
Abstract
Despite rapid progress in characterizing the role of host genetics in SARS-Cov-2 infection, there is limited understanding of genes and pathways that contribute to COVID-19. Here, we integrate a genome-wide association study of COVID-19 hospitalization (7,885 cases and 961,804 controls from COVID-19 Host Genetics Initiative) with mRNA expression, splicing, and protein levels (n = 18,502). We identify 27 genes related to inflammation and coagulation pathways whose genetically predicted expression was associated with COVID-19 hospitalization. We functionally characterize the 27 genes using phenome- and laboratory-wide association scans in Vanderbilt Biobank (n = 85,460) and identified coagulation-related clinical symptoms, immunologic, and blood-cell-related biomarkers. We replicate these findings across trans-ethnic studies and observed consistent effects in individuals of diverse ancestral backgrounds in Vanderbilt Biobank, pan-UK Biobank, and Biobank Japan. Our study highlights and reconfirms putative causal genes impacting COVID-19 severity and symptomology through the host inflammatory response.
Similar content being viewed by others


Introduction
Coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was first reported in December 2019 and rapidly progressed into a global pandemic1. Approximately 10–20% of patients known to be infected with the respiratory virus SARS-CoV-2 need hospitalization2, and among them, a fraction face significant morbidity and mortality3. In addition to social determinants of health, the host’s genetic background is likely to contribute in explaining such diverse clinical outcomes. While previous efforts have demonstrated the role of ACE2 and TMPRSS2 in host defense against COVID-194, there remains limited understanding for the role of host genetics contributing to severe COVID-19 outcome variability.
Genome-wide association studies (GWAS) have provided an opportunity to characterize the role of host genetics underlying COVID-19 risk and severity5,6,7,8,9. Initial efforts8 identified variants associated with COVID-19 related respiratory failure at the 3p21.31 and 9q34.2 regions, providing evidence for immune- and blood-group-related mechanisms. Subsequent studies leveraging much larger preexisting genetic cohorts have since replicated these results and identified more than 15 genomic regions associated with severe COVID-19 outcomes5,6,7,9. Due to extensive linkage-disequilibrium patterns, the association signals at identified risk regions typically span multiple genes (e.g., SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6, and XCR1 at 3p21.31), which makes identifying target genes challenging. To this end, previous work9 integrated lung and whole-blood transcriptomic data together with GWAS to identify genes associated with COVID-19 severity, however, this study had limited statistical power due to the relatively small sample size (2244 critically ill patients).
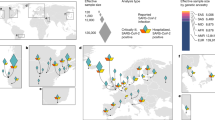
Here, to map genes and pathways involved in COVID-19 severity we integrate mRNA expression, splicing, and protein abundance data (n = 18,502) with data from a GWAS of COVID-19 related hospitalization (n = 7885 cases, 961,804 controls; Freeze 4 COVID-19 HGI excluding 23andMe participants5,6,7,8,9). We perform mRNA/splicing/protein transcriptome-wide association studies (TWAS/spTWAS/PWAS) to identify 27 genes across 13 genomic regions whose genetically predicted activity is associated with COVID-19 related hospitalization. We further investigate the clinical and functional role of these 27 genes using phenome-wide (PheWAS) and laboratory-wide (LabWAS) association scans to map their role in immunity and blood biomarkers in European and African ancestry patients from the Vanderbilt University Medical Center biobank (BioVU; n = 85,460). We replicate phenotypes identified from BioVU in secondary cohorts of multiethnic individuals from the Pan-UK Biobank (980 Admixed American, 6636 African, 8876 Central/South Asian, 2709 East Asian, 420,531 European, and 1599 Middle Eastern) and Biobank Japan (up to 200,000 Japanese participants). Taken together, our results suggest multiple molecular mechanisms contributing to severe COVID-19 outcomes and highlight potential therapeutic targets (Fig. 1).

We performed a multilevel transcriptome-wide association study (TWAS) of genetically regulated expression (GReX) by integrating gene, splicing, and proteome expression data with genome-wide summary statistics of COVID-19 hospitalization. For the significant genes identified, we performed pathway analysis, allele-specific imbalance, and gene-based PheWAS of clinical phenotypes and LabWAS of clinical laboratory measures using individual-level GReX values in Vanderbilt Biobank (BioVU). For the significant traits identified, we performed a second, SNP-based PheWAS in multi-ancestry Pan-UKBiobank and Biobank Japan.
Results
TWAS identifies genes for COVID-19 related hospitalization
To identify genes underlying COVID-19 related hospitalization, we tested the predicted expression of 22,207 genes across 49 tissues for association with COVID-19 related hospitalization (see Methods). We identified 123 associations representing 21 genes across 45 tissues at eight independent genomic regions (p value <2.3E-6; Fig. 2 and Supplementary Data 1, 2). Next, to improve statistical power, we tested for association between predicted gene expression levels from multiple tissues simultaneously with COVID-19 related hospitalization GWAS. Of the 22,207 tested genes, we identified 14 genes across ten genomic regions, which consisted of two additional genes—XCR1 and DNAH3 (p value <1.4E-06; Supplementary Fig. 1 and Supplementary Data 3). Overall, we found 23 TWAS-based gene associations across ten genomic regions.

A Manhattan plot of genes associated via multiple-tissue TWAS. Each data point represents a gene grouped by chromosome (x-axis) and lowest p value (y-axis) of the gene across significant tissues. B Distribution of z-scores across significant gene-tissue pairs. Genes are grouped based on chromosomes (y-axis) and respective tissues (x-axis). Significant genes are shown as pink triangles, wherein triangles facing up and down represent positive and negative z-scores, respectively.
To find additional support for genetic regulation of identified susceptibility genes by SNPs at risk regions, we tested the 123 gene/tissue pairs identified in single tissue scans for allelic imbalance within 1 Mb GWAS regions (see Methods). We identified nine genes (ABO, CCR2, CXCR6, FYCO1, IFNAR2, IL10RB, LZTFL1, OAS1, and OAS3) with evidence of allelic imbalance at COVID-19 GWAS risk variants, with three genes (ABO, OAS3, and IL10RB; see Supplementary Fig. 2 and Supplementary Data 4) when restricted to leading GWAS index variants (p value <0.05/21). Together, these results further support a model where risk is conferred through transcriptomic dysregulation of key target genes. Next, we focused on the impact of alternative splicing regulation for COVID-19 severity and performed a multi-tissue splicing transcriptome-wide association study (spTWAS; see Methods). Overall, we tested 131,376 splice sites of predicted alternative-splicing expression across 49 tissues for association with COVID19 related hospitalization and identified 420 associations representing 43 splice variants for 11 genes across 49 tissues and five genomic regions (see Fig. 3 and Supplementary Data 5, 6). Next, we performed a multi-tissue analysis (see Methods) and identified 34 splice variants for 12 genes (two genes—IFIT3 and GNL3 not identified in single-tissue scans) across 40 tissues (p value <3.7E-07; see Supplementary Fig. 3 and Supplementary Data 7).

A Manhattan plot of genes associated via multiple tissue spTWAS. Each data point represents a splice site grouped by chromosome (x-axis) and lowest p value (y-axis) of the splice site for each gene across significant tissues. The annotated genes to the splice site are labeled. Significant splice sites are shown as pink diamonds. B Distribution of splice sites across significant site-tissue pairs. The genes annotated to splice sites are grouped based on chromosomes (y-axis) and respective tissues (x-axis).
Comparing genes identified from TWAS (23 genes) and spTWAS (13 genes), nine genes were implicated by both approaches—LZTFL1, DPP9, IL10RB, IFNAR2, OAS3, FYCO1, ABO, OAS1, and XCR1. Alternative splicing had stronger overall association signals at the nine genes in common (p value = 2.17E-09), with 5/9 genes showing greater signals on average (p value <0.05/9; see Supplementary Data 8).
Next, we interrogated the role of genetic regulation of protein abundances and performed a proteome-wide association study (PWAS) using 1031 predictive models of plasma proteins fitted from population data in the INTERVAL study (N = 3301; see Methods)10. Of the 1031 tests performed, two genes (ABO and OAS1) were significantly associated with COVID-19 related hospitalization (p value <4.85E-5; Fig. 1 and Supplementary Data 9).
We employed gene set enrichment analysis (GSEA)11 to identify statistically overrepresented pathways for the 27 genes identified through multilevel transcriptomic integration. Significant pathways, among others, include cytokine–cytokine receptor interaction (pFDR = 3.13E-10; Supplementary Fig. 4), chemokine signaling pathway (pFDR = 9.89E-9), and JAK-STAT signaling pathway (pFDR = 1.87E-2) (Supplementary Table 1). Analyzing lung and whole-blood tissue-specific TWAS z-scores of genes that belong to the identified pathway set, we found consistent signals of downregulation, suggesting that decreased expression levels at these genes increase the severity of COVID19 outcomes (Supplementary Fig. 4). We also performed gene regulatory network (GRN) identification, connecting transcription factors (TFs) to gene targets. Retaining TFs with most of the number of gene targets, we observed 30 TFs that target 17 genes (Supplementary Fig. 5).
Studies have reported decreased lung function in COVID-19 affected individuals. Therefore, we investigated lung traits that share a locus (colocalize) with the novel genes identified in this study, and observed GNL3 colocalized with measures of lung function i.e., forced expiratory volume (FEV; see Methods: H4 PP: 93%) and forced vital capacity (FVC; H4 PP:89.4% Supplementary Data 10).
Phenome- and laboratory-wide association scans highlight functional role for the 27 genes
We investigated the potential functional role of the 27 TWAS genes using data of 1404 clinical phenotypes for N = 70,439 individuals of European ancestry using the Vanderbilt Biobank, BioVU (see Methods; Fig. 4 and Supplementary Data 11). Overall, 40 clinical phenotypes were significantly associated with genetically predicted ABO, IFNAR2, and CCR1 expression levels; ABO accounted for the majority (30 out of 40) of the associations. Top associations with genetically-derived ABO gene expression were driven by circulatory system phenotypes, including acute pulmonary heart disease, deep vein thrombosis, other venous embolism and thrombosis, pulmonary heart disease, and acute pulmonary thrombosis and infarction (OR = 1.47; p value = 3.97E-11). IFNAR2 was associated with migraine (OR = 1.35; p value = 4.10E-06) and with throat pain (OR = 2.05; p value = 2.62E-05; Supplementary Table 2). Across the 17 phenotype categories, we found circulatory system-related phenotypes were enriched for association (see Methods; 7.23-fold enrichment, p value = 8.62E-22, Supplementary Data 11). We repeated an enrichment analysis using all association data, regardless of statistical significance, and observed circulatory- and infectious disease-related phenotypes strongly enriched for association signal on average (p value <3.9E-44; Supplementary Table 3).

Each data point represents phenotypic associations with the genetically regulated expression of gene-tissue pairs. The data points are grouped and color-coded by phenotype groups (x-axis) and −log10(p value) (y-axis). The dashed line represents the Bonferroni threshold, and the most significant gene-phenotype associations across all significant tissues are text-labeled.
Next, we focused on laboratory results for N = 70,337 individuals of European ancestry using the Vanderbilt Biobank, BioVU (see Methods). For the 323 laboratory traits tested, we found 32 labs significantly associated with four genes (ABO, IFNAR2, KEAP1, and SLC6A20; p value <1.55E-04; see Fig. 5 and Supplementary Data 12). Of these, ABO captured 27/32 significant associations (mean OR = 1.33; p value = 8.02E-14 < 1.40E-04). Genetically-predicted ABO expression was associated with various measures of blood and platelet count, coagulation factors, and ferritin in blood, as well as labs measuring immune and metabolic function (Supplementary Data 12). Genetically-predicted IFNAR2 expression was negatively associated with creatine kinase (OR = 0.89; p = 5.85E-05; Supplementary Data 12). Genetically-predicted KEAP1 expression was positively associated with total cholesterol, and non-high-density lipoprotein levels (beta = 0.50, p = 3.72E-05). SLC6A20 genetically-predicted expression was negatively associated with basophil volume in the blood (beta = −0.19, p = 4.04e-5) and magnesium volume in serum/plasma (beta = −0.26, p = 1.04e-4). Across the 12 broad lab definitions in our data, among significant LabWAS findings we observed enrichment for blood-related lab measurements (see Methods; 4.21-fold enrichment, p value = 1.23E-11; Supplementary Table 4). When extending enrichment analyses to all associations, we found blood- (see Methods; p value = 9.23E-22; Supplementary Table 5) and immune-related labs (p value = 2.81E-14) displayed the greatest enrichment, with toxicology-, urinary-, and cancer-related labs exhibiting significant depletion of signal (Supplementary Table 5).

Each data point represents laboratory-trait associations with genetically regulated expression of gene-tissue pairs. The data points are grouped and color-coded by clinical laboratory-test groups (x-axis) and −log10(p value) (y-axis). The dashed line represents the Bonferroni threshold, and the most significant gene-laboratory trait associations across all significant tissues are text-labeled.
Cross-ancestry phenotypic comparisons
To perform cross-ancestry validation of the clinical and laboratory phenotypes implicated in the European-based results, we performed phenome-wide association study (PheWAS) and LabWAS in the N = 15,123 individuals of African ancestry in the BioVU records. Of the 32 identified laboratory measures, we found 22 ABO-associated labs replicated at nominal levels (p value <0.05) with five replicating after adjusting for the number of tests performed (p value <0.05/32). We attribute a lack of statistical power to the 17 phenotypes that did not replicate after multiple testing corrections. Effect sizes across ancestries were highly concordant (slope = 0.87, 95CI [0.73, 1.01], p value = 1.66-13; see Methods; Supplementary Data 12 and Supplementary Fig. 6), with no individual gene/lab pair demonstrating evidence of significant heterogeneity (p value <0.05/32). Considering the 40 clinical phenotypes identified in participants of European ancestry, we found none that replicated considering a Bonferroni-corrected significance in African Ancestry individuals. This was largely due to reduced statistical power from smaller sample sizes, as estimated effect sizes were similar across ancestries (slope = 0.33, 95CI [0.25, 0.41], p value = 6.72E-10; see Methods; Supplementary Data 11 and Supplementary Fig. 6). These cross-ancestry analyses suggest similar effects of predicted expression on relevant clinical and laboratory phenotypes.
We next sought to test cross-ancestry replication of identified phenotypes in the Pan-UK Biobank and Biobank Japan. Briefly, we performed PheWAS using LD-independent eQTL/sQTL SNPs (see Methods) of TWAS-identified genes with significant phenotype associations in GReX-based PheWAS from BioVU. We identified 233 FDR significant SNP-phenotype results dominated by the associations between ABO (214/233, 91.8%) SNPs and blood differential tests such as basophils and monocytes. A subset of 80/233 (34.3%) FDR significant associations also were nominally significant in at least one population of non-European ancestry (p value <0.05; Supplementary Data 13, 14). There were six instances of FDR significant effect estimate heterogeneity across ancestries, all of which involved ABO SNPs and the biomarker alkaline phosphatase or erythrocyte properties. We next tested how significant EUR effect estimates reflect SNP effects across ancestries. We found that EUR SNP effects significantly predicted SNP effects in six global ancestry groups (maximum prediction in AMR; Supplementary Data 15 and Supplementary Fig. 7).
Discussion
COVID-19 disease is characterized by wide variability in presentations and severity. We integrated multitiered regulatory information with publicly available variant-level data to identify genes associated with COVID-19 related hospitalization. To investigate the potential clinical relevance of these findings, we performed a phenome-wide and lab-wide assessment of the genetically predicted mRNA expression value of each gene that was significantly associated with COVID-19 related hospitalization. We further examined these associations across diverse ancestries and found nominal replication of blood cell traits in diverse ancestral cohorts of Pan-UKBB, BBJ, and an African American population in BioVU.
All three TWAS approaches (mRNA expression, splicing, and protein expression) identified two genes—ABO at 9q34.2 and OAS1 at 12q24.13 (Supplementary Fig. 8) PheWAS results implicated ABO in several thrombotic and coagulation-related phenotypes. Thrombotic complications are reported to be both risk factors and sequelae to COVID-19 diagnosis. For example, coagulopathic conditions such as venous thromboembolism12,13, deep vein thrombosis14, and pulmonary heart disease and embolism15,16 constitute of more than 30% prevalent disorders in hospitalized COVID-19 patients. Abnormal blood cell indices are a common denominator shared by severe COVID-1917 and thrombotic disorders18. These phenotypic observations are further supported by lab-trait associations which showed predicted expression of ABO was associated with coagulation factor VIII. This factor is critical for thrombotic homeostasis regulated by its carrier protein—von Willebrand factor19,20. Analysis in individuals of both European and African ancestry supported predicted ABO expression associating with blood differential tests including mean corpuscle volume, monocyte count, and erythrocytes. ABO gene encodes for blood type, and several genes have reported association of blood groups with COVID-19 infection risk21,22,23,24,25,26. ABO variants affect the von Willebrand factor and factor VIII levels27. Studies have also reported differing prevalence of thrombotic and vascular dysfunction in different blood groups, making the relationship of ABO with COVID-19 difficult to ascertain28,29,30. Furthermore, we also observed KEAP1, which is a regulator of cholesterol synthesis31. The ACE2 receptor for SARS-Cov-2 viral binding and uses lipids as docking sites32. Increased viral binding affects the instability of the arterial vasculature. Our identification of KEAP1 through splicing regulation in heart tissue and transcriptomic-phenotype association with cholesterol traits aligns with the hypothesized role of cholesterol in COVID-19 pathogenicity32. Furthermore, several studies have reported obesity as a causal risk factor to COVID-19 severity33,34,35.
We observed that associated phenotypes and gene functions converged on cytokine–cytokine receptor signaling involved in inflammatory response (e.g., CXCR6, CCR9, CCR5, XCR1, IFNAR2, IL10RB), and on JAK-STAT signaling pathways involved in antiviral host response (e.g., IFNAR2, OAS1, OAS3). IFNAR2 encodes the interferon-alpha/beta receptor beta chain and is responsible for stimulating interferon response which is critical for antiviral immunity previously observed in influenza viral infection36. IFNAR2 is hypothesized to modulate the immune response to COVID-1937,38,39 and interferon deficiency is reportedly associated with severe symptoms of COVID-1940,41,42. These findings are reinforced by results showing that reduced expression (observed in 14 of the 16 significant tissues) of IFNAR2 is associated with COVID-19 related hospitalization. Given its known function43, evidence supports a direct role of IFNAR2 in an innate antiviral response to COVID-1937,38,39,40,41,42. Genetically predicted expression of IFNAR2 was associated with migraine in BioVU patients without severe COVID-19. Given that more than 10% of the COVID-19 diagnosed individuals requiring hospitalization reported migraine and headache symptoms44,45, reduced host expression of IFNAR2 may also modulate risk for migraine symptoms in the context of severe COVID-19 infection.
For the pathways identified, we observed a negative TWAS association signal on average for genes in lung tissues, reflecting downregulation for all 13 pathways except the JAK-STAT signaling pathway. The IL10RB and IFNAR2 share an inverse relationship for viral infection, wherein macrophages have been reported to secrete higher IL10 in comparison to IFNAR246. Interestingly, we found cytokine and chemokine genes (CCR2, CCR3, IFNAR2, and OAS1)47 were downregulated48 in lung and whole blood, while IL10RB and OAS3 presented the opposite directions of dysregulation (Supplementary Fig. 4). These inflammatory pathways are further supported by a gene-regulatory network of immunomodulating TFs—NRF1 and IRF that target identified genes (Supplementary Fig. 5). Furthermore, we found evidence for GNL3 eQTL colocalizing with pulmonary function traits such as FEV and FVC which are decreased in COVID-19 patients49. GNL3 is involved in Wnt-B-catenin signaling and affects epithelial-mesenchymal transition, and increased expression is associated with lower survival in lung carcinoma50.
In summary, our findings are consistent with a model where genetic variations in immunogenetic loci reported by the previous GWAS6,8,9, are associated with hospitalization of COVID-19 via their impact on transcriptomic regulation and its downstream consequences. Furthermore, the transcriptome-wide analysis identified six novel loci, among others, consisting of genes—IFIT3, KEAP1, and GNL3 demonstrating their putative association with COVID-19 hospitalization outcome. We further validated previous and novel associations in a multi-ancestry cohort from Vanderbilt BioVU, showing that regulatory variation of identified targets is associated with clinical markers of inflammation, muscle weakness, and pulmonary events. Our cross-ancestry analysis showed how regulatory variants present correlated effect sizes among COVID-19-related phenotypes, expanding our understanding of the interpopulation variability in the susceptibility to the disease.
Previous studies have identified ACE2 and TMPRSS2 as targets of SARS-Cov-2 entry and pathogenesis4. While we did not observe a significant association between genetically regulated expression of ACE2 and TMPRSS2 with COVID-19 hospitalization in our data, this difference can be explained by the different phenotypic definitions. Our study focused on the severity in the host response to SARS-Cov-2 infection instead of the presence/absence of COVID-19 diagnosis. Furthermore, we investigated expression attributed to QTLs, instead of overall gene expression as captured by RNA-sequencing or microarray studies. This may indicate ACE2 and TMPRSS2 transcriptomic changes are related to the consequences of SARS-Cov2 and not to the molecular predisposition to develop severe COVID-19 symptoms.
Our results are consistent with previous studies investigating the impact of inflammation on severe COVID-19 outcomes; however, we note there are limitations. First, TWAS analyses rely on SNP-based predictive models of mRNA and alternative splicing trained using mostly European-ancestry individuals in GTEx v851,52,53. While consistent with the ancestry makeup of COVID-19 HGI GWAS5 (https://www.covid19hg.org/), applying these models to non-European individuals (e.g., African Americans in BioVU) will result in loss of power or bias due to different underlying linkage disequilibrium patterns. Second, TWAS uses mRNA, alternative splicing, or protein levels in bulk tissue, with cell-type effects likely to be missed54. Third, TWAS assumes additivity of SNP effects on gene expression and downstream hospitalization risk, which ignores the possibility of epistatic and gene-environment interactions contributing to COVID-19 related hospitalization risk. Fourth, while it would be ideal to compare risk and severity prediction using the measured expression, our study leverages large-scale functional datasets to maximize statistical power for TWAS using predicted gene expression. Fifth, our work integrates eQTL from multiple tissues in a joint model to increase statistical power55,56,57. Multiple lines of evidence have demonstrated shared regulatory architectures across tissues;58,59,60 however, we note that tissues unrelated to the pathophysiology of COVID-19 related hospitalization do not necessarily harbor risk, but reflect expression patterns correlated with those found in disease-relevant tissues. Finally, our study focuses on the host genetic factors that contribute to severe COVID-19 but did not incorporate the social determinants of health that are known to influence risk for severe COVID-19. The biological insights identified here should not be interpreted as explanatory factors for the disparity, but instead as key genomic pathways potentially modulating host response to SARS-Cov-2 across populations.
Functional studies of key genes identified are needed to identify mechanisms through which these genes influence COVID-19 related hospitalization. Additionally, while well-powered molecular genetic datasets in diverse populations often lag behind European-ancestry counterparts, massive collaborative science efforts such as the HGI-19 continue to accumulate new datasets to address this discrepancy. Leveraging population eQTL/spQTL/pQTL data with ancestry-matched COVID-19 GWAS will be crucial in identifying and understanding mechanisms underlying COVID-19 related hospitalization. In conclusion, our work raises specific hypotheses relating host genetic variation to the symptom and lab-trait profiles thereby focusing efforts for future drug repurposing and therapeutic discovery research.
Methods
COVID19-HGI genome-wide association summary statistics
We downloaded GWAS summary statistics for severe COVID-19 outcomes meta-analyzed across 21 studies (hospitalized N = 7885; population N = 961,804). A detailed description of the contributing studies, meta-analyses, and primary GWAS results for several COVID-19-related phenotypes are presented at https://www.covid19hg.org/results/, specifically the Freeze 4-October 2020 results File: COVID19_HGI_B2_ALL_leave_23andme_20201020.txt.gz. Summary statistics did not include 23&Me cohort results, and their sample size was removed from the final sample reported. Genome-wide association statistics consisted of inverse-variance meta-analyzed log-odds ratios and their standard errors to compute a final Wald statistic and p value. Most of the individuals of the contributing studies to meta-analysis genomic study were of European descent (93%). We performed strict quality control on GWAS data, by filtering statistics at palindromic variants, harmonizing variants with GTEx v8 European-panel genotypes51. Our quality control procedure resulted in a final count of 10,340,768 autosomal genetic variants with summary statistics.
TWAS and spTWAS using models of predicted gene and alternative splicing expression
To perform TWAS and spTWAS we leveraged pretrained prediction models fitted in GTEx v8 data (49 tissues, N = 838) using the fine-mapping software DAP-G with a biologically informed prior, Multivariate Adaptive Shrinkage in R (MASHR). For detailed information regarding molecular, genetic, and phenotypic data in GTEx v8, please see ref. 51. Prediction models for each tissue were integrated with COVID19-HGI GWAS data using the software S-PrediXcan56. In total, we tested 655,563 and 1,728,429 models of total expression and alternative splicing, respectively for association with severe COVID19 outcomes, however, owing to the significant amount of correlation across tissues, we used a per-tissue Bonferroni correction threshold in our multi-tissue analyses (see Supplemental Data 2). To combine association statistics across all tissues while adjusting for tissue–tissue correlation, we used S-MultiXcan55. For 22,206 genes, we also performed a joint multi-tissue approach using S-MultiXcan which accounts for tissue correlation and boosts statistical power. Here, we applied a single Bonferroni correction of 0.05/22,206.
PWAS using models of predicted expression
To perform a protein-wide association study (PWAS) using predicted protein expression, we fitted predictive models using genetic and plasma proteins from European-ancestry individuals in the INTERVAL study (N = 3301)10. We performed quality control on genotype data and kept only biallelic SNPs with MAF ≧0.01, HWE p > 5e-5, imputation quality INFO >0.6, and were annotated in HapMap3. Plasma proteins had undergone strict quality control and adjustment in the original study10. We fit predictive 3222 predictive models for 3170 proteins using genotypes within 1 Mb flanking the gene body (i.e., ±500 kb gene start and stop). For measured proteins consisting of multiple monomers (i.e., dimer, trimer, etc.), we fit multiple predictors, each restricted contributing gene’s region. We included the following covariates into all downstream models of protein abundance: age, sex, duration of blood processing, the top three genotyping PCs, contributing cohort, and top four protein PCs). To reduce the number of tests and increase statistical power, we restricted to genes whose protein levels exhibited evidence of genetic control by testing for nonzero cis-heritability (p value <0.05) using GCTA. Our final set of local/cis-based predictors resulted in 1031 models of protein with significant cis-SNP heritability (p value <0.05). We fit penalized linear models using SuSiE61 and performed downstream PWAS using the tool FUSION52 using quality-controlled COVID19-HGI genome-wide association statistics.
Allelic specific expression
To determine the allelic effect of GWAS SNPs (p value <5e-5) on identified susceptibility genes, we used haplotype-level ASE data with WASP filtering62 from the GTEx v8, containing 15,253 samples spanning over 49 human tissues and 838 individuals51,63. We used haplotype-aggregated allelic expression generated by phASER64. To assess the cis-acting regulatory effect of expression imbalance between the alleles in heterozygous individuals, we compared allelic imbalance between the individuals homozygous and heterozygous for each SNP. All individuals with minimum coverage of eight reads (with one pseudocount added) were included. An allelic imbalance was quantified as the log-ratio between the two allelic counts or log allelic fold change (log aFC)65 and to ensure robustness to rare variant effects and phasing errors the absolute value of log aFCs are compared, using a one-sided rank-sum test. We used a gene-level Bonferonni correction for the total number of genes tested (p value <0.05/21).
Pathway identification using GSEA and TF-gene network
We tested 27 genes for statistical overrepresentation of pathways using GSEA11. We analyzed gene sets of KEGG pathways using ShinyGO66. Significance was determined based on a false discovery rate of 5%. To identify the regulatory network, we extracted TF-gene targets using RegNetwork67 and created a minimum network, which connects most of the query genes by computing the shortest path between each pair of TF nodes with R packages—igraph and network.
Colocalization of identified eQTL loci with traits
We investigated novel genes identified in this study and colocalized traits using OpenTarget Genetics68, wherein posterior probability (PP) of hypothesis (H4) that two loci are shared under a single causal variant. We report colocalizing traits, only if the phenotype had the keyword “lung” among traits, and H4:PP of >80%. The colocalization was performed using a coloc-R package and described elsewhere68.
Calculating predicted expression in BioVU
To examine the clinical implications of the genes identified in our TWAS analysis in an independent population, we calculated genetically regulated expression for 85,613 individuals in the Vanderbilt biobank, BioVU. The BioVU population consists of individuals who receive care at Vanderbilt University Medical Center and choose to opt-in to the BioVU research study. A detailed description of program operations, ethical considerations, and continuing oversight and patient engagement have been published69. Individuals within our BioVU population have an average age of 55.17 years old (median = 59, range = 3 to 112) and 56.9% are female (43.1% male). Genotype information, as well as de-identified electronic medical records, are available for research purposes for these individuals, including information such as International Classification of Diseases, ninth and tenth editions (ICD9/10) billing codes, physician notes, and lab results. Genotype data for the BioVU population was generated using the Illumina MultiEthnic Genotype Array (MEGAEX) for 94,474 individuals. The genotype data were imputed into the HRC reference panel using the Michigan imputation server. Imputed data and the 1000 Genome Project data were combined to carry out principal component analysis (PCA) to identify individuals of European and African ancestry for analysis. We used the best-performance models from PrediXcan, UTMOST, and JTI approaches to impute the expression of 27 genes across 49 tissues53,70,71. These models were trained using the GTEx version 8 data51.
BioVU PheWAS
To better understand the phenotypic consequences of dysregulated mRNA expression across our genes of interest, we performed a PheWAS72 including patients in the Vanderbilt EHR and linked biobank, BioVU. Phenotypes in BioVU are represented as phecodes, which are assigned as a dichotomous trait and are a hierarchical clustering of the International Classification of Diseases (ICD9/ICD10) codes. For each phenotype, we required a minimum number of 100 cases for inclusion in our PheWAS analyses, which resulted in testing 1404 phecodes in 70,439 individuals of European ancestry and 740 phecodes in 15,174 individuals of African ancestry. We used the PheWAS package in R to perform logistic regression to identify the phecodes that are significantly associated with imputed gene expression after adjusting for sex, age, and the top ten principal components from genetic data to control for population stratification (Denny et al. 2010, 2013). We corrected for the number of tests (i.e., 0.05/1404 = 3.56e-05) to determine statistical significance.
BioVU biomarker LabWAS
The lab-wide association scan (LabWAS)73 allows us to screen clinical lab tests from the Vanderbilt University Medical Center EHR. For each gene identified in the TWAS analyses, we tested the association between its predicted gene expression and all clinical labs. We applied the QualityLab cleaning pipeline73 with settings to yield median age-adjusted (residual taken after regressing the cubic splines of age with four knots) inverse normal quantile transformed lab values (to control for skewness and non-normality). We screened across all labs with measurements for at least 100 individuals, which resulted in testing 323 labs in 70,337 individuals of European ancestry and 241 labs in 15,123 individuals of African ancestry. The lab tests are divided into 12 subcategories; blood, metabolic, endocrine, kidney, immune, liver, urinary, OB/gyn, toxicology, cardiovascular, and cancer. Our analyses included the covariates age, sex, and top ten principal components from genetic data to adjust for genetic ancestry. We used a Bonferroni-corrected threshold accounting for the number of labs present in the associations tested (i.e., 0.05/323 = 1.55e-04).
PheWAS and LabWAS category enrichment analyses
We tested for enrichment of association signals across the clinical categories of phenotypes (laboratories) in PheWAS (LabWAS) in two ways. First, we performed a hypergeometric test using phenotypes/labs that were labeled as significant/not-significant per category. Second, we performed a relaxed test that considers the average magnitude of the association signal in a given category. Here, we computed the mean \({\chi }^{2}\) association statistic per phenotype (laboratory) category and bootstrapped its standard error using 2000 bootstraps. Our enrichment (depletion) statistics for a phenotype (laboratory) category were the difference between its mean \({\chi }^{2}\) from 1 (the expected \({\chi }^{2}\) under the null) divided by the bootstrapped standard error. We used a Bonferroni adjusted p value <0.05/k to determine enrichment or depletion for either aproach, where k = 17 for PheWAS category enrichment tests and k = 12 for LabWAS category enrichment tests.
Cross-ancestry gene effects
We compared effect-sizes of predicted expression on phenotypes and laboratories estimated in European-ancestry patients from the BioVU with estimates obtained from of African-ancestry records. To do so, we performed a weighted linear regression, \({\hat{\beta }}_{A,i}={\hat{\beta }}_{E,i}\alpha +{\epsilon }_{i}\), where \({\hat{\beta }}_{\cdot ,i}\) are the effect-sizes estimated for the ith gene/phenotype or gene/laboratory pair in African- (A) or European-ancestry (E) patients, \(\alpha\) is the cross-ancestry relationship, and \({\epsilon }_{i}\sim N\left(0,{\sigma }^{2}\cdot {{{{{{\mathrm{SE}}}}}}}{\left({\hat{\beta }}_{A,i}\right)}^{2}\right)\) is the gene/phenotype-specific (gene/laboratory) noise parameterized by the overall variance \({\sigma }^{2}\) and squared standard-error around the African-ancestry-based estimate. We report estimates of \(\hat{\alpha }\) and its 95% confidence intervals assuming normality.
Cross-ancestry SNP effects
We investigated BioVU phenotypes and laboratory measures significantly associated with TWAS-associated loci for heterogeneous effects in the trans-ethnic Pan-UK Biobank (Pan-UKB). The Pan-UKB represents a multi-ancestry analysis of 7221 phenotypes in six continental ancestry groups: African (AFR N = 6636), Admixed American (AMR N = 980), Centra/South Asian (CSA N = 8876), East Asian (EAS N = 2709), European (EUR N = 420,531), and Middle Eastern (MID N = 1599). The Pan-UKB consists of 16,119 GWAS of biological assays, health status, behavioral information, and lifestyle factors. We clumped SNPs in PLINK using eQTL/sQTL p values (q value ≦0.05) reported in GTEx v8 and limited pair-wise SNP correlations to r2 = 0.1 over 250 kb windows. Per-tissue clumping resulted in 27 SNPs (ABO, IFNAR2, CCR1, and SLC6A20) that were tested with respect to 1571 Pan-UKB phenotypes from the same trait domains detected by PheWAS in BioVU (e.g., circulatory system, digestive disorder, neurological). We used linear models to test for consistency of EUR effect estimates at FDR significant and high confidence SNPs with those estimated in AFR, AMR, CSA, EAS (Pan-UKB and Japan Biobank), and MID populations. Additionally, we used Biobank Japan to verify associations between EUR and EAS-based SNP effects in the Pan-UKB due to increased sample size in the latter. Biobank Japan consists of genetic data for over 200,000 participants and ~120 disease states and quantitative measures (cell type percentage, body mass index, etc.).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Complete TWAS, spTWAS, PWAS, and allelic imbalance summary statistics results can be accessed here: https://doi.org/10.5281/zenodo.4292567. Covid19 HGI summary statistics: https://www.covid19hg.org/results/r4/. PrediXcan predictive models: https://github.com/hakyimlab/MetaXcan/blob/master/README.md. FUSION PWAS predictive models: https://mancusolab.com/projects/pwas/. Pan-UK Biobank summary statistics: https://pan.ukbb.broadinstitute.org/downloads. Biobank Japan summary statistics: http://jenger.riken.jp/en/result.
References
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020).
Richardson, S. et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA 323, 2052–2059 (2020).
Mao, L. et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurology 77.6, 683–690 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020).
COVID-19 Host Genetics Initiative. The COVID-19 host genetics initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718 (2020).
Roberts, G. H. L. et al. AncestryDNA COVID-19 host genetic study identifies three novel loci. Preprint at medRxiv https://doi.org/10.1101/2020.10.06.20205864 (2020).
Shelton, J. F. et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nature Genetics 53.6, 801–808 (2021).
Severe Covid-19 GWAS Group. et al. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 383, 1522–1534 (2020).
Pairo-Castineira, E. et al. Genetic mechanisms of critical illness in Covid-19. Nature 591.7848, 92–98 (2021).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Tal, S., Spectre, G., Kornowski, R. & Perl, L. Venous thromboembolism complicated with COVID-19: what do we know so far? Acta Haematol. 143, 417–424 (2020).
Frydman, G. H., Boyer, E. W., Nazarian, R. M., Van Cott, E. M. & Piazza, G. Coagulation status and venous thromboembolism risk in African Americans: a potential risk factor in COVID-19. Clin. Appl. Thromb. Hemost. 26, 1076029620943671 (2020).
Porfidia, A. et al. Incidence of Deep Vein Thrombosis among non-ICU Patients Hospitalized for COVID-19 Despite Pharmacological Thromboprophylaxis. J. Thromb. Haemost.: JTH (2020).
Klok, F. A. et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 191, 145–147 (2020).
Grillet, F., Behr, J., Calame, P., Aubry, S. & Delabrousse, E. Acute pulmonary embolism associated with COVID-19 pneumonia detected with pulmonary CT angiography. Radiology 296, E186–E188 (2020).
Lu, G. & Wang, J. Dynamic changes in routine blood parameters of a severe COVID-19 case. Clin. Chim. Acta 508, 98–102 (2020).
Rezende, S. M., Lijfering, W. M., Rosendaal, F. R. & Cannegieter, S. C. Hematologic variables and venous thrombosis: red cell distribution width and blood monocyte count are associated with an increased risk. Haematologica 99, 194–200 (2014).
Algahtani, F. H. & Stuckey, R. High factor VIII levels and arterial thrombosis: illustrative case and literature review. Ther. Adv. Hematol. 10, 2040620719886685 (2019).
Sabater-Lleal, M. et al. Genome-wide association transethnic meta-analyses identifies novel associations regulating coagulation factor VIII and von Willebrand factor plasma levels. Circulation 139, 620–635 (2019).
Ray, J. G. et al. Association between ABO and Rh blood groups and SARS-CoV-2 infection or severe COVID-19 illness: a population-based cohort study. Ann. Intern. Med. 174.3, 308–315 (2021).
Barnkob, M. B. et al. Reduced prevalence of SARS-CoV-2 infection in ABO blood group O. Blood Adv. 4, 4990–4993 (2020).
Hoiland, R. L. et al. The association of ABO blood group with indices of disease severity and multiorgan dysfunction in COVID-19. Blood Adv. 4, 4981–4989 (2020).
Zhao, J. et al. Relationship between the ABO blood group and the coronavirus disease 2019 (COVID-19) susceptibility. Clin. Infect. Dis. 73.2, 328–331 (2021).
Latz, C. A. et al. Blood type and outcomes in patients with COVID-19. Ann. Hematol. 99, 2113–2118 (2020).
Li, J. et al. Association between ABO blood groups and risk of SARS-CoV-2 pneumonia. Br. J. Haematol. 190, 24–27 (2020).
Goel, R. et al. ABO blood group and COVID‐19: a review on behalf of the ISBT COVID‐19 working group. Vox Sanguinis (2021).
Le Pendu, J. et al. ABO Blood Types and COVID-19: Spurious, Anecdotal, or Truly Important Relationships? A Reasoned Review of Available Data. Viruses 13.2, (2021).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Wiersinga, W. J., Rhodes, A., Cheng, A. C., Peacock, S. J. & Prescott, H. C. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA 324, 782–793 (2020).
da Costa, R. M. et al. Nrf2 as a potential mediator of cardiovascular risk in metabolic diseases. Front. Pharmacol. 10, 382 (2019).
Radenkovic, D. et al. Cholesterol in relation to COVID-19: should we care about it? J. Clin. Med. 9, 1909 (2020).
Batabyal, R. et al. Metabolic dysfunction and immunometabolism in COVID-19 pathophysiology and therapeutics. Int. J. Obes. 45.6, 1163–1169 (2021).
Aung, N., Khanji, M. Y., Munroe, P. B. & Petersen, S. E. Causal inference for genetic obesity, cardiometabolic profile and COVID-19 susceptibility: a Mendelian randomization study. Front. Genet. 11, 586308 (2020).
Motaib, I. et al. Obesity and disease severity among patients with COVID-19. Cureus 13, e13165 (2021).
Shepardson, K. M. et al. IFNAR2 is required for anti-influenza immunity and alters susceptibility to post-influenza bacterial superinfections. Front. Immunol. 9, 2589 (2018).
Prokunina-Olsson, L. et al. COVID-19 and emerging viral infections: The case for interferon lambda. J. Exp. Med. 217.5 (2020).
Sa Ribero, M., Jouvenet, N., Dreux, M. & Nisole, S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 16, e1008737 (2020).
Ziegler, C. G. K. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035.e19 (2020).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370.6515 (2020).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370.6515 (2020).
Meffre, E. & Iwasaki, A. Interferon deficiency can lead to severe COVID. Nature 587, 374–376 (2020).
Novick, D., Cohen, B. & Rubinstein, M. The human interferon alpha/beta receptor: characterization and molecular cloning. Cell 77, 391–400 (1994).
Membrilla, J. A., de Lorenzo, Í., Sastre, M. & Díaz de Terán, J. Headache as a cardinal symptom of coronavirus disease 2019: a cross-sectional study. Headache 60, 2176–2191 (2020).
García-Moncó, J. C. et al. Neurological reasons for consultation and hospitalization during the COVID-19 pandemic. Neurol. Sci. 41, 3031–3038 (2020).
Zdrenghea, M. T., Makrinioti, H., Muresan, A., Johnston, S. L. & Stanciu, L. A. The role of macrophage IL-10/innate IFN interplay during virus-induced asthma. Rev. Med. Virol. 25, 33–49 (2015).
Alon, R. et al. Leukocyte trafficking to the lungs and beyond: lessons from influenza for COVID-19. Nat. Rev. Immunol. 21.1, 49–64 (2021).
Zhou, S. et al. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat. Med. 27.4, 659–667 (2021).
Lv, D. et al. Pulmonary function of patients with 2019 novel coronavirus induced-pneumonia: a retrospective cohort study. Ann. Palliat. Med., 9.5, 3447–3452 (2020).
Li, W., Li, N., Gao, L. & You, C. Integrated analysis of the roles and prognostic value of RNA binding proteins in lung adenocarcinoma. PeerJ 8, e8509 (2020).
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Marigorta, U. M. et al. Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn’s disease. Nat. Genet. 49, 1517–1521 (2017).
Barbeira, A. N. et al. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 15, e1007889 (2019).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Feng, H. et al. Leveraging expression from multiple tissues using sparse canonical correlation analysis and aggregate tests improves the power of transcriptome-wide association studies. PLoS Genetics 17.4, e1008973 (2021).
Flutre, T., Wen, X., Pritchard, J. & Stephens, M. A statistical framework for joint eQTL analysis in multiple tissues. PLoS Genet. 9, e1003486 (2013).
Kim-Hellmuth, S., et al. Cell type–specific genetic regulation of gene expression across human tissues. Science 369.6509 (2020).
Sul, J. H., Han, B., Ye, C., Choi, T. & Eskin, E. Effectively identifying eQTLs from multiple tissues by combining mixed model and meta-analytic approaches. PLoS Genet. 9, e1003491 (2013).
Wang, G. et al. A simple new approach to variable selection in regression, with application to genetic fine mapping. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 82.5, 1273–1300 (2020).
van de Geijn, B., McVicker, G., Gilad, Y. & Pritchard, J. K. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods 12, 1061–1063 (2015).
Castel, S. E. et al. A vast resource of allelic expression data spanning human tissues. Genome Biol. 21, 234 (2020).
Castel, S. E., Mohammadi, P., Chung, W. K., Shen, Y. & Lappalainen, T. Rare variant phasing and haplotypic expression from RNA sequencing with phASER. Nat. Commun. 7, 12817 (2016).
Mohammadi, P., Castel, S. E., Brown, A. A. & Lappalainen, T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 27, 1872–1884 (2017).
Ge, S. X., Jung, D. & Yao, R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629 (2020).
Liu, Z.-P., Wu, C., Miao, H. & Wu, H. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015, bav095 (2015).
Ghoussaini, M. et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic. Acids. Res. 49.D1, D1311–D1320 (2021).
Roden, D. M. et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 84, 362–369 (2008).
Hu, Y. et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet. 51, 568–576 (2019).
Zhou, D. et al. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat. Genet. 52, 1239–1246 (2020).
Denny, J. C. et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26, 1205–1210 (2010).
Dennis, J. K., et al. Clinical laboratory test-wide association scan of polygenic scores identifies biomarkers of complex disease. Genome. Med. 13.1, 1–16 (2021).
Acknowledgements
We thank the COVID-19 Host Genetics Initiative (https://www.covid19hg.org/acknowledgements/) for providing open access to genetic association data, and all the studies comprising of the meta-analysis (https://www.covid19hg.org/publications/). We acknowledge openly available summary statistics from Pan-UK Biobank, and Biobank Japan. The BioVU projects at Vanderbilt University Medical Center are supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH-funded Shared Instrumentation Grant S10OD017985 and S10RR025141; CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/. Yale investigators acknowledge support from the National Institutes of Health (R21 280 DC018098, R21 DA047527, and F32 MH122058). L.K.D. is supported by grants from the National Institutes of Health including U54MD010722-04, R01MH113362, R01MH118223, and R56MH120736. T.W.M.-F. was supported by the National Human Genome Research Institute, NHGRI (T32 HG008341). N.E., P.M., and N.M. are supported by National Institute of Health, NIGMS (R01GM140287).
Author information
Authors and Affiliations
Contributions
G.A.P., K.S., and N.M. contributed to study design, analysis, writing, and editing the manuscript. G.A.P., K.S., T.W.M.-F., F.R.W., N.E., Z.L., and S.G. contributed to the study design and analysis. K.H. and R.J. analyzed and prepared summary data. G.A.P., K.S., T.W.M.-F., F.R.W., N.E., K.H., R.J., Z.L., S.G., L.Y., P.M., B.P., R.P., L.K.D., and N.M. provided critical feedback on the manuscript, study design, and expertize of multiple methods contributed to the manuscript. B.P., R.P., L.K.D., and N.M. co-supervised analyses and finalized the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pathak, G.A., Singh, K., Miller-Fleming, T.W. et al. Integrative genomic analyses identify susceptibility genes underlying COVID-19 hospitalization. Nat Commun 12, 4569 (2021). https://doi.org/10.1038/s41467-021-24824-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-24824-z
This article is cited by
-
Associations between genetically predicted plasma protein levels and Alzheimer’s disease risk: a study using genetic prediction models
Alzheimer's Research & Therapy (2024)
-
Computational network analysis of host genetic risk variants of severe COVID-19
Human Genomics (2023)
-
Phenome-wide association study and precision medicine of cardiovascular diseases in the post-COVID-19 era
Acta Pharmacologica Sinica (2023)
-
Headache in COVID-19 and Long COVID: to Know Facts for Clinical Practice
Current Neurology and Neuroscience Reports (2023)
-
SparkGC: Spark based genome compression for large collections of genomes
BMC Bioinformatics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
