
Abstract
Understanding how human genetics influence infectious disease susceptibility offers the opportunity for new insights into pathogenesis, potential drug targets, risk stratification, response to therapy and vaccination. As new infectious diseases continue to emerge, together with growing levels of antimicrobial resistance and an increasing awareness of substantial differences between populations in genetic associations, the need for such work is expanding. In this Review, we illustrate how our understanding of the host–pathogen relationship is advancing through holistic approaches, describing current strategies to investigate the role of host genetic variation in established and emerging infections, including COVID-19, the need for wider application to diverse global populations mirroring the burden of disease, the impact of pathogen and vector genetic diversity and a broad array of immune and inflammation phenotypes that can be mapped as traits in health and disease. Insights from study of inborn errors of immunity and multi-omics profiling together with developments in analytical methods are further advancing our knowledge of this important area.
Similar content being viewed by others

Introduction
Disease syndromes caused by infectious agents have occurred throughout the history of modern humans1. As a result of our continued interactions with pathogens, our genomes have been shaped through processes of co-evolution, with pathogen-imposed selection pressures leading to selection signatures in ancient and modern human genomes2. As one of several illustrative examples, genetic diversity involving human red blood cell structure and function is being impacted by an evolutionary arms race with malaria that is reciprocally seen in the parasite genome3. In the era of modern medicine with antimicrobial drugs and powerful organ support systems, we are curbing the selection pressure exerted through the traditional high mortality associated with many of these infectious agents in resource-rich settings, but the worldwide health-care burden of infectious diseases remains substantial4. This is exemplified by the emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)5,6 and the rapid evolution into a global COVID-19 pandemic requiring coordinated global responses. Additionally, the ongoing and dynamic challenges of managing malaria, tuberculosis (TB) and human immunodeficiency virus (HIV) infection alongside the constant threat of global influenza and other outbreaks such as Ebola virus disease, all particularly afflicting underserved populations, remind us of the importance of advancing infectious disease medicine.
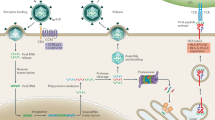
A seminal study of adoptees in the 1980s reported increased risk of death from infectious disease in children whose biological parents succumbed to an infectious disease7, highlighting the significance of human genetic background in susceptibility to infections. As genetic technology has developed, mirroring other disease syndromes, a raft of genetic loci influencing susceptibility to infectious diseases have been discovered through genome-wide association studies (GWAS), including for HIV infection, TB, hepatitis and malaria8 (Table 1). Such studies aim to identify common variants on the basis of a polygenic model of complex multifactorial traits. However, it has become clear that, although human genetics play a role in disease susceptibility, in contrast to other syndromes, GWAS of case–control design may not be the most powerful method to tease out complex host–pathogen relationships. Alongside developments in genotyping technologies enabling GWAS, the in-depth understanding of single-gene disorders facilitated by next-generation sequencing technologies has enabled us to better understand the functional mechanisms of host defence to infection through experiments of nature9 resulting in loss of function (LOF) or, in increasingly recognized instances, gain of function (GOF) of discrete genes (Fig. 1). Here, rare mutations of variable penetrance result in single-gene inborn errors of immunity and lead to life-threatening infectious disease (Table 2). More recently, the development and availability of multi-omics experimental techniques promise to deliver similar mechanistic understanding of the functional importance of genome-level variation at genomic loci implicated in disease pathogenesis.

Examples of specific proteins are shown (highlighted in colour), which when present as mutants give rise to monogenic inborn errors of immunity, with the main infectious disease phenotypes noted. a | Pattern recognition receptors (PRRs) responsible for detecting pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) with examples of PRR pathways illustrated for retinoic acid-inducible gene I protein (RIG-I)-like receptor (RLR) and Toll-like receptor (TLR). b | Receptor-interacting protein kinase (RIPK) signalling. RIPK1 and RIPK3 regulate inflammation and cell death (necroptosis). c | Interferon pathways. Type I interferons (for example, interferon-α (IFNα) and IFNβ) and type II interferons (for example, IFNγ) regulate the immune response to viral and bacterial infections. d | Antigen presentation pathways. Major histocompatibility (MHC) class I molecules present antigens (derived from intracellular proteins such as viruses and some bacteria) to cytotoxic CD8+ T cells via the endogenous pathway (left). MHC class II molecules present antigens from bacteria, parasites and other extracellular pathogens endocytosed into antigen-presenting cells to CD4+ T cells via the exogenous pathway (right). CARD, caspase activation and recruitment domain; CLIP, class II-associated invariant chain peptide; CMV, cytomegalovirus; CTD, carboxy-terminal domain; ERK, extracellular signal-regulated kinase; GAS, gamma-activated sequence; HSV, herpes simplex virus; IFNAR, interferon-α/β receptor; IKK, IκB kinase; IRAK, interleukin-1 receptor-associated kinase; IRF, interferon response factor; ISGs, interferon-stimulated genes; ISRE, interferon-stimulated response element; JNK, JUN amino-terminal kinase; MAPK, mitogen-activated protein kinase; MAPKKK, mitogen-activated protein kinase kinase kinase; NF-κB, nuclear factor-κB; TAB, transforming growth factor-β-activated kinase 1 (MAP3K7)-binding protein; TAK, transforming growth factor-β-activated kinase; TAP, transporter associated with antigen processing; TCR, T cell receptor; TIRAP, Toll–interleukin-1 receptor domain-containing adaptor protein; TRAFs, tumour necrosis factor receptor-associated factors; TRAM, Toll–interleukin-1 receptor domain-containing adapter-inducing interferon-β (TRIF)-related adaptor molecule; TRIF, Toll–interleukin-1 receptor domain-containing adapter-inducing interferon-β.
This Review aims to summarize the insights gained from the wide range of -omics approaches being used to understand genetic susceptibility to infectious disease. We begin by discussing recent findings and limitations of GWAS applied to infectious diseases, in particular the need for application in diverse global populations. We focus on lessons learnt from different infections and on a region of the genome consistently linked to infectious disease susceptibility: the major histocompatibility (MHC) locus encoding the vital immune human leukocyte antigen (HLA) genes (Box 1). We then proceed to discuss novel approaches, many of which involve large-scale sequencing, and highlight the importance of monogenic architecture leading to infectious disease susceptibility, although only selected examples will be covered as this topic was reviewed comprehensively recently9. This is followed by exploration of the potential of future integrated analyses of human–pathogen variation and application of multi-omics platforms. We postulate how these findings may inform our understanding of the genetic determinants of infectious disease susceptibility, how this knowledge may in turn improve our understanding of individual immune and vaccine responses, and how this might be directed more universally for the benefit of human health. Finally, we discuss strategic approaches to dissecting host genetic factors in COVID-19 as a case study in how such work is being implemented as part of global efforts to address this emerging public health threat.
Common-variant associations: HLA and beyond
For major infectious diseases, including HIV-1 infection, TB, malaria, leprosy and viral hepatitis, genetic evidence has been accrued primarily through GWAS, aiming to identify disease-associated common variants in populations on the basis of a polygenic model of complex multifactorial traits. Recent work involving these traits has highlighted the progress and challenges that continue to be associated with mapping and functionally interrogating genetic associations through GWAS.
Genetic factors are hypothesized to contribute to the observed heterogeneity in response to infectious disease, and twin studies are an important source of information to estimate heritability while recognizing the risks of overestimation10. It is also important to note that while ongoing work investigating rare and structural genomic variants may help further address the ‘missing heritability’ conundrum — the disparity between overall trait heritability and observed effects through GWAS — this has been reconciled mostly by the recognition that genetic predisposition in most common traits is spread and shared across large numbers of variants (in the thousands), each with a typically modest effect11. Understanding of the genetic architecture of disease predisposition continues to develop, with an omnigenic model for complex traits proposing genetic contribution from both core genes with strong genetic evidence and very many networked peripheral genes of very low effect size involving regulatory circuits12,13.
Of particular interest in infectious disease GWAS has been the role of genetic variation at the locus encoding HLA genes in determining the individual response to infection, given the role of encoded MHC molecules in binding and presenting antigens and thus determining which antigens are ‘seen’ by the immune system (Box 1). However, the extreme polymorphism of this region, high gene density, complex linkage disequilibrium and regions of homology make fine mapping genetic associations involving HLA very challenging14.
Across the genome, a substantial majority of genetic variants identified by GWAS are located in non-coding regions of the genome, requiring complementary functional genomic annotations to link such variants to modulation of individual genes or pathways. This is key to understanding the functional basis of such associations, in particular the specific modulated genes or pathways that may represent potential drug targets. Knowledge of network connectivity substantially enhances the ability to then identify nodal genes that may be the best therapeutic targets15. Prior genetic evidence for targets substantially increases the success rate in late-stage clinical trials16. As well as supporting the development of new drugs, it provides important opportunities to support repurposing existing drugs where a genetic association with a target is shared across traits, indicating new indications for such approved drugs17.
Genetics can also enable analytical approaches such as Mendelian randomization that leverage genetic variation as instrumental variables to establish causal relationships between non-genetic (modifiable) risk factors and disease18. A further example of translational relevance is the opportunity for genetic evidence to potentially stratify risk; for example, to identify high-risk individuals for whom early or pre-emptive intervention may be appropriate. Aggregated genetic risk through polygenic risk scores and consideration of large-effect rare variants are gaining traction as useful clinical tools19, with reports, for example, of application to dengue fever20.
HIV-1 infection
The two genomic regions most consistently associated with HIV infection outcomes are the MHC21 and the CCR5 locus (CCR5 is a coreceptor for the virus)22,23,24. Arguably, HIV-1 infection is the disease with the greatest insights into HLA–disease association pathogenesis, including seminal early advances supporting the hypothesis of ‘heterozygous advantage’, with individuals with greater heterozygosity of class I alleles (HLA-A, HLA-B and HLA-C) having delayed progression to AIDS25. A more recent discovery has demonstrated that differential expression of HLA-A contributes to disease progression through HLA-E-mediated effects, implicating the interaction between HLA-E and the NKG2A natural killer cell inhibitory receptor as a therapeutic target26. Both HLA-B and HLA-C have been consistently implicated in HIV control, but strong coinheritance or linkage disequilibrium in the MHC region has made disentangling the contributions of each difficult.
The repeated identification of HLA associations in HIV-1 GWAS has contributed, in part, to a catalogue of work focusing on the killer immunoglobulin-like receptor (KIR) genes that encode a family of highly polymorphic activating and inhibitory receptors expressed mainly by natural killer cells that interact with HLA. There has been a surprising lack of KIR gene associations in GWAS to date, which may be due to a multitude of reasons, including substantial diversification of KIR gene haplotypes, incomplete knowledge of the nature and diversity of such haplotypes across populations, and poor coverage of current genotyping arrays for this region, making it difficult to pinpoint specific genetic variants across populations. Thus, this region could represent an archetypal complex region that we have yet to accurately tag in even the most comprehensive of modern GWAS. Moreover, the effects of KIR gene variants may be detected only in the presence of their specific HLA ligand. As evidence for this effect, KIR3DL1 allotypes expressed highly in combination with specific HLA-B alleles (Bw4) were found to slow disease progression when compared with a low-expression allotype or lack of the Bw4 allele27. By contrast, homozygosity for the CCR5Δ32 mutation has been the most robustly confirmed genetic background for protection against HIV28, and antisense long non-coding RNA CCR5AS, which affects CCR5 mRNA stability and cell surface expression, has recently been shown to influence HIV infection susceptibility29. Furthermore, knowledge of the CCR5 pathway has led to the successful development of a class of antiviral agents targeting the entry of HIV-1 into target host cells.
Tuberculosis
Despite some of the earliest evidence for TB susceptibility being driven by genetic heterogeneity30,31, a consistent genetic signal of association in any one locus has yet to be consistently demonstrated. The largest study to date was undertaken in a combined sequencing and genotyping effort including 8,162 cases of combined presentations of TB in the Icelandic population32. This study identified variants in the MHC class II region as being associated with TB susceptibility, perhaps emphasizing the need to undertake GWAS in highly homogeneous populations when tackling ancient pathogens such as Mycobacterium tuberculosis. By contrast, other GWAS applied to TB of varied organ involvement in diverse populations have demonstrated association of pulmonary TB with variants in ESRRB and TGM6 in a Chinese population33 and ASAP1 in a Russian population34, although the biological basis by which these genes mediate an effect in TB is not always clear. For example, TB mouse models with CRISPR–Cas9 deletion of TGM6 lead to increased cytokine transcripts in the lungs, greater tissue damage and higher bacterial burden, but whether the cytokine levels are directly due to altered TGM6 levels is yet to be established.
The lack of consistent signals in the TB GWAS highlight the enormous challenges in undertaking multinational studies, where the observations may be attributed to differences in populations because of population structure either at the host level or the pathogen level or may be owing to technical differences, such as the genotyping array used. A meta-analysis of these studies that might help explain the observed incompatibilities and may inform future research in the area is eagerly awaited.
Leprosy
By contrast, many genes have been identified through GWAS for susceptibility to leprosy due to the related species Mycobacterium leprae35 that unsurprisingly implicate the immune system, including, most notably, the HLA-DR region36. Whereas a GWAS in an Indian cohort found HLA-DR, HLA-DQ and TLR1 (encoding Toll-like receptor 1) to be associated with susceptibility to leprosy37, a further GWAS of Chinese patients failed to replicate the TLR1 association despite adequate power, implicating population-specific effects38. This is again in line with the hypothesis of the genetic variation responsible for variability in the infectious disease trait (human genetic architecture) being subjected to selection pressure from pathogens, as different populations would have been exposed to various infective agents. The large size of the case–control cohorts has enabled the identification and downstream functional characterization of some of the non-HLA loci; for example, for NOD2, where the single-nucleotide polymorphism (SNP) rs1981760 is associated with differential NOD2 gene expression by being an expression quantitative trait locus (eQTL) in neutrophils39. The SNP acts by affecting binding of the transcription factor STAT3 and influencing neutrophil interferon-β (IFNβ) responses39, an effect that has been also been reported in TB40.
Malaria
This parasitic infection is perhaps the most famous example of host genetics influencing infectious disease susceptibility, with haemoglobinopathies — such as sickle haemoglobin (HbS), thalassaemia and glucose 6-phosphate dehydrogenase (G6PD) deficiency — long being associated with protection against malaria. As one example of an underlying biological mechanism, haemoglobin oxidation products in HbS affect the ability of the Plasmodium falciparum parasite to hijack red blood cell cytoskeletal remodelling41. Balancing selection, illustrated by the heterozygote advantage conferred by HbS, provides an important mechanism for maintaining advantageous genetic diversity in human populations42.
Malaria is an example where modern GWAS have confirmed several loci previously identified largely by candidate gene or protein studies, albeit still not most of them; most notably, despite many early observations of associations with HLA, GWAS projects have not found equivalent evidence. The Malaria Genomic Epidemiology Network (MalariaGEN)43 has successfully identified or confirmed genetic loci associated with severe Plasmodium falciparum malaria across Africa, Asia and Australasia, including HBB (β-globin gene), ABO (gene for glycosyltransferase enzyme that determines ABO blood groups), ATP2B4 (gene for a calcium transporter on the erythrocyte membrane), G6PD (activity levels of the enzyme affect oxidative stress in red blood cells) and CD40LG (a key gene in B cell immune responses). A notable finding was that among both different phenotypes and different populations, the heterogeneity of effect of risk alleles was substantial, possibly explaining the inability to replicate some of the previous genetic findings, together with pathogen diversity. The consortium further identified a locus neighbouring glycophorin genes (encoding erythrocyte cell surface receptors that allow Plasmodium falciparum invasion)44 to be implicated in malaria susceptibility. This finding highlights the possibility that loci conferring resistance to malaria involve polymorphisms with features of ancient balancing selection based on haplotype sharing between humans and chimpanzees45, in keeping with the hypothesis of a protracted evolutionary arms race between host and pathogen leading to the host and pathogen genetic diversity that we see today. The link between glycophorin genes and malaria resistance was further elucidated in a study using next-generation sequencing data and reference haplotypes that identified a complex structural rearrangement involving GYPA and GYPB gene copy number that reduced the risk of severe malaria by 40% and that explained the previous association signal46.
Viral hepatitis
Whereas three-quarters of patients with acute hepatitis C virus (HCV) infections eventually reach chronicity, the remaining quarter clear the virus spontaneously. As a key example of a strong GWAS signal not related to HLA with major translational impact, polymorphisms in the gene encoding IFNλ3 (IFNL3; formerly known as IL28B) have been associated with resolution of HCV infection47 and with differences in response to HCV drug treatment48 (viral clearance following treatment with pegylated interferon and ribavirin) in individuals of both European and African ancestry, enabling development of a precision medicine approach to therapy49 (Fig. 2). HLA class II associations with spontaneous HCV clearance have been reported by GWAS50. In a separate GWAS recently of African, European and Hispanic populations, HLA class II signals were replicated, while loci around the IFNλ genes and GPR158 were also implicated in spontaneous viral clearance51.

Examples of how understanding of human genetic information can be or has begun to be leveraged for improved patient care. Strategies include identifying specific molecular targets based on genetic understanding, stratifying patients to decide on use of certain drugs and using genetic knowledge to predict severe adverse reactions to medications. GWAS, genome-wide association studies; HCV, hepatitis C virus; HIV, human immunodeficiency virus; JAK, Janus kinase.
Novel approaches and technologies
Human population diversity
Lessons learnt in infectious disease susceptibility GWAS to date highlight several principles for increasing the chances of success. A key emerging area is the extent of population-specific associations genome-wide (Table 1). This in turn emphasizes the relative paucity of GWAS undertaken in diverse worldwide populations, many of which have the highest burden of infectious disease. Greater efforts will be needed to focus GWAS and other genetic studies on such populations, especially where ethnic background may be associated with differential risk52. It is thus imperative that genotyping arrays are designed for worldwide populations and that the availability of large imputation reference panels from diverse populations enables effective statistical inference of additional non-genotyped SNPs (and hence substantially increasing the informativeness of GWAS at no additional cost)53,54,55. New technologies such as those based on low-coverage whole-genome sequencing may provide an alternative cost-effective approach for genotyping common and rarer genetic variants56.
As is apparent from the studies reported so far herein, infections can afflict populations limited by geography and local ecology, and it is therefore crucial that genetic associations are tested for in non-European cohorts. However, such analyses come with technical challenges owing to the need to adjust the data for potential confounding by differences in population structure or relatedness that may cause an excess of type I errors. These aspects have been appreciated for some time, and many methods have been developed to facilitate such adjustments. One method is principal component analysis (a mathematical method to summarize the main sources of variance in data) to model ancestry differences and account for differing allele frequencies in different populations57. A further development is in statistical modelling using linear mixed model algorithms that can account for population structure and cryptic relatedness simultaneously58.
The Population Architecture Using Genomics and Epidemiology (PAGE) Consortium has demonstrated both the power and the importance in accounting for multi-ethnic and diverse populations in various non-infectious disease and physical trait settings; a GWAS conducted by the consortium demonstrated that in different populations, the effect size of implicated loci differed, exemplifying the need to account for population structure59. Any initiatives planning to develop personalized medicine in infectious diseases based on GWAS will therefore have to depend on the availability of genetic studies that are commensurately precise in a population context and that include an appreciation of the genomic complexity of regions such as the MHC, KIR and immunoglobulin heavy chain (IGH) regions.
Leveraging self-report
The maturation of methods for undertaking GWAS has been paralleled by improvements in methods for recruiting and testing associations. A key study from the personal genetic provider 23andMe used self-report to assess the influence of host genetic background on susceptibility to a range of common infectious diseases of low mortality (for example, common cold, rubella and cold sores)60. Including more than 200,000 individuals of European descent, this study reported many genetic loci related to the immune system and embryonic development, including genes both within and outside the MHC. Despite the large number of associations identified, the use of surveys requiring retrospective recall to identify cases will require follow-up replication and validation. A similar method was used for whooping cough, where self-report of the characteristic cough in childhood was associated with variation across the MHC61.
Phenome-wide association studies
Another development in recent years has been to establish the phenome-wide association study (PheWAS) approach. This has been facilitated by the use of electronic health records for clinical phenotyping. Instead of taking a single disease phenotype and scanning genome-wide as in GWAS, this approach takes a number of genetic loci (usually based on a priori biological knowledge) and looks for associations with any of a large number of phenotypes. Although revealing for many immune-related diseases, it has had limited use thus far in the field of infectious diseases. A PheWAS on HLA predictably highlighted autoimmune diseases, but found only a relatively small number of associations with infectious diseases62. Given the known key role of HLA genes in infectious diseases, it is likely that this was due to a lack of statistical power or challenges in defining cases from controls using hospital data.
Nevertheless, one example of a PheWAS in infectious disease genetics identified an association between the rs3211783 SNP in the factor X gene, encoding an enzyme of the coagulation cascade, with infection-related phenotypes63. Recent evidence for the role of factor X in antibacterial immunity has emerged64, and more studies will be needed to confirm this role, but the study provides an example of what may come in the field of infectious disease genetics through future PheWAS.
Genome sequencing and large-scale bioresources
Standard GWAS and imputation methods are unable to test the effects of rare, large-effect variants, as these variants are not covered by genotyping arrays and are not common enough in haplotype references. To overcome this challenge, direct whole-genome sequencing (WGS) or whole-exome sequencing (WES) approaches have been used. In one example, WES was first performed to identify rare alleles that are relevant in West Nile neuroinvasive disease, enabling subsequent imputation of the rare alleles into a primary cohort. The findings were then extended to larger cohorts for genome-wide association analysis. Loci revealed as being important included HERC5, an interferon-stimulated gene, and an intergenic region between CD83 and JARID2, which included a site for STAT5A transcription factor binding. With very few other such examples in the literature for any trait, it is becoming increasingly clear that the successful application of these methods will require hundreds of thousands to millions of individuals65.
To this end, several population-level bioresources and personal genetic services such as the UK Biobank, the China Kadoorie Biobank and the 100,000 Genomes Project — which are large repositories of samples and data — are enabling the use of novel approaches and enhancing the quality of data analysis using existing methods. In light of early discoveries afforded by the availability of genetic data from these studies66, it will be exciting to see how they may provide further insights into infectious disease as they mature.
Long-read sequencing technologies and highly polymorphic loci
A GWAS of rheumatic heart disease in Pacific Island populations recently identified a signal in the IGH locus67. This disease, which occurs following Streptococcus pyogenes infection, used to have a more widespread worldwide prevalence and is now rather limited to underserved populations across the Pacific. This intriguing finding in the IGH region has long been suspected for many diseases but has been infrequently observed. Studying the IGH region has been challenging owing to its high level of complexity (including substantial copy number variation68) and the sparse coverage of this locus on genotyping arrays. However, with the advent of new long-read sequencing and other technologies, it is likely that we will better capture the extent of IGH diversity and possible disease associations, similarly to what has been achieved for regions such as the MHC and KIR regions69.
New insights from inborn errors of immunity
Another key route to understanding infectious disease susceptibility is through the study of inborn errors of immunity that disrupt host defence against infection. In contrast to more common variants with small to moderate effect sizes identified by GWAS, rare mutations (of variable penetrance) typically result in a large phenotypic effect with substantial infections for the individual that may involve multiple pathogens. Such analysis has been highly informative in revealing mechanisms of infectious disease pathogenesis (Fig. 1; Table 2). Intersection with GWAS is seen: for example, single-gene defects in complement components result in increased susceptibility to meningococcal disease70,71, while GWAS identified SNPs within complement factor H (CFH) and CFH-related protein 3, consolidating evidence for the role of complement in host defence against meningococcal infection72. Application of WGS and WES has now identified more than 400 genetic causes73 of inborn errors of immunity, many of which cause primary immunodeficiencies (PIDs), including genes of known and novel mediators of innate and adaptive immunity (Table 2) mutated in various manners leading to the full range of mechanisms for Mendelian inheritance patterns. One notable advancement in this area has been the recognition of disease-causing genes that do not necessarily lead to overt immunological abnormalities, with differences in penetrance and specificities in clinical infection phenotype among other features9 (Table 2).
The link between certain immune pathways with specific clinical infection phenotypes has been well documented. For example, the TLR1/2/6–Toll–interleukin-1 receptor domain-containing adaptor protein (TIRAP)–nuclear factor-κB (NF-κB) pathway influences invasive pneumococcal disease (Fig. 1a), whereas the TLR3–interferon pathway affects susceptibility to herpes simplex virus encephalitis8,74,75,76,77,78 (Fig. 1b,c). However, other studies are highlighting non-canonical immune pathways.
As an example of a PID, recent studies include demonstration of a critical role for RIPK1 in susceptibility to infections. RIPK1 is a key regulator of cell death and survival and, in the case of cell death, mediation of either apoptosis or necroptosis. In a study of a small group of patients with recurrent infections, early-onset inflammatory bowel disease and progressive polyarthritis, WES identified homozygous LOF mutations79, while biallelic mutations in RIPK1 were found in a further series of patients with very early onset inflammatory bowel disease — the patients all had recurrent bacterial and viral infections and in some instances sepsis80.
Haploinsufficient mechanisms have also been identified, for example, involving BACH2 (ref.81), which is key in controlling B and T cell maturation and functional specification. In a case of a PID with recurrent upper respiratory tract infections and early-onset colitis, a heterozygous non-synonymous mutation in BACH2 resulted in reduced ability of the protein to dimerize or localize to the nucleus. On the other hand, GOF mutations leading to infectious disease susceptibility highlight opportunities for precision medicine. In patients with heterozygous GOF mutations in STAT1 — who have fungal (persistent oral candidiasis) and viral (CMV) infection and in one case TB (Fig. 1c) — the resultant excessive CD4+ T cell STAT1 phosphorylation after IFNβ stimulation highlighted the opportunity for therapeutic suppression by the Janus kinase (JAK) inhibitor ruxolitinib82. A separate study of patients affected by STAT1 GOF mutations and chronic mucocutaneous candidiasis showed successful treatment with ruxolitinib83 (Fig. 2).
Chronic granulomatous disease arises owing to the inability of phagocytes to mount a reactive oxygen species burst in response to pathogens and is characterized by severe recurrent bacterial and fungal infections84. This PID is often caused by mutations in the genes encoding the components of the NADPH oxidase complex. A homozygous LOF mutation in CYBC1 was identified as a further cause85, revealing a novel function for CYBC1 as a chaperone for one of the components of the NADPH oxidase complex.
Studies of patients with extremes of response to viral infections have provided mechanistic insights. In a case of recurrent viral infections requiring intensive care admission86, WES revealed a homozygous mutation in IFIH1 (encoding a retinoic acid-inducible gene I protein (RIG-I)-like helicase receptor responsible for sensing viral double-stranded RNA in the cytosol), whereas, in cases of life-threatening influenza, WES identified compound heterozygous mutations in IRF7, which reduces both type I and type III interferon production87. Influenza virus pneumonia may also arise owing to rare inborn errors of immunity involving IRF9 and TLR3 deficiencies88,89 whereas homozygous CCR5-null and FUT2-null alleles protect against CCR5-tropic HIV and norovirus, respectively22,23,24,90.
Mendelian susceptibility to mycobacterial disease can arise owing to mutations in genes encoding proteins involved in the production of, or response to, the cytokine IFNγ. Such mutations have important treatment implications; for example, the deficiency of IFNγ signalling can be ameliorated by therapeutic supplementation with IFNγ or haematopoietic stem cell transplantation, dependent on the degree of functional pathway responsiveness91. WES has revealed a novel cause of Mendelian susceptibility to mycobacterial disease in a case of homozygous splice-site mutations in SPPL2A (required to cleave CD74, the invariant chain of HLA class II); the mutation is proposed to disrupt functional polarization of T cells owing to altered priming by dendritic cells after mycobacterial antigen presentation92 (Fig. 1d).
One hurdle in monogenic disorders is incomplete penetrance. Polymorphism of TIRAP is known to influence susceptibility to invasive pneumococcal disease, TB, malaria and other bacterial infections93,94,95,96. In a study of a family with TIRAP deficiency, seven of eight individuals with the genetic defect did not have the severe staphylococcal infection that the index case did97. A possible explanation was the lack of antibodies to lipoteichoic acid found in the index patient compared with other family members that may allow monocytes in the unaffected family members to respond to lipoteichoic acid stimulation of TLR2 despite deficiency in TIRAP.
Integrating host and pathogen genetic variation
Although the impact of host genetic variation on susceptibility to infection is often viewed separately from that of pathogen genetics, it is highly likely that a major limitation in identifying novel genetic signals in GWAS applied to common infections will be attributable to heterogeneity at the level of the infectious agent. Approaches attempting to simultaneously interrogate host and pathogen transcriptomic responses (dual RNA sequencing) have been developed and applied to infectious diseases98,99,100; this joint characterization has also occurred at the genetic level to account for variability in both the host genome and the pathogen genome. In a Dutch cohort, a GWAS of both the host and the pathogen in pneumococcal meningitis allowed delineation of the contributions of host genetic background to the susceptibility and severity of disease versus the effects of pneumococcal genetic variation on invasive potential but not disease severity101. The study revealed an intronic SNP in a host ubiquitin-converting enzyme gene, UBE2U, to be significantly associated, with possible interaction with other host genes, including PGM1 (encoding phosphoglucomutase 1) and ROR1 (encoding a tyrosine-protein kinase transmembrane receptor). Similarly, a joint genetic study of HCV infection with human genome-wide genotyping and genome sequencing of HCV genotype 3 infections led to more biological insights, including understanding of reciprocal effects (for example, of human genetic variation in driving viral genome polymorphism)102. Rather than association tests using disease status, this study examined the associations between the host genetic background and variable sites of the viral proteome. Host HLA alleles were found to be associated with HCV amino acids leaving multiple ‘footprints’ in the HCV genome; and, in addition, a specific interaction between the HCV NS5A protein and human IFNL4 (encoding IFNλ4) genotype was found to affect viral load.
Simultaneous evaluation of host and pathogen genetics was also used in HIV datasets to reveal the influence of HLA genes. By considering how a virus mutates throughout the course of its infection, the relevance of the host genetic background can be considered. A recent study analysed a large number of existing HIV datasets, using host HLA class I genotypes and pathogen reverse transcriptase and protease sequences and modelling the processes of within-host evolution of the virus103. Known allele–epitope combinations (for example, escape at codons 173/195 of reverse transcriptase when paired with the HLA-B*51 allele) were replicated, but many epitopes highlighted to be HLA associated were not previously reported. Deeper analysis of viral sequence subtypes showed that differential selection pressures between the same HLA allele (HLA-B*58) and the different viral subtypes occurred at a reverse transcriptase codon that differed between the two subtypes. HLA-B*58 selection pressure may have consolidated the divergence in viral sequence subtypes.
The selective pressure of other pathogens on human populations has been delineated by genome-wide analyses of natural selection. For example, for cholera, targets of positive selection were identified in a population from the Ganges River Delta, supporting strong selective pressure by the pathogen on innate immune pathways, including NF-κB and inflammasome signalling104.
Studies of dual-pronged interrogation of host and infectious agent genetics in malaria have yet to be conducted at a large scale, but there have been projects to characterize heterogeneity in both Plasmodium falciparum, Plasmodium knowlesi and Plasmodium malariae and the malarial vector Anopheles gambiae at genetic and gene expression levels105,106,107. These studies will be informative for drug resistance and vector control; for example, one study highlighted the need to collect vector genomic data to establish which strategies lead to insecticide resistance107.
Several major challenges remain for studies considering both host and pathogen genetic variation. Most notably, the multiple testing burden means that substantially larger sample sizes will be needed in future studies, although an important analytical strategy is dimensionality reduction, which takes advantage of genome-wide correlation between variants101. Additionally, understanding the impact of complex spatio-temporal and ecological scales requires careful acquisition of comprehensive metadata that can be used with genomic data108.
Genetics of immune function phenotypes
In the evaluation of any disease, accounting for phenotype heterogeneity is of paramount importance. The use of various -omics technologies is a strategy to address this (Fig. 3). For example, transcriptomic analysis in sepsis has been able to stratify patients into different disease endotypes regardless of the source of infection109,110,111,112. Crucially, these endotypes have been shown to have interaction effects with steroid treatment, with evidence that patients of a relatively immunocompetent endotype have increased mortality on being given steroids (Fig. 2). This finding highlights the need for accurate phenotyping in future clinical trials where the effect of treatment may be diluted by heterogeneity in patient cohorts, and such improved phenotyping will also enable more successful application of GWAS.

a | Traditional case–control genome-wide association study (GWAS) approaches compare allele frequencies of genetic variants in cases versus controls. b | Mendelian disease mapping with pedigree analysis (including case–parent trio analyses) and use of whole-exome or whole-genome sequencing. c | Multi-omics approaches, which enable intermediate phenotypes to be quantified by various -omics technologies. d | Leveraging genetic information to interrogate or leverage intermediate phenotypes. Differences in intermediate phenotypes such as gene expression can be mapped to genetic variation by quantitative trait locus (QTL) mapping. Mendelian randomization methods can use intermediate phenotypes that are risk factors for disease, with genetic variants that affect the intermediate phenotype allocated randomly to allow confounders to also be randomly distributed.
Thus, aside from clinical disease phenotypes, innovative approaches to examining infectious disease genetics have investigated intermediate phenotypes that may be relevant to the disease (Fig. 3). For example differential blood cell types are under substantial genetic influence113. Furthermore, levels of immunoglobulin are also genetically influenced not only by genetic variants in IGH genes and related immunoglobulin genes and the MHC but also by RUNX3 variants altering isoform shifting and FCGR2B variants impacting IgG binding to its receptor114. These approaches are now also being translated in the context of antigen specificity, including the role of genetic variation in agent-specific antibody responses, with a role for MHC identified in various responses, including anti-Epstein–Barr virus and anti-rubella virus IgG levels115,116. It will be interesting to determine what findings these markers of previous exposure may contribute in the context of acute infection risk in the future.
In a different attempt to examine the role of genetic variants in controlling the human immune system, Roederer et al. performed detailed immunophenotyping in 669 female twins117. The top 151 heritable immune traits from extensive flow cytometry were analysed. For example, FCGR2A variants were associated with multiple immune phenotypes such as expression levels of various T cell markers such as CD27 or CD161, and FCGR2A has been associated with HIV infection progression.
The use of microbial stimuli to produce intermediate phenotypes has also led to interesting insights. One study used 528 lymphoblastoid cell lines and subjected them to challenges with eight different kinds of pathogens, selected carefully for their high health-care burden and the wide range of host responses elicited against them118. Genome-wide association tests revealed 17 significant SNPs; for example, a SNP in ZBTB20 (which functions as a transcriptional repressor) was associated with both increased salmonella-induced pyroptosis and chlamydia replication. A PheWAS approach was further used with clinical phenotypes from the eMERGE dataset119, which highlighted the same SNP, this time relevant in viral hepatitis.
Ultimately, the link from gene to biological function requires characterization of candidate loci for mechanistic understanding of genetic contributions. The vast majority of loci found in GWAS reside in non-coding regions, implicating gene regulation in disease pathogenesis. Much effort in recent years has therefore been focused on understanding the genetic basis of various forms of molecular traits (molecular quantitative trait locus (QTL) mapping) (Fig. 3). Such associations are often highly context specific, dependent, for example, on cell type120 and activation state121,122,123, with effects specific to exposure to particular pathogens124, and dependent on the disease state; for example, in patients with sepsis due to community acquired pneumonia109. This characterization of molecular traits can inform GWAS interpretation; for example, in a GWAS of non-typhoidal Salmonella bacteraemia, a SNP acting as an eQTL for STAT4 was found, with its effect reported to be a reduction in IFNγ production by natural killer cells125. Genetic modulators of gene expression may also operate at the level of alternative splicing126, including after stimulation by influenza A virus127, with evidence that genetic regulation of isoform use was mostly distinct from that of gene expression. Immune cell-specific protein QTLs have also been demonstrated128, including effects on expression levels of HLA-DRB1 in specific cell types that may be relevant in diseases such as sepsis, where reduced HLA-DR expression is known to correlate with poor outcome129. A further study mapped cytokine production to genetic variants (cQTL mapping) in response to microbial stimuli, identifying QTLs involving pathways containing pattern recognition receptors, cytokine and complement inhibitors, and the kallikrein system130.
The maturation of technologies has seen multi-omics approaches applied to increasing numbers of diseases to identify and link genomic alterations to biological mechanisms. The infectious disease field has seen recent examples emerge: the combination of the plasma proteome, metabolome and lipidome and peripheral blood mononuclear cell transcriptome in analysing Ebola virus disease pathogenesis is one such example131. Macrophages and neutrophils were found to be particularly relevant cell types. Although this study itself did not tie the findings to the host genetic background, it is nevertheless an important example of the use of multi-omics functional characterization, and it is likely that we will soon be seeing the linking of multi-omics data back to genetic background.
Genetics of responses to vaccinations
As vast amounts of data continue to accrue on the relationship between the human host, its immune responses and infectious agents, the question as to how these data are best used is timely. Arguably, vaccination has offered one of the most effective public health efforts of modern medicine and is the most cost-effective opportunity to substantially reduce the incidence of many important infections, such as malaria, HIV infection and TB. Thus, given the large amount of evidence of human genetics influencing both non-specific and specific immune responses and susceptibility to disease, it is curious that there is still a relative lack of investigations looking at the effect of host genetics on vaccine response132, especially as the study of responses against conserved epitopes offers a means to control for pathogen diversity. Here, we review the state of the field in terms of the genetics of response to vaccination and its potential utility.
The vaccine targeting hepatitis B virus (HBV) remains the most studied, with many genetic associations identified in the MHC region, and there is controversy over whether it is HLA-DR, HLA-DP or class III MHC (involving the C4A gene) that plays independent or linked roles133,134,135,136. It is clear, however, that while HLA-DRB1*03 and HLA-DRB1*07 are linked to lower antibody responses, the opposite is true of HLA-DRB1*01, HLA-DRB1*13 and HLA-DRB1*15 (refs134,137,138), and the associations between HLA-DP and vaccine response against HBV are robust, as they are for other clinical phenotypes associated with the infection, particularly in Asian populations.
Rubella vaccination has seen similar HLA associations, although evidence is again conflicting. The HLA-DPB1 locus has been associated with rubella vaccination antibody responses139,140, but SNPs in the region have been associated in opposite directions, highlighting the need for further replication and functional studies to identify plausible biological mechanisms. Increased antibody responses to measles vaccination has been associated with the HLA-DQA1*0201 allele141.
Although a twin study has shown that the contribution of HLA genes is relatively small compared with non-HLA genes in antibody-inducing vaccines142, there has been relatively little robust and replicated evidence of what the non-HLA genes may be. A recent two-stage GWAS looking at data from more than 3,600 children revealed both HLA and non-HLA associations on studying a combination of capsular group C meningococcal, Haemophilus influenzae type b and tetanus toxoid vaccines143. This study found SNPs in the locus coding for signal-regulatory proteins SIRPA, SIRPB and SIRPG were associated with greater antibody titres in group C meningococcal vaccines. Notably, the association was present only for serum bactericidal antibodies (functional antibodies as assessed by rabbit/human complement assays, and not with total meningitis C-specific IgG), emphasizing the need for caution about the agent-specific readout used. The study also identified four HLA class II alleles associated with tetanus toxoid vaccine IgG concentrations, with the lead SNP being an eQTL for HLA-DRB1 and HLA-DRB5 (ref.144).
Inborn errors of immunity have been reported to occasionally result in life-threatening disease following administration of live attenuated vaccines, for example, live poliovirus vaccine (vaccine-associated paralytic polio in patients with agammaglobulinaemia145), whereas defects in interferon immunity may result in severe illness following administration of the yellow fever or MMR vaccine (measles strain) (IFNAR1 (ref.146) and IFNAR2 (ref.147), and STAT1 (ref.148) and STAT2 (ref.149) deficiencies, respectively) (Fig. 1c).
Overall, current work highlights that new cohorts and mechanistic studies to further explore the effect of host genetics on vaccine response are needed to address this important area.
Genetic strategies for a new disease: COVID-19
Emerging infections such as SARS-CoV-2 pose a major threat to human health, and the early observation of striking heterogeneity in clinical presentations and outcomes following infection has highlighted the ongoing need to apply genetic and genomic approaches to understand drivers of individual susceptibility, severity and outcomes to this disease. Only a small minority of those infected with SARS-CoV-2 develop severe disease, with recognized risk factors including older age, male sex and co-morbidities, including obesity150. Genetic factors are hypothesized to contribute to the observed heterogeneity in response, and early reports such as from TwinsUK indicate relatively high heritability estimates for self-reported COVID-19 symptoms such as anosmia151. Genetic approaches provide the opportunity to gain novel insights into disease pathogenesis through the identification of specific disease associations involving particular genes or pathways, to define novel drug targets or to develop personalized medicine approaches with early intervention or therapy tailored to the individual. The opportunity to identify repurposing opportunities for approved medications based on shared genetic associations across traits is a particularly important consideration in a pandemic situation such as COVID-19, where, while not based on genetic evidence, the RECOVERY trial has demonstrated the therapeutic utility of existing drugs such as dexamethasone for treatment of severe disease152.
The international response to answering the question of whether human genetics alters susceptibility to or outcome from SARS-CoV-2 infection is an exemplar of modern genomic collaboration. The establishment of large appropriately phenotyped cohorts for GWAS in a pandemic situation has been enabled by prospective collection through existing or hibernating studies, rapid deployment of new studies and leveraging existing population biobank studies with large numbers of already genotyped individuals. Monthly data releases from the UK Biobank of COVID-19 results together with outcome and phenotypic information early in the pandemic highlight the opportunity provided by such population biobanks, while application of meta-analysis methods across cohorts will maximize power for genetic discovery. International collaborative efforts are key to facilitate genetic studies in infectious diseases such as COVID-19. For example, the COVID-19 Host Genetics Initiative153 was rapidly established to support sharing of relevant tools and data with an emphasis on GWAS. The COVID Human Genetic Effort154, by contrast, is focused on identifying monogenic cases with rare, highly penetrant mutations through analysis of young patients (less than 50 years of age) who were previously well and developed life-threatening disease, as well as those naturally resistant despite repeated exposure. Integrative analysis, considering both host and viral genetic variation, will likely be highly informative, although early reports suggest that viral genetic variation did not significantly affect outcomes in COVID-19 (ref.155).
An early exemplar of GWAS during the COVID-19 pandemic is a study by the Severe Covid-19 GWAS Group, who conducted a GWAS of 1,980 patients with severe disease (hospitalized with respiratory failure requiring oxygenation or mechanical ventilation) in Italian and Spanish epicentres of the pandemic versus population controls. The European meta-analysis revealed genome-wide significant associations at 3p21.31 and 9q34.2 (ref.156). The limitations of self-reported data notwithstanding, a large GWAS of more than 1.05 million individuals from the 23andMe research platform replicated the associations at these two loci for disease severity157, while the GenOMICC investigators recruited patients with COVID-19 from intensive care units only and robustly reproduced the 3p21.31 signal in addition to finding associations at other loci, including ones with genes of well-known antiviral function such as 12q24.13 (OAS1, OAS2 and OAS3) and 21q22.1 (IFNAR2)158.
Despite the important achievements, these results exemplify current challenges in GWAS biology. First, the 3p21.31 association, which was also supported by preliminary results from the COVID-19 Host Genetics Initiative, spans multiple genes, including SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6 and XCR1, but will require careful functional genomic and other annotations to establish the causal genes underlying the genomic signal, as non-coding variants may modulate gene expression and other regulatory mechanisms at a distance159. Intriguingly, there is evidence that the risk haplotype, spanning ~50 kb at 3p21.31, was inherited from Neanderthals and shows substantial variation in frequency between populations, being carried by ~50% of people in South Asia, while being almost absent in East Asia, suggesting it may have been affected by selection in the past160. Second, the association at 9q34.2 coincides with the ABO blood group locus, and increased risk with blood group A was shown, but the mechanism for this association remains unclear and will require further study especially as this locus is frequently a site of type I error resulting from population stratification. Despite the apparent lack of significant signal of association at the locus encoding HLA genes in these initial GWAS of COVID-19, there is considerable interest in the importance of such variation in the response to infection, with evidence, for example, of differing capacity between HLA alleles in presentation of highly conserved SARS-CoV-2 peptides to immune cells161.
There is also early evidence from the COVID Human Genetic Effort to support the role of rare variants in the risk of severe disease on the basis of a study of 659 patients (0.1–99 years of age) hospitalized with critical disease due to COVID-19 and requiring mechanical ventilation or organ support in intensive care units162. Following genome and exome sequencing, the study authors tested the hypothesis that inborn errors of TLR3- and IRF7-dependent type I interferon immunity contribute to critical disease risk, analysing 13 genomic regions comprising loci previously shown to be mutated in critical influenza pneumonia and connected loci mutated in patients with other viral illnesses. Significant enrichment of rare variants predicted to result in LOF was found at these loci relative to people with asymptomatic or benign infection, with experimental validation including demonstration of inborn errors at eight loci in up to 23 patients (3.5%) of different ages (17–77 years) and population ancestries. The importance of type I interferons in protective immunity against SARS-CoV-2 was underlined by an accompanying article showing evidence for a high level of neutralizing autoantibodies to type I interferons in other patients with critical COVID-19, together highlighting the opportunities for therapeutic intervention based on screening at-risk individuals and developing targeted interventions163. Indeed, in the GenOMICC study described above, beyond uncovering key potential genetic contributions to COVID-19 severity, the group further used Mendelian randomization to demonstrate a link between low IFNAR2 expression and high TYK2 expression and life-threatening disease, the former further evidencing the importance of type I interferons in COVID-19 pathogenesis158. Altogether, the emerging insights from this fast-moving field of research exemplify the complex nature of the genetic architecture of susceptibility to infectious disease that may have relevance not only for the agent inflicting the largest pandemic of our generation but also potentially for a range of other infections afflicting our public health worldwide.
Conclusions and perspectives
A huge wealth of data is becoming available that confirms the influence of our host genetic variation on how we become susceptible to and respond to diseases caused by infectious agents. Although traditional case–control GWAS have yielded many insights, a large amount of information is also coming from other methods, such as intermediate phenotype mapping and multi-omics approaches. Issues of heterogeneity in host and pathogen genetic diversity and difficulty in arriving at consensus for disease case definition both hamper power in GWAS approaches. However, as recent studies emphasize, these problems are not insurmountable with appropriate power and will be aided by greater precision in defining disease phenotypes and application in a range of populations.
The challenge now comes with bringing all of these findings and approaches together to translate them into a clinical benefit. Moving forwards, it will be critical to consider context specificity. The cellular studies and vaccine responses emphasize the importance of considering pathogen-specific immune responses in the context of both time and appropriate sample type. The extent and nature of pathogen diversity is a key context, with large gaps remaining in our understanding. Our ability to leverage genetic information will also improve as we further appreciate the overlaps between monogenic syndromes with a high infection risk and the more common infectious diseases with similar pathogens. All of these opportunities are likely to come as data from cohorts such as the UK Biobank and the China Kadoorie Biobank mature.
The clinical benefit of the large body of genetic work has already been demonstrated through associations translated to clinical implementation, such as abacavir hypersensitivity eliminated by screening for HLA-B*5701 (Fig. 2), and HCV treatment influenced by IL28B. We should be inspired by these lessons learnt as we move forwards into an era where large-scale genomics may help predict our risks of disease or facilitate future vaccine development and deployment through an understanding of genetic diversity at both the individual level and the population level to enable more tailored application in a precision medicine approach that maximizes effectiveness for a given person or population group.
References
Mühlemann, B. et al. Ancient hepatitis B viruses from the Bronze Age to the medieval period. Nature 557, 418–423 (2018).
Quintana-Murci, L. Human immunology through the lens of evolutionary genetics. Cell 177, 184–199 (2019).
Karlsson, E. K., Kwiatkowski, D. P. & Sabeti, P. C. Natural selection and infectious disease in human populations. Nat. Rev. Genet. 15, 379–393 (2014).
Stanaway, J. D. et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1923–1994 (2018).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Sørensen, T. I. A., Nielsen, G. G., Andersen, P. K. & Teasdale, T. W. Genetic and Environmental influences on premature death in adult adoptees. N. Engl. J. Med. 318, 727–732 (1988). This is a key study showing that premature death due to infections in adults has a strong genetic background.
Chapman, S. J. & Hill, A. V. S. Human genetic susceptibility to infectious disease. Nat. Rev. Genet. 13, 175–188 (2012).
Casanova, J.-L. & Abel, L. Lethal infectious diseases as inborn errors of immunity: toward a synthesis of the germ and genetic theories. Annu. Rev. Pathol. Mech. Dis. https://doi.org/10.1146/annurev-pathol-031920-101429 (2020).
Young, A. I. Solving the missing heritability problem. PLoS Genet. 15, e1008222 (2019).
Claussnitzer, M. et al. A brief history of human disease genetics. Nature 577, 179–189 (2020).
Boyle, E. A., Li, Y. I. & Pritchard, J. K. An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017). This article proposes the omnigenic model with most heritability in complex traits explained by effects on genes outside core pathways.
Liu, X., Li, Y. I. & Pritchard, J. K. Trans effects on gene expression can drive omnigenic inheritance. Cell 177, 1022–1034.e6 (2019).
Dendrou, C. A., Petersen, J., Rossjohn, J. & Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 18, 325–339 (2018).
Fang, H. et al. A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat. Genet. 51, 1082–1091 (2019).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015). This study provides important evidence showing how genetically supported targets have a higher success rate in clinical development.
Nabirotchkin, S. et al. Next-generation drug repurposing using human genetics and network biology. Curr. Opin. Pharmacol. https://doi.org/10.1016/j.coph.2019.12.004 (2020).
Emdin, C. A., Khera, A. V. & Kathiresan, S. Mendelian randomization. JAMA 318, 1925–1926 (2017).
Torkamani, A., Wineinger, N. E. & Topol, E. J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19, 581–590 (2018).
Pare, G. et al. Genetic risk for dengue hemorrhagic fever and dengue fever in multiple ancestries. EBioMedicine 51, 102584 (2020).
Kaslow, R. A. et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2, 405–411 (1996).
Deng, H. K. et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381, 661–666 (1996).
Alkhatib, G. et al. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272, 1955–1958 (1996).
Dragic, T. et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381, 667–673 (1996).
Carrington, M. et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283, 1748–1752 (1999). This is a seminal article showing evidence to support selective advantage for heterozygosity in the HLA.
Ramsuran, V. et al. Elevated HLA-A expression impairs HIV control through inhibition of NKG2A-expressing cells. Science 359, 86–90 (2018).
Jiang, Y. et al. KIR3DS1/L1 and HLA-Bw4-80I are associated with HIV disease progression among HIV typical progressors and long-term nonprogressors. BMC Infect. Dis. 13, 405 (2013).
McLaren, P. J. & Carrington, M. The impact of host genetic variation on infection with HIV-1. Nat. Immunol. 16, 577–583 (2015).
Kulkarni, S. et al. CCR5AS lncRNA variation differentially regulates CCR5, influencing HIV disease outcome. Nat. Immunol. 20, 824–834 (2019).
Comstock, G. W. Tuberculosis in twins: a re-analysis of the Prophit survey. Am. Rev. Respir. Dis. 117, 621–624 (1978).
van der Eijk, E. A., van de Vosse, E., Vandenbroucke, J. P. & van Dissel, J. T. Heredity versus environment in tuberculosis in twins. Am. J. Respir. Crit. Care Med. 176, 1281–1288 (2007).
Sveinbjornsson, G. et al. HLA class II sequence variants influence tuberculosis risk in populations of European ancestry. Nat. Genet. 48, 318–322 (2016). This article shows through WGS evidence of HLA association with Mycobacterium tuberculosis infection.
Zheng, R. et al. Genome-wide association study identifies two risk loci for tuberculosis in Han Chinese. Nat. Commun. 9, 4072 (2018).
Curtis, J. et al. Susceptibility to tuberculosis is associated with variants in the ASAP1 gene encoding a regulator of dendritic cell migration. Nat. Genet. 47, 523–527 (2015).
Fava, V. M., Dallmann-Sauer, M. & Schurr, E. Genetics of leprosy: today and beyond. Hum. Genet. 139, 835–846 (2020).
Wang, Z. et al. A large-scale genome-wide association and meta-analysis identified four novel susceptibility loci for leprosy. Nat. Commun. 7, 13760 (2016).
Wong, S. H. et al. Leprosy and the adaptation of human toll-like receptor 1. PLoS Pathog. 6, e1000979 (2010).
Zhang, F. et al. Identification of two new loci at IL23R and RAB32 that influence susceptibility to leprosy. Nat. Genet. 43, 1247–1251 (2011).
Naranbhai, V. et al. Genomic modulators of gene expression in human neutrophils. Nat. Commun. 6, 7545 (2015).
Mayer-Barber, K. D. et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511, 99–103 (2014).
Cyrklaff, M. et al. Hemoglobins S and C interfere with actin remodeling in Plasmodium falciparum-infected erythrocytes. Science 334, 1283–1286 (2011).
Key, F. M., Teixeira, J. C., de Filippo, C. & Andrés, A. M. Advantageous diversity maintained by balancing selection in humans. Curr. Opin. Genet. Dev. 29, 45–51 (2014).
Malaria Genomic Epidemiology Network. Reappraisal of known malaria resistance loci in a large multicenter study. Nat. Genet. 46, 1197–1204 (2014).
Malaria Genomic Epidemiology Network. A novel locus of resistance to severe malaria in a region of ancient balancing selection. Nature 526, 253–257 (2015). This GWAS of severe malaria identifies a novel resistance locus close to genes encoding glycophorins.
Leffler, E. M. et al. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 340, 1578–1582 (2013).
Leffler, E. M. et al. Resistance to malaria through structural variation of red blood cell invasion receptors. Science 356, 1140–1152 (2017).
Thomas, D. L. et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461, 798–801 (2009).
Ge, D. et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461, 399–401 (2009). This article provides evidence for a precision medicine approach with a substantial difference in response to therapy predicted by human genetic variation in the region of IFNL4 (also known as IL28B).
Jones, C. R., Flower, B. F., Barber, E., Simmons, B. & Cooke, G. S. Treatment optimisation for hepatitis C in the era of combination direct-acting antiviral therapy: a systematic review and meta-analysis. Wellcome Open Res. https://doi.org/10.12688/wellcomeopenres.15411.1 (2019).
Duggal, P. et al. Genome-wide association study of spontaneous resolution of hepatitis C virus infection: data from multiple cohorts. Ann. Intern. Med. 158, 235–245 (2013).
Vergara, C. et al. Multi-ancestry genome-wide association study of spontaneous clearance of hepatitis C virus. Gastroenterology 156, 1496–1507.e7 (2019).
Harrison, E. M. et al. Ethnicity and outcomes from COVID-19: the ISARIC CCP-UK prospective observational cohort study of hospitalised patients. SSRN Electron. J. https://doi.org/10.2139/ssrn.3618215 (2020).
Kowalski, M. H. et al. Use of > 100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 15, e1008500 (2019).
Loh, P. R. et al. Reference-based phasing using the haplotype reference consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Gurdasani, D. et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 517, 327–332 (2015). This article describes genetic diversity in African individuals, demonstrating the importance of population-informative genotype array design and imputation.
Gilly, A. et al. Very low-depth whole-genome sequencing in complex trait association studies. Bioinformatics 35, 2555–2561 (2019).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Yang, J., Zaitlen, N. A., Goddard, M. E., Visscher, P. M. & Price, A. L. Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 46, 100–106 (2014).
Wojcik, G. L. et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570, 514–518 (2019).
Tian, C. et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat. Commun. 8, 599 (2017). This article presents a GWAS for 23 common infections, identifying HLA and non-HLA loci.
McMahon, G., Ring, S. M., Davey-Smith, G. & Timpson, N. J. Genome-wide association study identifies SNPs in the MHC class II loci that are associated with self-reported history of whooping cough. Hum. Mol. Genet. 24, 5930–5939 (2015).
Karnes, J. H. et al. Phenome-wide scanning identifies multiple diseases and disease severity phenotypes associated with HLA variants. Sci. Transl Med. 9, eaai8708 (2017).
Choby, J. E. et al. A phenome-wide association study uncovers a pathological role of coagulation factor X during Acinetobacter baumannii infection. Infect. Immun. 87, e00031–19 (2019).
Chen, J. et al. Coagulation factors VII, IX and X are effective antibacterial proteins against drug-resistant Gram-negative bacteria. Cell Res. 29, 711–724 (2019).
Locke, A. E. et al. Exome sequencing of Finnish isolates enhances rare-variant association power. Nature 572, 323–328 (2019). This is an exome sequencing study showing informativeness for specific populations dependent on population genetic history.
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Parks, T. et al. Association between a common immunoglobulin heavy chain allele and rheumatic heart disease risk in Oceania. Nat. Commun. 8, 14946 (2017). This article provides an important example of genetic association involving the IGH locus.
Bashford-Rogers, R. J. M. et al. Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature 574, 122–126 (2019).
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018).
Lewis, L. A. & Ram, S. Meningococcal disease and the complement system. Virulence 5, 98–126 (2014).
Schröder-Braunstein, J. & Kirschfink, M. Complement deficiencies and dysregulation: pathophysiological consequences, modern analysis, and clinical management. Mol. Immunol. 114, 299–311 (2019).
Davila, S. et al. Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat. Genet. 42, 772–776 (2010). This article shows using GWAS the importance of genetic variation involving CFH in invasive meningococcal disease.
Bousfiha, A. et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J. Clin. Immunol. 40, 66–81 (2020).
Zhang, S. Y. et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317, 1522–1527 (2007).
Jain, A. et al. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat. Immunol. 2, 223–228 (2001).
Picard, C. et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 299, 2076–2079 (2003).
Von Bernuth, H. et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321, 691–696 (2008).
Döffinger, R. et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB signaling. Nat. Genet. 27, 277–285 (2001).
Cuchet-Lourenço, D. et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813 (2018). This is an exemplar of the informativeness of studying rare inborn errors of immunity to understand the immune system.
Li, Y. et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl Acad. Sci. USA 116, 970–975 (2019).
Afzali, B. et al. BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Nat. Immunol. 18, 813–823 (2017).
Baris, S. et al. Severe early-onset combined immunodeficiency due to heterozygous gain-of-function mutations in STAT1. J. Clin. Immunol. 36, 641–648 (2016).
Higgins, E. et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J. Allergy Clin. Immunol. 135, 551–553.e3 (2015).
Arnold, D. E. & Heimall, J. R. A review of chronic granulomatous disease. Adv. Ther. 34, 2543–2557 (2017).
Arnadottir, G. A. et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat. Commun. 9, 4447 (2018).
Lamborn, I. T. et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J. Exp. Med. 214, 1949–1972 (2017).
Ciancanelli, M. J. et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 348, 448–453 (2015). This is an exemplar of how single-gene inborn errors of immunity can result in severe infection.
Hernandez, N. et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J. Exp. Med. 215, 2567–2585 (2018).
Lim, H. K. et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 216, 2038–2056 (2019).
Lindesmith, L. et al. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 9, 548–553 (2003).
Rosain, J. et al. Mendelian susceptibility to mycobacterial disease: 2014–2018 update. Immunol. Cell Biol. 97, 360–367 (2019).
Kong, X.-F. et al. Disruption of an antimycobacterial circuit between dendritic and helper T cells in human SPPL2a deficiency. Nat. Immunol. 19, 973–985 (2018).
Kumpf, O. et al. Influence of genetic variations in TLR4 and TIRAP/Mal on the course of sepsis and pneumonia and cytokine release: an observational study in three cohorts. Crit. Care 14, R103 (2010).
Khor, C. C. et al. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat. Genet. 39, 523–528 (2007).
Hawn, T. R. et al. A polymorphism in toll-interleukin 1 receptor domain containing adaptor protein is associated with susceptibility to meningeal tuberculosis. J. Infect. Dis. 194, 1127–1134 (2006).
Ladhani, S. N. et al. Association between single-nucleotide polymorphisms in Mal/TIRAP and Interleukin-10 genes and susceptibility to invasive Haemophilus influenzae serotype b infection in immunized children. Clin. Infect. Dis. 51, 761–767 (2010).
Israel, L. et al. Human adaptive immunity rescues an inborn error of innate immunity. Cell 168, 789–800.e10 (2017). This article demonstrates incomplete penetrance in monogenic disorders of inborn errors of immunity with an example of functional characterization of TIRAP deficiency.
Westermann, A. J. et al. Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature 529, 496–501 (2016).
Nuss, A. M. et al. Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host-pathogen transcriptomes. Proc. Natl Acad. Sci. USA 114, E791–E800 (2017).
Montoya, D. J. et al. Dual RNA-Seq of human leprosy lesions identifies bacterial determinants linked to host immune response. Cell Rep. 26, 3574–3585.e3 (2019).
Lees, J. A. et al. Joint sequencing of human and pathogen genomes reveals the genetics of pneumococcal meningitis. Nat. Commun. 10, 2176 (2019).
Ansari, M. A. et al. Genome-to-genome analysis highlights the effect of the human innate and adaptive immune systems on the hepatitis C virus. Nat. Genet. 49, 666–673 (2017). This article illustrates the successful application of the genome-to-genome approach, highlighting the interplay between innate immunity and the viral genome in hepatitis C control.
Palmer, D. S. et al. Mapping the drivers of within-host pathogen evolution using massive data sets. Nat. Commun. 10, 3017 (2019).
Karlsson, E. K. et al. Natural selection in a Bangladeshi population from the cholera-endemic Ganges River Delta. Sci. Transl Med. 5, 192ra86 (2013). This work demonstrates genetic association with cholera resistance involving genes with strong evidence of selection, including innate immunity genes.
Amambua-Ngwa, A. et al. Major subpopulations of Plasmodium falciparum in sub-Saharan Africa. Science 365, 813–816 (2019).
Howick, V. M. et al. The Malaria Cell Atlas: single parasite transcriptomes across the complete plasmodium life cycle. Science 365, eaaw2619 (2019).
Miles, A. et al. Genetic diversity of the African malaria vector anopheles gambiae. Nature 552, 96–100 (2017).
Näpflin, K. et al. Genomics of host-pathogen interactions: challenges and opportunities across ecological and spatiotemporal scales. PeerJ 7, e8013 (2019).
Davenport, E. E. et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir. Med. 4, 259–271 (2016). This article provides evidence for transcriptomically defined subphenotypes in sepsis that are informative for outcome and immune response state.
Burnham, K. L. et al. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. Am. J. Respir. Crit. Care Med. 196, 328–339 (2017).
Antcliffe, D. B. et al. Transcriptomic signatures in sepsis and a differential response to steroids from the VaNISH randomized trial. Am. J. Respir. Crit. Care Med. 199, 980–986 (2019). This article illustrates how sepsis subphenotypes are associated with differential response to therapy.
Scicluna, B. P. et al. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir. Med. 5, 816–826 (2017).
Astle, W. J. et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415–1429.e19 (2016).
Jonsson, S. et al. Identification of sequence variants influencing immunoglobulin levels. Nat. Genet. 49, 1182–1191 (2017).
Scepanovic, P. et al. Human genetic variants and age are the strongest predictors of humoral immune responses to common pathogens and vaccines. Genome Med. 10, 59 (2018).
Hammer, C. et al. Amino acid variation in HLA class II proteins is a major determinant of humoral response to common viruses. Am. J. Hum. Genet. 97, 738–743 (2015).
Roederer, M. et al. The genetic architecture of the human immune system: a bioresource for autoimmunity and disease pathogenesis. Cell 161, 387–403 (2015).
Wang, L. et al. An atlas of genetic variation linking pathogen-induced cellular traits to human disease. Cell Host Microbe 24, 308–323.e6 (2018).
Denny, J. C. et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 31, 1102–1111 (2013).
Fairfax, B. P. et al. Genetics of gene expression in primary immune cells identifies cell type–specific master regulators and roles of HLA alleles. Nat. Genet. 44, 502–510 (2012).
Quach, H. et al. Genetic adaptation and neandertal admixture shaped the immune system of human populations. Cell 167, 643–656.e17 (2016).
Fairfax, B. P. et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014). This article provides evidence of context-specific eQTLs on innate immune activation.
Lee, M. N. et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980 (2014).
Nédélec, Y. et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 167, 657–669.e21 (2016). This article shows evidence for pathogen- and population-specific genetic associations with differential gene expression.
Gilchrist, J. J. et al. Risk of nontyphoidal Salmonella bacteraemia in African children is modified by STAT4. Nat. Commun. 9, 1014 (2018).
Kornblihtt, A. R. et al. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 14, 153–165 (2013).
Rotival, M., Quach, H. & Quintana-Murci, L. Defining the genetic and evolutionary architecture of alternative splicing in response to infection. Nat. Commun. 10, 1671 (2019).
Patin, E. et al. Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors resource. Nat. Immunol. 19, 302–314 (2018).
Venet, F., Lukaszewicz, A. C., Payen, D., Hotchkiss, R. & Monneret, G. Monitoring the immune response in sepsis: a rational approach to administration of immunoadjuvant therapies. Curr. Opin. Immunol. 25, 477–483 (2013).
Li, Y. et al. A functional genomics approach to understand variation in cytokine production in humans. Cell 167, 1099–1110.e14 (2016).
Eisfeld, A. J. et al. Multi-platform ’omics analysis of human Ebola virus disease pathogenesis. Cell Host Microbe 22, 817–829.e8 (2017).
Mentzer, A. J., O’Connor, D., Pollard, A. J. & Hill, A. V. S. Searching for the human genetic factors standing in the way of universally effective vaccines. Philos. Trans. R. Soc. B Biol. Sci. 370, 20140341 (2015).
Png, E. et al. A genome-wide association study of hepatitis B vaccine response in an Indonesian population reveals multiple independent risk variants in the HLA region. Hum. Mol. Genet. 20, 3893–3898 (2011).
Desombere, I., Willems, A. & Leroux-Roels, G. Response to hepatitis B vaccine: multiple HLA genes are involved. Tissue Antigens 51, 593–604 (1998).
Kruger, A. et al. Hepatitis B surface antigen presentation and HLA-DRB1*- lessons from twins and peptide binding studies. Clin. Exp. Immunol. 140, 325–332 (2005).
Salazar, M. et al. Normal HBsAg presentation and T-cell defect in the immune response of nonresponders. Immunogenetics 41, 366–374 (1995).
McDermott, A. B., Zuckerman, J. N., Sabin, C. A., Marsh, S. G. E. & Madrigal, J. A. Contribution of human leukocyte antigens to the antibody response to hepatitis B vaccination. Tissue Antigens 50, 8–14 (1997).
Höhler, T. et al. The influence of major histocompatibility complex class II genes and T-cell Vbeta repertoire on response to immunization with HBsAg. Hum. Immunol. 59, 212–218 (1998).
Lambert, N. D. et al. Polymorphisms in HLA-DPB1 Are associated with differences in rubella virus-specific humoral immunity after vaccination. J. Infect. Dis. 211, 898–905 (2015).
Ovsyannikova, I. G., Pankratz, V. S., Larrabee, B. R., Jacobson, R. M. & Poland, G. A. HLA genotypes and rubella vaccine immune response: additional evidence. Vaccine 32, 4206–4213 (2014).
Jacobson, R. M., Ovsyannikova, I. G., Vierkant, R. A., Shane Pankratz, V. & Poland, G. A. Human leukocyte antigen associations with humoral and cellular immunity following a second dose of measles-containing vaccine: persistence, dampening, and extinction of associations found after a first dose. Vaccine 29, 7982–7991 (2011).
Newport, M. J. et al. Genetic regulation of immune responses to vaccines in early life. Genes. Immun. 5, 122–129 (2004).
O’Connor, D. et al. Common genetic variations associated with the persistence of immunity following childhood immunization. Cell Rep. 27, 3241–3253.e4 (2019). This article shows how GWAS can be applied to study persistence of immunity following childhood vaccination.
Zeller, T. et al. Genetics and beyond – the transcriptome of human monocytes and disease susceptibility. PLoS ONE 5, e10693 (2010).
Shaghaghi, M. et al. Combined immunodeficiency presenting with vaccine-associated paralytic poliomyelitis: a case report and narrative review of literature. Immunol. Invest. 43, 292–298 (2014).
Hernandez, N. et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J. Exp. Med. 216, 2057–2070 (2019). This article shows how rare IFNAR1 deficiency can lead to life-threatening complications following vaccination with live attenuated viruses.
Duncan, C. J. A. et al. Human IFNAR2 deficiency: lessons for antiviral immunity. Sci. Transl Med. 7, 307ra154 (2015).
Burns, C. et al. A novel presentation of homozygous loss-of-function STAT-1 mutation in an infant with hyperinflammation — a case report and review of the literature. J. Allergy Clin. Immunol. Pract. 4, 777–779 (2016).
Moens, L. et al. A novel kindred with inherited STAT2 deficiency and severe viral illness. J. Allergy Clin. Immunol. 139, 1995–1997.e9 (2017).
Docherty, A. B. et al. Features of 20 133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: prospective observational cohort study. BMJ 369, m1985 (2020).
Williams, F. M. et al. Self-reported symptoms of covid-19 including symptoms most predictive of SARS-CoV-2 infection, are heritable. Preprint at medRxiv https://doi.org/10.1101/2020.04.22.20072124 (2020).
RECOVERY Collaborative Group. Dexamethasone in hospitalized patients with covid-19 — preliminary report. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2021436 (2020).
COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718 (2020).
Casanova, J. L. et al. A global effort to define the human genetics of protective immunity to SARS-CoV-2 infection. Cell 181, 1194–1199 (2020).
Zhang, X. et al. Viral and host factors related to the clinical outcome of COVID-19. Nature https://doi.org/10.1038/s41586-020-2355-0 (2020).
Severe Covid-19 GWAS Group. Genomewide association study of severe covid-19 with respiratory failure. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2020283 (2020). This study illustrates the early successful application of GWAS in the COVID-19 pandemic, identifying the 3p21.31 locus and a potential role for the ABO blood-group system.
Shelton, J. F. et al. Trans-ethnic analysis reveals genetic and non-genetic associations with COVID-19 susceptibility and severity. Preprint at medRxiv https://doi.org/10.1101/2020.09.04.20188318 (2020).
Pairo-Castineira, E. et al. Genetic mechanisms of critical illness in Covid-19. Preprint at medRxiv https://doi.org/10.1101/2020.09.24.20200048 (2020).
Smemo, S. et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 (2014). This is a key article showing how GWAS variants may act at a distance.
Zeberg, H. & Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature https://doi.org/10.1038/s41586-020-2818-3 (2020).
Nguyen, A. et al. Human leukocyte antigen susceptibility map for SARS-CoV-2. J. Virol. https://doi.org/10.1128/JVI.00510-20 (2020).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Bastard, P. et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science 65, eabd4585 (2020).
Fellay, J. et al. A whole-genome association study of major determinants for host control of HIV-1. Science 317, 944–947 (2007).
Fellay, J. et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 5, e1000791 (2009).
Pereyra, F. et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330, 1551–1557 (2010).
Johnson, E. O. et al. Novel genetic locus implicated for HIV-1 acquisition with putative regulatory links to HIV replication and infectivity: a genome-wide association study. PLoS ONE 10, e0118149 (2015).
Wei, Z. et al. Genome-wide association studies of HIV-1 host control in ethnically diverse Chinese populations. Sci. Rep. 5, 10879 (2015).
McLaren, P. J. et al. Association study of common genetic variants and HIV-1 acquisition in 6,300 infected cases and 7,200 controls. PLoS Pathog. 9, e1003515 (2013).
McLaren, P. J. et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl Acad. Sci. USA 112, 14658–14663 (2015). This study provides high-resolution HLA association mapping for HIV viral load.
Ekenberg, C. et al. Association between single-nucleotide polymorphisms in HLA alleles and human immunodeficiency virus type 1 viral load in demographically diverse, antiretroviral therapy-naive participants from the strategic timing of antiretroviral treatment trial. J. Infect. Dis. 220, 1325–1334 (2019).
Sobota, R. S. et al. A chromosome 5q31.1 locus associates with tuberculin skin test reactivity in HIV-positive individuals from tuberculosis hyper-endemic regions in east Africa. PLoS Genet. 13, e1006710 (2017).
Luo, Y. et al. Early progression to active tuberculosis is a highly heritable trait driven by 3q23 in Peruvians. Nat. Commun. 10, 3765 (2019). This is a genetic study of TB in Peru showing significant heritability and novel genetic associations.
Qi, H. et al. Discovery of susceptibility loci associated with tuberculosis in Han Chinese. Hum. Mol. Genet. 26, 4752–4763 (2017).
Thye, T. et al. Common variants at 11p13 are associated with susceptibility to tuberculosis. Nat. Genet. 44, 257–259 (2012).
Grant, A. V. et al. A genome-wide association study of pulmonary tuberculosis in Morocco. Hum. Genet. 135, 299–307 (2016).
Timmann, C. et al. Genome-wide association study indicates two novel resistance loci for severe malaria. Nature 489, 443–446 (2012).
Liu, H. et al. Discovery of six new susceptibility loci and analysis of pleiotropic effects in leprosy. Nat. Genet. 47, 267–271 (2015).
Hu, Z. et al. New loci associated with chronic hepatitis B virus infection in Han Chinese. Nat. Genet. 45, 1499–1503 (2013).
Li, Y. et al. Genome-wide association study identifies 8p21.3 associated with persistent hepatitis B virus infection among Chinese. Nat. Commun. 7, 11664 (2016).
Miki, D. et al. HLA-DQB1*03 confers susceptibility to chronic hepatitis C in Japanese: a genome-wide association study. PLoS ONE 8, e84226 (2013).
Khor, C. C. et al. Genome-wide association study identifies susceptibility loci for dengue shock syndrome at MICB and PLCE1. Nat. Genet. 43, 1139–1141 (2011).
Dunstan, S. J. et al. Variation at HLA-DRB1 is associated with resistance to enteric fever. Nat. Genet. 46, 1333–1336 (2014).
Rivers, L. & Gaspar, H. B. Severe combined immunodeficiency: recent developments and guidance on clinical management. Arch. Dis. Child. 100, 667–672 (2015).
Amirifar, P., Ranjouri, M. R., Yazdani, R., Abolhassani, H. & Aghamohammadi, A. Ataxia-telangiectasia: a review of clinical features and molecular pathology. Pediatr. Allergy Immunol. 30, 277–288 (2019).
Chan, A. C. et al. ZAP-70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science 264, 1599–1601 (1994). This work provides important early evidence from a genetic study of patients with severe combined immunodeficiency supporting a functional role for ZAP70 in T cell receptor signal transduction.
Bonilla, F. A. et al. International consensus document (ICON): common variable immunodeficiency disorders. J. Allergy Clin. Immunol. Pract. 4, 38–59 (2016).
Suri, D., Rawat, A. & Singh, S. X-linked agammaglobulinemia. Indian J. Pediatr. 83, 331–337 (2016).
Yazdani, R. et al. The hyper IgM syndromes: epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin. Immunol. 198, 19–30 (2019).
Dinauer, M. C. Disorders of neutrophil function: an overview. Methods Mol. Biol. 1124, 501–515 (2014).
Ram, S., Lewis, L. A. & Rice, P. A. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin. Microbiol. Rev. 23, 740–780 (2010).
Tangye, T. & XLP, S. G. Clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J. Clin. Immunol. 34, 772–779 (2014).
Nahum, A. Chronic mucocutaneous candidiasis: a spectrum of genetic disorders. LymphoSign J. https://doi.org/10.14785/lymphosign-2017-0011 (2017).
Lanternier, F. et al. Deep dermatophytosis and inherited CARD9 deficiency. N. Engl. J. Med. 369, 1704–1714 (2013).
Parham, P. & Moffett, A. Variable NK cell receptors and their MHC class i ligands in immunity, reproduction and human evolution. Nat. Rev. Immunol. 13, 133–144 (2013).
Dawkins, R. et al. Genomics of the major histocompatibility complex: haplotypes, duplication, retroviruses and disease. Immunol. Rev. 167, 275–304 (1999).
Trowsdale, J. & Knight, J. C. Major histocompatibility complex genomics and human disease. Annu. Rev. Genomics. Hum. Genet. 14, 301–323 (2013).
Parham, P. & Ohta, T. Population biology of antigen presentation by MHC class I molecules. Science 272, 67–74 (1996).
Hedrick, P. W. Pathogen resistance and genetic variation at MHC loci. Evolution 56, 1902–1908 (2002).
Radwan, J., Babik, W., Kaufman, J., Lenz, T. L. & Winternitz, J. Advances in the evolutionary understanding of MHC polymorphism. Trends Genet. 36, 298–311 (2020).
Acknowledgements
Funding for this work was received from the UK National Institute for Health Research Oxford Biomedical Research Centre (to J.C.K. and A.M.), a Wellcome Trust Investigator Award (204969/Z/16/Z to J.C.K.), the Chinese Academy of Medical Sciences Innovation Fund for Medical Science (grant number 2018-I2M-2-002 to J.C.K.), the Croucher Foundation (to A.J.K.) and a Wellcome Trust Grant (090532/Z/09/Z) to core facilities at the Wellcome Centre for Human Genetics.
Author information
Authors and Affiliations
Contributions
The authors contributed to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
COVID-19 Host Genetics Initiative: https://www.covid19hg.org/
COVID Human Genetic Effort: https://www.covidhge.com/
NHGRI–EBI GWAS Catalog: https://www.ebi.ac.uk/gwas/
UK Biobank of COVID-19: https://www.ukbiobank.ac.uk/learn-more-about-uk-biobank/covid-19-hub
Glossary
- Penetrance
-
The proportion of individuals with a particular genotype that also has an associated phenotype.
- Linkage disequilibrium
-
The non-random association of alleles at different loci in a population, often owing to physical proximity of the loci on a chromosome.
- Heterozygous advantage
-
Where heterozygotes have fitness advantages over homozygotes.
- Haplotypes
-
Sets of genetic variants that tend to be inherited together.
- Allotypes
-
A group of alleles that encode a unique protein sequence.
- Population structure
-
Also referred to as population stratification. The presence of differences in allele frequencies between subpopulations in a population.
- Expression quantitative trait locus
-
(eQTL). A genomic region that is associated with a proportion of variation in mRNA expression levels.
- Balancing selection
-
A selection process that maintains genetic diversity.
- Imputation
-
The process of replacing missing information on the basis of known information. In genetics, this refers to the statistical inference of unobserved genotypes based on known genotypes.
- Type I errors
-
The rejection of true null hypotheses in hypothesis testing (that is, false positives).
- Principal component analysis
-
A mathematical technique that summarizes data into components that explain the variance in the data. It is used in genome-wide association studies to infer cryptic population structure and/or outlier individuals.
- Linear mixed model algorithms
-
A statistical model that contains both fixed and random effects. This is in contrast to linear models, which contain only fixed effects.
- Complement
-
A system of plasma proteins in the innate immune system that react with each other to opsonize pathogens and induce inflammatory responses.
- Haploinsufficient
-
A model of dominant gene action where a single functional copy of the gene is inadequate for normal cellular function.
- Pyroptosis
-
An inflammatory form of programmed cell death occurring most commonly following infection with intracellular pathogens.
Rights and permissions
About this article

Cite this article
Kwok, A.J., Mentzer, A. & Knight, J.C. Host genetics and infectious disease: new tools, insights and translational opportunities. Nat Rev Genet 22, 137–153 (2021). https://doi.org/10.1038/s41576-020-00297-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-020-00297-6
This article is cited by
-
Increased risk of hearing loss associated with macrolide use: a systematic review and meta-analysis
Scientific Reports (2024)
-
Genetic characterisation of the Connemara pony and the Warmblood horse using a within-breed clustering approach
Genetics Selection Evolution (2023)
-
Mapping the epigenomic landscape of human monocytes following innate immune activation reveals context-specific mechanisms driving endotoxin tolerance
BMC Genomics (2023)
-
The dynamic interface of genetics and immunity: toward future horizons in health & disease
Genes & Immunity (2023)
-
Combinatorial design of nanoparticles for pulmonary mRNA delivery and genome editing
Nature Biotechnology (2023)
