
Abstract
Diverse inflammatory diseases, infections and malignancies are associated with wasting syndromes. In many of these conditions, the standards for diagnosis and treatment are lacking due to our limited understanding of the causative molecular mechanisms. Here, we discuss the complex immunological context of cachexia, a systemic catabolic syndrome that depletes both fat and muscle mass with profound consequences for patient prognosis. We highlight the main cytokine and immune cell-driven pathways that have been linked to weight loss and tissue wasting in the context of cancer-associated and infection-associated cachexia. Moreover, we discuss the potential immunometabolic consequences of cachexia on the basis of newly identified pathways and explore the multilayered area of immunometabolic crosstalk both upstream and downstream of tissue catabolism. Collectively, this Review highlights the intricate relationship of the immune system with cachexia in the context of malignant and infectious diseases.
Similar content being viewed by others
Introduction
Unintentional excessive weight loss has been documented in association with chronic illness throughout medical history. Dating back to ancient Greece (460–377 BC), Hippocrates wrote “the flesh is consumed and becomes water … the abdomen fills with water; the feet and legs swell, the shoulders, clavicles, chest and thighs melt away”1. This description contains most of the main elements by which cachexia was defined in the early medical records2,3. Cachexia typically manifests itself alongside chronic inflammatory illnesses, various aggressive cancers (such as gastrointestinal and lung cancers) and chronic infections, including HIV infection and Mycobacterium tuberculosis infection4,5,6. The severe emaciation resulting from cachexia not only has significant mental health implications but can eventually reach a debilitating state that renders the patient incapable of fulfilling basic daily needs. Depleted muscle mass and strength may result in cardiac arrhythmias, respiratory weakness and other complications that lead to premature death4. The latest international consensus defined cancer-associated cachexia (CAC) as “a multifactorial syndrome characterized by an ongoing loss of skeletal muscle mass (with or without loss of fat mass) that cannot be fully reversed by conventional nutritional support and is driven by a variable combination of reduced food intake and abnormal metabolism”7. This definition is relevant for both CAC and infection-associated cachexia (IAC) and provides a clear framework representing the most consistent features of cachexia from a clinical and metabolic perspective. Notably, this definition does not address the immunological aspects of the syndrome, likely due to the limited data that are currently available in this area. However, while we still do not fully understand why cachexia occurs, it is clear that the onset of cachexia is driven by the inflammatory programme triggered by the underlying illness. In this Review, we focus on the immunological aspects of cachexia within cancer and infection to better understand the commonalities and distinctions within the elements involved in the cachexia programme. Additionally, we will discuss the immunometabolic consequences of cachexia by highlighting the pathways through which immune cell activation can influence and be influenced by altered metabolic environments.
Cytokines and cachexia
Much of the immunological data available from patients with cachexia are derived from measurements of circulating cytokines and acute-phase proteins. Tumour necrosis factor (TNF), IL-1β, IL-6 and interferon-γ (IFNγ) are among the most consistently upregulated cytokines in cachexia4. These cytokines are abundantly expressed by both immune and non-immune cell types downstream of pattern-recognition receptor activation by pathogen-associated molecular patterns and/or damage-associated molecular patterns8. This leads to activation of JAK–STAT and NF-κB signalling pathways and downstream transcriptional regulation that can induce various catabolic pathways in muscles and adipose tissue9,10. More studies are required to clarify the precise roles of pattern-recognition receptor activation in cachexia-inducing diseases; in the following sections we discuss our current understanding of cytokines in cachexia associated with cancer or infection.
Classical cachexia-associated cytokines
The link between immunology and cachexia first became a point of interest after the isolation of TNF11, which was known as ‘cachectin’ at that time12. In 1985, Beutler et al. showed that adipocytes exposed to TNF suppress their expression of lipoprotein lipase (LPL), thereby reducing lipid uptake11. This built on the observations made by Rouzer and Cerami 5 years earlier, which showed that rabbits infected with Trypanosoma brucie brucie exhibit hypertriglyceridaemia as a result of reduced triglyceride clearance from the circulation13. These animals were also emaciated, a phenotype that was later shown in murine models to be a manifestation of cachexia14. Subsequent studies in adipocyte cultures showed that TNF promotes adipose triglyceride lipase (ATGL)-mediated lipolysis by depleting the ATGL-inhibitory protein G0/G1 switch protein 2 (G0S2)15. In myoblast cultures, exposure to TNF, IL-1β, IL-6 and IFNγ inhibits myoblast differentiation and induces protein loss through STAT3-mediated NF-κB activation16,17,18,19,20. These results were further validated in mouse models, where mice into which tumour cells overexpressing TNF, IL-1α, IL-6 or IFNγ had been implanted exhibited severe weight loss, adipose tissue depletion and muscle atrophy21,22,23,24 (Fig. 1a). In models where tumour cell lines overexpressing IL-6 or IFNγ were injected into mice, blockade of IL-6 or IFNγ, respectively, ameliorated the cachectic phenotype. Notably, the overexpression of IL-1α and IL-6 in these models also reduced overall survival and led to more aggressive tumour progression22,23. In a different approach, mice into which pancreatic tumour cells lacking IL-6 expression had been injected showed reduced loss of fat tissue and protection against muscle atrophy25. Further analysis of adipose tissue and muscles, both in vivo and in vitro, revealed a catabolic feedforward loop of IL-6 signalling between the tumour, the adipose tissue and the muscles25.

a | Tumorigenesis is associated with the release of a wide range of cytokines by tumour cells, by the surrounding tissue and from innate and adaptive immune cells downstream of pattern-recognition receptor (PRR) activation by tumour cell-associated damage-associated molecular patterns. Tumour necrosis factor (TNF), interferon-γ (IFNγ), IL-6 and IL-1β are able to induce tissue catabolism by modulating gene-expression profiles in both adipose tissue and muscle cells. IL-1β also contributes to cachexia through the central nervous system (CNS), where it modulates food intake and activates the hypothalamic–pituitary–adrenal (HPA) axis. The subsequent release of glucocorticoids contributes to tissue catabolism. GDF15 release induces weight loss through effects on the CNS that modulate food intake and by increasing adipose tissue lipolysis through sympathetic nervous system (SNS) signalling. IL-20 also contributes to adipose tissue depletion, while IL-4 shows protective effects on muscle cells. Transforming growth factor-β (TGFβ) released from tumour cells and/or bone (during bone metastasis-induced osteoclastic resorption) can induce adipose tissue fibrosis and compromise muscle strength. b | During parasitic infection, TNF and IL-1 release downstream of PRRs supports the production of IFNγ, which is important for controlling the pathogen load. IL-1 is also involved in the catabolism of adipose tissues and muscles. During viral infection, type I interferon (IFN) signalling to CD8+ T cells during antigen recognition and T cell activation is an important step in triggering cachexia.
In addition to promoting catabolism through NF-κB activation, cytokines can promote cachexia through NF-κB-independent mechanisms. For instance, IL-1β signalling in the hypothalamus can trigger the activation of the hypothalamic–pituitary–adrenal axis, which ramps up glucocorticoid production, resulting in catabolic effects on both muscle tissue and adipose tissue26,27 (Fig. 1a). Administration of IL-1β through intracerebroventricular injection was sufficient to elicit muscle atrophy. This was associated with increased expression of factors involved in muscle catabolism, such as muscle atrophy F-box protein (MAFBX; also known as FBXO32 and atrogin 1), MURF1 (also known as E3 ubiquitin-protein ligase TRIM63) and forkhead box protein O1 (FOXO1), reduced muscle mass and reduced muscle fibre cross-sectional area27 (Fig. 2a). Intraperitoneal administration of IL-1β did not induce muscle loss27, suggesting that IL-1β acts predominantly by directly stimulating the hypothalamic–pituitary–adrenal axis. Of note, tissue mass comparison with a pair-fed group demonstrated that the loss of fat mass subsequent to IL-1β administration was coupled to food intake, whereas the loss of muscle mass occurred independently of food intake27.

a | Cytokines induce muscle catabolism by regulating the transcription of various genes. NF-κB signalling suppresses Myod1 expression, which leads to inhibition of myoblast differentiation. Forkhead box protein O (FOXO) activation results in its translocation to the nucleus, where it upregulates the expression of genes involved in proteasomal degradation (for example, Murf1 and Mafbx) and autophagy (for example, Becn1 and Map1lc3b2, which encode beclin 1 and LC3B, respectively). Transforming growth factor-β (TGFβ)–SMAD signalling increases Nox4 expression, and NADPH oxidase 4 (NOX4) oxidizes and destabilizes the RYR1 calcium channel, leading to calcium leakage and compromised muscle contraction. b | Adipose tissue depletion is induced through the binding of catecholamines to β-adrenergic receptors, which activates cAMP phosphorylation of protein kinase A (PKA) downstream of GNAS and adenylyl cyclase. PKA is then able to phosphorylate adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL), resulting in their translocation to the surface of lipid droplets, where lipolysis occurs. ATGL-mediated lipolysis is modulated through its interaction with G0/G1 switch protein 2 (G0S2) and CGI58. Cytokine signalling in the adipose tissue induces transcriptional changes downstream of NF-κB and STAT signalling, resulting in an increase in the levels of lipolytic enzymes and a suppression of genes involved in lipogenesis. Perilipin 1 (PLIN1; encoded by Plin1) coats the surface of the lipid droplet and is also transcriptionally suppressed during lipolysis to expose a wider surface area to lipase activity. IFNγ, interferon-γ; IFNγR, interferon-γ receptor; IL-6R, IL-6 receptor; NEFA, non-esterified fatty acid; p-HSL, phosphorylated HSL; p-SMAD, phosphorylated SMAD; SR, sarcoplasmic reticulum; TG, triacylglycerol; TGFβR, TGFβ receptor; TNF, tumour necrosis factor; TNFR, tumour necrosis factor receptor.
In summary, these data show that pro-inflammatory cytokines such as TNF, IL-1, IL-6 and IFNγ are able to activate diverse catabolic processes across multiple organs in experimental models of CAC (Figs 1,2). However, targeting these cytokines in a clinical setting has shown underwhelming results. Over the years, a number of cytokine inhibitors, mainly those targeting TNF, have been tested in patients with cancer who were undergoing cachexia. Most of these treatments showed no significant increase in body weight, strength or survival28,29. In two small randomized controlled trials, patients with CAC were treated with thalidomide for its wide immunomodulatory functions and its ability to inhibit cytokines, including TNF30,31. In these trials, thalidomide treatment resulted in a small increase of body weight and muscle mass, with no increase in hand-grip strength or survival30,31. This highlights the possibility that cytokines are not singularly sufficient to induce cachexia, but can be elements of a more complex coordinated immune and metabolic response leading to cachexia.
Mechanistic data examining the role of cytokines during IAC are scarce, and available experimental models come with their own intrinsic limitations (Box 1). Mice chronically infected with Toxoplasma gondii, an obligatory intracellular parasite, develop cachexia as evident by their severe weight loss and depletion of fat and lean mass32. This occurs within the first week of infection and coincides with elevated serum levels of cytokines, including TNF, IL-1β, IL-6 and IFNγ32. In this context, TNF and IL-1β support the production of IFNγ, which is essential in controlling T. gondii infection and limiting parasite replication33,34 (Fig. 1b). Notably, animals deficient in TNF or IFNγ show accelerated death and an increase in parasite burdens35,36,37. On the other hand, studies that inhibit IL-1 signalling through genetic disruption of the IL-1 receptor (IL-1R) pathway have reported conflicting results. For instance, one study showed that disruption of IL-1 signalling through the abrogation of inflammasome activation in mice (using animals that lacked caspase 1 and caspase 11, NLRP3, NLRP1 or IL-1R) led to increased parasite burdens and reduced survival38. Meanwhile, a more recent study found that, compared with wild-type mice, IL-1R-deficient mice show increased survival, reduced weight loss and increased lean and fat mass following T. gondii infection39. These effects were independent of pathogen clearance, brain inflammation, immune cell infiltration and anorexia. Notably, IL-1β signalling through the gut–brain signalling axis has been associated with the induction of anorexia in other models of infection40 (Box 2).
In a model of IAC that involves chronic lymphocytic choriomeningitis virus (LCMV) infection with LCMV strain clone 13 (LCMV-Cl13) (Box 1), the depletion of TNF, IFNγ or both cytokines together had no effect on weight loss even though the levels of both cytokines were increased in the serum during infection41. By contrast, abrogation of type I interferon signalling ameliorated weight loss in this model41. Importantly, type I interferons are the first line of innate immune defence against viral infections and exert a wide range of influences on both immunity and metabolism42,43.
Other cytokines associated with cachexia
In the preceding section, we discussed some of the key inflammatory cytokines that have been associated with cachexia. However, in both cancer and infection the concentration levels of numerous other cytokines are increased in both the circulation and within local tissue environments. Many of these cytokines have not been systematically probed for their role in the pathophysiology of cachexia. In the following subsections we discuss a few additional examples of cytokines that have been implicated in the development of cachexia.
Il-20
IL-20 is a pro-inflammatory cytokine produced mainly by haematopoietic cells, facilitating their communication with epithelial cells. It has an important role in enhancing innate defence mechanisms and tissue repair processes at epithelial surfaces44. IL-20 is highly expressed in the tumours of patients with pancreatic cancer. In mouse models of pancreatic ductal adenocarcinoma (PDAC), levels of IL-20 correlate with increased tumour fibrosis, PDL1 expression and an overall reduction in survival45. In the cachectic mouse models of PDAC and Lewis lung carcinoma (LLC), anti-IL-20 treatment ameliorated body weight loss and prevented loss of adipose tissue mass by decreasing ATGL and hormone-sensitive lipase (HSL) expression. However, this treatment had no effect on muscle atrophy, suggesting that the procachectic function of IL-20 is linked to its influence on the adipose tissue45 (Fig. 1a).
IL-4
IL-4 is produced by both innate and adaptive immune cells during type 2 immune responses46. A recent study demonstrated a protective role for IL-4 in the C26 colon cancer model47. Daily administration of IL-4 to C26 tumour-bearing mice prevented excessive weight loss, improved physical performance and muscle protein synthesis and regeneration, and increased survival47. Moreover, IL-4 administration increased tumour necrosis and promoted CD8+ T cell and macrophage infiltration into tumours. In the context of muscle injury induced by cardiotoxin administration, IL-4 secretion activates STAT6 signalling in fibroadipogenic progenitor (FAP) cells. This leads to their differentiation into fibroblast cells that are capable of phagocytosing cellular debris, thereby promoting muscle regeneration48,49. However, FAPs may also differentiate into adipocytes and, in this context, can be detrimental to muscle growth50. For instance, glycerol-mediated muscle damage results in adipogenic differentiation of FAPs, a process that is also inhibited by administration of IL-4 (ref.48). These data opens up an interesting avenue for investigations both into the fate of FAPs during different models of CAC and for understanding the potential protective effect of IL-4 administration in CAC (Fig. 1a). Similarly to IL-4, other cytokines with anabolic effects on muscles warrant further study. This includes IL-15, which is known to induce myogenesis and mitigate the catabolic effect of TNF in cultured myotubes51.
TGFβ family cytokines
Transforming growth factor-β (TGFβ) signalling is involved in multiple aspects of cancer, including tumour suppression, cell-cycle arrest, modulation of the tumour microenvironment and tumour metastasis52. In the context of cachexia, biopsies of tumours and adipose tissue from patients with cancer with cachexia showed an association between high levels of TGFβ and tissue fibrosis that is not seen in weight-stable patients with cancer53,54. In mouse models of bone metastasis, bone-derived TGFβ directly affected muscle strength and mediated the phosphorylation of SMAD signalling factors. This resulted in increased levels of NADPH oxidase 4 (NOX4) and the oxidation of the Ca2+ sarcoplasmic reticulum channel RYR1, causing Ca2+ leakage and compromised muscle contraction55 (Figs 1a,2a).
GDF15 is another member of the TGFβ family and was initially described as macrophage inhibitory cytokine 1 (MIC1). It modulates energy homeostasis during inflammation, ageing and nutritional imbalance56,57,58. GDF15 modulates those functions by binding to its receptor GDNF family receptor α-like (GFRAL), which is exclusively expressed by neuronal cells within the area postrema and nucleus tractus solitarius in the brainstem59. GDF15 is ubiquitously expressed and found at relatively high basal levels in the circulation. Its link to cachexia is derived from its high circulating levels in patients with cancer with cachexia compared with patients without cachexia with cancer56 (Fig. 1a). Additionally, mice into which recombinant GDF15 had been injected or GDF15-overexpressing tumours had been implanted exhibited a marked increase in weight loss secondary to reduced food intake and increased sympathetic stimulation of adipose tissue lipolysis through ATGL60,61,62.
In summary, accumulating evidence suggests that various cytokines are induced in a context-dependent manner in cachexia and may contribute to its development. More research is needed to understand the mechanistic roles of both pro-inflammatory and anti-inflammatory cytokines in the pathophysiology of cachexia. In the following section we consider how different types of immune cells may influence the development of cachexia.
Immune cells in cachexia
In response to tumorigenesis or pathogenic infections, the immune system launches cellular responses that are specifically tailored to each illness with various degrees of overlap63,64. Although data on the link between immune cell function and cachexia are currently limited, a number of recent studies indicate the involvement of multiple immune cell types, including macrophages, neutrophils, myeloid-derived suppressor cells (MDSCs) and T cells (Fig. 3).

A schematic summary of the immune cell types that correlate with cachexia in the setting of cancer or infection is shown. The mechanisms by which immune cells influence cachexia are not yet clear, and correlations do not necessarily imply causality between immune cell function and cachexia due to the entanglement of direct and/or indirect effects of the underlying disease and the cachectic processes. a | Experimental models of cancer-associated cachexia (CAC) demonstrate that an increased number of microglial cells in the brain has a protective effect against severe cachexia, potentially owing to microglial cell-mediated depletion of neutrophils, as high neutrophil numbers correlate with a higher degree of cachexia. b,c | In adipose tissue, macrophages protect against lipolysis, whereas in muscle tissue, macrophage numbers correlate with a higher degree of atrophy, and this is potentially affected by the macrophage polarization. In muscle, increased numbers of CD8+ T cells correlate negatively with cachexia, whereas the numbers of CD4+ T cells show both positive and negative correlations with cachexia, which is likely dependant on the specific subtype of T cell being measured in the population. d | The numbers of myeloid-derived suppressor cells (MDSCs) in the tumour, bone marrow and spleen positively correlate with cachexia. e | In parasite infection-associated cachexia, the numbers of CD4+FOXP3+ T cells negatively correlate with cachexia. f | During viral infection, antigen-specific CD8+ T cell responses show procachectic effects. Red and blue circles beside cell populations indicate procachetic or anticachectic effects of that population, respectively. IAC, infection-associated cachexia.
Innate immune cells
Studies using the PDAC mouse model of cachexia demonstrated a significant increase in myeloid immune cell infiltration into the brain65,66. These infiltrates targeted specific brain regions that influence feeding behaviour and/or energy metabolism, such as the hypothalamus. This region is particularly sensitive to inflammation and is highly responsive to changes in the systemic nutritional state, making it a likely target of immune-mediated metabolic regulation during cachexia67,68. In this tumour model, an increased numbers of microglial cells in the hypothalamus was found to be associated with increased IL-1β and arginase 1 secretion66. Reducing microglial cell numbers through treatment with a colony-stimulating factor 1 receptor (CSF1R) inhibitor exacerbated the cachectic phenotype, affecting food intake, activity and muscle atrophy, with no measured effects on tumour size66. This suggests a protective role for microglial cells in mitigating cachexia during pancreatic tumour development. Interestingly, microglial cells were shown to phagocytose infiltrating CC-chemokine receptor 2 (CCR2)-positive neutrophils in the central nervous system parenchyma65. In the absence of CCR2, tumour-bearing mice exhibited reduced anorexia and muscle atrophy that correlated with reduced levels of neutrophil infiltrates. Hence, it has been speculated that phagocytosis of neutrophils by microglial cells could provide a degree of protection against cachexia (Fig. 3a).
A similar protective effect of macrophages in cachexia was observed in adipose tissue in a mouse model of hepatocellular carcinoma69. In that study, mice with a myeloid-specific deficiency in hypoxia-inducible factor 1α (HIF1α) — which show defective myeloid cell activation — had increased adipose tissue depletion during genetically induced hepatocellular carcinoma, and this phenotype was coupled to reduced macrophage infiltration into adipose tissue. Importantly, this defective myeloid cell activity had no influence on muscles, liver tissue or tumour size. By contrast, macrophages seemed to contribute to the tissue catabolism observed in cachexia associated with the mouse PDAC and LLC models45,70. In both models the adipose tissue-protective effect observed following anti-IL-20 treatment (discussed earlier) was associated with reduced macrophage infiltration in adipose tissue45. Moreover, the depletion of macrophages in the PDAC model using clodronate-filled liposomes resulted in reduced tumour sizes and increased body weight, muscle mass and strength coupled with a reduction in the levels of markers of proteasomal degradation. This apparent discrepancy could potentially be explained by differences in macrophage polarization within each tissue environment (Fig. 3b,c).
Macrophages are well known for their roles in adipose tissue inflammation and metabolism during ageing and obesity71, and the nature of their interaction with the tissue is highly influenced by their polarization. In the context of obesity, there is a shift in the abundance of ‘M2-like’ macrophages to ‘M1-like’ macrophages72. In this setting, macrophages sequester noradrenaline from adipocytes by increasing noradrenaline import and degradation, which results in downregulated lipolysis and adipose tissue enlargement73,74. On the other hand, M2-like macrophages promoted lipolysis in a mouse model of cold-induced thermogenesis via the production of noradrenaline downstream of IL-4 stimulation75. Muscle biopsy samples taken from patients with pancreatic cancer showed an inverse correlation between the density of CD163+ M2-like macrophages and muscle fibre in cross-sectional areas70. In line with this, myotubes (derived from the C2C12 cell line) co-cultured with M2-polarized macrophages and exposed to tumour-conditioned medium showed a reduction in myotube thickness and protein content and an increase in the levels of proteasomal degradation markers.
In a mouse model of peritonitis-induced sepsis with concomitant loss of adipose tissue and muscle mass, single-cell RNA sequencing analysis revealed that macrophage populations increase in muscles and decrease in adipose tissue 1 day after intraperitoneal injection of faecal slurry76. This was coupled with an increase in the levels of muscle-infiltrating neutrophils, natural killer cells and T cells that remained elevated in both the muscle and the adipose tissue 1 month after injection. Although the transcriptional profiles of these cell populations showed, as expected, a role of pathways involved in infection and in the tissue damage response, further analysis focused on metabolic regulation could reveal valuable information with regard to tissue catabolism.
MDSCs constitute a heterogeneous population of myeloid cells with immunosuppressive functions77. They are found in large numbers in the tumours of patients with gastric and pancreatic cancers78,79 and in several mouse models of CAC, such as 4T1 mammary carcinoma, C26 colon adenocarcinoma and LLC80. Levels of MDSCs in the spleen and bone marrow correlate with total body weight loss, adipose tissue depletion and increased oxygen consumption rate78,80. However, the exact mechanism by which MDSC population expansion could contribute to cachexia is still unknown (Fig. 3d), and the currently available models for MDSC depletion, such as clodronate-mediated liposomal depletion, are not specific to MDSCs but also eliminate macrophages and other myeloid cells80.
Adaptive immune cells
The role of T cells has been extensively studied in the context of antitumour and infectious immunology, but whether and how they influence systemic metabolic reprogramming in the context of cachexia remains elusive. Muscle biopsies of patients with different cancers, most of whom had gastrointestinal cancers, showed a positive correlation between the abundance of CD3+CD4− cells (presumably CD8+ T cells) and muscle fibre cross-sectional area81. In a separate cohort of patients with cancer who showed early signs of muscle impairment, circulating levels of recent thymic migrant and effector memory CD8+ T cells were associated with increased muscle mass82. Meanwhile, the abundance of regulatory T (Treg) cells and central memory T cells showed a negative correlation with muscle mass. These studies suggest that an efficient and robust antitumour CD8+ T cell response can protect against muscle catabolism during CAC, while immune suppression may have the opposite effect (Fig. 3). The reason for this protection is unclear as there is no evidence so far that uncouples the antitumour activity of the T cells from their direct catabolic effect on muscles. T cell-mediated tumour clearance may well be the predominant mechanism preventing muscle atrophy. However, another and not mutually exclusive mechanism may be that the immune cell composition within the local tissue environment alters the functionality and secretory profile of each cell type, with consequences for muscle atrophy. An indication of this could be derived from measurements of circulating immune cells, where the abundance of immune cell populations in relation to one another may also have an impact on the prognosis of patients with cancer. A high ratio of the number of circulating neutrophils to the number of circulating lymphocytes, for instance, correlates with the development of cachexia. The reason for this is unknown, and ongoing efforts aim to better understand the role of neutrophils within the circulation and the tumour microenvironement83,84.
In the mouse model of LLC, increasing the numbers of CD4+CD44+ T cells in the spleen and lymph nodes through the adoptive transfer of these cells delayed the onset of cachexia and prevented muscle loss85. The adoptive transfer of CD4+FOXP3+ Treg cells to mice infected with T. gondii reduced weight loss and prolonged survival, highlighting the regulatory function of CD4+ T cells as a potential mechanism for reducing cachexia during parasitic infection86 (Fig. 3). By contrast, in the chronic LCMV-Cl13 infection model, loss of CD4+ T cells had no effect on the initiation of cachexia, while the activation of antigen-specific CD8+ T cells was essential to trigger IAC41 (Fig. 3f). These differences could be related to the different types of inflammatory milieus, kinetics and immune responses that are associated with tumours, parasitic infections and viral infections. More studies are required to systematically investigate and compare how T cells affect the course of cachexia in both IAC and CAC (Box 1).
Interestingly, IAC during LCMV infection was both viral dose and strain dependent, which may be linked to differences in the associated T cell responses41. Infection with LCMV-Cl13 (which causes a chronic viral infection) but not with LCMV strain Armstrong (LCMV-Arm; which causes an acute infection) results in cachexia and induces T cell exhaustion, which is characterized by low levels of T cell proliferation, reduced cytokine potential and high expression of inhibitory receptors, including PD1, CTLA4 and LAG3 (ref.87). The exhausted phenotype is established early on during T cell priming and is seen in chronic infections and other chronic diseases such as cancer88,89. The expression of inhibitory receptors such as PD1 and CTLA4 has been shown to alter the metabolic reliance of CD8+ T cells by promoting oxidative phosphorylation and suppressing glycolysis90. This raises the question of whether the T cell exhaustion programme may be connected to the systemic immunometabolic changes preceding the development of cachexia. Likewise, cachexia-associated metabolic changes may also impact the differentiation of exhausted T cells.
In the LCMV-Cl13 IAC model, the muscle microenvironment is depleted of certain cytokines, such as IL-12, IFNβ and IFNγ, which provides an environment for CD8+ cells to thrive91. These cells exhibited a lower degree of exhaustion in the muscle compared with that seen in the splenic compartment, and had a higher proliferative and cytokine-production capacity. The reduced exhaustion phenotype is facilitated by IL-15 within the muscle microenvironment91. It is unclear how these immunologically distinct zones are formed and how they interplay with muscle atrophy at this stage of infection. However, one may speculate that these functional CD8+ T cells may directly take up muscle-derived amino acids to support their cytokine production and/or proliferation. In a different model of LCMV infection using LCMV strain Traub, infection of IFNγ-deficient mice, but not wild-type mice, resulted in severe weight loss that was CD8+ T cell dependent92. Although it remains unclear whether the weight loss induced by LCMV-Traub infection is a manifestation of cachexia or anorexia (Box 2), it will be of interest to reconcile this observation with recent results relating to the IFNγ-shielded muscle microenvironment in the LCMV-Cl13 model91. In yet another variation of the LCMV model, the intracranial injection of LCMV-Arm in mice lacking MHC class I molecules results in a chronic weight loss attributed solely to anorexia93.
Overall, the current evidence indicates that activation of immune cells can have both anticachectic and procachectic effects, and aspects other than immune cell population expansion and cytokine production can be involved. Other pertinent questions that require investigation include the pathophysiological role of cachexia itself in regulating processes relevant to infection, tumorigenesis and immune regulation, and the pathological consequences that come with chronic non-resolving cachexia (Box 3).
Tissue catabolism downstream of immune responses
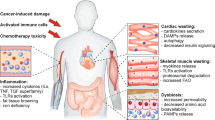
As highlighted by the consensus definition7, the key pathophysiological characteristic of cachexia is the severe weight loss that occurs as a result of muscle atrophy and adipose tissue depletion. Muscle atrophy is one of the most extensively studied aspects of cachexia as it is a main contributor to functional impairment and death94. It is identified by a reduction of overall muscle mass, reduced muscle fibre cross-sectional area and weakened grip strength in both humans and mice. There are three main cellular processes that facilitate the depletion of skeletal and cardiac muscle mass downstream of FOXO and NF-κB signalling9, namely proteasomal degradation (measured through markers such as MURF1 and MAFBX)95, autophagy (measured by expression of beclin 1 and LC3B)96,97 and reduced myocyte regeneration (measured by myoblast determination protein 1 (MYOD1) expression)98 (Fig. 2a). Skeletal muscle atrophy can lead to severe functional impairment with a reduced quality of life, and cardiac atrophy increases the likelihood of life-threatening cardiac arrhythmias99. Moreover, the compromised cardiac function results in elevated heart rate and oxygen consumption, which further increases energy expenditure100.
During cachexia, adipose tissue undergoes depletion of its lipid droplets as a consequence of increased lipolysis coupled with a suppression of lipid uptake and lipogenesis101,102. Adipose tissue wasting can be evaluated by measuring adipose tissue mass, adipocyte diameter to evaluate the size of the lipid droplets and circulating levels of non-esterified fatty acids (NEFAs) and glycerol as a result of triglyceride breakdown41,69,101. These changes in adipose tissue metabolism occur either through transcriptional modulation or via ATGL-mediated and HSL-mediated lipolysis101 (Fig. 2b).
More studies are needed to understand how the local and infiltrating immune cell populations influence tissue metabolism and vice versa. Moreover, the altered circulatory environment that results from cachexia and the underlying pathology could indirectly affect the local tissue environment through central regulatory pathways, for instance by modulating sympathetic neuronal signalling. Finally, it is well known that immunological and metabolic processes converge on overlapping signalling cascades63, highlighting yet another crucial area to probe the immunological mechanisms of cachexia. In the following section we consider potential immunometabolic consequences of cachexia.
Immunometabolic consequences of cachexia
The onset of cachexia and the subsequent depletion of the energetic tissue reserves enrich the circulation with metabolic substrates that are likely to impact systemic metabolism, inflammation and immune cell activation103,104. Using current knowledge derived from the field of immunometabolism, we discuss potential aspects of immunometabolic crosstalk during cachexia with a focus on fatty acids and amino acids as the main products of adipose tissue and muscle catabolism, respectively.
Fatty acids and cachexia
During cachexia, excessive adipose tissue lipolysis increases circulating levels of NEFAs and glycerol, which are then taken up and utilized by other organs105,106. In mouse models of cachexia, metabolic cage measurements of the respiratory exchange ratio of the animal indicate a shift in the metabolic reliance from carbohydrates towards fat utilization41,107,108. From the data available thus far, it is not possible to deduce whether this shift in metabolism is reactionary and occurs as result of increased exposure of cells to lipid substrates or whether it serves as a controlled step in an adaptive programme. Potentially both of these scenarios could be true depending on the cell type in question.
NEFAs can be categorized on the basis of their degree of saturation and their fatty acid chain length, both of which are properties that influence their activity and immunomodulatory function. In vitro treatment of macrophages or primary Kupffer cells with palmitate, a saturated fatty acid, shifted the cells to an M1-like phenotype engaging PPARγ–ΝF-κB signalling and increasing pro-inflammatory cytokine production109. On the other hand, treatment with polyunsaturated fatty acids promoted an M2-like phenotype, and the cells no longer increased TNF and IL-6 production in response to saturated NEFA treatment110. Along similar lines, intravenous infusion of long-chain saturated fatty acids in wild-type mice increased TNF levels in the circulation and led to the accumulation of macrophages and dendritic cells in the liver111. This suggests that the composition of fatty acids released during cachexia-induced adipose tissue lipolysis could have a significant role in altering the state of innate immune cell activity, which could have profound effects on the immunopathology associated with chronic illnesses.
NEFA-mediated modulation of cell function extends beyond immune cells to parenchymal cells, such as adipocytes and hepatocytes. In vitro studies demonstrated that treating adipocyte and hepatocyte cell lines with saturated fatty acids activates Toll-like receptor 4 (TLR4) signalling, which results in NF-κB activation and increased TNF and IL-6 production110. This TLR4-mediated NF-κB activation was also seen in the adipose tissue of mice into which fatty acids had been infused and was abrogated in TLR4-deficient mice. TLR4 activation of the NF-κB pathway has been shown to play a major role in the development of cachexia in the LLC model. In this model, genetic or pharmacological blockade of TLR4 signalling protected tumour-bearing mice from weight loss and reduced adipose tissue and muscle depletion112,113. Interestingly, the protective effect seen in the adipose tissue was mainly localized to the subcutaneous fat pad. This is likely due to the abrogated adipose tissue browning seen in TLR4-deficient mice112. In the muscle tissue, TLR4-mediated NF-κB activation resulted in a direct modulation of pathways involved in muscle degradation, inducing proteasomal degradation and autophagy113. This suggests that adipose tissue lipolysis and NEFA release may further aggravate tissue catabolism through the activation of TLR4.
Aside from NEFAs acting as upstream signalling modulators, there are various examples of other factors shifting cellular metabolism towards fat utilization. As discussed before, PD1 signalling in human CD4+ T cells shifts their metabolic reliance towards fatty acid oxidation and inhibits glucose and glutamine metabolism90. This contributes to limiting effector T cell function and promoting an ‘exhausted’ phenotype. During chronic LCMV-Cl13 infection, CD8+ T cells show impaired energy production and cell proliferation 8 days after infection, whereas CD8+ T cells primed during LCMV-Arm infection are able to engage in glycolysis and successfully reach maximal population expansion114. In both chronic viral infection and tumour models, CD8+ T cells exhibit mitochondrial damage, as reflected by decreased oxygen consumption rates, decreased mitochondrial membrane potential and increased production of mitochondrial reactive oxygen species114. The combination of anti-PD1 and bezafibrate (a mitochondrial activator) increased antitumour immunity in a mouse model of colon cancer by increasing proliferation of CD8+ T cells and their tumour infiltration115. This treatment also promoted glycolysis and fatty acid oxidation. Systematic profiling of T cell function upon inhibition of lipolysis is expected to offer valuable insights into the directionality of the relationship between high NEFA levels, T cell exhaustion and cachexia.
Overall, research in the area of immunometabolism indicates that a local and systemic high lipid content can directly influence immune cell function, as either an energy substrate or a biosynthetic substrate. Alternatively, metabolites derived from fatty acid metabolism, such as arachidonic acid and docosahexaenoic acid, can act as signalling molecules by binding to G-protein-coupled receptors and/or through the regulation of transcription factors116,117. However, there is little evidence so far indicating whether lipids derived from wasted adipose tissue may exert such effects on immune activation during cachexia.
Amino acids and cachexia
Alterations in amino acid profiles have been observed in the tumour microenvironment and in the circulation of cachexia-inducing tumours118,119,120. For example, the tumours of patients with colon and stomach cancers were enriched in several amino acids, including serine, tryptophan, arginine and glutamate118. In the case of pancreatic tumours, the amino acid profile and abundance seem to depend on the tumour stage. Tumour samples collected from patients with PDAC showed low abundance of most amino acids evaluated, with the exception of taurine, arginine and hydroxyproline120. Several other studies reported high abundance of amino acids in the tumour and low abundance in the serum of patients with gastric cancer119. Patients with colorectal cancer have high circulating concentrations of phenylalanine and low concentrations of glutamine and histidine, which correlates with systemic inflammation and poor prognosis121. Such varied concentrations of circulating amino acids could result from the rate of amino acid release or could be due to amino acid uptake by tumour or immune cells, such as is the case for glutamine119,121.
Immune cells and tumour cells compete for available amino acids to support their high anabolic demands122, which modulates the activation status of immune cells. For instance, tumour cells take up arginine and glutamine to support their growth, thereby limiting the availability of these amino acids for T cells118,123. Blocking glutamine uptake using an antagonist that targets the tumour microenvironment impairs tumour growth and metabolism while enabling a robust activation of antitumour T cell responses124.
T cell priming and IL-2-mediated activation induces the expression of amino acid transporters, including SLC7A5, which mediates the import of large neutral amino acids such as phenylalanine and leucine125. Lack of SLC7A5 impairs CD4+ effector T cell and CD8+ effector T cell proliferation and differentiation, but does not affect the development of CD4+ Treg cells125. This is thought to occur as a result of disrupted mTORC1 activity and therefore the failure of mTOR to tune T cell responses to nutrient availability. The uptake of other amino acids, such as serine, glutamine and arginine, is important for T cell receptor signalling and T cell proliferation and differentiation126,127,128,129,130. The availability of arginine for T cells within the tumour microenvironment is limited as a result of its uptake by MDSCs131. Decreased circulating arginine levels are commonly observed during many virus infections, including LCMV and SARS-CoV-2 infection, leading to reduced CD8+ T cell responses and reduced tissue damage132,133. These and other studies showcase the effect of amino acid availability on T cell function. It will be interesting to examine the bidirectional nature of this relationship in animal models, for instance by altering the T cell activation state and examining how the amino acid profile is affected. This could be particularly relevant following anti-PD1 treatment to counter T cell exhaustion.
In summary, cachexia-associated muscle depletion releases a wide range of amino acids into the circulation. Comprehensive profiling and dissection of systemic and local amino acid levels in different models of cachexia, as well as in patients with cachexia, will contribute to a better appreciation of their pathophysiological effect on inflammation, metabolism and disease outcomes.
Concluding remarks
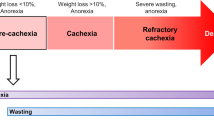
Recent years have seen a substantial improvement in our understanding of cachexia. Yet, when viewing cachexia in the light of metabolic adaptation, the available evidence remains insufficient to definitively classify it as a maladaptive syndrome. The cachexia-associated complications that could result in fatality occur at progressive stages of disease, which renders it difficult to disentangle what occurs as a result of cachexia as opposed to the underlying illness. Moreover, cachexia occurs in different stages4,134, termed ‘precachexia’, ‘cachexia’ and ‘refractory cachexia’. From an evolutionary perspective135, it is tempting to speculate that these stages could reflect the adaptive states of cachexia as the interplay between various trade-off mechanisms and immune responses attempting to resolve the underlying illness.
Many of the models used in the study of cachexia are also employed to examine immune cell activation and function relevant to the underlying disease. However, there is currently an information gap that disconnects what we know about the immune aspects and the metabolic aspects of these diseases. Notably, many valuable patient studies examining immune cell infiltration within the muscles and adipose tissue of patients with cachexia are limited by the variability within their selected cohorts, including different tumour types at variable stages of muscle impairment and cachexia. Such factors can influence the antitumour immune response as well as the metabolic environment in which this response occurs. Considering the difficulty of acquiring biopsy samples from patients with cachexia, such studies could benefit from new technologies that are capable of more thoroughly characterizing immune cells within a single sample. This could be achieved by single-cell RNA sequencing, which allows an unbiased characterization of the immune cell repertoire, and spatial metabolomics using imaging mass spectrometry136.
In addition to the importance of cachexia in the context of disease and patient prognosis, cachexia provides a rich arena for immunological research that is full of unanswered questions. For instance, the role of tissue-resident immune cells in the development of cachexia and how these populations adapt to the altered nutrient release and inflammatory signals within their local tissue environment are largely unknown. These bidirectional immune system–tissue interactions create continuous feedback loops that allow the surveillance and modulation of supply and demand. The energy and substrate supply is derived from a finite pool that must be monitored. Supplier cells (for example, adipocytes) require the ability to modulate the level of demand by influencing the molecular and cellular pathways generating the demand137. Moreover, the supply sources (lipids, carbohydrates and proteins) are not entirely interchangeable as they produce a specific repertoire of breakdown products. This results in substrate competition within the tissue microenvironment, and systemic energy redistribution138. Finally, the immune responses combating the underlying illness can result in collateral damage that must be regulated to avoid excessive tissue damage139. In this case, regulation of energy and substrate availability may provide an efficient means for immune modulation. Additionally, the differential energy and substrate pool within the circulation and in the tissue microenvironment provides compartmentalized immune niches such as that seen for T cell populations in muscle tissue91.
Overall, future studies of cachexia will benefit from a more rigorous integration of immunology and the use of tools and knowledge from systems biology and evolutionary medicine140. Moreover, it will be critical to pursue strategies that bridge the, at times, apparent divide between experimental animal models of cachexia and patient studies and realize the great synergistic potential of such complementary approaches. Immune cell functionality is an understudied area of cachexia research and likely a missing link that will support the development of effective therapeutic strategies against cachexia (Box 3). Studying immune cell function in relation to the metabolic environment induced by cachexia will also enhance our general understanding of the metabolic regulation of immunity independently of cachexia (Box 3). This and various other questions require a more holistic and interconnected understanding of the immunology of cachexia (Box 3).
References
Katz, A. M. & Katz, P. B. Diseases of the heart in the works of Hippocrates. Br. Heart J. 24, 257–264 (1962).
Slane, J. Malarial cachexia in the Garo Hills. Ind. Med. Gaz. 12, 123–124 (1877).
Buchanan, W. J. Case of general tuberculosis simulating malarial cachexia. Ind. Med. Gaz. 33, 139–140 (1898).
Baracos, V. E., Martin, L., Korc, M., Guttridge, D. C. & Fearon, K. C. H. H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 4, 1–18 (2018).
Keithley, J. K. & Swanson, B. HIV-associated wasting. J. Assoc. Nurses AIDS Care 24, S103–S111 (2013).
Cheung, W. W., Paik, K. H. & Mak, R. H. Inflammation and cachexia in chronic kidney disease. Pediatr. Nephrol. 25, 711–724 (2010).
Fearon, K. et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495 (2011). This consensus process set a benchmark definition for CAC, and provided guidelines for the diagnosis and classification of cachexia in patients with cancer.
Bianchi, M. E. DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5 (2007).
Guttridge, D. C., Mayo, M. W., Madrid, L. V., Wang, C. Y. & Baldwin, J. NF-κB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2365 (2000).
Fearon, K. C. H., Glass, D. J. & Guttridge, D. C. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 16, 153–166 (2012).
Beutler, B., Mahoney, J., Le Trang, N., Pekala, P. & Cerami, A. Purification of cachectin, a lipoprotein lipase-suppressing hormone secreted by endotoxin-induced raw 264.7 cells. J. Exp. Med. 161, 984–995 (1985).
Beutler, B. et al. Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature 316, 552–554 (1985). Both studies by Beutler et al. (1985) were among the first to identify TNF as a mediator of wasting in chronic disease.
Rouzer, C. A. & Cerami, A. Hypertriglyceridemia associated with Trypanosoma brucei brucei infection in rabbits: role of defective triglyceride removal. Mol. Biochem. Parasitol. 2, 31–38 (1980).
Truyens, C. et al. The endogenous balance of soluble tumor necrosis factor receptors and tumor necrosis factor modulates cachexia and mortality in mice acutely infected with Trypanosoma cruzi. Inf. Immun. 67, 5579–5586 (1999).
Yang, X., Zhang, X., Heckmann, B. L., Lu, X. & Liu, J. Relative contribution of adipose triglyceride lipase and hormone-sensitive lipase to tumor necrosis factor-α (TNF-α)-induced lipolysis in adipocytes. J. Biol. Chem. 286, 40477–40485 (2011).
Trendelenburg, A. U., Meyer, A., Jacobi, C., Feige, J. N. & Glass, D. J. TAK-1/p38/nNFκB signaling inhibits myoblast differentiation by increasing levels of activin A. Skelet. Muscle 2, 3 (2012).
Li, Y. P. & Reid, M. B. NF-κB mediates the protein loss induced by TNF-α in differentiated skeletal muscle myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1165–R1170 (2000).
Zhao, Q. et al. TNF alpha inhibits myogenic differentiation of C2C12 cells through NF-κB activation and impairment of IGF-1 signaling pathway. Biochem. Biophys. Res. Commun. 458, 790–795 (2015).
Fong, Y. et al. Cachectin/TNF or IL-1α induced cachexia with redistribution of body proteins. Am. J. Physiol. Regul. Integr. Comp. Physiol. 256, R659–65 (1989).
Ma, J. F. et al. STAT 3 promotes IFN γ/TNF α-induced muscle wasting in an NF-κB-dependent and IL-6-independent manner. EMBO Mol. Med. 9, 622–637 (2017).
Oliff, A. et al. Tumors secreting human TNF/cachectin induce cachexia in mice. Cell 50, 555–563 (1987).
Kumar, S. et al. Interleukin-1α promotes tumor growth and cachexia in MCF-7 xenograft model of breast cancer. Am. J. Pathol. 163, 2531–2541 (2003).
Miller, A. et al. Blockade of the IL-6 trans-signalling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma. Oncogene 36, 3059–3066 (2017).
Matthys, P. et al. Severe cachexia in mice inoculated with interferon-γ-producing tumor cells. Int. J. Cancer 49, 77–82 (1991).
Rupert, J. E. et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J. Exp. Med. 218, e20190450 (2021).
Katsuura, G., Gottschall, P. E., Dahl, R. R. & Arimura, A. Adrenocorticotropin release induced by intracerebroventricular injection of recombinant human interleukin-1 in rats: possible involvement of prostaglandin. Endocrinology 122, 1773–1779 (1988).
Braun, T. P. et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis. J. Exp. Med. 208, 2449–2463 (2011).
Roeland, E. J. et al. Management of cancer cachexia: ASCO guideline. J. Clin. Oncol. 38, 2438–2453 (2020).
Advani, S. M., Advani, P. G., Vonville, H. M. & Jafri, S. H. Pharmacological management of cachexia in adult cancer patients: a systematic review of clinical trials. BMC Cancer 18, 1174 (2018).
Gordon, J. N. et al. Thalidomide in the treatment of cancer cachexia: a randomised placebo controlled trial. Gut 54, 540–545 (2005).
Yennurajalingam, S. et al. The role of thalidomide and placebo for the treatment of cancer-related anorexia-cachexia symptoms: results of a double-blind placebo-controlled randomized study. J. Palliat. Med. 15, 1059–1064 (2012).
Hatter, J. A. et al. Toxoplasma gondii infection triggers chronic cachexia and sustained commensal dysbiosis in mice. PLoS ONE 13, e0204895 (2018). This study characterizes the pathophysiological changes in adipose tissue and muscle following T. gondii infection, thereby consolidating its value as a model for sustained cachexia.
Hunter, C. A. & Sibley, L. D. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10, 766–778 (2012).
Yarovinsky, F. Innate immunity to Toxoplasma gondii infection. Nat. Rev. Immunol. 14, 109–121 (2014).
Sturge, C. R. et al. TLR-independent neutrophil-derived IFN-γ is important for host resistance to intracellular pathogens. Proc. Natl Acad. Sci. USA 110, 10711–10716 (2013).
Suzuki, Y., Orellana, M. A., Schreiber, R. D. & Remington, J. S. Interferon-γ: the major mediator of resistance against Toxoplasma gondii. Science 240, 516–518 (1988).
Schlüter, D. et al. Both lymphotoxin-α and TNF are crucial for control of toxoplasma gondii in the central nervous system. J. Immunol. 170, 6172–6182 (2003).
Gorfu, G. et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. mBio 5, e01117-13 (2014).
Melchor, S. J. et al. IL-1R regulates disease tolerance and cachexia in toxoplasma gondii infection. J. Immunol. 204, 3329–3338 (2020).
Rao, S. et al. Pathogen-mediated inhibition of anorexia promotes host survival and transmission. Cell 168, 503–516.e12 (2017). This study using a mouse model of Salmonella enterica subsp. enterica serovar Typhimurium infection highlights how pathogens can actively suppress sickness-induced anorexia to allow transmission through faecal shedding.
Baazim, H. et al. CD8+ T cells induce cachexia during chronic viral infection. Nat. Immunol. 20, 701–710 (2019). This study establishes chronic LCMV infection as a model for infection-associated cachexia and identifies antigen-specific CD8+ T cells as a trigger for cachexia.
McNab, F., Mayer-Barber, K., Sher, A., Wack, A. & O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103 (2015).
Fritsch, S. D. & Weichhart, T. Effects of interferons and viruses on metabolism. Front. Immunol. 7, 1–13 (2016).
Rutz, S., Wang, X. & Ouyang, W. The IL-20 subfamily of cytokines-from host defence to tissue homeostasis. Nat. Rev. Immunol. 14, 783–795 (2014).
Lu, S. W. et al. IL-20 antagonist suppresses PD-L1 expression and prolongs survival in pancreatic cancer models. Nat. Commun. 11, 1–13 (2020).
Ho, I. C. & Miaw, S. C. Regulation of IL-4 expression in immunity and diseases. Adv. Exp. Med. Biol. 941, 31–77 (2016).
Costamagna, D. et al. Interleukin-4 administration improves muscle function, adult myogenesis, and lifespan of colon carcinoma-bearing mice. J. Cachexia Sarcopenia Muscle 11, 783–801 (2020). This study shows for the first time a connection between IL-4 administration and increased body weight and muscle strength during CAC.
Heredia, J. E. et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388 (2013).
Meng, J. et al. Accelerated regeneration of the skeletal muscle in RNF13-knockout mice is mediated by macrophage-secreted IL-4/IL-6. Protein Cell 5, 235–247 (2014).
Joe, A. W. B. et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153–163 (2010).
O’Leary, M. F., Wallace, G. R., Bennett, A. J., Tsintzas, K. & Jones, S. W. IL-15 promotes human myogenesis and mitigates the detrimental effects of TNFα on myotube development. Sci. Rep. 7, 12997 (2017).
Colak, S. & ten Dijke, P. Targeting TGF-β signaling in cancer. Trends Cancer 3, 56–71 (2017).
Lima, J. D. C. C. et al. Tumour-derived transforming growth factor-β signalling contributes to fibrosis in patients with cancer cachexia. J. Cachexia Sarcopenia Muscle 10, 1045–1059 (2019).
Alves, M. J. et al. Adipose tissue fibrosis in human cancer cachexia: the role of TGFβ pathway. BMC Cancer 17, 190 (2017).
Waning, D. L. et al. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 21, 1262–1271 (2015). This study identifies a mechanism by which TGFβ released as a consequence of bone metastasis induces Ca2+ leakage from the sarcoplasmic reticulum and compromises muscle contraction.
Tsai, V. W. W., Husaini, Y., Sainsbury, A., Brown, D. A. & Breit, S. N. The MIC-1/GDF15-GFRAL pathway in energy homeostasis: implications for obesity, cachexia, and other associated diseases. Cell Metab. 28, 353–368 (2018).
Patel, S. et al. GDF15 provides an endocrine signal of nutritional stress in mice and humans. Cell Metab. 29, 707–718.e8 (2019).
Wiklund, F. E. et al. Macrophage inhibitory cytokine-1 (MIC-1/GDF15): a new marker of all-cause mortality. Aging Cell 9, 1057–1064 (2010).
Hsu, J. Y. et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 550, 255–259 (2017).
Johnen, H. et al. Tumor-induced anorexia and weight loss are mediated by the TGF-β superfamily cytokine MIC-1. Nat. Med. 13, 1333–1340 (2007).
Suriben, R. et al. Antibody-mediated inhibition of GDF15–GFRAL activity reverses cancer cachexia in mice. Nat. Med. 26, 1264–1270 (2020).
Macia, L. et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) decreases food intake, body weight and improves glucose tolerance in mice on normal & obesogenic diets. PLoS ONE 7, e34868 (2012).
Hotamisligil, G. S. Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 (2017).
Kotas, M. E. & Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827 (2015).
Burfeind, K. G. et al. Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancer. eLife 9, 1–27 (2020).
Burfeind, K. G. et al. Microglia in the hypothalamus respond to tumor-derived factors and are protective against cachexia during pancreatic cancer. Glia 68, 1479–1494 (2020). This study characterizes myeloid cell infiltration in the central nervous system and the protective effects of microglial cells against cachexia.
Burfeind, K. G., Michaelis, K. A. & Marks, D. L. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin. Cell Dev. Biol. 54, 42–52 (2016).
Geller, S. et al. Tanycytes regulate lipid homeostasis by sensing free fatty acids and signaling to key hypothalamic neuronal populations via FGF21 secretion. Cell Metab. 30, 833–844.e7 (2019).
Erdem, M. et al. Macrophages protect against loss of adipose tissue during cancer cachexia. J. Cachexia Sarcopenia Muscle 10, 1128–1142 (2019).
Shukla, S. K. et al. Macrophages potentiate STAT3 signaling in skeletal muscles and regulate pancreatic cancer cachexia. Cancer Lett. 484, 29–39 (2020).
Schäffler, A. & Schölmerich, J. Innate immunity and adipose tissue biology. Trends Immunol. 31, 228–235 (2010).
McNelis, J. C. & Olefsky, J. M. Macrophages, immunity, and metabolic disease. Immunity 41, 36–48 (2014).
Pirzgalska, R. M. et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat. Med. 23, 1309–1318 (2017).
Camell, C. D. et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550, 119–123 (2017).
Nguyen, K. D. et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480, 104–108 (2011).
Cho, D. S., Schmitt, R. E., Dasgupta, A., Ducharme, A. M. & Doles, J. D. Single-cell deconstruction of post-sepsis skeletal muscle and adipose tissue microenvironments. J. Cachexia Sarcopenia Muscle 11, 1351–1363 (2020).
Gabrilovich, D. I. & Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174 (2009).
Khaled, Y. S., Ammori, B. J. & Elkord, E. Increased levels of granulocytic myeloid-derived suppressor cells in peripheral blood and tumour tissue of pancreatic cancer patients. J. Immunol. Res. 2014, 879897 (2014).
Ohki, S. et al. Circulating myeloid-derived suppressor cells are increased and correlate to immune suppression, inflammation and hypoproteinemia in patients with cancer. Oncol. Rep. 28, 453–458 (2012).
Cuenca, A. G. A. L. et al. Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J. Immunol. 192, 6111–6119 (2014).
Anoveros-Barrera, A. et al. Immunohistochemical phenotyping of T cells, granulocytes, and phagocytes in the muscle of cancer patients: association with radiologically defined muscle mass and gene expression. Skelet. Muscle 9, 1–13 (2019).
Narsale, A. et al. Cancer-driven changes link T cell frequency to muscle strength in people with cancer: a pilot study. J. Cachexia Sarcopenia Muscle 10, 827–843 (2019).
Barker, T., Fulde, G., Moulton, B., Nadauld, L. D. & Rhodes, T. An elevated neutrophil-to-lymphocyte ratio associates with weight loss and cachexia in cancer. Sci. Rep. 10, 1–6 (2020).
Szczerba, B. M. et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 566, 553–557 (2019).
Wang, Z., Zhao, C., Moya, R. & Davies, J. D. A novel role for CD4+ T cells in the control of cachexia. J. Immunol. 181, 4676–4684 (2008).
Olguín, J. E. et al. Adoptive transfer of CD4+ Foxp3+ regulatory T cells to C57BL/6J mice during acute infection with Toxoplasma gondii down modulates the exacerbated Th1 immune response. Microbes Infect. 17, 586–595 (2015).
Wherry, E. J. & Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015).
Sullivan, B. M., Teijaro, J. R., De La Torre, J. C. & Oldstone, M. B. A. Early virus-host interactions dictate the course of a persistent infection. PLoS Pathog. 11, 1004588 (2015).
McLane, L. M., Abdel-Hakeem, M. S. & Wherry, E. J. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37, 457–495 (2019).
Patsoukis, N. et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6, 6692 (2015).
Wu, J. et al. Skeletal muscle antagonizes antiviral CD8+ T cell exhaustion. Sci. Adv. 6, eaba3458 (2020).
Nansen, A. et al. Compromised virus control and augmented perforin-mediated immunopathology in IFN-gamma-deficient mice infected with lymphocytic choriomeningitis virus. J. Immunol. 163, 6114–6122 (1999).
Hildeman, D. & Muller, D. Immunopathologic weight loss in intracranial LCMV infection initiated by the anorexigenic effects of IL-1β. Viral Immunol. 13, 273–285 (2000).
Cohen, S., Nathan, J. A. & Goldberg, A. L. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 14, 58–74 (2014).
Sandri, M. Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 54, 11–19 (2016).
Aversa, Z. et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 6, 1–11 (2016).
Penna, F. et al. Autophagy exacerbates muscle wasting in cancer cachexia and impairs mitochondrial function. J. Mol. Biol. 431, 2674–2686 (2019).
Bossola, M., Marzetti, E., Rosa, F. & Pacelli, F. Skeletal muscle regeneration in cancer cachexia. Clin. Exp. Pharmacol. Physiol. 43, 522–527 (2016).
Murphy, K. T. The pathogenesis and treatment of cardiac atrophy in cancer cachexia. Am. J. Physiol. Heart Circ. Physiol. 310, H466–H477 (2016).
Hyltander, A., Drott, C., Körner, U., Sandström, R. & Lundholm, K. Elevated energy expenditure in cancer patients with solid tumours. Eur. J. Cancer Clin. Oncol. 27, 9–15 (1991).
Zechner, R. et al. FAT SIGNALS — lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 15, 279–291 (2012).
Sun, X. et al. Fat wasting is damaging: role of adipose tissue in cancer-associated cachexia. Front. Cell Dev. Biol. 8, 33 (2020).
Rohm, M., Zeigerer, A., Machado, J. & Herzig, S. Energy metabolism in cachexia. EMBO Rep. 20, 1–13 (2019).
Lercher, A., Baazim, H. & Bergthaler, A. Systemic immunometabolism: challenges and opportunities. Immunity 53, 496–509 (2020).
Liu, K. J. M., Drucker, Y. & Jarad, J. Hepatic glycerol metabolism in tumorous rats: a 13C nuclear magnetic resonance study. Cancer Res. 55, 761–766 (1995).
Fukawa, T. et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 22, 666–671 (2016).
Rohm, M. et al. An AMP-activated protein kinase-stabilizing peptide ameliorates adipose tissue wasting in cancer cachexia in mice. Nat. Med. 22, 1120–1130 (2016).
Petruzzelli, M. et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 20, 433–447 (2014).
Luo, W., Xu, Q., Wang, Q., Wu, H. & Hua, J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 7, 44612 (2017).
Shi, H. et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 116, 3015–3025 (2006).
Harmon, D. B. et al. Adipose tissue-derived free fatty acids initiate myeloid cell accumulation in mouse liver in states of lipid oversupply. Am. J. Physiol. Endocrinol. Metab. 315, E758–E770 (2018).
Henriques, F. et al. Toll-like receptor-4 disruption suppresses adipose tissue remodeling and increases survival in cancer cachexia syndrome. Sci. Rep. 8, 1–14 (2018).
Zhang, G. et al. Toll-like receptor 4 mediates Lewis lung carcinoma-induced muscle wasting via coordinate activation of protein degradation pathways. Sci. Rep. 7, 1–8 (2017).
Bengsch, B. et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 45, 358–373 (2016). This study demonstrates how metabolic changes in the early phase of chronic viral infection control the exhaustion of CD8+ T cells.
Chowdhury, P. S., Chamoto, K., Kumar, A. & Honjo, T. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+T cells and facilitates anti–PD-1 therapy. Cancer Immunol. Res. 6, 1375–1387 (2018).
Chiurchiù, V. et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci. Transl Med. 8, 353ra111 (2016).
Harizi, H., Corcuff, J. B. & Gualde, N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol. Med. 14, 461–469 (2008).
Hirayama, A. et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 69, 4918–4925 (2009).
Huang, S. et al. A systematic review of metabolomic profiling of gastric cancer and esophageal cancer. Cancer Biol. Med. 17, 181–198 (2020).
Hiraoka, N. et al. Tissue amino acid profiles are characteristic of tumor type, malignant phenotype, and tumor progression in pancreatic tumors. Sci. Rep. 9, 9816 (2019).
Sirniö, P. et al. Alterations in serum amino-acid profile in the progression of colorectal cancer: associations with systemic inflammation, tumour stage and patient survival. Br. J. Cancer 120, 238–246 (2019).
Lemos, H., Huang, L., Prendergast, G. C. & Mellor, A. L. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat. Rev. Cancer 19, 162–175 (2019).
Czystowska-Kuzmicz, M. et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 10, 3000 (2019).
Leone, R. D. et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019).
Sinclair, L. V. et al. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 14, 500–508 (2013).
Ma, E. H. et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 25, 345–357 (2017).
Klysz, D. et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 8, ra97 (2015).
Wang, K. et al. Glutamine supplementation suppresses herpes simplex virus reactivation. J. Clin. Invest. 127, 2626–2630 (2017).
Rodriguez, P. C. et al. Regulation of T cell receptor CD3ζ chain expression by l-arginine. J. Biol. Chem. 277, 21123–21129 (2002).
Rodriguez, P. C., Quiceno, D. G. & Ochoa, A. C. l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 109, 1568–1573 (2007).
Fletcher, M. et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 75, 275–283 (2015).
Shen, B. et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 182, 59–72.e15 (2020).
Lercher, A. et al. Type I interferon signaling disrupts the hepatic urea cycle and alters systemic metabolism to suppress T cell function. Immunity 51, 1074–1087.e9 (2019).
Tito Fojo, A. W. Lo Price, value, and the cost of cancer drugs. Lancet Oncol. 17, 927–960 (2010).
Wang, A., Luan, H. H. & Medzhitov, R. An evolutionary perspective on immunometabolism. Science 363, eaar3932 (2019).
Alexandrov, T. Spatial metabolomics and imaging mass spectrometry in the age of artificial intelligence. Annu. Rev. Biomed. Data Sci. 3, 61–87 (2020).
Kahn, C. R., Wang, G. & Lee, K. Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Invest. 129, 3990–4000 (2019).
Goodpaster, B. H. & Sparks, L. M. Metabolic flexibility in health and disease. Cell Metab. 25, 1027–1036 (2017).
McCarville, J. L. & Ayres, J. S. Disease tolerance: concept and mechanisms. Curr. Opin. Immunol. 50, 88–93 (2018).
Gluckman, P., Beedle, A., Buklijas, T., Low, F. & Hanson, M. Principles of Evolutionary Medicine (Oxford Univ. Press, 2016).
Penna, F., Busquets, S. & Argilés, J. M. Experimental cancer cachexia: evolving strategies for getting closer to the human scenario. Semin. Cell Dev.Biol. 54, 20–27 (2016).
Corbett, T. H., Griswold, D. P., Roberts, B. J., Peckham, J. C. & Schabel, F. M. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 35, 2434–2439 (1975).
Matsuyama, T. et al. Tumor inoculation site affects the development of cancer cachexia and muscle wasting. Int. J. Cancer 137, 2558–2565 (2015).
Ito, S. et al. Enhanced cancer metastasis in mice deficient in vasohibin-1 gene. PLoS ONE 8, 73931 (2013).
Bernardo, B. et al. Characterization of cachexia in the human fibrosarcoma HT-1080 mouse tumour model. J. Cachexia Sarcopenia Muscle 11, 1813–1829 (2020).
Hingorani, S. R. et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 (2005).
Baltgalvis, K. A. et al. Interleukin-6 and cachexia in ApcMin/+ mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, 2705–2720 (2008).
Talbert, E. E. et al. Modeling human cancer-induced cachexia. Cell Rep. 28, 1612–1622.e4 (2019).
Teijaro, J. R. et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013).
Althaus, C. L., Ganusov, V. V. & De Boer, R. J. Dynamics of CD8+ T cell responses during acute and chronic lymphocytic choriomeningitis virus infection. J. Immunol. 179, 2944–2951 (2007).
Rouse, B.T. & Sehrawat, S. Immunity and immunopathology to viruses: what decides the outcome? Nat. Rev. Immunol. 10, 514–526 (2010).
Kelley, K. W. et al. Cytokine-induced sickness behavior. Brain Behav. Immun. 17, 112–118 (2003).
Filiano, A. J. et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 535, 425–429 (2016).
Dantzer, R., O’Connor, J. C., Freund, G. G., Johnson, R. W. & Kelley, K. W. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56 (2008).
Wang, A. et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell 166, 1512–1525.e12 (2016). This study highlights how the host mounts pathogen-specific metabolic adaptations.
Kir, S. et al. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab. 23, 315–323 (2016).
Butler, M. J., Perrini, A. A. & Eckel, L. A. The role of the gut microbiome, immunity, and neuroinflammation in the pathophysiology of eating disorders. Nutrients 13, 1–19 (2021).
Belmonte, L. et al. A role for intestinal TLR4-driven inflammatory response during activity-based anorexia. Sci. Rep. 6, 35813 (2016).
Acknowledgements
This work was supported by the European Research Council under the European Union’s Seventh Framework Programme and Horizon 2020 research and innovation programme (grant agreement no. 677006, ‘CMIL’, to A.B.).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Myeloid-derived suppressor cells
-
(MDSCs). A heterogeneous population of immune cells that develop from myeloid lineage cells. MDSCs are expanded in chronic infection and cancer and show immunosuppressive activity.
- ‘M1-like’ macrophages
-
‘M1’ and ‘M2’ are classifications historically used to define macrophages activated in vitro as pro-inflammatory (when ‘classically’ activated with interferon-γ (IFNγ) and lipopolysaccharide) or anti-inflammatory (when ‘alternatively’ activated with IL-4 or IL-10), respectively. However, in vivo macrophages are highly specialized, transcriptomically dynamic and extremely heterogeneous with regard to their phenotypes and functions, which are continuously shaped by their tissue microenvironment. Therefore, the M1 or M2 classification is too simplistic to explain the true nature of in vivo macrophages, although these terms are still often used to indicate whether the macrophages in question are more pro-inflammatory or anti-inflammatory.
Rights and permissions
About this article

Cite this article
Baazim, H., Antonio-Herrera, L. & Bergthaler, A. The interplay of immunology and cachexia in infection and cancer. Nat Rev Immunol 22, 309–321 (2022). https://doi.org/10.1038/s41577-021-00624-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-021-00624-w
This article is cited by
-
Extracellular vesicles in cancer cachexia: deciphering pathogenic roles and exploring therapeutic horizons
Journal of Translational Medicine (2024)
-
Exosomes in the pathogenesis and treatment of cancer-related cachexia
Journal of Translational Medicine (2024)
-
Impact on survival of sarcopenia, systemic inflammatory response and anthropometric factors after pancreatectomy for resectable pancreatic adenocarcinoma
World Journal of Surgical Oncology (2024)
-
Leukemia inhibitory factor suppresses hepatic de novo lipogenesis and induces cachexia in mice
Nature Communications (2024)
-
Mycobacterium tuberculosis cough aerosol culture status associates with host characteristics and inflammatory profiles
Nature Communications (2024)
