adv. 6, eaba4353 (2020).
Shentu, Y. P. et al. CIP2A-promoted astrogliosis induces AD-like synaptic degeneration and cognitive deficits. Neurobiol. Aging 75, 198–208 (2019).
Muppirala, M., Gupta, V. & Swarup, G. Emerging role of tyrosine phosphatase, TCPTP, in the organelles of the early secretory pathway. Biochim. Biophys. Acta 1833, 1125–1132 (2013).
Pike, K. A. & Tremblay, M. L. TC-PTP and PTP1B: regulating JAK-STAT signaling, controlling lymphoid malignancies. Cytokine 82, 52–57 (2016).
Hao, L., Tiganis, T., Tonks, N. K. & Charbonneau, H. The noncatalytic C-terminal segment of the T cell protein tyrosine phosphatase regulates activity via an intramolecular mechanism. J. Biol. Chem. 272, 29322–29329 (1997).
Singh, J. P. et al. The catalytic activity of TCPTP is auto-regulated by its intrinsically disordered tail and activated by Integrin alpha-1. Nat. Commun. 13, 94 (2022).
Mattila, E. et al. Negative regulation of EGFR signalling through integrin-α1β1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 7, 78–85 (2005).
Wiede, F., Sacirbegovic, F., Leong, Y. A., Yu, D. & Tiganis, T. PTPN2-deficiency exacerbates T follicular helper cell and B cell responses and promotes the development of autoimmunity. J. Autoimmun. 76, 85–100 (2017).
Wiede, F. et al. T-cell-specific PTPN2 deficiency in NOD mice accelerates the development of type 1 diabetes and autoimmune comorbidities. Diabetes 68, 1251–1266 (2019). This report demonstrates that selective deletion of TC-PTP in T cells of NOD mice on the non-obese diabetic background leads to accelerated diabetes onset and increased symptoms of colitis and Sjögren’s syndrome.
Wiede, F., Ziegler, A., Zehn, D. & Tiganis, T. PTPN2 restrains CD8+ T cell responses after antigen cross-presentation for the maintenance of peripheral tolerance in mice. J. Autoimmun. 53, 105–114 (2014).
Hassan, S. W. et al. Increased susceptibility to dextran sulfate sodium induced colitis in the T cell protein tyrosine phosphatase heterozygous mouse. PLoS ONE 5, e8868 (2010).
Spalinger, M. R. et al. PTPN2 controls differentiation of CD4(+) T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol. 8, 918–929 (2015). This report shows that selective loss of TC-PTP in T cells enhances colitis in the DSS and CD4+ T cell transfer mouse models.
Spalinger, M. R. et al. PTPN2 regulates inflammasome activation and controls onset of intestinal inflammation and colon cancer. Cell Rep. 22, 1835–1848 (2018).
Spalinger, M. R. et al. PTPN2 regulates interactions between macrophages and intestinal epithelial cells to promote intestinal barrier function. Gastroenterology 159, 1763–1777 (2020). This publication demonstrates that selective loss of TC-PTP in macrophages increases permeability of the colon epithelium in mice.
Bussieres-Marmen, S. et al. Loss of T-cell protein tyrosine phosphatase in the intestinal epithelium promotes local inflammation by increasing colonic stem cell proliferation. Cell Mol. Immunol. 15, 367–376 (2018). This publication demonstrates that selective deletion of TC-PTP in IECs exacerbates DSS-induced colitis in mice.
Kasper, S. H. et al. Deficiency of protein tyrosine phosphatase non-receptor type 2 in intestinal epithelial cells has no appreciable impact on dextran sulphate sodium colitis severity but promotes wound healing. Digestion 93, 249–259 (2016).
Svensson, M. N. et al. Reduced expression of phosphatase PTPN2 promotes pathogenic conversion of Tregs in autoimmunity. J. Clin. Invest. 129, 1193–1210 (2019). This report shows that global Ptpn2 haloinsufficiency or haploinsufficiency in T cells or Treg cells enhances arthritis in the SKG mouse model by promoting conversion of Treg cells into pathogenic IL-17-producing ‘exTreg’ cells.
Hsieh, W. C. et al. PTPN2 links colonic and joint inflammation in experimental autoimmune arthritis. JCI Insight 5, e141868 (2020).
Zaiss, M. M., Joyce Wu, H. J., Mauro, D., Schett, G. & Ciccia, F. The gut–joint axis in rheumatoid arthritis. Nat. Rev. Rheumatol. 17, 224–237 (2021).
Moron, B. et al. Activation of protein tyrosine phosphatase non-receptor type 2 by spermidine exerts anti-inflammatory effects in human THP-1 monocytes and in a mouse model of acute colitis. PLoS ONE 8, e73703 (2013).
LaFleur, M. W. et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat. Immunol. 20, 1335–1347 (2019). This report shows that loss of TC-PTP in CD8+ T cells enhances their anti-tumour immunity when transferred into mice carrying xenografted tumours.
Katkeviciute, E. et al. Protein tyrosine phosphatase nonreceptor type 2 controls colorectal cancer development. J. Clin. Investig. 131, e140281 (2021).
Manguso, R. T. et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418 (2017). This report of an in vivo genetic screen using genome-editing identifies PTPN2 as a candidate target for sensitizing B16 melanoma tumour cells to immunotherapy.
Lawson, K. A. et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126 (2020).
Spalinger, M. R., McCole, D. F., Rogler, G. & Scharl, M. Role of protein tyrosine phosphatases in regulating the immune system: implications for chronic intestinal inflammation. Inflamm. Bowel Dis. 21, 645–655 (2015).
Long, S. A. et al. An autoimmune-associated variant in PTPN2 reveals an impairment of IL-2R signaling in CD4+ T cells. Genes Immun. 12, 116–125 (2011).
Scharl, M. et al. Protein tyrosine phosphatase nonreceptor type 2 regulates autophagosome formation in human intestinal cells. Inflamm. Bowel Dis. 18, 1287–1302 (2012).
You-Ten, K. E. et al. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J. Exp. Med. 186, 683–693 (1997).
Mattila, E. et al. Inhibition of receptor tyrosine kinase signalling by small molecule agonist of T-cell protein tyrosine phosphatase. BMC cancer 10, 7 (2010).
Ylilauri, M. et al. Molecular mechanism of T-cell protein tyrosine phosphatase (TCPTP) activation by mitoxantrone. Biochim. Biophys. Acta 1834, 1988–1997 (2013).
Luo, N. et al. Melanoma response to anti-PD-L1 immunotherapy requires JAK1 signaling, but not JAK2. Oncoimmunology 7, e1438106 (2018).
Krishnan, N. et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 10, 558–566 (2014).
Wiede, F. et al. PTP1B is an intracellular checkpoint that limits T cell and CAR T cell anti-tumor immunity. Cancer Discov. 12, 752–773 (2022).
Montalibet, J. & Kennedy, B. P. Using yeast to screen for inhibitors of protein tyrosine phosphatase 1B. Biochem. Pharmacol. 68, 1807–1814 (2004).
Penafuerte, C. et al. Downregulation of PTP1B and TC-PTP phosphatases potentiate dendritic cell-based immunotherapy through IL-12/IFNγ signaling. Oncoimmunology 6, e1321185 (2017).
Hof, P., Pluskey, S., Dhe-Paganon, S., Eck, M. J. & Shoelson, S. E. Crystal structure of the tyrosine phosphatase SHP-2. Cell 92, 441–450 (1998).
Barford, D. & Neel, B. G. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure 6, 249–254 (1998).
Poole, A. W. & Jones, M. L. A SHPing tale: perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell Signal. 17, 1323–1332 (2005).
Tao, Y. et al. A novel partially-open state of SHP2 points to a “multiple gear” regulation mechanism. J. Biol. Chem. 296, 100538 (2021).
Bollu, L. R., Mazumdar, A., Savage, M. I. & Brown, P. H. Molecular pathways: targeting protein tyrosine phosphatases in cancer. Clin. Cancer Res. 23, 2136–2142 (2017).
Chan, G., Kalaitzidis, D. & Neel, B. G. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 27, 179–192 (2008).
Yuan, X., Bu, H., Zhou, J., Yang, C. Y. & Zhang, H. Recent advances of SHP2 inhibitors in cancer therapy: current development and clinical application. J. Med. Chem. 63, 11368–11396 (2020).
Zhernakova, A. et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. 7, e1002004 (2011).
Maeshima, K. et al. Abnormal PTPN11 enhancer methylation promotes rheumatoid arthritis fibroblast-like synoviocyte aggressiveness and joint inflammation. JCI Insight 1, e86580 (2016).
Wang, J. et al. Inhibition of SHP2 ameliorates the pathogenesis of systemic lupus erythematosus. J. Clin. Invest. 126, 2077–2092 (2016).
Kerr, D. L., Haderk, F. & Bivona, T. G. Allosteric SHP2 inhibitors in cancer: targeting the intersection of RAS, resistance, and the immune microenvironment. Curr. Opin. Chem. Biol. 62, 1–12 (2021).
Quintana, E. et al. Allosteric Inhibition of SHP2 stimulates antitumor immunity by transforming the immunosuppressive environment. Cancer Res. 80, 2889–2902 (2020). This report shows that allosteric inhibition of SHP2 in mice with RMC-4550 slows growth of multiple tumour models through a CD8+ T cell-mediated action.
Stanley, E. R. & Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 6, a021857 (2014).
Niogret, C., Birchmeier, W. & Guarda, G. SHP-2 in lymphocytes’ cytokine and inhibitory receptor signaling. Front. Immunol. 10, 2468 (2019).
Hui, E. et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 (2017).
Celis-Gutierrez, J. et al. Quantitative interactomics in primary T cells provides a rationale for concomitant PD-1 and BTLA coinhibitor blockade in cancer immunotherapy. Cell Rep. 27, 3315–3330 (2019).
Peled, M. et al. Affinity purification mass spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Proc. Natl Acad. Sci. USA 115, E468–E477 (2018).
Patsoukis, N. et al. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun. Biol. 3, 128 (2020).
Marasco, M. et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci. Adv. 6, eaay4458 (2020).
Marasco, M., Kirkpatrick, J. P. & Carlomagno, T. 1H, 13C, 15N chemical shift assignments of SHP2 SH2 domains in complex with PD-1 immune-tyrosine motifs. Biomol. NMR Assign. 14, 179–188 (2020).
Xu, X. et al. PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. J. Cell Biol. 219, e201905085 (2020).
Zhang, T. et al. Loss of SHP-2 activity in CD4+ T cells promotes melanoma progression and metastasis. Sci. Rep. 3, 2845 (2013).
Rota, G. et al. Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Rep. 23, 39–49 (2018).
Xiao, P. et al. Myeloid-restricted ablation of Shp2 restrains melanoma growth by amplifying the reciprocal promotion of CXCL9 and IFN-γ production in tumor microenvironment. Oncogene 37, 5088–5100 (2018).
Strazza, M. et al. SHP2 targets ITK downstream of PD-1 to inhibit T cell function. Inflammation 44, 1529–1539 (2021).
Chen, D. et al. SHP-2 and PD-L1 inhibition combined with radiotherapy enhances systemic antitumor effects in an anti-PD-1-resistant model of non-small cell lung cancer. Cancer Immunol. Res. 8, 883–894 (2020).
Lorenz, U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol. Rev. 228, 342–359 (2009).
Varone, A., Spano, D. & Corda, D. Shp1 in solid cancers and their therapy. Front. Oncol. 10, 935 (2020).
Pei, D., Wang, J. & Walsh, C. T. Differential functions of the two Src homology 2 domains in protein tyrosine phosphatase SH-PTP1. Proc. Natl Acad. Sci. USA 93, 1141–1145 (1996).
Pao, L. I., Badour, K., Siminovitch, K. A. & Neel, B. G. Nonreceptor protein-tyrosine phosphatases in immune cell signaling. Annu. Rev. Immunol. 25, 473–523 (2007).
Singh, K. et al. ERK-dependent T cell receptor threshold calibration in rheumatoid arthritis. J. Immunol. 183, 8258–8267 (2009).
Christophi, G. P. et al. SHP-1 deficiency and increased inflammatory gene expression in PBMCs of multiple sclerosis patients. Lab. Invest. 88, 243–255 (2008).
Christophi, G. P. et al. Macrophages of multiple sclerosis patients display deficient SHP-1 expression and enhanced inflammatory phenotype. Lab. Invest. 89, 742–759 (2009).
Christophi, G. P. et al. Interferon-β treatment in multiple sclerosis attenuates inflammatory gene expression through inducible activity of the phosphatase SHP-1. Clin. Immunol. 133, 27–44 (2009).
Deng, C. et al. Expression of the tyrosine phosphatase SRC homology 2 domain-containing protein tyrosine phosphatase 1 determines T cell activation threshold and severity of experimental autoimmune encephalomyelitis. J. Immunol. 168, 4511–4518 (2002).
Fowler, C. C., Pao, L. I., Blattman, J. N. & Greenberg, P. D. SHP-1 in T cells limits the production of CD8 effector cells without impacting the formation of long-lived central memory cells. J. Immunol. 185, 3256–3267 (2010).
Mercadante, E. R. & Lorenz, U. M. T Cells deficient in the tyrosine phosphatase SHP-1 resist suppression by regulatory T cells. J. Immunol. 199, 129–137 (2017).
Johnson, D. J. et al. Shp1 regulates T cell homeostasis by limiting IL-4 signals. J. Exp. Med. 210, 1419–1431 (2013).
Pao, L. I. et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity 27, 35–48 (2007).
Khalil, A. M., Cambier, J. C. & Shlomchik, M. J. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science 336, 1178–1181 (2012).
Kaneko, T. et al. Dendritic cell-specific ablation of the protein tyrosine phosphatase Shp1 promotes Th1 cell differentiation and induces autoimmunity. J. Immunol. 188, 5397–5407 (2012).
Abram, C. L., Roberge, G. L., Pao, L. I., Neel, B. G. & Lowell, C. A. Distinct roles for neutrophils and dendritic cells in inflammation and autoimmunity in motheaten mice. Immunity 38, 489–501 (2013).
Speir, M. et al. Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation. Nat. Immunol. 21, 54–64 (2020).
Markovics, A., Toth, D. M., Glant, T. T. & Mikecz, K. Regulation of autoimmune arthritis by the SHP-1 tyrosine phosphatase. Arthritis Res. Ther. 22, 160 (2020). This publication demonstrates that treatment with SHP1 activator regorafenib reduces arthritis incidence and severity in the proteoglycan-induced arthritis model.
Tai, W. T. et al. Nintedanib (BIBF-1120) inhibits hepatocellular carcinoma growth independent of angiokinase activity. J. Hepatol. 61, 89–97 (2014).
Fan, L. C. et al. SHP-1 is a target of regorafenib in colorectal cancer. Oncotarget 5, 6243–6251 (2014).
Tai, W. T. et al. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3. Mol. Cancer Ther. 11, 452–463 (2012).
Huang, C. Y. et al. Dovitinib acts as a novel radiosensitizer in hepatocellular carcinoma by targeting SHP-1/STAT3 signaling. Int. J. Radiat. Oncol. Biol. Phys. 95, 761–771 (2016).
Monu, N. & Frey, A. B. Suppression of proximal T cell receptor signaling and lytic function in CD8+ tumor-infiltrating T cells. Cancer Res. 67, 11447–11454 (2007).
Watson, H. A. et al. Purity of transferred CD8+ T cells is crucial for safety and efficacy of combinatorial tumor immunotherapy in the absence of SHP-1. Immunol. Cell Biol. 94, 802–808 (2016).
Snook, J. P., Soedel, A. J., Ekiz, H. A., O’Connell, R. M. & Williams, M. A. Inhibition of SHP-1 expands the repertoire of antitumor T cells available to respond to immune checkpoint blockade. Cancer Immunol. Res. 8, 506–517 (2020).
Hoffmann, M. M. & Slansky, J. E. T-cell receptor affinity in the age of cancer immunotherapy. Mol. Carcinog. 59, 862–870 (2020).
Myers, D. R. et al. Shp1 loss enhances macrophage effector function and promotes anti-tumor immunity. Front. Immunol. 11, 576310 (2020). This reports shows that global, inducible SHP1 deletion in mice leads to enhanced control of immune-rich adenocarcinoma tumours.
Kundu, S. et al. Novel SHP-1 inhibitors tyrosine phosphatase inhibitor-1 and analogs with preclinical anti-tumor activities as tolerated oral agents. J. Immunol. 184, 6529–6536 (2010).
Sathish, J. G. et al. Constitutive association of SHP-1 with leukocyte-associated Ig-like receptor-1 in human T cells. J. Immunol. 166, 1763–1770 (2001).
Teng, R. et al. Hypoxia impairs NK cell cytotoxicity through SHP-1-mediated attenuation of STAT3 and ERK signaling pathways. J. Immunol. Res. 2020, 4598476 (2020).
Tai, W. T. et al. Discovery of novel Src homology region 2 domain-containing phosphatase 1 agonists from sorafenib for the treatment of hepatocellular carcinoma. Hepatology 59, 190–201 (2014).
Kauko, O. & Westermarck, J. Non-genomic mechanisms of protein phosphatase 2A (PP2A) regulation in cancer. Int. J. Biochem. Cell Biol. 96, 157–164 (2018).
Huang, W., Leonard, D. & Taylor, D. J. Pack a STRIPAK with hubs inside a hub. Nat. Struct. Mol. Biol. 28, 232–233 (2021).
Pagano, M. A. et al. Mitochondrial apoptosis is induced by alkoxy phenyl-1-propanone derivatives through PP2A-mediated dephosphorylation of Bad and Foxo3A in CLL. Leukemia 33, 1148–1160 (2019). Report of the discovery of non-phosphorylatable FTY720 analogue CC11, which activates PP2A by displacing negative regulator SET and inhibits chronic lymphocytic leukaemia cell growth.
Vicente, C. et al. A novel FTY720 analogue targets SET-PP2A interaction and inhibits growth of acute myeloid leukemia cells without inducing cardiac toxicity. Cancer Lett. 468, 1–13 (2020). Report of the discovery of PP2A activator CM-1231, a non-phosphorylatable FTY720 analogue that displaces SET from PP2A and inhibits AML cell growth.
Gutierrez, A. et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J. Clin. Invest. 124, 644–655 (2014).
Kastrinsky, D. B. et al. Reengineered tricyclic anti-cancer agents. Bioorg. Med. Chem. 23, 6528–6534 (2015).
Leonard, D. et al. Selective PP2A enhancement through biased heterotrimer stabilization. Cell 181, 688–701 (2020).
Vit, G. et al. Chemogenetic profiling reveals PP2A-independent cytotoxicity of proposed PP2A activators iHAP1 and DT-061. EMBO J. 41, e110611 (2022).
Mazhar, S., Taylor, S. E., Sangodkar, J. & Narla, G. Targeting PP2A in cancer: combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 1866, 51–63 (2019).
Katsiari, C. G., Kyttaris, V. C., Juang, Y. T. & Tsokos, G. C. Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. J. Clin. Invest. 115, 3193–3204 (2005).
Come, C. et al. CIP2A promotes T-cell activation and immune response to Listeria monocytogenes infection. PLoS ONE 11, e0152996 (2016).
Apostolidis, S. A. et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat. Immunol. 17, 556–564 (2016).
Ding, Y., Yu, A., Tsokos, G. C. & Malek, T. R. CD25 and protein phosphatase 2A cooperate to enhance IL-2R signaling in human regulatory T cells. J. Immunol. 203, 93–104 (2019).
Sharabi, A. et al. PP2A enables IL-2 signaling by preserving IL-2Rβ chain expression during Treg development. JCI Insight 5, e126294 (2019).
Crispin, J. C. et al. Cutting edge: protein phosphatase 2A confers susceptibility to autoimmune disease through an IL-17-dependent mechanism. J. Immunol. 188, 3567–3571 (2012).
Apostolidis, S. A., Rauen, T., Hedrich, C. M., Tsokos, G. C. & Crispin, J. C. Protein phosphatase 2A enables expression of interleukin 17 (IL-17) through chromatin remodeling. J. Biol. Chem. 288, 26775–26784 (2013).
Xu, Q. et al. Phosphatase PP2A is essential for TH17 differentiation. Proc. Natl Acad. Sci. USA 116, 982–987 (2019). This publication demonstrates that catalytic inactivation of PP2A in T cells or global PP2A inhibition is protective against EAE and reduces TH17 cell differentiation in vitro and in vivo.
Khan, M. M. et al. CIP2A constrains Th17 differentiation by modulating STAT3 signaling. iScience 23, 100947 (2020).
Meidan, E. et al. Serine/threonine phosphatase PP2A is essential for optimal B cell function. JCI Insight 5, e130655 (2020). This report shows that B cell PP2A deficiency impairs B cell development and function and leads to reduced lupus-like features in the pristine-induced lupus mouse model.
Tan, W. et al. Association of PPP2CA polymorphisms with systemic lupus erythematosus susceptibility in multiple ethnic groups. Arthritis Rheum. 63, 2755–2763 (2011).
Sunahori, K., Juang, Y. T., Kyttaris, V. C. & Tsokos, G. C. Promoter hypomethylation results in increased expression of protein phosphatase 2A in T cells from patients with systemic lupus erythematosus. J. Immunol. 186, 4508–4517 (2011).
Pan, W. et al. The regulatory subunit PPP2R2A of PP2A enhances Th1 and Th17 differentiation through activation of the GEF-H1/RhoA/ROCK signaling pathway. J. Immunol. 206, 1719–1728 (2021).
Kitabatake, M. et al. JNK regulatory molecule G5PR induces IgG autoantibody-producing plasmablasts from peritoneal B1a cells. J. Immunol. 194, 1480–1488 (2015).
Zhou, P. et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 506, 52–57 (2014). Report of an in vivo pooled short hairpin RNA screen that identifes the gene encoding the PP2A regulatory subunit (PPP2R2D) as a target for T cell-mediated tumour immunotherapy.
Eil, R. et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 537, 539–543 (2016).
D’Arcy, B. M. et al. The antitumor drug LB-100 is a catalytic inhibitor of protein phosphatase 2A (PPP2CA) and 5 (PPP5C) coordinating with the active-site catalytic metals in PPP5C. Mol. Cancer Ther. 18, 556–566 (2019).
Zhang, C. et al. A synthetic cantharidin analog for the enhancement of doxorubicin suppression of stem cell-derived aggressive sarcoma. Biomaterials 31, 9535–9543 (2010).
Lu, J. et al. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc. Natl Acad. Sci. USA 106, 11697–11702 (2009).
Gregorieff, A., Cloutier, J. F. & Veillette, A. Sequence requirements for association of protein-tyrosine phosphatase PEP with the Src homology 3 domain of inhibitory tyrosine protein kinase p50. J. Biol. Chem. 273, 13217–13222 (1998).
Mustelin, T., Bottini, N. & Stanford, S. M. The contribution of PTPN22 to rheumatic disease. Arthritis Rheumatol. 71, 486–495 (2019).
Tizaoui, K. et al. The role of PTPN22 in the pathogenesis of autoimmune diseases: a comprehensive review. Semin. Arthritis Rheum. 51, 513–522 (2021).
Armitage, L. H., Wallet, M. A. & Mathews, C. E. Influence of PTPN22 allotypes on innate and adaptive immune function in health and disease. Front. Immunol. 12, 636618 (2021).
Abbasifard, M., Imani, D. & Bagheri-Hosseinabadi, Z. PTPN22 gene polymorphism and susceptibility to rheumatoid arthritis (RA): updated systematic review and meta-analysis. J. Gene Med. 22, e3204 (2020).
Carmona, F. D. & Martin, J. The potential of PTPN22 as a therapeutic target for rheumatoid arthritis. Expert. Opin. Ther. Targets 22, 879–891 (2018).
Brownlie, R. J., Zamoyska, R. & Salmond, R. J. Regulation of autoimmune and anti-tumour T-cell responses by PTPN22. Immunology 154, 377–382 (2018).
Vang, T., Nielsen, J. & Burn, G. L. A switch-variant model integrates the functions of an autoimmune variant of the phosphatase PTPN22. Sci. Signal. 11, eaat0936 (2018).
Bottini, N. et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 36, 337–338 (2004).
Bottini, N. & Peterson, E. J. Tyrosine phosphatase PTPN22: multifunctional regulator of immune signaling, development, and disease. Annu. Rev. Immunol. 32, 83–119 (2014).
Rieck, M. et al. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J. Immunol. 179, 4704–4710 (2007).
Arechiga, A. F. et al. Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J. Immunol. 182, 3343–3347 (2009).
Menard, L. et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J. Clin. Invest. 121, 3635–3644 (2011).
Schickel, J. N. et al. PTPN22 inhibition resets defective human central B cell tolerance. Sci. Immunol. 1, aaf7153 (2016).
Rodriguez-Rodriguez, L. et al. The PTPN22 R263Q polymorphism is a risk factor for rheumatoid arthritis in Caucasian case-control samples. Arthritis Rheum. 63, 365–372 (2011).
Orru, V. et al. A loss-of-function variant of PTPN22 is associated with reduced risk of systemic lupus erythematosus. Hum. Mol. Genet. 18, 569–579 (2009).
Hasegawa, K. et al. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science 303, 685–689 (2004).
Maine, C. J., Marquardt, K., Cheung, J. & Sherman, L. A. PTPN22 controls the germinal center by influencing the numbers and activity of T follicular helper cells. J. Immunol. 192, 1415–1424 (2014).
Brownlie, R. J. et al. Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Sci. Signal. 5, ra87 (2012).
Sood, S. et al. Loss of the protein tyrosine phosphatase ptpn22 reduces mannan-induced autoimmune arthritis in SKG mice. J. Immunol. 197, 429–440 (2016).
Obiri, D. D. et al. PEST-domain-enriched tyrosine phosphatase and glucocorticoids as regulators of anaphylaxis in mice. Allergy 67, 175–182 (2012).
Chang, H. H. et al. PTPN22 modulates macrophage polarization and susceptibility to dextran sulfate sodium-induced colitis. J. Immunol. 191, 2134–2143 (2013).
Spalinger, M. R. et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Invest. 126, 4388 (2016).
Wang, Y. et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity 39, 111–122 (2013).
Lin, X. et al. CRISPR-Cas9-mediated modification of the NOD mouse genome with Ptpn22R619W mutation increases autoimmune diabetes. Diabetes 65, 2134–2138 (2016).
Holmes, D. A. et al. Autoimmunity-associated protein tyrosine phosphatase PEP negatively regulates IFN-α receptor signaling. J. Exp. Med. 212, 1081–1093 (2015).
Maine, C. J. et al. The effect of the autoimmunity-associated gene, PTPN22, on a BXSB-derived model of lupus. Clin. Immunol. 156, 65–73 (2015).
Vang, T. et al. LYP inhibits T-cell activation when dissociated from CSK. Nat. Chem. Biol. 8, 437–446 (2012).
Brownlie, R. J. et al. Resistance to TGFβ suppression and improved anti-tumor responses in CD8+ T cells lacking PTPN22. Nat. Commun. 8, 1343 (2017).
Brownlie, R. J., Wright, D., Zamoyska, R. & Salmond, R. J. Deletion of PTPN22 improves effector and memory CD8+ T cell responses to tumors. JCI Insight 5, e127847 (2019).
Du, X., Darcy, P. K., Wiede, F. & Tiganis, T. Targeting PTPN22 does not enhance the efficacy of CAR T cells in solid tumours. Mol. Cell. Biol. 42, e0044921 (2022).
Cubas, R. et al. Autoimmunity linked protein phosphatase PTPN22 as a target for cancer immunotherapy. J. Immunother. Cancer 8, e001439 (2020).
Orozco, R. C., Marquardt, K., Mowen, K. & Sherman, L. A. Proautoimmune allele of tyrosine phosphatase, PTPN22, enhances tumor immunity. J. Immunol. 207, 1662–1671 (2021).
Krishnan, N. et al. Harnessing insulin- and leptin-induced oxidation of PTP1B for therapeutic development. Nat. Commun. 9, 283 (2018).
Schmaier, A. A. et al. Tie2 activation protects against prothrombotic endothelial dysfunction in COVID-19. JCI Insight 6, e151527 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04511650 (2020).
Zhang, S. et al. Acquisition of a potent and selective TC-PTP inhibitor via a stepwise fluorophore-tagged combinatorial synthesis and screening strategy. J. Am. Chem. Soc. 131, 13072–13079 (2009).
Loh, K. et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 14, 684–699 (2011).
Dodd, G. T. et al. Insulin regulates POMC neuronal plasticity to control glucose metabolism. eLife 7, e38704 (2018).
Vickers, C. F. et al. Structure-based design of MptpB inhibitors that reduce multidrug-resistant Mycobacterium tuberculosis survival and infection burden in vivo. J. Med. Chem. 61, 8337–8352 (2018).
Salamoun, J. M. et al. Photooxygenation of an amino-thienopyridone yields a more potent PTP4A3 inhibitor. Org. Biomol. Chem. 14, 6398–6402 (2016).
Zhang, Z., Kozlov, G., Chen, Y. S. & Gehring, K. Mechanism of thienopyridone and iminothienopyridinedione inhibition of protein phosphatases. Medchemcomm 10, 791–799 (2019).
McQueeney, K. E. et al. Targeting ovarian cancer and endothelium with an allosteric PTP4A3 phosphatase inhibitor. Oncotarget 9, 8223–8240 (2018).
Kabakci, Z. et al. Pharmacophore-guided discovery of CDC25 inhibitors causing cell cycle arrest and tumor regression. Sci. Rep. 9, 1335 (2019).
Lee, Z. F., Huang, T. H., Chen, S. P. & Cheng, I. H. Altered nociception in Alzheimer disease is associated with striatal-enriched protein tyrosine phosphatase signaling. Pain 162, 1669–1680 (2021).
Chatterjee, M. et al. STEP inhibition prevents Aβ-mediated damage in dendritic complexity and spine density in Alzheimer’s disease. Exp. Brain Res. 239, 881–890 (2021).
Castonguay, D. et al. The tyrosine phosphatase STEP is involved in age-related memory decline. Curr. Biol. 28, 1079–1089 (2018).
Chatterjee, M. et al. STEP inhibition reverses behavioral, electrophysiologic, and synaptic abnormalities in Fmr1 KO mice. Neuropharmacology 128, 43–53 (2018).
Xu, J. et al. Inhibitor of the tyrosine phosphatase STEP reverses cognitive deficits in a mouse model of Alzheimer’s disease. PLoS Biol. 12, e1001923 (2014).
Krueger, A. B. et al. Allosteric inhibitors of the Eya2 phosphatase are selective and inhibit Eya2-mediated cell migration. J. Biol. Chem. 289, 16349–16361 (2014).
Wang, M., Lu, J., Wang, M., Yang, C. Y. & Wang, S. Discovery of SHP2-D26 as a first, potent, and effective PROTAC degrader of SHP2 protein. J. Med. Chem. 63, 7510–7528 (2020).
Tang, K., Jia, Y. N., Yu, B. & Liu, H. M. Medicinal chemistry strategies for the development of protein tyrosine phosphatase SHP2 inhibitors and PROTAC degraders. Eur. J. Med. Chem. 204, 112657 (2020).
Yang, X. et al. Discovery of thalidomide-based PROTAC small molecules as the highly efficient SHP2 degraders. Eur. J. Med. Chem. 218, 113341 (2021).
Salowe, S. P. & Hermes, J. D. Competitive and slow-binding inhibition of calcineurin by drug x immunophilin complexes. Arch. Biochem. Biophys. 355, 165–174 (1998).
Grassberger, M. et al. A novel anti-inflammatory drug, SDZ ASM 981, for the treatment of skin diseases: in vitro pharmacology. Br. J. Dermatol. 141, 264–273 (1999).
Rovin, B. H. et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 397, 2070–2080 (2021).
Zhou, H. et al. Identification of a small-molecule inhibitor that disrupts the SIX1/EYA2 complex, EMT, and metastasis. Cancer Res. 80, 2689–2702 (2020).
Das, I. et al. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348, 239–242 (2015).
Way, S. W. et al. Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat. Commun. 6, 6532 (2015).
Krzyzosiak, A. et al. Target-based discovery of an inhibitor of the regulatory phosphatase PPP1R15B. Cell 174, 1216–1228 (2018).
Neviani, P. et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Invest. 123, 4144–4157 (2013).
Neviani, P. et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J. Clin. Invest. 117, 2408–2421 (2007).
Roberts, K. G. et al. Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+cancers. Cancer Res. 70, 5438–5447 (2010).
Nagaoka, Y., Otsuki, K., Fujita, T. & Uesato, S. Effects of phosphorylation of immunomodulatory agent FTY720 (fingolimod) on antiproliferative activity against breast and colon cancer cells. Biol. Pharm. Bull. 31, 1177–1181 (2008).
De Palma, R. M. et al. The NMR-based characterization of the FTY720-SET complex reveals an alternative mechanism for the attenuation of the inhibitory SET-PP2A interaction. FASEB J. 33, 7647–7666 (2019).
Perrotti, D. & Neviani, P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 14, e229–e238 (2013).
Sangodkar, J. et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Invest. 127, 2081–2090 (2017).
Thura, M. et al. PRL3-zumab as an immunotherapy to inhibit tumors expressing PRL3 oncoprotein. Nat. Commun. 10, 2484 (2019). This publication describes expression of PRL3 on the surface of cancer cells and the anti-tumour efficacy of PRL3-zumab, which targets the cancer cell surface-expressed PRL3.
Thura, M. et al. PRL3-zumab, a first-in-class humanized antibody for cancer therapy. JCI Insight 1, e87607 (2016).
Lang, B. T. et al. Modulation of the proteoglycan receptor PTPsigma promotes recovery after spinal cord injury. Nature 518, 404–408 (2015).
Li, H. et al. Enhanced regeneration and functional recovery after spinal root avulsion by manipulation of the proteoglycan receptor PTPsigma. Sci. Rep. 5, 14923 (2015).
Gardner, R. T. et al. Targeting protein tyrosine phosphatase sigma after myocardial infarction restores cardiac sympathetic innervation and prevents arrhythmias. Nat. Commun. 6, 6235 (2015).
Hardy, S. et al. Physiological and oncogenic roles of the PRL phosphatases. FEBS J. 285, 3886–3908 (2018).
Tasker, N. R. et al. Tapping the therapeutic potential of protein tyrosine phosphatase 4A with small molecule inhibitors. Bioorg. Med. Chem. Lett. 29, 2008–2015 (2019).
Duciel, L., Monraz Gomez, L. C., Kondratova, M., Kuperstein, I. & Saule, S. The phosphatase PRL-3 is involved in key steps of cancer metastasis. J. Mol. Biol. 431, 3056–3067 (2019).
Guo, K., Tang, J. P., Tan, C. P., Wang, H. & Zeng, Q. Monoclonal antibodies target intracellular PRL phosphatases to inhibit cancer metastases in mice. Cancer Biol. Ther. 7, 750–757 (2008).
Park, J. E. et al. Oncogenic roles of PRL-3 in FLT3-ITD induced acute myeloid leukaemia. EMBO Mol. Med. 5, 1351–1366 (2013).
Chuang, H. C. et al. Downregulation of the phosphatase JKAP/DUSP22 in T cells as a potential new biomarker of systemic lupus erythematosus nephritis. Oncotarget 7, 57593–57605 (2016).
Zhou, R. et al. JNK pathway-associated phosphatase/DUSP22 suppresses CD4+ T-cell activation and Th1/Th17-cell differentiation and negatively correlates with clinical activity in inflammatory bowel disease. Front. Immunol. 8, 781 (2017).
Mou, X. Y., Jin, D., Zhang, Q., Guan, J. T. & Jin, Y. JKAP correlates with lower disease risk and inflammation, and its increment during etanercept treatment associates with commendable treatment efficiency in rheumatoid arthritis patients. Eur. Rev. Med. Pharmacol. Sci. 25, 2654–2661 (2021).
Sun, L. et al. JNK pathway-associated phosphatase associates with rheumatoid arthritis risk, disease activity, and its longitudinal elevation relates to etanercept treatment response. J. Clin. Lab. Anal. 35, e23709 (2021).
Zhu, S., Lv, H., Luo, Y., Huang, Q. & Shen, J. JNK pathway-associated phosphatase as a serum marker for disease activity and treatment outcome of juvenile idiopathic arthritis. Tohoku J. Exp. Med. 253, 19–28 (2021).
Li, J. P. et al. The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat. Commun. 5, 3618 (2014).
Tsygankov, A. Y. TULA-family proteins: jacks of many trades and then some. J. Cell Physiol. 234, 274–288 (2018).
Dong, G. et al. STS-1 promotes IFN-α induced autophagy by activating the JAK1-STAT1 signaling pathway in B cells. Eur. J. Immunol. 45, 2377–2388 (2015).
Carpino, N. et al. Regulation of ZAP-70 activation and TCR signaling by two related proteins, Sts-1 and Sts-2. Immunity 20, 37–46 (2004).
Newman, T. N. et al. Members of the novel UBASH3/STS/TULA family of cellular regulators suppress T-cell-driven inflammatory responses in vivo. Immunol. Cell Biol. 92, 837–850 (2014).
Okabe, N. et al. Suppressor of TCR signaling-2 (STS-2) suppresses arthritis development in mice. Mod. Rheumatol. 28, 626–636 (2018).
Yang, C. Y. et al. Dual-specificity phosphatase 14 (DUSP14/MKP6) negatively regulates TCR signaling by inhibiting TAB1 activation. J. Immunol. 192, 1547–1557 (2014).
Hofmann, S. R. et al. cAMP response element modulator α induces dual specificity protein phosphatase 4 to promote effector T cells in juvenile-onset lupus. J. Immunol. 203, 2807–2816 (2019).
Hsiao, W. Y., Lin, Y. C., Liao, F. H., Chan, Y. C. & Huang, C. Y. Dual-specificity phosphatase 4 regulates STAT5 protein stability and helper T cell polarization. PLoS ONE 10, e0145880 (2015).
Barbour, M., Plevin, R. & Jiang, H. R. MAP kinase phosphatase 2 deficient mice develop attenuated experimental autoimmune encephalomyelitis through regulating dendritic cells and T cells. Sci. Rep. 6, 38999 (2016).
Fujimura, A. et al. PTPN3 expressed in activated T lymphocytes is a candidate for a non-antibody-type immune checkpoint inhibitor. Cancer Immunol. Immunother. 68, 1649–1660 (2019).
Stromnes, I. M. et al. Abrogation of SRC homology region 2 domain-containing phosphatase 1 in tumor-specific T cells improves efficacy of adoptive immunotherapy by enhancing the effector function and accumulation of short-lived effector T cells in vivo. J. Immunol. 189, 1812–1825 (2012).
Dan, L. et al. The phosphatase PAC1 acts as a T cell suppressor and attenuates host antitumor immunity. Nat. Immunol. 21, 287–297 (2020).
Guo, K. et al. Engineering the first chimeric antibody in targeting intracellular PRL-3 oncoprotein for cancer therapy in mice. Oncotarget 3, 158–171 (2012).
Vartuli, R. L. et al. Eya3 promotes breast tumor-associated immune suppression via threonine phosphatase-mediated PD-L1 upregulation. J. Clin. Invest. 128, 2535–2550 (2018).
Acknowledgements
The authors were supported by grants R01AI148073, R01HL151306, R01AR066053, R01DK106233, R01HL152717 and R01AI070544 to N.B. and R21CA245621 to S.M.S. from the NIH, and Pathway to Stop Diabetes Grant 1-15-INI-13 to S.M.S. from the American Diabetes Association.
Author information
Authors and Affiliations
Contributions
N.B. and S.M.S. contributed equally to researching data for the article, discussing its content, writing the article and to the review and/or editing of the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
N.B. and S.M.S. have a relationship with Nerio Therapeutics, Inc., which consists of being scientific founders with stock options and acting as consultants with income. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Dimerization model
-
A model that depicts the interaction between SHP2 and PD1, in which SHP2 induces PD1 dimerization through N-SH2 and C-SH2 binding to phosphorylated immunoreceptor tyrosine-based switch motifs on two PD1 molecules.
- Nucleophilic substrate attack during catalysis
-
Covalent bond formation resulting from attack by an enzyme nucleophile on a substrate electrophile. Dephosphorylation by PTPs involves nucleophilic attack on the substrate phosphate by the catalytic Cys or His, forming a phospho-enzyme intermediate and releasing the dephosphorylated substrate. An active-site Asp serves as an acid, donating a proton to the tyrosyl group. The Asp (Glu in some PTPs) acts as a base to deprotonate a water molecule, which acts as a nucleophile to hydrolyse the phospho-enzyme intermediate.
- Orthosteric targeting
-
Development of modulators that target the enzyme active site.
- OT-I T cell
-
CD8+ T cell that carries a transgenic T cell receptor that recognizes ovalbumin peptide residues 257–264.
- OT-I–OVA mouse system
-
The OT-I mouse model bears a transgenic T cell receptor expressed on CD8+ T cells designed to recognize MHC class I-restricted ovalbumin (OVA) peptide residues 257–264. Transplantation of T cells from the OT-I mouse into mice bearing this fragment of OVA results in immune destruction of these cells.
- Two-step activation model
-
A model that depicts the activation of SHP2 by binding to PD1, in which T cell PD1 ligand binding recruits SHP2 to phosphorylated PD1 immunoreceptor tyrosine-based switch motif, and further PD1 phosphorylation on the immunoreceptor tyrosine-based inhibitory motif is needed to unfold SHP2 into its active conformation and propagate inhibitory PD1 signalling.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Stanford, S.M., Bottini, N. Targeting protein phosphatases in cancer immunotherapy and autoimmune disorders. Nat Rev Drug Discov 22, 273–294 (2023). https://doi.org/10.1038/s41573-022-00618-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-022-00618-w
This article is cited by
-
An atlas of cell-type-specific interactome networks across 44 human tumor types
Genome Medicine (2024)
-
The phosphatase inhibitor LB-100 creates neoantigens in colon cancer cells through perturbation of mRNA splicing
EMBO Reports (2024)
-
INPP5A phosphatase is a synthetic lethal target in GNAQ and GNA11-mutant melanomas
Nature Cancer (2024)
