
Abstract
Galectins are a family of endogenous glycan-binding proteins that have crucial roles in a broad range of physiological and pathological processes. As a group, these proteins use both extracellular and intracellular mechanisms as well as glycan-dependent and independent pathways to reprogramme the fate and function of numerous cell types. Given their multifunctional roles in both tissue fibrosis and cancer, galectins have been identified as potential therapeutic targets for these disorders. Here, we focus on the therapeutic relevance of galectins, particularly galectin 1 (GAL1), GAL3 and GAL9 to tumour progression and fibrotic diseases. We consider an array of galectin-targeted strategies, including small-molecule carbohydrate inhibitors, natural polysaccharides and their derivatives, peptides, peptidomimetics and biological agents (notably, neutralizing monoclonal antibodies and truncated galectins) and discuss their mechanisms of action, selectivity and therapeutic potential in preclinical models of fibrosis and cancer. We also review the results of clinical trials that aim to evaluate the efficacy of galectin inhibitors in patients with idiopathic pulmonary fibrosis, nonalcoholic steatohepatitis and cancer. The rapid pace of glycobiology research, combined with the acute need for drugs to alleviate fibrotic inflammation and overcome resistance to anticancer therapies, will accelerate the translation of anti-galectin therapeutics into clinical practice.
Similar content being viewed by others

Introduction
The complex repertoire of glycan structures present in cells and tissues (that is, the glycome) stores crucial biological information that contributes to the reprogramming of cellular fate and function and thus has a profound influence on the delicate balance between health and disease1,2,3,4. The diversity and spatiotemporal regulation of glycans within glycoconjugates rely on the synchronized action of glycan-modifying enzymes, including glycosyltransferases and glycosylhydrolases. Glycan remodelling is regulated by intracellular and environmental signals, such as metabolic stress, oxygen and nutrient availability, growth factors and cytokines2,5. Dysregulation of many cellular processes, including cellular communication, proliferation, differentiation and survival has been linked to aberrant glycosylation. This finding implicates the glycome in the pathophysiology of nearly every major disease, with notable impact on cancer, inflammation and fibrosis5,6. Changes in the glycosylation signature of tumour, immune and endothelial cells (ECs) are among the common hallmarks of the tumorigenic process, as these signatures can influence cell adhesion, epithelial-to-mesenchymal transition (EMT), angiogenesis, immunoediting and metastasis1,5,7,8,9,10,11,12. Moreover, selective glycan profiles may also help to control the initiation, persistence and resolution of inflammatory and fibrotic processes1,13. Thus, an aberrant glycome might alter cellular functions by regulating the exposure or masking of specific glycoepitopes. These aberrations can ultimately lead to the development of pathological responses.
Converting glycan-encoded information into biological programmes relies, at least in part, on the contributions of endogenous glycan-binding proteins or lectins6. The three major lectin families that have decisive roles in shaping both inflammatory and tumour microenvironments are sialic acid-binding immunoglobulin-like lectins (Siglecs), C-type lectin receptors (including selectins14) and galectins. Galectins are a family of soluble lectins that share affinity for β-galactoside-containing saccharides15. Examples of some natural ligands for mammalian galectins are summarized in Box 1. Galectins might be involved in the transition from healthy to neoplastic or inflamed tissues and contribute to the persistence of these pathological conditions via both intracellular and extracellular mechanisms16,17. Galectins also influence most hallmarks of tumour progression17 and modulate resistance to numerous anticancer treatments, including immunotherapy, chemotherapy, radiotherapy, targeted therapies and anti-angiogenic therapy18. Furthermore, galectins might contribute to regulatory circuits that amplify, sustain or alleviate tissue fibrosis and inflammation by selectively targeting different cell types and their microenvironments16,19,20,21. Accordingly, these glycan-binding proteins have been proposed as therapeutic targets in a broad range of pathological conditions and are currently under clinical evaluation.
In this Review, we consider the pathophysiological relevance of specific galectin–glycan interactions from a translational perspective and discuss the design, mechanisms of action, selectivity and therapeutic relevance of galectin-targeted agents. We focus on the results of preclinical studies and clinical trials and highlight lessons learned from targeting galectins in a diverse group of disease states, including nonalcoholic steatohepatitis (NASH)19, idiopathic pulmonary fibrosis (IPF)22,23,24, as well as various malignancies11,17. These examples underscore the numerous potential opportunities to capitalize on galectin–glycan interactions for therapeutic purposes.
We focus in particular on agents designed to target galectin–glycan interactions, including small-molecule inhibitors, natural polysaccharides and their synthetic derivatives, as well as peptides and peptidomimetics. We also discuss the relevance of biological agents such as neutralizing monoclonal antibodies (mAbs), aptamers and truncated galectins, which have emerged as potential therapeutic modalities to target galectin-regulated circuits. Finally, we discuss various galectin-targeted strategies, highlight new translational applications and consider the current challenges and prospects for using these agents to modulate treatments for cancer, IPF and NASH. However, we do not discuss other targets of the translational potential of galectins, which include allergic inflammation25,26, autoimmune inflammation16,27,28,29, neuroinflammation30,31, cardiovascular disorders and atherosclerosis32,33,34, sepsis35 and failing pregnancies36.

Cellular roles of galectins
Galectins regulate cell signalling and recalibrate the nature and magnitude of cellular responses via their impact on both intracellular and extracellular mechanisms16,37,38,39,40,41. Galectins were originally defined as β-galactoside-binding proteins, as they recognize epitopes containing Galβ(1–4)GlcNAc — also known as N-acetyllactosamine, LacNAc — present in various glycoconjugates. However, recent biochemical studies on mammalian galectins have revealed subtle differences in their carbohydrate recognition domains (CRDs) that can explain some of their divergent biological activities15,42,43,44 (Box 1). Members of the galectin family have been categorized into three subfamilies (Fig. 1), which includes prototype galectins (GAL1, GAL2, GAL5, GAL7, GAL10, GAL11, GAL13, GAL14 and GAL15) that function as monomers or non-covalent homodimers with identical CRDs; chimera-type galectins (GAL3), which contain only one CRD and can oligomerize via a non-lectin N-terminal domain; and tandem repeat-type galectins (GAL4, GAL6, GAL8, GAL9 and GAL12), which contain two CRDs each with a distinct specificity15,43,44 joined by a flexible linker peptide.

Galectins are classified into three different subfamilies on the basis of their structure: a, prototype galectins (GAL1, GAL2, GAL5, GAL7, GAL10, GAL11, GAL13, GAL14 and GAL15) that function as monomers or non-covalent homodimers with identical carbohydrate recognition domains (CRDs); b, chimera-type galectins (GAL3) that contain only one CRD and have the capacity to oligomerize via a non-lectin N-terminal domain; and c, tandem repeat-type galectins (GAL4, GAL6, GAL8, GAL9 and GAL12) that contain two CRDs each with a distinct specificity joined by a flexible linker peptide. Depending on distinct carbohydrate-binding specificities and oligomerization status, these proteins can form lattices by interacting with multiple glycosylated ligands on the cell surface and/or with extracellular matrix glycoproteins. In doing so, they elicit different biological responses via mechanisms that include assembly of multivalent glycan–galectin complexes and modulation of transmembrane signalling events by selective receptor segregation, retention and endocytosis.
The expression and subcellular localization of galectins differ considerably in individual cell types. These properties are dynamically regulated by cell activation, differentiation and anatomical distribution and can undergo dramatic modulation in response to specific pathological conditions8,15. Galectins are synthesized as cytosolic proteins and in response to environmental signals can shuttle between the cytosol and the nucleus. They control intracellular processes via specific protein–protein or protein–glycan interactions45,46. For example, GAL1 and GAL3 interact with RAS GTPases in the cytoplasm, whereas through binding to Gemin4 (ref. 47) they have a role in spliceosome assembly in the nucleus48. Moreover, intracellular partners of GAL3 include β-catenin45 and hnRNPA2B1 (ref. 49) in the nucleus, and Alix, a component of the endosomal sorting complex required for intracellular transport50,51. Interestingly, cytosolic galectins might act as ‘danger signal sensors’ that detect abnormal exposure of glycan moieties within the cytosolic compartment52,53.
Galectins are also secreted from cells via a non-canonical pathway that is independent of the endoplasmic reticulum and Golgi35,37. In the extracellular space, galectins can cross-link glycosylated receptors on the cell surface (Fig. 1), thereby establishing multivalent lattices that convert glycan-based information into diverse signalling programmes40,54,55. The relatively low affinity of galectins for most, but not all, small oligosaccharidic structures43,56,57 can be compensated by the cluster glycoside effect (Box 2), which explains the apparent increase in affinity due to multivalent interactions between a ligand and its target. Segregation of glycoconjugates into membrane microdomains and the assembly of galectin–glycan complexes can control the retention of cell surface glycosylated receptors, thereby modulating the threshold for signalling and amplifying or attenuating cellular responses9,58,59. The responses generated by galectin–glycan interactions — typically resulting from intracellular and extracellular activities — modulate a broad range of cellular responses, including proliferation, differentiation and survival60.
Dysregulated galectin expression occurs in a range of malignancies and is frequently associated with poor prognosis and limited responses to anticancer therapies17,18,61,62. Moreover, galectin levels are frequently elevated at various stages of the fibrotic cascade21,63. These data highlight the emerging roles of galectins as possible biomarkers and therapeutic targets in these pathological conditions. In the next sections we discuss the role of galectins in cancer and fibrosis, with a focus on GAL1, GAL3 and GAL9.
Galectins in cancer
Galectins have a broad influence on tumour progression via glycosylation-dependent or independent mechanisms that modulate proliferation, evasion of growth suppressors and immune responses, resistance to cell death, induction of angiogenesis, inflammation and metastasis17,64. These galectin-influenced circuits can shape tumour, endothelial and immune compartments in individual tumour microenvironments (Fig. 2). These actions have both local and systemic impact and might contribute to processes that influence clinical outcomes in patients with cancer. To substantiate the therapeutic relevance of these glycan-binding proteins, we provide several examples that highlight the impact of galectins, most notably GAL1, GAL3 and GAL9, on the reprogramming of the tumour, vascular and immune landscapes and their contributions to tumour growth and progression (Fig. 2).

a, Galectins (for example, GAL1, GAL3 and GAL9) use both intracellular and extracellular mechanisms to reprogramme the fate of tumour cells. They interact with receptors such as mucin 1 (MUC1), epidermal growth factor receptor (EGFR) and αvβ3 integrin and with intracellular proteins such as RAS to modulate signalling pathways that promote epithelial–mesenchymal transition (EMT), migration and invasion. b, Galectins control endothelial cell responses and promote aberrant angiogenesis by engaging various glycosylated cell surface receptors including neuropilin 1 (NRP1; ligand for GAL1), vascular endothelial growth factor receptor 2 (VEGFR2; ligand for GAL1 and GAL3) and αvβ3 integrin (ligand for GAL3), thereby modulating cell surface retention, endocytosis and signalling. GAL1 also modulates angiogenesis by binding to the mRNA of angiogenesis-related factors such as early growth response protein 1 (EGR1). c, Galectins shape the immune landscape of the tumour microenvironment by acting on both lymphoid and myeloid cells. Additionally, they suppress antitumour responses by engaging immune checkpoint or co-inhibitory molecules including CD45 and CD43 (GAL1), CTLA4 and LAG3 (GAL3) and PD1 and TIM3 (GAL9) leading to inhibition of lymphocyte cell-specific protein-tyrosine kinase (LCK)-mediated T cell activation, or promotion of STAT3-mediated myeloid cell reprogramming. Therefore, galectins facilitate tumour immune escape and mediate resistance to immunotherapeutic strategies. BAT3, HLA-B-associated transcript 3; ECAD, E-cadherin; GLI1, glioma-associated oncogene homologue 1; MMP, matrix metalloproteinase; PP2A, protein phosphatase 2 phosphatase activator; PTP, protein tyrosine phosphatase; SHP2, protein tyrosine phosphatase non-receptor type 11.
Galectins reprogramme tumour cell fate
GAL1 is upregulated in most tumour tissues and their associated stroma and has been proposed as a biomarker of poor prognosis in breast, colon, lung, prostate adenocarcinoma and melanoma65. Tumours of the digestive tract and urinary system, as well as thyroid cancer and melanomas, express high levels of GAL3; by contrast, GAL3 expression is downregulated in tumours of the reproductive tract. Subcellular compartmentalization of GAL3 is a crucial factor contributing to its pro- or antitumoural roles. For example, increased GAL3 within the nuclear compartment has been associated with a better outcome among patients with neuroblastoma66. On the other hand, GAL9 expression can be up- or downregulated in association with neoplastic transformation depending on the specific tumour type65,67. As the expression of these lectins can change during tumour progression, they have been proposed as biomarkers for diagnosis, prognosis and/or intervention in various cancer stages65.
Extracellular GAL1 and GAL3 directly modulate tumour cell fitness, migration, EMT and stemness through their interactions with glycosylated tumour-associated receptors, including epidermal growth factor (EGF), transforming growth factor β (TGFβ) receptors and cell surface integrins68,69,70,71,72,73,74, that contribute to cancer progression and metastasis. GAL1 can trigger EMT in gastric cancer75 and hepatocellular carcinoma72,76 via non-canonical activation of the Hedgehog pathway75, downregulation of E-cadherins72 and induction of αvβ3 integrin-dependent AKT signalling76. Collectively, these actions have an impact on clinical outcomes and therapeutic responses to both sorafenib and doxorubicin77,78. GAL1 also regulates events that promote the growth of pancreatic adenocarcinoma, including tumour cell proliferation, invasion and metastasis78, and GAL1 silencing inhibits migration and invasion of metastatic castration-resistant prostate cancer cells via suppression of androgen receptor and AKT-mediated signalling79. In turn, interactions between GAL3 and the glycosylated transmembrane protein mucin 1 (MUC1) enhance EGF receptor dimerization and activation (Fig. 2), and thus promote proliferation and motility of epithelial cancer cells by activating ERK1/2 and AKT signalling pathways80,81. Notably, complex N-glycans linked to EGF receptors on breast carcinoma cells control EMT, cell motility and tumour metastasis by facilitating the exposure of GAL3-specific glycoepitopes68 (Fig. 2). Tumour-derived GAL3 also promotes invasion via its capacity to impair interactions between adhesion molecules expressed on the surface of malignant cells and N-glycosylated proteins within the extracellular matrix, such as laminin and fibronectin74. Furthermore, GAL3 promotes the establishment of metastatic niches by binding to the Thomsen–Friedenreich antigen (Tf antigen) expressed by metastatic lung tumour cells70 and facilitating homotypic and heterotypic aggregation and emboli formation82. Similarly, overexpression of GAL3 promotes proliferation, migration and invasion of oral squamous cell carcinoma (OSCC) cells via enhanced WNT–β-catenin signalling and EMT83. The interaction of GAL3 with αvβ3 integrin promotes KRAS addiction84; these findings identified GAL3 as a potential druggable target for ‘KRAS-addicted’ lung and pancreatic cancers (Fig. 2).
GAL1 and GAL3 also promote oncogenic signalling, apoptosis resistance and cell cycle progression when present in the intracellular compartment85,86. GAL1 associates with oncogenic HRAS (Fig. 2) and enhances HRAS membrane anchorage and oncogenesis by increasing signalling to the kinase RAF1 (refs. 87,88). Likewise, GAL3 promotes cancer cell proliferation and survival via its interactions with activated KRAS and enhances colorectal tumour cell invasion via constitutive activation of RAF–MEK–ERK signalling88,89 (Fig. 2). Interestingly, in vitro studies revealed that a combination of the KRAS inhibitor salirasib and the GAL3 inhibitor modified citrus pectin (MCP) induced cell cycle arrest and apoptosis in anaplastic thyroid cells90. Other studies showed that GAL3 expression was suppressed by p53. This response was largely eliminated in cells with TP53 mutations, thus explaining the elevated levels of GAL3 expression in TP53-mutant tumours91. Remarkably, galectin-mediated mechanisms also promote tumour resistance to most anticancer therapies18, including chemotherapy77, immunotherapy92, anti-angiogenic therapy9, radiotherapy93 and targeted therapies94.
Galectins induce aberrant angiogenesis
Abnormal angiogenesis and diversion of pre-existing blood vessels are crucial in neoplastic as well as inflammatory and fibrotic conditions, as these processes provide the nutrients and oxygen needed to sustain otherwise hypoxic environments95. Interestingly, the glycome of ECs varies considerably in response to immunosuppressive or hypoxic stimuli, which favour exposure of galectin-specific glycan epitopes9. Several galectin family members have pro-angiogenic activity as a result of their capacity to bind distinct glycosylated receptors on the EC surface96. For example, GAL1 interacts with complex N-glycans on vascular endothelial growth factor (VEGF) receptor 2 (VEGFR2) and promotes receptor phosphorylation followed by activation of the AKT and ERK1/2 signalling pathways, thus preserving vascularization in tumours that are resistant to anti-VEGF treatment9 (Fig. 2). These actions link tumour hypoxia to aberrant angiogenesis pathways identified in melanoma as well as lung, prostate and pancreatic adenocarcinoma9,78,97,98,99,100,101. Likewise, interactions with the transmembrane glycoprotein, neuropilin 1 (NRP1) are essential for GAL1-driven angiogenesis and induction of vascular permeability102,103 (Fig. 2). Recently, an alternative mechanism was proposed whereby GAL1 regulates angiogenesis through binding to the mRNAs of angiogenesis-related factors, including VEGF-A, early growth response 1 protein (EGR1) and α5 laminin104.
GAL3 promotes vascularization via interactions with complex branched N-glycans present on αvβ3 integrin and VEGFR2 (Fig. 2) that promote retention of these receptors on the EC surface105,106. Interestingly, this pro-angiogenic effect involves cleavage of the N terminus of GAL3 by matrix metalloproteinase 2 (MMP2) and MMP9 (ref. 107). By contrast, GAL9 mediates angiogenesis through different mechanisms108. In vitro studies with individual GAL9 CRDs revealed opposing anti- and pro-angiogenic effects, suggesting a dual role for this lectin in regulating vascular networks109. Finally, in addition to their role in promoting angiogenesis, interactions of GAL3 with ECs stimulate the secretion of metastasis-promoting cytokines110 and the expression of cell adhesion molecules111. Thus, agents that target galectins and/or their glycosylated receptors may prevent aberrant vascularization, interrupt the metastasis cascade and circumvent resistance to anti-angiogenic therapies.
Galectins shape immune landscapes
Galectins dampen antitumour immune responses by targeting both lymphoid and myeloid cells38 (Fig. 2). Notably, galectin–glycan interactions alter the immune landscape in several cancer types112,113,114,115,116,117,118. Moreover, galectins modulate cancer-associated fibroblasts and can influence their pro-tumorigenic and pro-metastatic activities119.
The role of galectins in suppressing antitumour immunity involves multiple pathways linked to innate and adaptive immune responses. For example, the induction of tolerogenic dendritic cells is driven by GAL1 (refs. 120,121), and the recruitment and differentiation of immunosuppressive (M2-type) macrophages is triggered by GAL1 (ref. 31), GAL3 (refs. 122,123) or GAL9 (ref. 124). Differentiation and expansion of CD4+ and CD8+ regulatory T cells (Treg cells) is induced by GAL1 (refs. 103,125,126) and GAL9 (ref. 127); whereas apoptosis of effector T helper 1 (TH1), TH17 and CD8+ T cells is mediated by GAL1 (refs. 128,129,130,131,132) and GAL9 (ref. 133); synthesis of anti-inflammatory cytokines such as IL-10 and IL-27 is elicited by GAL1 (refs. 126,134); inhibition of natural killer (NK) cell-mediated cytotoxicity135 and expansion of monocytic myeloid-derived suppressor cells (MDSCs) is induced by GAL3 (ref. 136); T cell exclusion is orchestrated by GAL1 (ref. 92), and the inhibition of chemokine gradients for T cell infiltration occurs in response to GAL3 (ref. 137).
To add further complexity, galectins can also impair T cell activation by forming multivalent lattices that restrict the mobility of relevant immune receptors50,138. For example, GAL3 dampens antitumour responses by decreasing T cell receptor (TCR) mobility through formation of a glycoprotein lattice that limits interactions between the TCR and CD8 coreceptor, thus promoting dysfunction and anergy of tumour-infiltrating lymphocytes138 (Fig. 2). Interestingly, galectins may also support transmission of immune inhibitory signals by acting as ligands of co-inhibitory and immune checkpoint molecules, illustrated by the association of GAL1 with CD45 and CD43 (ref. 131), GAL3 with LAG3 and CTLA4 (refs. 139,140) and GAL9 with TIM3 and PD1 (refs. 133,141) (Fig. 2).
By interrupting immune-activating mechanisms or triggering immune inhibitory pathways, galectins thwart various immunotherapeutic modalities. For example, GAL1 can reprogramme the tumour endothelium to upregulate both PDL1 and GAL9; blockade of this pathway increased T cell infiltration into the tumour and improved the response to anti-PD1 therapy92. Moreover, GAL9-mediated cross-talk between PD1 and TIM3 controls T cell exhaustion programmes and sensitivity to anti-PD1 therapy133. Thus, targeting galectins and/or their glycosylated ligands might counteract both local and systemic immunosuppressive circuits, dismantle tumour immune escape mechanisms and recalibrate responses to immunotherapeutic modalities.
Galectins in fibrosis
Pathological fibrosis is the consequence of abnormal mechanisms of tissue repair that are typically associated with cellular stress, chronic inflammation and/or severe tissue damage. It can lead to organ failure. Inadequate tissue regeneration has been described in several chronic fibroproliferative diseases such as IPF24 and chronic inflammatory diseases such as NASH19. The abnormal wound-healing process results in scar formation with excessive collagen deposition via mechanisms that involve fibroblasts, epithelial and endothelial cells, and immune cells, notably macrophages19. Given the high prevalence of fibrotic diseases and the limitations of current drugs, which mainly limit the rate of organ dysfunction, the quest for effective therapies is clearly important.
GAL3 expression is upregulated in fibrotic lesions in human subjects63, and the severity of hepatic and lung fibrosis is reduced in GAL3-deficient mice33,142,143,144,145. Thus, GAL3 has emerged as a promising therapeutic target for fibrotic diseases. Mechanistically, GAL3 induces a pro-fibrotic macrophage phenotype by interacting with the neutral amino acid transporter CD98 (ref. 123). Likewise, GAL3-secreting macrophages drive myofibroblast differentiation, which ultimately results in scar formation146. Moreover, ECs and myofibroblasts upregulate GAL3 expression upon their activation142,147,148; this facilitates EMT, apoptosis, myofibroblast proliferation and enhanced production of fibronectin and other proteins found in the extracellular matrix149,150,151.
Similarly to GAL3, GAL1 and GAL9 are also thought to contribute to the fibrotic process, largely on the basis of studies in galectin-deficient mice152,153. Increased levels of GAL1 mRNA were detected in the lungs of patients with IPF154, and GAL1 and GAL9 were prominently expressed in fibrotic liver155,156. The pro-fibrotic role of GAL1 is associated with TGFβ-driven fibroblast differentiation157. Accordingly, silencing GAL1 prevented hypoxia-induced pulmonary fibrosis and blocked the anticipated decline in lung function154. The role of GAL9 in fibrotic diseases of the lung remains under debate. Matsumoto et al.158 proposed that GAL9 may have a protective role on the basis of its ability to suppress growth and induce dose-dependent apoptosis of human lung fibroblasts. However, GAL9 has also been highlighted as a mediator of lung fibrosis that acts via mechanisms that involve TGFβ signalling153. Thus, modulation of GAL1, GAL3 and/or GAL9 might control fibrotic processes by targeting fibroblasts, macrophages and ECs in several prominent pathological conditions, including IPF and NASH.
Galectin-targeted strategies
As our understanding of the role of galectins in cancer and fibrosis has improved, various chemical and biological agents that target GAL1, GAL3 and GAL9 have been developed. These agents include small-molecule chemical inhibitors, natural polysaccharides and their synthetic derivatives, peptides and peptidomimetics, and biological agents such as small interfering RNA (siRNA), aptamers, truncated galectins and neutralizing mAbs. In the next section, we describe both the advances and the difficulties in the development and clinical evaluation of current galectin-targeted agents.
Small-molecule carbohydrate inhibitors
The binding of galectins to β-galactosides such as LacNAc (1, Fig. 3) is determined by several distinct non-covalent interactions, including a hydrophilic hydrogen-bond network between the sugar hydroxyl groups and the conserved amino acids Arg, His, Asn and Glu in the galectin. Other interactions include hydrophobic CH–π bonds between the galactoside and a Trp-containing side chain in the galectin CRD159 (Fig. 3a). The first rational approach to design galectin-antagonizing agents focused on small β-galactoside derivatives160. However, single carbohydrate–lectin interactions are typically low affinity, selectivity is not easily achieved, and carbohydrate molecules are cleared rapidly from the systemic circulation and are susceptible to degradation by glycosidases. Modifications at the C1 and C3 positions of monosaccharide β-galactose-based antagonists resulted in new molecules with improved interaction profiles within the galectin ligand-binding groove. These modified β-galactoside-based antagonists had enhanced affinity for galectins as determined by NMR and X-ray crystallography161,162,163, but remained susceptible to hydrolytic degradation in vivo because of their labile O-glycosidic bonds.

a, Representation of human GAL3 in complex with LacNAc (1). Details of GAL3–LacNAc interactions, with GAL3 shown in silver (representation in new cartoon) and the key residues for LacNAc recognition in the same colour (licorice representation). LacNAc dissacharide is shown in yellow, also in licorice representation. On the right, the chemical structure of LacNAc disaccharide is also shown. b, GAL3 selective inhibitors, α-thiogalactosides 2 (also named GB1107) and 3. c, GAL9C and GAL9N selective inhibitors, α-thiogalactoside 4 and α-thioguloside 5. d, LacNAc-derived GAL3 inhibitor (6). e, Lactulose (7). f, TDG (8) and TDG derivatives (9, 10, 11 and 12). g, Multivalent inhibitors, including glycoclusters (13), glycodendrimers (14) and neoglycoproteins (15). Kd, dissociation constant; NB, no binding observed; ND, not determined.
In this context, N-, C- and S-glycosides were developed and evaluated as potentially hydrolytically stable galectin ligands163,164,165,166, as well as novel α-thiogalactosides, which have an unexpectedly high affinity for GAL3. Zetterberg et al.167 reported that C1 and C3 substitution with chlorophenylthiols and fluoroaryl triazoles, respectively, resulted in an α-thiogalactoside derivative (2) (3,4-dichlorophenyl 3-deoxy-3-[4-(3,4,5-trifluorophenyl)-1H-1,2,3-triazol-1-yl]-1-thio-α-d-galactopyranoside) with a dissociation constant (Kd) of 37 nM for GAL3 and 100-fold selectivity compared with GAL1 (Fig. 3b). Oral administration of compound (2), named GB1107 by Galecto (formerly Galecto Biotech), reduced the rate of human (A549) and mouse (LLC1) lung adenocarcinoma growth and blocked metastasis in experimental models168. Furthermore, treatment with GB1107 resulted in increased M1 polarization of intratumoural macrophages and favoured CD8+ T cell infiltration, thereby enhancing the immunostimulatory impact of an anti-PDL1 mAb and inducing higher levels of expression of interferon-γ (IFNγ), granzyme B, perforin, Fas ligand and caspase 3 (ref. 168). GB1107 also inhibited tumour growth in orthotopic mouse models of gastric cancer, thus substantiating the therapeutic relevance of this inhibitor under relevant neoplastic conditions169.
Studies also documented the design of aminopyrimidinyl α-thiogalactoside (3), which, despite its comparatively low affinity for GAL3 (Kd = 1.7 μM) compared with GB1107 (2), proved to be a more selective GAL3 inhibitor. Specifically, it showed >500-fold selectivity compared with GAL1 and a range of 34- to >500-fold selectivity compared with other galectins tested as determined by fluorescence polarization assays170 (Fig. 3b). Furthermore, a patent assigned to Bristol Myers Squibb reporting α-thiogalactosides derived from GB1107 bearing additional chemical modifications, such as alkylation of HO-2 and oxidation of the anomeric thiol group, was recently published171. Moreover, Idorsia Pharmaceuticals patented α-C-galactopyranosides bearing modifications on HO-2, HO-3 and the anomeric group172. Galecto developed GB1211, an α-thiogalactoside that is closely related to GB1107 with excellent affinity for GAL3 (Kd = 23 nM)173. GB1211 was characterized as a highly selective, orally available GAL3 inhibitor, and its therapeutic potential was demonstrated in mouse models of CCl4-induced liver fibrosis and bleomycin-induced lung fibrosis174. Orally administered GB1211 was well tolerated in phase I clinical studies in healthy subjects (NCT03809052, Table 1). A phase I/II study of this compound in patients with confirmed NASH and liver fibrosis was withdrawn (NCT04607655, Table 1). However, a phase I/II trial of GB1211 in participants with hepatic impairment (Child–Pugh class B and C) is ongoing (NCT05009680, Table 1). Galecto recently presented initial results from this phase I/II trial indicating that GB1211 is safe and well tolerated175, and early signs of clinical effect in patients with moderate and severe hepatic impairment were noted176. A phase I/II trial to assess the combination of GB1211 with the anti-PDL1 mAb atezolizumab in patients with non-small-cell lung cancer (NSCLC) has started recruitment (NCT05240131, Table 1).
To achieve higher selectivity towards GAL3, interactions between galectins and alternative monosaccharides, including d-mannose, d-talose and d-gulose derivatives have been evaluated, although these compounds exhibited no significant functional activity in in vitro or in vivo assays177,178,179,180,181,182. However, a recent study reported that GAL9C and GAL9N CRDs demonstrated good affinity towards synthetic 3-deoxy-3-N-arylated-α-d-galactoside (4) and α-guloside (5) derivatives, respectively, compared with other galectins, as determined by fluorescence anisotropy assays183 (Fig. 3c).
Lactose (Lac) and LacNAc disaccharides, the natural galectin ligands in glycoconjugates, have been evaluated as galectin inhibitors37,184. Substitution of LacNAc at O3′ with aromatic moieties has been explored extensively as a means to generate new and favourable π–arene interactions with the conserved Arg144 residue; this effort has resulted in the development of several effective GAL3 ligands, including a LacNAc derivative163,185,186 (6, Fig. 3d). Alternatively, synthetic lactulose amines evaluated as galectin modulators were found to induce tumour cell apoptosis and inhibit homotypic cell aggregation and EC morphogenesis187. More recently, Kishor and colleagues188 explored interactions of GAL1 and GAL3 with lactulose (7, Fig. 3e) by crystallography and proposed this disaccharide as another scaffold for the design of novel galectin inhibitors.
S-, C- and N-disaccharides have been investigated as stable galectin inhibitors. The synthetic non-reducing disaccharide thiodigalactoside (TDG) (8), a compound that binds to galectins with similar affinity to LacNAc but lacks labile glycosidic bonds, emerged as a valuable building block for the preparation of stable galectin ligands189,190 (Fig. 3f). Similarly, previous work on galactose and lactose derivatives led to the symmetrical substitution of TDG at O3 and O3′ with aromatic esters, amides or triazole groups, which ultimately resulted in several galectin inhibitors with nanomolar affinities164,189,190,191 (Fig. 3f). Bis-[3-O-(3-methoxybenzoyl)-β-d-galactopyranosyl] sulfane (named Td131_1) (9) and bis-(3-O-1-naphthoyl-β-d-galactopyranosyl) sulfane (10) (Fig. 3f) inhibited the migration of human prostate and NSCLC cell lines189. Td131_1 (9) was also tested in papillary thyroid cancer cells in vitro, and, despite the high concentrations required, it promoted cancer cell apoptosis and synergized with doxorubicin192. These successful results led to the evaluation of 3,3′-disubstituted TDG derivatives as GAL3 inhibitors in chronic inflammation and fibrosis193. For example, bis-(3-deoxy-3-(4-(3-fluorophenyl)-1H-1,2,3-triazol-1-yl)-β-d-galactopyranosyl) sulfane (11, Fig. 3f; also named TD139, 33DFTG, and later renamed GB0139 by Galecto), is a selective potent GAL1 and GAL3 inhibitor145 with low affinity for GAL2, GAL4N, GAL4C, GAL8N and GAL9N145 and low or no significant affinity for GAL7 (refs. 144,145). Inhibition of GAL3 with GB0139 (11) in a model of bleomycin-induced lung fibrosis resulted in decreased TGFβ receptor at the cell surface, together with suppressed β-catenin activation and attenuated severity of lung fibrosis145. This result was obtained when GB0139 was administered at much lower concentrations than pirfenidone, an anti-inflammatory drug approved for the treatment of IPF194. Bratteby et al.195 recently developed surrogate positron emission tomography (PET) radiotracers for GB0139 and GB1107 (2) and described their pharmacokinetics. The 18F-radiolabelled GB0139 surrogate was cleared much more rapidly than the GB1107-derived compound, suggesting that although systemic administration may be favourable for monosaccharide-based galectin inhibitors, the rapid excretion of disaccharide derivatives could limit their activity. A phase I/II clinical trial initiated in 2014 to assess the safety, pharmacokinetics and tolerability of GB0139 via a dry-powder inhaler in human volunteers and patients with IPF (NCT02257177) showed that this compound was safe and well tolerated. Furthermore, administration via this route inhibited GAL3 expression by bronchoalveolar lavage macrophages and decreased the concentration of plasma biomarkers associated with IPF progression23. A phase IIb clinical trial to investigate the safety and efficacy of GB0139 (11) in patients with IPF has completed enrolment (NCT03832946). As TDG-based ligands progressed towards clinical trials, further studies to improve selectivity highlighted that their substitution with bulky aromatic groups favours interactions with GAL3 over GAL1 (ref. 196). Consistent with these results, a coumarin-derived TDG (12) showed a 175-fold increase in selectivity over GAL1 with similar efficacy to GB0139 in the bleomycin-induced model of lung fibrosis197 (Fig. 3f). Recently, tetrahydropyran-based thiodisaccharide mimics with a reduced number of H-bond donors to improve permeability and bioavailability were also described198.
Natural multimeric ligands such as multi-antennary N-glycans have inspired the design of multivalent glycosylated architectures to achieve tighter binding and overcome the naturally weak lectin–glycan interactions based on the cluster glycoside effect199. Research over the past decade has identified a wide variety of galectin-targeted multivalent ligands of distinctive nature and valency, including glycoclusters, glycodendrimers, glyconanoparticles, glycopolymers and neoglycoproteins. Multivalent glycoclusters and low-valency multivalent ligands have been designed with strict control of valency, geometry and conformation, and with scaffolds ranging in size from oligosaccharides and cyclodextrins (13, Fig. 3g) to calixarenes and macrocycles165,200,201,202. Poly(amidoamine) lactose or LacNAc-bearing glycodendrimers with valencies up to 95 have also been evaluated as potential GAL3 inhibitors and shown to inhibit (when small dendrimers) or enhance GAL3-mediated cell clustering in vitro203 (14, Fig. 3g). Moreover, conjugation of LacdiNAc (GalNAcβ(1,4)GlcNAc) disaccharides or TDG derivatives to bovine serum albumin generated neoglycoproteins with good affinity and moderate selectivity for GAL3 over GAL1 (refs. 204,205). Notably, a natural glycopeptide rich in Galβ(1,3)GalNAc structures isolated from the cod antifreeze glycoprotein binds to GAL3 with high affinity (Kd 97 pM) and inhibits adhesion of PC-3 prostate cancer cells to ECs as well as GAL3-mediated T cell apoptosis206. Moreover, complex type N-glycans prepared from chicken eggs and coupled by click chemistry to human serum albumin (HSA) as scaffold led to multivalent GAL3 ligands207. More recently, the chemoenzymatic synthesis of neoglycoproteins based on coupling of C3-substituted LacdiNAc glycomimetics to HSA (15, Fig. 3g) has been reported; these derivatives protected T lymphocytes against GAL3-induced apoptosis in vitro208.
Given that the functions of galectins vary between the intracellular and extracellular compartments16,46, Stegmayr et al.209 investigated the intracellular activity and membrane permeability of selected small-molecule GAL3 inhibitors including compounds GB1107 (2), GB0139 (11) and a 1H-1,2,3-triazol-1-yl TDG derivative210. Inhibitor uptake and intracellular potency correlated with the polar surface area of the compounds, which is a determinant for passive membrane permeability. Of the compounds investigated, GB1107 was the most potent, although GB0139 had increased intracellular activity following pre-incubation, suggesting that it can reach the cytosolic compartment given sufficient time to cross the plasma membrane209. Thus, small-molecule galectin inhibitors that can cross the cell membrane, such as GB0139, are important tools to discern extracellular and intracellular functions of galectins, and could potentially be used to target intracellular galectins.
Polysaccharides and derivatives
Complex polysaccharides derived from natural sources such as pectins and galactomannans, pH- and heat-modified pectins (for example, MCPs), and galactomannan-derived compounds have shown antitumoural, antimetastatic and anti-inflammatory activities138,211,212,213,214.
Pectins are a class of complex polysaccharides found in the primary cell walls of plants and are composed of an anionic galacturonan backbone (a linear chain of α(1–4)-linked d-galacturonic acids) and neutral sugar side chains that contain primarily d-galactose, l-rhamnose and l-arabinose215,216. Underivatized pectins have shown beneficial health effects, primarily when introduced as dietary fibre to prevent colon cancer217; however, the most promising anticancer effects were observed in response to MCPs. Treatment of pectins to obtain MCPs eliminates galacturonic acid esters from the homogalacturonan regions and exposes galactose-containing side chains, which have been proposed as major determinants for galectin binding. However, the affinity and selectivity of these compounds is still under debate, as little or no inhibitory activity was detected by fluorescence anisotropy218. Nevertheless, MCPs have been extensively studied for potential antitumour and antimetastatic activities in several cancer types211,212,219,220. For example, PectaSol and PectaSol-C (16) (Fig. 4) are commercial MCPs developed by EcoNugenics that have cytotoxic activity against murine and human prostate cancer cell lines via suppression of MAPK signalling and induction of caspase 3 cleavage213. PectaSol-C functions synergistically with paclitaxel to promote cytotoxicity in ovarian cancer cells by modulating GAL3-driven activation of signal transducer and activator of transcription 3 (STAT3)221. In 2003, a phase II pilot study222 evaluated tolerability and efficacy of PectaSol-C in patients diagnosed with prostate cancer. Oral administration of PectaSol-C resulted in a statistically significant increase in the prostate-specific antigen (PSA) doubling time, potentially indicating a slowing of tumour progression. In a subsequent phase II clinical trial in patients with biochemical relapse of prostate cancer, oral administration of PectaSol-C was evaluated by measuring PSA kinetics as a potential marker for cancer progression (NCT01681823, Table 1). Results showed that patients treated with PectaSol-C exhibited no PSA progression or lengthening of PSA doubling time compared with historical data, potentially indicating slower disease progression223.

a, Structures of modified citrus pectin (MCP) polysaccharides PectaSol (16) and GCS-100 (17), galactoarabino-rhamnogalacturonate GR-MD-02 (belapectin, 18), galactomannan Davanat (19) and arabinogalactan RN1 (20). n, m, o and p denote the number of repetitive residues in the polysaccharide structure. Dissociation constant (Kd) values were not measured for these compounds. b, Non-canonical binding site of Davanat on GAL3. The S- and F-faces of the GAL3 carbohydrate-recognition domain (CRD) are shown. Amino acids that interact with Davanat are shown in greenish yellow (low interaction), orange (moderate interaction) or red (high interaction). Kd values were not measured for these compounds. Glycans are depicted following guidelines from the Symbol Nomenclature for Glycans Group305.
GCS-100 (17, Fig. 4), developed by La Jolla Pharmaceutical Company, was initially developed as a potential inhibitor of GAL3. GCS-100 has broad antitumour activity, including inducing apoptosis of multiple myeloma cells212,224,225,226. Moreover, GCS-100 alone or in combination with BCL-2 homology domain 3 (BH3) mimetics induced apoptosis in acute myeloid leukaemia cells227. A phase II clinical trial (NCT00514696) evaluated intravenous administration of GCS-100 in patients with chronic lymphocytic leukaemia. The inhibitor showed excellent tolerability and led to partial remission in 25% of patients and >50% shrinkage of lymph node lesions in 16% of patients.
GR-MD-02 (18) (renamed belapectin by Galectin Therapeutics) (Fig. 4), a pectin-derived galactoarabino-rhamnogalacturonate substituted with predominantly β(1–4)-d-galactose and α(1–5)-l-arabinose side chains, has been proposed as a GAL3 inhibitor. The antitumour effects of belapectin were explored in immunocompetent cancer models in combination with an agonist anti-OX40 antibody. This drug combination promoted tumour regression and increased survival in mouse models by mitigating MDSC-driven immunosuppression, increasing CD8+ T cell recruitment and reducing the frequency of CD4+FOXP3+ Treg cells122. This compound was also tested in two phase I clinical trials in combination with mAbs that inhibit the immune checkpoint proteins CTLA4 (ipilimumab) or PD1 (pembrolizumab) in patients diagnosed with melanoma (NCT02117362 and NCT02575404). A combination of belapectin and pembrolizumab administered intravenously to patients with advanced metastatic melanoma or head and neck squamous cell carcinoma (HNSCC) (NCT02575404) was well tolerated and generated fewer immune-mediated adverse events than anticipated, based on findings from pembrolizumab monotherapy. Moreover, belapectin and pembrolizumab treatment enhanced the activation of effector memory T cells and decreased the frequency of MDSCs in patients responding to this regimen (50% of patients with metastasic melanoma and 33% of patients with HNSCC)228.
GM-CT-01 (19), also known as Davanat (Galectin Therapeutics; Fig. 4), is a chemically modified galactomannan from the plant Cyamopsis tetragonoloba (guar gum) that has been shown to enhance the efficacy of the chemotherapeutic agent 5-fluorouracil (5-FU) in models of colon and breast cancer229. These results led to phase I (NCT00054977) and phase II (NCT00110721) clinical trials that demonstrated a lack of toxicity and a 46% increase in survival of patients with colorectal cancer who received this drug combination214,230. A comparison of GCS-100 (17) and Davanat (19) suggests that these two polysaccharides might have different mechanisms of action. Although the binding site for PectaSol or GCS-100 is still not clear218, Davanat binds to the F-face of the GAL3 β-sandwich and not to the canonical site for β/α-galactosides231 (Fig. 4b).
Plant-derived polysaccharides have also been proposed as therapeutics for liver, kidney and lung fibrosis, mainly via mechanisms that involve inhibition of GAL3 (refs. 142,146). Inhibition of GAL3 with belapectin (18, Fig. 4) and Davanat (19, Fig. 4) was first evaluated in a toxin-induced model of liver fibrosis232. Intraperitoneal administration of these polysaccharides resulted in decreased collagen content, attenuated liver fibrosis, diminished cirrhosis and a reduced percentage of GAL3-expressing macrophages232. These two inhibitors were also tested in a murine model of NASH233. Intravenous administration of belapectin resulted in a substantial reduction in collagen deposition, hepatocellular damage, NASH activity and fibrosis — features that were associated with reduced markers of inflammation that included inducible nitric oxide synthase (iNOS) and CD36+ pro-inflammatory macrophages. By contrast, administration of Davanat had no effect233. A phase I clinical trial of belapectin (18) in patients with NASH with advanced hepatic fibrosis revealed no toxicity and good tolerability234 (NCT01899859, Table 1). Two phase II clinical trials evaluated the efficacy of this compound in liver fibrosis. In patients with NASH with advanced fibrosis (NCT02421094, Table 1), belapectin had no significant effects on levels of non-invasive biomarkers of liver inflammation or fibrosis over a 4-month period234. In liver fibrosis and resultant portal hypertension in patients with NASH cirrhosis235 (NCT02462967, Table 1), belapectin had no impact on fibrosis or nonalcoholic fatty liver disease activity score. However, in a patient subgroup it showed a significant effect on portal pressure and prevented the development of oesophageal varices, which is an early sign of serious complications in patients with cirrhosis. This led to the development of a phase IIb/III trial designed to evaluate its safety and efficacy specifically in patients with NASH-associated cirrhosis for the prevention of oesophageal varices (NCT04365868, Table 1).
Other polysaccharides have also emerged as potential galectin inhibitors. For example, RN1 (20, Fig. 4), an arabinogalactan polysaccharide isolated from the flowers of the Chinese ginseng plant (Panax notoginseng), was proposed as a GAL3 inhibitor. This compound showed antitumoural activity in pancreatic ductal adenocarcinoma both in vitro and in vivo236. Other ginseng-derived pectins prevented GAL3-driven T cell apoptosis and reduced tumour growth in mouse models of sarcoma237.
Peptides and peptidomimetics
A peptide known as Anginex (βpep-25, 21, Fig. 5) that was designed based on the structure of anti-angiogenic proteins was later found to be a potential anti-galectin therapeutic238,239. Anginex is a 33-mer with an amphipathic β-sheet that initially lacked a specific molecular target. However, several studies revealed that this compound had potent anti-angiogenic activity via its capacity to block EC adhesion and migration, ultimately leading to inhibition of angiogenesis and tumour growth240,241,242. GAL1 was subsequently identified as the molecular target of Anginex using fluorescence microscopy, NMR and surface plasmon resonance (SPR) analysis, with a strong binding constant (Kd 90 nM) (ref. 243). However, despite these initially promising results, Anginex was beset by several drawbacks that hampered its translation to clinical settings; these include its complex chemical synthesis, short half-life and poor stability. Aiming at a more affordable alternative than peptide synthesis, an artificial gene encoding Anginex was developed, offering a recombinant peptide with the same structure and slightly decreased activity244. To overcome its rapid clearance and sensitivity to endogenous endopeptidases, an adeno-associated virus vehicle delivery system was evaluated245. Using this delivery system, Anginex suppressed proliferation, migration and invasion of human umbilical vein endothelial cells (HUVECs) and inhibited angiogenesis and tumour growth in an ovarian cancer xenograft model. Finally, the Anginex derivative 6DBF7 (22, Fig. 5), a dibenzofuran (DBF)-based partial peptidomimetic, demonstrated improved stability246. 6DBF7 retained anti-angiogenic and tumour growth inhibitory activity in ovarian cancer mouse models and was even more effective in vivo than Anginex246. Dings et al.247 designed and tested new optimized analogues of 6DBF7 (22) and introduced DB21 (23, Fig. 5), a derivative capable of increased inhibition of angiogenesis and tumour growth in melanoma, lung adenocarcinoma and ovarian cancer models. Both 6DBF7 and DB21 were identified as GAL1 inhibitors, although no data on their interactions with other members of the galectin family are available.

Structures of: a, Anginex (21); b, 6DBF7 (22); c, DB21 (23); d, OTX008 (24); e, PTX013 (25); f, G3-A9 (26); and g, G3-C12 (27). h, GAL1 binding site for OTX008 (24). Amino acid residues that interact with OTX008 are highlighted in orange (moderate interaction) or red (high interaction). NB, no binding observed; ND, not determined.
More recently, OTX008 (or 0118, also called PTX008; 24, Fig. 5) was developed as a non-peptidic calixarene-based Anginex topomimetic with GAL1 binding capacity248. OTX008 inhibited angiogenesis in both in vitro and mouse model systems99,249. It also synergized with the tyrosine kinase inhibitor sunitinib in ovarian carcinoma and glioblastoma xenograft models250. A phase I clinical trial sponsored by Oncoethix assessed the impact of OTX008 administered subcutaneously to patients with advanced solid tumours and documented its rapid absorption and urinary excretion (NCT01724320)251. More recently, Leung et al.252 demonstrated that OTX008 combined with sorafenib reduced tumour growth and enhanced the therapeutic effects of sorafenib alone in experimental models of hepatocellular carcinoma. OTX008 was also effective against thyroid cancer in in vitro and in vivo studies253. Other calixarene derivatives include the polycationic calixarene-based compound PTX013 (25), which displayed 50-fold enhanced activity over that of OTX008 in an experimental melanoma model254. Furthermore, new analogues showed increased cell growth inhibitory activity in HUVECs and MA148 ovarian cancer cells, when compared with the parent calixarene compound 24 (ref. 255). However, the quest for improved activity led to ‘off-target’ side effects: PTX013 was toxic in mice, and considering that, in vitro, PTX013 could be considered a cytotoxic drug more than a cytostatic agent (like parental PTX008), the authors postulated that GAL1 might not be the only target of this compound254,256. In this sense, PTX013 has recently been shown to bind to both GAL3 and GAL1; this was suggested as a potential mechanism underlying its increased activity257. The development of PTX013 is an important example of how structural modifications aimed at improving biological activity can result in increased toxicity via potentially different mechanisms of action254.
An alternative approach to designing galectin-targeting peptides was developed by the Deutscher group258, which generated two small synthetic 15-mer peptides, G3-A9 (26, Fig. 5) and G3-C12 (27, Fig. 5), which were identified by combinatorial bacteriophage display. These peptides were selective for GAL3, but not for GAL1 or GAL4. They blocked the interaction of GAL3 with the Tf antigen and inhibited the adhesion of MDA-MD-435 human breast carcinoma cells to ECs in vitro. Peptide G3-C12 (27) is antimetastatic via its capacity to modulate GAL3 activity258; its administration resulted in a 72% reduction in MDA-MB-231 cell adhesion to vasculature in nude mice. However, despite its therapeutic potential, in vivo studies showed that G3-C12 accumulated in the kidneys259.
To overcome this limitation, control its renal clearance and improve its therapeutic efficacy, G3-C12 (27, Fig. 5) was conjugated to an N-(2-hydroxypropyl)methacrylamide polymeric carrier. This substitution increased its molecular size and generated a larger GAL3-targeted copolymer that was proposed as an anticancer agent260,261. Conjugation of HMPA–G3-C12 copolymer to 5-FU led to increased cytotoxicity, tumour cell apoptosis and inhibition of migration in vitro compared with 5-FU, as well as greater reductions in tumour volume in a PC-3 prostate cancer mouse model262. Micellar nanoparticles assembled by poly(oligo(ethylene glycol)) monomethyl ether methacrylate (POEGMA) and poly(ε-caprolactone) (PCL) copolymers conjugated to G3-C12 peptide were designed to deliver G3-C12 (27, Fig. 5) to the tumour microenvironment. This copolymer formed nanoparticles that were loaded with the anticancer drug bufalin (BUF) and tested as a drug delivery system for castration-resistant prostate cancer (CRPC). Biodegradable BUF-G3-C12 nanoparticles exhibited controlled drug release and enhanced tumour reduction in mice bearing DU145 prostate cancer cells263.
Biological agents
To avoid ‘off-target’ effects and achieve higher selectivity, efforts are underway to develop biological agents that target individual members of the galectin family, including galectin-specific neutralizing mAbs, decoys, aptamers and RNA silencing strategies using siRNA9,98,100,264,265,266,267. Both RNA interference (RNAi) and CRISPR technologies have been used to inhibit the expression and function of specific galectins in several experimental models268. GAL1 silencing with RNA technologies led to dramatic antitumour effects in several cancer models9,98,114,125,126,128,256 by unleashing antitumour immunity and suppressing aberrant vascularization9,98,266. More recently, administration of a GAL1-targeted DNA aptamer resulted in antitumour effects via its capacity to restore immune function in vivo267. Despite promising results in preclinical models, more investigations into the mechanisms that underlie nucleic acid-based targeting strategies will be required to translate these findings into clinically important therapies.
Over the past two decades, mAb-based therapies that target immune checkpoint or pro-angiogenic pathways have generated unprecedented success in treating a broad range of malignancies269. mAb-based therapeutics directed against galectins could be an important approach, as these agents are target specific and well tolerated in humans270. To date, several GAL1-neutralizing mAbs have been developed, characterized and functionally evaluated in in vitro and in vivo models9,98,100,271,272. For example, anti-GAL1 mAbs inhibit GAL1-mediated apoptosis of Epstein–Barr virus (EBV)-specific CD8+ T cells, suggesting that they could prevent EBV-induced immune evasion in post-transplant lymphoproliferative disorders (PTLDs)271. Moreover, these mAbs restrained tumour growth in models of Kaposi’s sarcoma, lung adenocarcinoma, melanoma and T cell lymphoma9,98. Because GAL1 is known to activate VEGFR2 signalling9, the ability of anti-GAL1 mAb to prevent resistance to VEGF blockade was assessed in various models. Blockade of GAL1–N-glycan interactions using a functional blocking mAb not only circumvented resistance to anti-VEGF but also normalized aberrant tumour vasculature and promoted an increased influx of immune cells to the tumour bed9. Given these promising findings, additional mAbs specific for GAL1 have been designed and found to have therapeutic potential100,272. Interestingly, mAb-mediated GAL1 blockade enhanced the immunostimulatory effect of anti-PD1 therapy in HNSCC by preventing T cell exclusion from tumour sites92. Moreover, a recombinant vaccine protein consisting of bacterial sequences fused to GAL1 was recently described to generate high anti-GAL1 antibody levels in experimental models of melanoma, associated with increased cytotoxic T cell infiltration and decreased tumour burden273.
Based on the oncogenic roles of GAL3 triggered upon interaction with N-glycans on MUC16 (ref. 274), Stasenko et al.275 evaluated the therapeutic potential of anti-GAL3 mAbs in high-grade serous ovarian cancer and other MUC16/CA-125-expressing malignancies. The GAL3-blocking mAb 14D11 recognizes both human and mouse GAL3, but not other galectin family members. Administration of this agent extended overall survival in tumour-bearing mice and delayed the occurrence of lung metastasis in breast cancer models275. Recently, anti-GAL3 mAbs developed by the company TrueBinding were patented for the treatment of liver fibrosis, pulmonary fibrosis, NASH and various neoplastic conditions276.
Furthermore, given the tolerogenic activities of GAL9 in Dectin-1-expressing macrophages124, a fully human IgG4 mAb to GAL9 known as LYT200 was developed by PureTech Health, and was the first anti-galectin mAb to reach clinical evaluation. This agent is in phase I/II clinical trials either alone or in combination with chemotherapy (gemcitabine/nab-paclitaxel) or anti-PD1 mAbs as a potential treatment for metastatic solid tumours (NCT04666688, Table 1). In 2021, the FDA designated LYT200 an orphan drug for the treatment of pancreatic cancer. Additionally, anti-GAL9-based therapies (including LYT200) exhibit cytotoxic effects in models of human T cell acute lymphoblastic leukaemia (T-ALL)277. Thus, GAL1-, GAL3- or GAL9-blocking mAbs, either alone or in combination, represent an array of multifunctional and selective strategies that can reprogramme the tumour, immune and vascular landscapes in a broad range of malignancies.
Another strategy for GAL3 blockade was the development of an N-terminally truncated form of this lectin (GAL3C); the N-terminal fragment is not involved in carbohydrate recognition but is essential for cooperative binding and cross-linking. As a consequence, GAL3C functions as a competitive inhibitor of GAL3 by preventing homophilic cross-linking following its interactions with glycan structures. Recombinant GAL3C has advantages such as a relatively simple and controlled production process and high selectivity compared with other synthetic inhibitors and natural polysaccharides278. GAL3C exhibited antitumour and antimetastatic activities in models of breast279 and ovarian278 cancer via mechanisms involving CD44 inactivation and inhibition of the effects of MUC1, including abrogation of AKT and nuclear factor-κB (NF-κB) downstream signalling pathways. Furthermore, intravenous administration of GAL3C in a multiple myeloma model resulted in diminished tumour growth and increased the therapeutic impact of the proteasome inhibitor bortezomib280. However, a major limitation of GAL3C is its rapid clearance; hence, efforts are underway to explore the effects of GAL3C conjugation with other molecules to increase its molecular weight and stability. In this regard, the fusion of GAL1 with the Fc region of human IgG1 (GAL1hFc) resulted in enhanced stability and lifespan of this lectin accompanied by more pronounced immunoregulatory activity134. Thus, structural modifications might improve galectin-mediated therapeutic efficacy, mitigate ‘off-target’ effects and amplify clinically relevant antagonistic or agonistic activities.
Future potential of galectin inhibitors
Several challenges need to be addressed to accelerate the progress of galectin inhibitors into clinical settings. Some challenges are specific to particular therapeutic modalities; for example, compared with peptides, pectins or mAbs, small-molecule inhibitors such as galactosides or lactosides are more challenging to synthesize, and the development of new synthetic methods for these types of compound is currently an active area of research281. Other challenges are common to some or all of the modalities, including thorough understanding of their selectivity, biodistribution and safety profiles, and dissection of specific mechanisms of action. Below, we compare the advantages and limitations of various galectin-targeted therapeutic modalities and consider their future clinical potential.
Understanding selectivity
A key challenge in the design of galectin inhibitors is to avoid ‘off-target’ effects by preventing binding to other cellular proteins, including other family members. Although GAL1, GAL3 and GAL9 seem to share a common pro-fibrotic and pro-tumoural function, they might have contrasting roles in other pathological conditions. Moreover, given their structural similarities and wide tissue distribution, selective targeting of specific galectins could also become relevant in fibrotic or neoplastic diseases. To achieve selective targeting, it might be necessary to exploit the differences in galectin expression pattern and biological activity as well as differences in their ligand-binding specificity, capacity to cross-link individual glycosylated receptors and extracellular or intracellular distribution. Moreover, biochemical factors (stability, redox status, pH and multimerization) that govern their glycan-dependent or independent interactions should also be considered282,283,284.
The extensive investigations into higher affinity and more selective small-molecule galectin inhibitors has led to an improved understanding of the crucial interactions that underlie their inhibitory activity and enhanced selectivity179. The use of GB0139 (11) for IPF and GB1211 for NASH laid the foundations for translational studies focused on small-molecule galectin inhibitors. GB0139 was first described as a potent inhibitor of GAL3 but also exhibited affinity for GAL1 (ref. 144). This finding highlighted the potential use of these compounds for therapeutic targeting of multiple galectins, including those with pro-fibrotic activity.
Regarding peptidomimetics, most of the beneficial effects of Anginex have been associated with GAL1 inhibition, although affinity for GAL2, GAL7, GAL8N and GAL9N has also been demonstrated285, suggesting that Anginex and related compounds have limited selectivity. Additional work is needed to explore the selectivity of peptide-based inhibitors towards different galectins and potential ‘off-target’ effects.
Modified natural complex polysaccharides such as MCPs or galactomannans are well tolerated in humans with few adverse effects. However, they vary substantially in molecular weight and degree of esterification, and their non-synthetic nature limits the thorough structural characterization required to understand their selectivity. Of particular note, the affinity of galectins for pectins and galactomannans has been questioned218. GCS-100 has been proposed as a GAL3 inhibitor, although its selectivity with respect to other galectins remains unknown. Further biochemical characterization of galectin–polysaccharide interactions is required, as well as in vivo studies on the biological activities of these compounds in galectin-null mice, as the possibility that polysaccharides could interact with other key mediators of disease pathogenesis has not been ruled out218.
Despite its enhanced selectivity when compared with other inhibitors, truncated GAL3C has not yet reached clinical trials. The specificity of anti-GAL1 and anti-GAL3 mAbs has been demonstrated in in vitro assays9,35,272,275, although further studies performed in vivo are needed to examine potential ‘off-target’ effects.
For these highly specific biological agents, it is crucial to recognize that neoplastic diseases present a case-by-case challenge; in some cases, selectivity for a specific galectin might not be crucial because two or more galectins exert similar biological activities (for example, immunosuppression driven by GAL1, GAL3 and/or GAL9). In these cases, overall galectin activities could be blocked using a pan-galectin inhibitor. The varied expression levels, diverse cell sources and possible opposing functions of galectins based on extracellular or intracellular localization should be examined in individual cancers. Recent advances in human organoids and humanized animal models, together with the development of single-cell technologies286,287,288 and machine learning algorithms, will be invaluable to discern the source and cell-specific function of these lectins in the tumour microenvironment. Furthermore, the exquisite regulation by glycan epitope density on specific receptors as well as the multivalent nature of these interactions should also be considered58. In this respect, multivalent glycodendrimers, sugar calixarenes and nanotechnology-based developments could be useful to understand the natural clustering and presentation of ligands on the target cell surface, providing crucial mechanistic insights into their multivalency. A low-affinity inhibitor might be presented in clusters, which could increase its potency. As an example, lactose calixarenes were described as selective GAL4 inhibitors in vitro, with 300-fold improvement in binding289. However, these large, polar and labile molecules could present a difficult challenge owing to their ADME(T) properties.
Overall, a deeper understanding of the multifunctionality and disease-specific expression profiles of the various galectins, improved characterization of the cellular glycome and further identification of galectin binding partners and receptors will be essential for the design of novel galectin inhibitors with various degrees of selectivity.
Identifying mechanisms of action
The mechanisms of action of currently available galectin inhibitors differ considerably from one another, and, in some cases, remain a matter of ongoing debate. Although plant-derived polysaccharides such as MCPs or galactomannans have reached clinical trials, their specific targets and/or mechanisms of action remain unclear218,290,291,292. The apparent contradiction between the limited interactions with galectins and the health-promoting effects of these polysaccharides was evaluated by Miller et al.231,293,294, who suggested that the biological effects do not involve interactions with canonical galectin CRDs. Indeed, Stegmayr et al.218 have proposed that Davanat inactivates GAL1 by an oxidative mechanism. Likewise, the negative charge of pectins suggests that their therapeutic effects could be dependent on charge-based interactions instead of specific intermolecular affinities. Other studies of pectin-derived polysaccharides focused on their direct cytotoxic effects involving apoptosis and autophagy213,295. Moreover, it is not clear whether these high molecular weight polysaccharides target the intracellular functions of galectins via internalization (as observed in the case of dextrans), followed by selective distribution to various subcellular compartments296. Additionally, these macromolecules might function by sequestering galectins in the extracellular space, thereby modifying their distribution between the intracellular and extracellular compartments. Although this is an attractive hypothesis, intracellular levels of GAL3 did not change upon the addition of MCPs218. Additional studies focused on the mechanisms that underlie the uptake, function and target engagement of plant-derived polysaccharides will be necessary to improve our understanding of their therapeutic potential.
Interestingly, regarding peptidomimetics, the DBF-based analogues 6DBF7 and DB21 inhibited tumour growth in mice to an even greater extent than Anginex, possibly owing to their increased bioavailability and/or potential differences in mechanisms of action242,247. Whereas Anginex paradoxically enhances GAL1 binding to several oligosaccharides and glycoproteins285, the peptidomimetics 6DBF7 and DB21 behave as non-competitive allosteric inhibitors of GAL1 that reduce its affinity for lactose247. Topomimetics OTX008 and PTX013 also function as allosteric inhibitors of GAL1, albeit via an interaction site that differs from the one used by Anginex derivatives (Fig. 5). Like 6DBF7 and DB21, and in contrast to parental Anginex, these compounds decrease the affinity of the GAL1 CRD for lactose254. Collectively, these results suggest that structural modification of these peptides results in diversification of their mechanisms of action. Interestingly, using a recombinant adeno-associated virus to deliver Anginex revealed that this compound inhibited VEGF expression and the AKT, JNK and NF-κB downstream signalling pathways245. These GAL1-independent effects, together with the ability of Anginex to disrupt EC membranes via electrostatic interactions297, point to alternative modes of action. Additional in vivo studies in GAL1-deficient mice might help to unravel these effects.
Clinical outlook
The development of small-molecule galectin inhibitors has progressed towards high-affinity agents, surmounting limitations such as low stability and lifespan and leading to two pioneering compounds currently under clinical evaluation: the TDG derivative GB0139 for IPF, and the α-thiogalactoside GB1211 for NASH, liver fibrosis and NSCLC. Given the results obtained in clinical trials, their therapeutic potential is promising in both fibrosis and cancer. For example, a phase II trial to evaluate the safety and efficacy of GB1211 in combination with pembrolizumab in HNSCC and melanoma is expected to begin in 2023.
Regarding polysaccharides, clinical evaluation of GCS-100 and galactomannan Davanat has not been resumed, whereas PectaSol-C is commercially available as a dietary supplement for health and oncology support. Future studies will be crucial to unveil the full potential of the galactoarabino-rhamnogalacturonate belapectin, which is currently in phase IIb/III clinical trials for the prevention of eosophageal varices in NASH, and in the early stages of evaluation for melanoma, NSCLC and HNSCC in combination with pembrolizumab.
Among peptide and peptidomimetics, only OTX008 has been clinically evaluated, and this was more than a decade ago. Although the results of this phase I study documented its safety and low toxicity, no further advances have been described. Despite considerable efforts, our understanding of the mechanistic basis of the antitumour effects of peptide and peptidomimetic drugs beyond their specific interactions with galectins remains incomplete252,253.
Anti-galectin mAbs have been introduced more recently to reinforce antitumour immunity, reprogramme pathological angiogenesis and restrain tumour metastasis9,98,124,272. This concept is based on findings that suggest that anti-galectin mAbs have multifunctional roles as cancer therapeutics and that their use helps to avoid the accumulated toxicity associated with administration of multiple mAbs. The anti-GAL9 mAb LYT200 is under clinical assessment and is an example of the emerging strategy of targeting glycosylation-related immune checkpoints in cancer14,298. The use of intrabodies designed to target the intracellular functions of galectins, or nanobodies299 capable of reaching areas within tumours and fibrotic foci, might be an important breakthrough in the coming years. Moreover, functionalized nanoparticles containing galectin-specific siRNA265, aptamers267 and scFv fragments300 might be alternative agents to target galectin–glycan interactions.
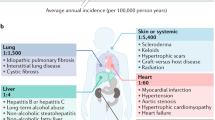
Finally, results obtained in clinical trials for NASH and IPF should facilitate the clinical translation of galectin blockers designed to treat other fibrotic conditions. For example, GAL3 has a crucial role in cardiac fibrosis leading to heart failure301, and clinical trials designed to evaluate PectaSol-C in patients with high blood pressure have been completed (NCT01960946)302. Moreover, inhibition of GAL3 with GB0139 (11) resulted in diminished VEGF-driven angiogenesis in a mouse model of corneal fibrosis303. In this regard, GAL1 has been proposed as a relevant target in renal fibrosis304, atherosclerosis and abdominal aortic aneurysms34, highlighting novel therapeutic opportunities for its inhibitors. Additionally, recognition of the central role of galectins in the pathogenesis of autoimmune inflammation16 and neurodegeneration30,31 has led to the design of galectin-based agonists and antagonists to treat these chronic inflammatory conditions. In this regard, TB006, an anti-GAL3 mAb developed by TrueBinding, is in a phase II clinical trial for treatment of acute ischaemic stroke (NCT05156827) and a phase I/II trial for Alzheimer disease (NCT05074498).
In conclusion, the rapid pace of advances in glycobiology, the promising performance of some galectin inhibitors in preclinical studies and clinical trials, and the urgent need for alternative therapies for cancer and fibrotic diseases should help to accelerate the translation of anti-galectin therapeutics into clinical practice.
Change history
20 February 2023
In the version of this article originally published, in Figure 3g, compound 13, galactose units were presented without methylene groups, as are now amended in the HTML and PDF versions of the article.
References
Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 15, 346–366 (2019).
Ohtsubo, K. & Marth, J. D. Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867 (2006).
Gagneux, P., Hennet, T. & Varki, A. Essentials of Glycobiology 4th edn. 79–92 (Cold Spring Harbor Laboratory Press, 2022).
Dube, D. H. & Bertozzi, C. R. Glycans in cancer and inflammation — potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 4, 477–488 (2005).
Pinho, S. S. & Reis, C. A. Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15, 540–555 (2015).
van Kooyk, Y. & Rabinovich, G. A. Protein–glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 9, 593–601 (2008).
McCarthy, C. et al. The role and importance of glycosylation of acute phase proteins with focus on alpha-1 antitrypsin in acute and chronic inflammatory conditions. J. Proteome Res. 13, 3131–3143 (2014).
Rabinovich, G. A. & Croci, D. O. Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity 36, 322–335 (2012).
Croci, D. O. et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156, 744–758 (2014). This study identifies the relevance of GAL1–glycan interactions as a mechanism of resistance to anti-VEGF therapies and reports the synergistic effects of anti-VEGF and anti-GAL1 mAbs in anti-angiogenic therapies.
Dewald, J. H., Colomb, F., Bobowski-Gerard, M., Groux-Degroote, S. & Delannoy, P. Role of cytokine-induced glycosylation changes in regulating cell interactions and cell signaling in inflammatory diseases and cancer. Cells 5, 43 (2016).
Cagnoni, A. J., Pérez Sáez, J. M., Rabinovich, G. A. & Mariño, K. V. Turning-off signaling by siglecs, selectins, and galectins: chemical inhibition of glycan-dependent interactions in cancer. Front. Oncol. 6, 1–21 (2016).
Kirwan, A., Utratna, M., O’Dwyer, M. E., Joshi, L. & Kilcoyne, M. Glycosylation-based serum biomarkers for cancer diagnostics and prognostics. Biomed. Res. Int. 2015, 490531 (2015).
Hanić, M., Lauc, G. & Trbojević-Akmačić, I. N-glycan analysis by ultra-performance liquid chromatography and capillary gel electrophoresis with fluorescent labeling. Curr. Protoc. Protein Sci. 97, e95 (2019).
Smith, B. A. H. & Bertozzi, C. R. The clinical impact of glycobiology: targeting selectins, siglecs and mammalian glycans. Nat. Rev. Drug Discov. 20, 217–243 (2021). This review highlights the role of selectins and siglecs as emerging glycocheckpoints and therapeutic targets.
Cerliani, J. P., Blidner, A. G., Toscano, M. A., Croci, D. O. & Rabinovich, G. A. Translating the ‘Sugar Code’ into immune and vascular signaling programs. Trends Biochem. Sci. 42, 255–273 (2017).
Toscano, M. A., Martínez Allo, V. C., Cutine, A. M., Rabinovich, G. A. & Mariño, K. V. Untangling galectin-driven regulatory circuits in autoimmune inflammation. Trends Mol. Med. 24, 348–363 (2018).
Girotti, M. R., Salatino, M., Dalotto-Moreno, T. & Rabinovich, G. A. Sweetening the hallmarks of cancer: galectins as multifunctional mediators of tumor progression. J. Exp. Med. 217, e20182041 (2020).
Navarro, P., Martínez-Bosch, N., Blidner, A. G. & Rabinovich, G. A. Impact of galectins in resistance to anticancer therapies. Clin. Cancer Res. 26, 6086–6101 (2020).
Loomba, R., Friedman, S. L. & Shulman, G. I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564 (2021).
Xu, W.-D., Huang, Q. & Huang, A.-F. Emerging role of galectin family in inflammatory autoimmune diseases. Autoimmun. Rev. 20, 102847 (2021).
Slack, R. J., Mills, R. & Mackinnon, A. C. The therapeutic potential of galectin-3 inhibition in fibrotic disease. Int. J. Biochem. Cell Biol. 130, 105881 (2021).
Somogyi, V. et al. The therapy of idiopathic pulmonary fibrosis: what is next? Eur. Respir. Rev. 28, 190021 (2019).
Hirani, N. et al. Target-inhibition of galectin-3 by inhaled TD139 in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 57, 2002559 (2021). This study reports a phase I/IIa study of inhaled TD139 in healthy subjects and patients with IPF.
Mora, A. L., Rojas, M., Pardo, A. & Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 16, 755–772 (2017).
Ge, X. N. et al. Regulation of eosinophilia and allergic airway inflammation by the glycan-binding protein galectin-1. Proc. Natl Acad. Sci. USA 113, E4837–E4846 (2016).
Ge, X. N. et al. Allergen-induced airway remodeling is impaired in galectin-3–deficient mice. J. Immunol. 185, 1205–1214 (2010).
Morosi, L. G. et al. Control of intestinal inflammation by glycosylation-dependent lectin-driven immunoregulatory circuits. Sci. Adv. 7, eabf8630 (2021).
Martínez Allo, V. C. et al. Suppression of age-related salivary gland autoimmunity by glycosylation-dependent galectin-1driven immune inhibitory circuits. Proc. Natl Acad. Sci. USA 117, 6630–6639 (2020).
Sundblad, V. et al. Galectin-1 impacts on glucose homeostasis by modulating pancreatic insulin release. Glycobiology 31, 908–915 (2021).
Burguillos, M. A. et al. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 10, 1626–1638 (2015).
Starossom, S. C. et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity 37, 249–263 (2012).
Seropian, I. M. et al. Galectin-1 controls cardiac inflammation and ventricular remodeling during acute myocardial infarction. Am. J. Pathol. 182, 29–40 (2013).
MacKinnon, A. C. et al. Inhibition of galectin-3 reduces atherosclerosis in apolipoprotein E-deficient mice. Glycobiology 23, 654–663 (2013).
Roldán-Montero, R. et al. Galectin-1 prevents pathological vascular remodeling in atherosclerosis and abdominal aortic aneurysm. Sci. Adv. 8, eabm7322 (2022).
Russo, A. J. et al. Intracellular immune sensing promotes inflammation via gasdermin D-driven release of a lectin alarmin. Nat. Immunol. 22, 154–165 (2021).
Ramhorst, R. E. et al. Galectin-1 confers immune privilege to human trophoblast: implications in recurrent fetal loss. Glycobiology 22, 1374–1386 (2012).
Barondes, S. H. et al. Galectins: a family of animal β-galactoside-binding lectins. Cell 76, 597–598 (1994).
Méndez-Huergo, S. P., Blidner, A. G. & Rabinovich, G. A. Galectins: emerging regulatory checkpoints linking tumor immunity and angiogenesis. Curr. Opin. Immunol. 45, 8–15 (2017).
Vasta, G. R. Roles of galectins in infection. Nat. Rev. Microbiol. 7, 424–438 (2009).
Thiemann, S. & Baum, L. G. Galectins and immune responses — just how do they do those things they do? Annu. Rev. Immunol. 34, 243–264 (2016).
Liu, F.-T. & Rabinovich, G. A. Galectins: regulators of acute and chronic inflammation. Ann. N. Y. Acad. Sci. 1183, 158–182 (2010).
Rabinovich, G. A. & Toscano, M. A. Turning ‘sweet’ on immunity: galectin–glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol. 9, 338–352 (2009).
Hirabayashi, J., Hashidate, T., Arata, Y. & Nishi, N. Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim. Biophys. Acta 1572, 1–23 (2002).
Blixt, O. et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc. Natl Acad. Sci. USA 101, 17033 (2004).
Shimura, T. et al. Galectin-3, a novel binding partner of β-catenin. Cancer Res. 64, 6363–6367 (2004).
Yang, R.-Y., Rabinovich, G. A. & Liu, F.-T. Galectins: structure, function and therapeutic potential. Expert Rev. Mol. Med. 10, e17 (2008).
Park, J. W., Voss, P. G., Grabski, S., Wang, J. L. & Patterson, R. J. Association of galectin-1 and galectin-3 with Gemin4 in complexes containing the SMN protein. Nucleic Acids Res. 29, 3595–3602 (2001).
Liu, F.-T., Patterson, R. J. & Wang, J. L. Intracellular functions of galectins. Biochim. Biophys. Acta Gen. Subj. 1572, 263–273 (2002).
Fritsch, K. et al. Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer 16, 502 (2016).
Chen, H.-Y. et al. Galectin-3 negatively regulates TCR-mediated CD4+ T-cell activation at the immunological synapse. Proc. Natl Acad. Sci. USA 106, 14496–14501 (2009). This study highlights the intracellular role of GAL3 in limiting TCR-dependent T cell activation.
Liu, W. et al. Galectin-3 regulates intracellular trafficking of EGFR through Alix and promotes keratinocyte migration. J. Invest. Dermatol. 132, 2828–2837 (2012).
Weng, I.-C. et al. Cytosolic galectin-3 and -8 regulate antibacterial autophagy through differential recognition of host glycans on damaged phagosomes. Glycobiology 28, 392–405 (2018).
Hong, M.-H., Weng, I.-C., Li, F.-Y., Lin, W.-H. & Liu, F.-T. Intracellular galectins sense cytosolically exposed glycans as danger and mediate cellular responses. J. Biomed. Sci. 28, 16 (2021).
Nabi, I. R., Shankar, J. & Dennis, J. W. The galectin lattice at a glance. J. Cell Sci. 128, 2213–2219 (2015).
Rabinovich, G. A., Toscano, M. A., Jackson, S. S. & Vasta, G. R. Functions of cell surface galectin-glycoprotein lattices. Curr. Opin. Struct. Biol. 17, 513–520 (2007).
Blenda, A. V. et al. Galectin-9 recognizes and exhibits antimicrobial activity toward microbes expressing blood group–like antigens. J. Biol. Chem. 298, 101704 (2022).
Wu, S.-C. et al. Full-length galectin-3 is required for high affinity microbial interactions and antimicrobial activity. Front. Microbiol. 12, 731026 (2021).
Dennis, J. W. & Brewer, C. F. Density-dependent lectin-glycan interactions as a paradigm for conditional regulation by posttranslational modifications. Mol. Cell. Proteom. 12, 913–920 (2013).
Nielsen, M. I. et al. Galectin binding to cells and glycoproteins with genetically modified glycosylation reveals galectin–glycan specificities in a natural context. J. Biol. Chem. 293, 20249–20262 (2018).
Johannes, L., Jacob, R. & Leffler, H. Galectins at a glance. J. Cell Sci. 131, jcs208884 (2018).
Mereiter, S., Balmaña, M., Campos, D., Gomes, J. & Reis, C. A. Glycosylation in the era of cancer-targeted therapy: where are we heading? Cancer Cell 36, 6–16 (2019).
Okoye, I. et al. Galectin-9 expression defines exhausted T cells and impaired cytotoxic NK cells in patients with virus-associated solid tumors. J. Immunother. Cancer 8, e001849 (2020).
Henderson, N. C. & Sethi, T. The regulation of inflammation by galectin-3. Immunol. Rev. 230, 160–171 (2009).
Liu, F.-T. & Rabinovich, G. A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 5, 29–41 (2005).
Thijssen, V. L., Heusschen, R., Caers, J. & Griffioen, A. W. Galectin expression in cancer diagnosis and prognosis: a systematic review. Biochim. Biophys. Acta Rev. Cancer 1855, 235–247 (2015).
Veschi, V. et al. Galectin-3 is a marker of favorable prognosis and a biologically relevant molecule in neuroblastic tumors. Cell Death Dis. 5, e1100 (2014).
Lahm, H. et al. Comprehensive galectin fingerprinting in a panel of 61 human tumor cell lines by RT-PCR and its implications for diagnostic and therapeutic procedures. J. Cancer Res. Clin. Oncol. 127, 375–386 (2001).
Partridge, E. A. et al. Regulation of cytokine receptors by golgi N-glycan processing and endocytosis. Science 306, 120–124 (2004). This study emphasizes the role of galectin–glycan interactions in modulating retention and endocytosis of N-glycosylated receptors.
Perrotta, R. M., Bach, C. A., Salatino, M. & Rabinovich, G. A. Reprogramming the tumor metastasis cascade by targeting galectin-driven networks. Biochem. J. 478, 597–617 (2021).
Reticker-Flynn, N. E. & Bhatia, S. N. Aberrant glycosylation promotes lung cancer metastasis through adhesion to galectins in the metastatic niche. Cancer Discov. 5, 168–181 (2015).
Nakajima, K. et al. Galectin-3 cleavage alters bone remodeling: different outcomes in breast and prostate cancer skeletal metastasis. Cancer Res. 76, 1391–1402 (2016).
Bacigalupo, M. L. et al. Galectin-1 triggers epithelial-mesenchymal transition in human hepatocellular carcinoma cells. J. Cell. Physiol. 230, 1298–1309 (2015).
Zhu, J. et al. Galectin-1 induces metastasis and epithelial-mesenchymal transition (EMT) in human ovarian cancer cells via activation of the MAPK JNK/p38 signalling pathway. Am. J. Transl. Res. 11, 3862–3878 (2019).
Nangia-Makker, P., Hogan, V. & Raz, A. Galectin-3 and cancer stemness. Glycobiology 28, 172–181 (2018).
Chong, Y. et al. Galectin-1 induces invasion and the epithelial-mesenchymal transition in human gastric cancer cells via non-canonical activation of the hedgehog signaling pathway. Oncotarget 7, 50 (2016).
Zhang, P.-F. et al. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 7, e2201–e2201 (2016).
Carabias, P. et al. Galectin-1 confers resistance to doxorubicin in hepatocellular carcinoma cells through modulation of P-glycoprotein expression. Cell Death Dis. 13, 79 (2022).
Orozco, C. A. et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor–stroma crosstalk. Proc. Natl Acad. Sci. USA 115, 3769–3778 (2018).
Shih, T.-C. et al. Targeting galectin-1 impairs castration-resistant prostate cancer progression and invasion. Clin. Cancer Res. 24, 4319–4331 (2018).
Piyush, T. et al. Interaction of galectin-3 with MUC1 on cell surface promotes EGFR dimerization and activation in human epithelial cancer cells. Cell Death Differ. 24, 1937–1947 (2017).
Mori, Y. et al. Binding of galectin-3, a β-galactoside-binding lectin, to MUC1 protein enhances phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) and Akt, promoting tumor cell malignancy. J. Biol. Chem. 290, 26125–26140 (2015).
Murugaesu, N. et al. An in vivo functional screen identifies ST6GalNAc2 sialyltransferase as a breast cancer metastasis suppressor. Cancer Discov. 4, 304–317 (2014).
Wang, L.-P., Chen, S.-W., Zhuang, S.-M., Li, H. & Song, M. Galectin-3 accelerates the progression of oral tongue squamous cell carcinoma via a Wnt/β-catenin-dependent pathway. Pathol. Oncol. Res. 19, 461–474 (2013).
Seguin, L. et al. Galectin-3, a druggable vulnerability for KRAS-addicted cancers. Cancer Discov. 7, 1464–1479 (2017).
Hsu, D. K., Yang, R.-Y., Saegusa, J. & Liu, F.-T. Analysis of the intracellular role of galectins in cell growth and apoptosis. Methods Mol. Biol. 1207, 451–463 (2015).
Belanis, L., Plowman, S. J., Rotblat, B., Hancock, J. F. & Kloog, Y. Galectin-1 is a novel structural component and a major regulator of h-ras nanoclusters. Mol. Biol. Cell 19, 1404–1414 (2008).
Paz, A., Haklai, R., Elad-Sfadia, G., Ballan, E. & Kloog, Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 20, 7486–7493 (2001). This work documents the intracellular role of GAL1 in mediating cellular tumorigenesis by interacting with oncogenic HRAS.
Elad-Sfadia, G., Haklai, R., Balan, E. & Kloog, Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J. Biol. Chem. 279, 34922–34930 (2004).
Wu, K.-L. et al. Overexpression of galectin-3 enhances migration of colon cancer cells related to activation of the K-Ras–Raf–Erk1/2 pathway. J. Gastroenterol. 48, 350–359 (2013).
Menachem, A., Bodner, O., Pastor, J., Raz, A. & Kloog, Y. Inhibition of malignant thyroid carcinoma cell proliferation by Ras and galectin-3 inhibitors. Cell Death Discov. 1, 15047 (2015).
Stiasny, A. et al. The involvement of E6, p53, p16, MDM2 and Gal-3 in the clinical outcome of patients with cervical cancer. Oncol. Lett. 14, 4467–4476 (2017).
Nambiar, D. K. et al. Galectin-1–driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. J. Clin. Invest. 129, 5553–5567 (2019). This study identifies a role of GAL1 in promoting T cell exclusion and modulating resistance to anti-PD1 immunotherapy in HNSCC.
Kuo, P. et al. Galectin-1 mediates radiation-related lymphopenia and attenuates NSCLC radiation response. Clin. Cancer Res. 20, 5558–5569 (2014).
Tseng, P.-C., Chen, C.-L., Shan, Y.-S. & Lin, C.-F. An increase in galectin-3 causes cellular unresponsiveness to IFN-γ-induced signal transduction and growth inhibition in gastric cancer cells. Oncotarget 7, 15150–15160 (2016).
Carmeliet, P. & Jain, R. K. Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307 (2011).
Thijssen, V. L., Rabinovich, G. A. & Griffioen, A. W. Vascular galectins: regulators of tumor progression and targets for cancer therapy. Cytokine Growth Factor. Rev. 24, 547–558 (2013).
Thijssen, V. L. et al. Tumor cells secrete galectin-1 to enhance endothelial cell activity. Cancer Res. 70, 6216–6224 (2010).
Croci, D. O. et al. Disrupting galectin-1 interactions with N-glycans suppresses hypoxia-driven angiogenesis and tumorigenesis in Kaposi’s sarcoma. J. Exp. Med. 209, 1985–2000 (2012).
Astorgues-Xerri, L. et al. OTX008, a selective small-molecule inhibitor of galectin-1, downregulates cancer cell proliferation, invasion and tumour angiogenesis. Eur. J. Cancer 50, 2463–2477 (2014).
van Beijnum, J. R. et al. A key role for galectin-1 in sprouting angiogenesis revealed by novel rationally designed antibodies. Int. J. Cancer 139, 824–835 (2016).
Laderach, D. J. et al. A unique galectin signature in human prostate cancer progression suggests galectin-1 as a key target for treatment of advanced disease. Cancer Res. 73, 86–96 (2013).
Hsieh, S. H. et al. Galectin-1, a novel ligand of neuropilin-1, activates VEGFR-2 signaling and modulates the migration of vascular endothelial cells. Oncogene 27, 3746–3753 (2008).
Wu, M.-H. et al. Galectin-1 induces vascular permeability through the neuropilin-1/vascular endothelial growth factor receptor-1 complex. Angiogenesis 17, 839–849 (2014).
Wei, J., Li, D. K., Hu, X., Cheng, C. & Zhang, Y. Galectin-1–RNA interaction map reveals potential regulatory roles in angiogenesis. FEBS Lett. 595, 623–636 (2021).
Markowska, A. I., Jefferies, K. C. & Panjwani, N. Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells. J. Biol. Chem. 286, 29913–29921 (2011).
Markowska, A. I., Liu, F.-T. & Panjwani, N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J. Exp. Med. 207, 1981–1993 (2010).
Nangia-Makker, P. et al. Cleavage of galectin-3 by matrix metalloproteases induces angiogenesis in breast cancer. Int. J. Cancer 127, 2530–2541 (2010).
Heusschen, R., Schulkens, I. A., van Beijnum, J., Griffioen, A. W. & Thijssen, V. L. Endothelial LGALS9 splice variant expression in endothelial cell biology and angiogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 1842, 284–292 (2014).
Aanhane, E. et al. Different angioregulatory activity of monovalent galectin-9 isoforms. Angiogenesis 21, 545–555 (2018).
Colomb, F. et al. Galectin-3 interacts with the cell-surface glycoprotein CD146 (MCAM, MUC18) and induces secretion of metastasis-promoting cytokines from vascular endothelial cells. J. Biol. Chem. 292, 8381–8389 (2017).
Chen, C. et al. Increased circulation of galectin-3 in cancer induces secretion of metastasis-promoting cytokines from blood vascular endothelium. Clin. Cancer Res. 19, 1693–1704 (2013).
Liebscher, L. et al. A minigene DNA vaccine encoding peptide epitopes derived from Galectin-1 has protective antitumoral effects in a model of neuroblastoma. Cancer Lett. 509, 105–114 (2021).
Shah, D. et al. A novel miR1983-TLR7-IFNβ circuit licenses NK cells to kill glioma cells, and is under the control of galectin-1. Oncoimmunology 10, 1939601 (2021).
Juszczynski, P. et al. The AP1-dependent secretion of galectin-1 by Reed-Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc. Natl Acad. Sci. USA 104, 13134–13139 (2007).
Croci, D. O. & Rabinovich, G. A. Linking tumor hypoxia with VEGFR2 signaling and compensatory angiogenesis: glycans make the difference. Oncoimmunology 3, e29380 (2014).
Rutkowski, M. R. et al. Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell 27, 27–40 (2015).
Tiraboschi, C. et al. Combining inhibition of galectin-3 with and before a therapeutic vaccination is critical for the prostate-tumor-free outcome. J. Immunother. Cancer 8, e001535 (2020).
de Mingo Pulido, Á. et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 54, 1154–1167 (2021).
Elola, M. T. et al. Galectins: multitask signaling molecules linking fibroblast, endothelial and immune cell programs in the tumor microenvironment. Cell. Immunol. 333, 34–45 (2018).
Ilarregui, J. M. et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat. Immunol. 10, 981–991 (2009).
Tesone, A. J. et al. Satb1 overexpression drives tumor-promoting activities in cancer-associated dendritic cells. Cell Rep. 14, 1774–1786 (2016).
Sturgill, E. R. et al. Galectin-3 inhibition with belapectin combined with anti-OX40 therapy reprograms the tumor microenvironment to favor anti-tumor immunity. Oncoimmunology 10, 1892265 (2021).
MacKinnon, A. C. et al. Regulation of alternative macrophage activation by galectin-3. J. Immunol. 180, 2650–2658 (2008).
Daley, D. et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat. Med. 23, 556–567 (2017). This study reports the role of GAL9 as a relevant immunotherapeutic target in pancreatic adenocarcinoma by reprogramming macrophage function.
Dalotto-Moreno, T. et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res. 73, 1107–1117 (2013).
Cagnoni, A. J. et al. Galectin-1 fosters an immunosuppressive microenvironment in colorectal cancer by reprogramming CD8+ regulatory T cells. Proc. Natl Acad. Sci. USA 118, e2102950118 (2021).
Pang, N. et al. Activated galectin-9/Tim3 promotes Treg and suppresses Th1 effector function in chronic lymphocytic leukemia. FASEB J. 35, e21556 (2021).
Rubinstein, N. et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection: a potential mechanism of tumor-immune privilege. Cancer Cell 5, 241–251 (2004). This study identifies the role of GAL1 in tumour cell evasion of immune responses in human and mouse melanoma.
Banh, A. et al. Tumor galectin-1 mediates tumor growth and metastasis through regulation of T-cell apoptosis. Cancer Res. 71, 4423–4431 (2011).
Liu, S. D. et al. Galectin-1 tunes TCR binding and signal transduction to regulate CD8 burst size. J. Immunol. 182, 5283–5295 (2009).
Stillman, B. N. et al. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J. Immunol. 176, 778–789 (2006).
Toscano, M. A. et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 8, 825–834 (2007).
Yang, R. et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 12, 832 (2021). This study unveils the role of GAL9 in cancer immunotherapy by interacting with PD1 and TIM3 immune checkpoints on T cells.
Cedeno-Laurent, F., Opperman, M., Barthel, S. R., Kuchroo, V. K. & Dimitroff, C. J. Galectin-1 triggers an immunoregulatory signature in Th cells functionally defined by IL-10 expression. J. Immunol. 188, 3127–3137 (2012).
Baker, G. J. et al. Natural killer cells eradicate galectin-1-deficient glioma in the absence of adaptive immunity. Cancer Res. 74, 5079–5090 (2014).
Zhang, C. et al. Galectin-9 promotes a suppressive microenvironment in human cancer by enhancing STING degradation. Oncogenesis 9, 65 (2020).
Gordon-Alonso, M., Hirsch, T., Wildmann, C. & van der Bruggen, P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat. Commun. 8, 793 (2017).
Demotte, N. et al. A galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Res. 70, 7476–7488 (2010). This study highlights the importance of targeting GAL3 to prevent T cell anergy and promote tumour rejection.
Kouo, T. et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol. Res. 3, 412–423 (2015).
Lau, K. S. et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129, 123–134 (2007).
Zhu, C. et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 6, 1245–1252 (2005).
Henderson, N. C. et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl Acad. Sci. USA 103, 5060–5065 (2006). This article underscores the relevance of GAL3 as a therapeutic target in liver fibrosis.
Iacobini, C. et al. Galectin-3 ablation protects mice from diet-induced NASH: a major scavenging role for galectin-3 in liver. J. Hepatol. 54, 975–983 (2011).
Hsieh, T. J. et al. Dual thio-digalactoside-binding modes of human galectins as the structural basis for the design of potent and selective inhibitors. Sci. Rep. 6, 29457 (2016).
Mackinnon, A. C. et al. Regulation of transforming growth factor-β1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 185, 537–546 (2012). This study validates the use of GAL3 inhibitors in lung fibrosis.
Henderson, N. C. et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 172, 288–298 (2008).
Argüeso, P., Mauris, J. & Uchino, Y. Galectin-3 as a regulator of the epithelial junction: Implications to wound repair and cancer. Tissue Barriers 3, e1026505–e1026505 (2015).
Kasper, M. & HUghes, R. C. Immunochemical evidence for a modulation of galectin-3 (Mac-2), a carbohydrate binding protein, in pulmonary fibrosis. J. Pathol. 179, 309–316 (1996).
Fichtner-Feigl, S., Strober, W., Kawakami, K., Puri, R. K. & Kitani, A. IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nat. Med. 12, 99–106 (2006).
Liu, J. C., Heilshorn, S. C. & Tirrell, D. A. Comparative cell response to artificial extracellular matrix proteins containing the RGD and CS5 cell-binding domains. Biomacromolecules 5, 497–504 (2004).
Rishikof, D. C., Ricupero, D. A., Kuang, P.-P., Liu, H. & Goldstein, R. H. Interleukin-4 regulates connective tissue growth factor expression in human lung fibroblasts. J. Cell. Biochem. 85, 496–504 (2002).
Jiang, Z. et al. Galectin-1 gene silencing inhibits the activation and proliferation but induces the apoptosis of hepatic stellate cells from mice with liver fibrosis. Int. J. Mol. Med. 43, 103–116 (2019).
Hsu, Y.-A. et al. Amelioration of bleomycin-induced pulmonary fibrosis via TGF-β-induced Smad and non-Smad signaling pathways in galectin-9-deficient mice and fibroblast cells. J. Biomed. Sci. 27, 24 (2020).
Kathiriya, J. J. et al. Galectin-1 inhibition attenuates profibrotic signaling in hypoxia-induced pulmonary fibrosis. Cell Death Discov. 3, 17010 (2017).
Fujita, K. et al. Correlation between serum galectin-9 levels and liver fibrosis. J. Gastroenterol. Hepatol. 33, 492–499 (2018).
Wu, M.-H. et al. Glycosylation-dependent galectin-1/neuropilin-1 interactions promote liver fibrosis through activation of TGF-β- and PDGF-like signals in hepatic stellate cells. Sci. Rep. 7, 11006 (2017).
Jin Lim, M. et al. Induction of galectin-1 by TGF-β1 accelerates fibrosis through enhancing nuclear retention of Smad2. Exp. Cell Res. 326, 125–135 (2014).
Matsumoto, N. et al. A possible role of galectin-9 in the pulmonary fibrosis of patients with interstitial pneumonia. Lung 191, 191–198 (2013).
Modenutti, C. P., Capurro, J. I. B., Di Lella, S. & Martí, M. A. The structural biology of galectin-ligand recognition: current advances in modeling tools, protein engineering, and inhibitor design. Front. Chem. 7, 823 (2019).
Leffler, H. & Nilsson, U. J. In Galectins and Disease Implications for Targeted Therapeutics vol. 1115, 2–47 (American Chemical Society, 2012).
Tejler, J., Salameh, B., Leffler, H. & Nilsson, U. J. Fragment-based development of triazole-substituted O-galactosyl aldoximes with fragment-induced affinity and selectivity for galectin-3. Org. Biomol. Chem. 7, 3982–3990 (2009).
Rajput, V. K., Leffler, H., Nilsson, U. J. & Mukhopadhyay, B. Synthesis and evaluation of iminocoumaryl and coumaryl derivatized glycosides as galectin antagonists. Bioorg. Med. Chem. Lett. 24, 3516–3520 (2014).
Blanchard, H., Bum-Erdene, K. & Hugo, M. W. Inhibitors of galectins and implications for structure-based design of galectin-specific therapeutics. Aust. J. Chem. 67, 1763–1779 (2014).
Mackinnon, A. et al. In Carbohydrates as Drugs (eds Seeberger, P. H. & Rademacher, C.) 95–121 (Springer International Publishing, 2014).
Cagnoni, A. J., Kovensky, J. & Uhrig, M. L. Design and synthesis of hydrolytically stable multivalent ligands bearing thiodigalactoside analogues for peanut lectin and human galectin-3 binding. J. Org. Chem. 79, 6456–6467 (2014).
Liu, C. et al. Identification of monosaccharide derivatives as potent, selective, and orally bioavailable inhibitors of human and mouse galectin-3. J. Med. Chem. 65, 11084–11099 (2022).
Zetterberg, F. R. et al. Monosaccharide derivatives with low-nanomolar lectin affinity and high selectivity based on combined fluorine–amide, phenyl–arginine, sulfur–π, and halogen bond interactions. ChemMedChem 13, 133–137 (2018).
Vuong, L. et al. An orally active galectin-3 antagonist inhibits lung adenocarcinoma growth and augments response to PD-L1 blockade. Cancer Res. 79, 1480–1493 (2019).
Kim, S.-J. et al. Crosstalk between WNT and STAT3 is mediated by galectin-3 in tumor progression. Gastric Cancer 24, 1050–1062 (2021).
Dahlqvist, A., Zetterberg, F. R., Leffler, H. & Nilsson, U. J. Aminopyrimidine–galactose hybrids are highly selective galectin-3 inhibitors. MedChemComm 10, 913–925 (2019).
Jalagam, P. R., Nair, S. K., Panda, M. & Regueiro-Ren, A. Small molecule inhibitors of galectin-3. Patent no. WO2019075045A1 (2022).
Bolli, M. et al. Alpha-d-galactopyranoside derivatives. Patent no. WO2021038068A1 (2021).
Brimert, T., Johnsson, R., Leffler, H., Nilsson, U. & Zetterberg, F. Alpha-d-galactoside inhibitors of galectins. Patent no. WO2016120403A1 (2016).
Zetterberg, F. R. et al. Discovery and optimization of the first highly effective and orally available galectin-3 inhibitors for treatment of fibrotic disease. J. Med. Chem. 65, 12626–12638 (2022).
Tonev, D. et al. Gulliver-2 is an innovative, hybrid, hepatic impairment trial of the oral galectin-3 inhibitor GB1211 [abstract 3627]. Hepatology 76, S1146–S1147 (2022).
Lindmark, B. GB1211, An oral galectin-3 inhibitor, in decompensated cirrhotic patients: initial findings from the phase 2 randomized, placebo-controlled Gulliver-2 trial [abstract]. Presented at the The Liver Meeting, AASLD (2022).
Tejler, J., Skogman, F., Leffler, H. & Nilsson, U. J. Synthesis of galactose-mimicking 1H-(1,2,3-triazol-1-yl)-mannosides as selective galectin-3 and 9N inhibitors. Carbohydr. Res. 342, 1869–1875 (2007).
Öberg, C. T., Leffler, H. & Nilsson, U. J. Arginine binding motifs: design and synthesis of galactose-derived arginine tweezers as galectin-3 inhibitors. J. Med. Chem. 51, 2297–2301 (2008).
Öberg, C. T., Noresson, A.-L., Leffler, H. & Nilsson, U. J. Arene–anion based arginine-binding motif on a galactose scaffold: structure–activity relationships of interactions with arginine-rich galectins. Chemistry 17, 8139–8144 (2011).
Collins, P. M., Öberg, C. T., Leffler, H., Nilsson, U. J. & Blanchard, H. Taloside inhibitors of galectin-1 and galectin-3. Chem. Biol. Drug Des. 79, 339–346 (2012).
Masuyer, G. et al. Inhibition mechanism of human galectin-7 by a novel galactose-benzylphosphate inhibitor. FEBS J. 279, 193–202 (2012).
Pal, K. B., Mahanti, M., Leffler, H. & Nilsson, U. J. A galactoside-binding protein tricked into binding unnatural pyranose derivatives: 3-deoxy-3-methyl-gulosides selectively inhibit galectin-1. Int. J. Mol. Sci. 20, 3786–3808 (2019).
Mahanti, M., Pal, K. B., Sundin, A. P., Leffler, H. & Nilsson, U. J. Epimers switch galectin-9 domain selectivity: 3N-aryl galactosides bind the C-terminal and gulosides bind the N-terminal. ACS Med. Chem. Lett. 11, 34–39 (2020).
Barondes, S. H., Cooper, D. N., Gitt, M. A. & Leffler, H. Galectins. Structure and function of a large family of animal lectins. J. Biol. Chem. 269, 20807–20810 (1994). This pioneering review provides a first definition of galectins and their glycan binding specificities and summarizes early studies on structure–function relationship.
Sörme, P. et al. Structural and thermodynamic studies on cation-Pi interactions in lectin-ligand complexes: high-affinity galectin-3 inhibitors through fine-tuning of an arginine-arene interaction. J. Am. Chem. Soc. 127, 1737–1743 (2005).
Campo, V. L., Marchiori, M. F., Rodrigues, L. C. & Dias-Baruffi, M. Synthetic glycoconjugates inhibitors of tumor-related galectin-3: an update. Glycoconj. J. 33, 853–876 (2016).
Rabinovich, G. A. et al. Synthetic lactulose amines: novel class of anticancer agents that induce tumor-cell apoptosis and inhibit galectin-mediated homotypic cell aggregation and endothelial cell morphogenesis. Glycobiology 16, 210–220 (2006).
Kishor, C., Ross, R. L. & Blanchard, H. Lactulose as a novel template for anticancer drug development targeting galectins. Chem. Biol. Drug Des. 92, 1801–1808 (2018).
Delaine, T. et al. Galectin-inhibitory thiodigalactoside ester derivatives have antimigratory effects in cultured lung and prostate cancer cells. J. Med. Chem. 51, 8109–8114 (2008).
Cumpstey, I., Carlsson, S., Leffler, H. & Nilsson, U. J. Synthesis of a phenyl thio-β-d-galactopyranoside library from 1,5-difluoro-2,4-dinitrobenzene: discovery of efficient and selective monosaccharide inhibitors of galectin-7. Org. Biomol. Chem. 3, 1922–1932 (2005).
Cumpstey, I., Salomonsson, E., Sundin, A., Leffler, H. & Nilsson, U. J. Double affinity amplification of galectin–ligand interactions through arginine–arene interactions: synthetic, thermodynamic, and computational studies with aromatic diamido thiodigalactosides. Chemistry 14, 4233–4245 (2008).
Lin, C.-I. et al. Galectin-3 targeted therapy with a small molecule inhibitor activates apoptosis and enhances both chemosensitivity and radiosensitivity in papillary thyroid cancer. Mol. Cancer Res. 7, 1655–1662 (2009).
Vašíček, T. et al. Regioselective 3-O-substitution of unprotected thiodigalactosides: direct route to galectin inhibitors. Chemistry 26, 9620–9631 (2020).
Delaine, T. et al. Galectin-3-binding glycomimetics that strongly reduce bleomycin-induced lung fibrosis and modulate intracellular glycan recognition. ChemBioChem 17, 1759–1770 (2016).
Bratteby, K. et al. In vivo veritas: 18F-radiolabeled glycomimetics allow insights into the pharmacological fate of galectin-3 inhibitors. J. Med. Chem. 63, 747–755 (2020). The pharmacokinetics for TD139 and GB1107 surrogate radiotracers is discussed, providing a rationale on different administration routes and efficacy of individual galectin inhibitors.
Hattum, H. Van et al. Tuning the preference of thiodigalactoside- and lactosamine-based ligands to galectin-3 over galectin-1. J. Med. Chem. 56, 1350–1354 (2013). This study reports the design of small molecules, based on TDG or lactosamine, with different selectivity for GAL3 and GAL1.
Rajput, V. K. et al. A selective galactose-coumarin-derived galectin-3 inhibitor demonstrates involvement of galectin-3-glycan interactions in a pulmonary fibrosis model. J. Med. Chem. 59, 8141–8147 (2016).
Xu, L. et al. Synthesis, structure–activity relationships, and in vivo evaluation of novel tetrahydropyran-based thiodisaccharide mimics as galectin-3 inhibitors. J. Med. Chem. 64, 6634–6655 (2021).
Cecioni, S., Imberty, A. & Vidal, S. Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 115, 525–561 (2015).
André, S., Kaltner, H., Furuike, T., Nishimura, S.-I. & Gabius, H.-J. Persubstituted cyclodextrin-based glycoclusters as inhibitors of protein−carbohydrate recognition using purified plant and mammalian lectins and wild-type and lectin-gene-transfected tumor cells as targets. Bioconjug. Chem. 15, 87–98 (2004).
André, S. et al. Calix[n]arene-based glycoclusters: bioactivity of thiourea-linked galactose/lactose moieties as inhibitors of binding of medically relevant lectins to a glycoprotein and cell-surface glycoconjugates and selectivity among human adhesion/growth-regulatory G. ChemBioChem 9, 1649–1661 (2008).
Gouin, S. G. et al. Multimeric lactoside “Click Clusters” as tools to investigate the effect of linker length in specific interactions with peanut lectin, galectin-1, and -3. ChemBioChem 11, 1430–1442 (2010).
Ennist, J. H., Termuehlen, H. R., Bernhard, S. P., Fricke, M. S. & Cloninger, M. J. Chemoenzymatic synthesis of galectin binding glycopolymers. Bioconjug. Chem. 29, 4030–4039 (2018).
Böcker, S., Laaf, D. & Elling, L. Galectin binding to neo-glycoproteins: LacDiNAc conjugated BSA as ligand for human galectin-3. Biomolecules 5, 1671–1696 (2015).
Zhang, H., Laaf, D., Elling, L. & Pieters, R. J. Thiodigalactoside-bovine serum albumin conjugates as high-potency inhibitors of galectin-3: an outstanding example of multivalent presentation of small molecule inhibitors. Bioconjug. Chem. 29, 1266–1275 (2018).
Guha, P. et al. Cod glycopeptide with picomolar affinity to galectin-3 suppresses T-cell apoptosis and prostate cancer metastasis. Proc. Natl Acad. Sci. USA 110, 5052–5057 (2013).
Wang, H. et al. Design and synthesis of glycoprotein-based multivalent glyco-ligands for influenza hemagglutinin and human galectin-3. Bioorg. Med. Chem. 21, 2037–2044 (2013).
Heine, V. et al. Immunoprotective neo-glycoproteins: chemoenzymatic synthesis of multivalent glycomimetics for inhibition of cancer-related galectin-3. Eur. J. Med. Chem. 220, 113500 (2021).
Stegmayr, J. et al. Extracellular and intracellular small-molecule galectin-3 inhibitors. Sci. Rep. 9, 1–12 (2019). Given the different function of galectins depending on their subcellular distribution, this study explores the cellular uptake of small-molecule carbohydrate GAL3 inhibitors and their function.
Salameh, B. A., Cumpstey, I., Sundin, A., Leffler, H. & Nilsson, U. J. 1H-1,2,3-triazol-1-yl thiodigalactoside derivatives as high affinity galectin-3 inhibitors. Bioorg. Med. Chem. 18, 5367–5378 (2010).
Nangia-Makker, P. et al. Inhibition of human cancer cell growth and metastasis in nude mice by oral intake of modified citrus pectin. J. Natl Cancer Inst. 94, 1854–1862 (2002). This early study reports the use of MCP in modulating tumour growth and metastasis through binding to GAL3.
Chauhan, D. et al. A novel carbohydrate-based therapeutic GCS-100 overcomes bortezomib resistance and enhances dexamethasone-induced apoptosis in multiple myeloma cells. Cancer Res. 65, 8350–8358 (2005).
Yan, J. & Katz, A. PectaSol-C modified citrus pectin induces apoptosis and inhibition of proliferation in human and mouse androgen-dependent and- independent prostate cancer cells. Integr. Cancer Ther. 9, 197–203 (2010).
Klyosov, A. A. & Traber, P. G. in Galectins and Disease Implications for Targeted Therapeutics (eds Klyosov, A. A. & Traber, P. G.) 1–3 ACS Symposium series 1115 (American Chemical Society, 2013).
Zhang, W., Xu, P. & Zhang, H. Pectin in cancer therapy: a review. Trends Food Sci. Technol. 44, 258–271 (2015).
Banerjee, S., Parasramka, M. & Paruthy, S. B. in Polysaccharides (eds Ramawat, K. G. & Mérillon, J.-M.) 2179–2214 (Springer International Publishing, 2015).
Bergman, M., Djaldetti, M., Salman, H. & Bessler, H. Effect of citrus pectin on malignant cell proliferation. Biomed. Pharmacother. 64, 44–47 (2010).
Stegmayr, J. et al. Low or no inhibitory potency of the canonical galectin carbohydrate-binding site by pectins and galactomannans. J. Biol. Chem. 291, 13318–13334 (2016).
Platt, D. & Raz, A. Modulation of the lung colonization of B16-F1 melanoma cells by citrus pectin. J. Natl Cancer Inst. 84, 438–442 (1992).
Inohara, H. & Raz, A. Effects of natural complex carbohydrate (citrus pectin) on murine melanoma cell properties related to galectin-3 functions. Glycoconj. J. 11, 527–532 (1994).
Hsieh, T. C. & Wu, J. M. Changes in cell growth, cyclin/kinase, endogenous phosphoproteins and nm23 gene expression in human prostatic JCA-1 cells treated with modified citrus pectin. Biochem. Mol. Biol. Int. 37, 833–841 (1995).
Guess, B. W. et al. Modified citrus pectin (MCP) increases the prostate-specific antigen doubling time in men with prostate cancer: a phase II pilot study. Prostate Cancer Prostatic Dis. 6, 301–304 (2003).
Keizman, D. et al. Modified citrus pectin treatment in non-metastatic biochemically relapsed prostate cancer: results of a prospective phase II study. Nutrients 13, 4295 (2021).
Grous, J. J., Redfern, C. H., Mahadevan, D. & Schindler, J. GCS-100, a galectin-3 antagonist, in refractory solid tumors: a phase I study. J. Clin. Oncol. 24, 13023 (2006).
Cotter, F. et al. Single-agent activity of GCS-100, a first-in-class galectin-3 antagonist, in elderly patients with relapsed chronic lymphocytic leukemia. J. Clin. Oncol. 27, 7006 (2009).
Streetly, M. J. et al. GCS-100, a novel galectin-3 antagonist, modulates MCL-1, NOXA, and cell cycle to induce myeloma cell death. Blood 115, 3939–3948 (2010).
Ruvolo, P. P. et al. Combination of galectin inhibitor GCS-100 and BH3 mimetics eliminates both p53 wild type and p53 null AML cells. Biochim. Biophys. Acta 1863, 562–571 (2016).
Curti, B. D. et al. Enhancing clinical and immunological effects of anti-PD-1 with belapectin, a galectin-3 inhibitor. J. Immunother. Cancer 9, e002371 (2021). This article reports the synergistic effect of GAL3 and PD1 blockade in augmenting antitumour immunity and restraining tumour progression.
Platt, D., Klyosov, A. A. & Zomer, E. in Carbohydrate Drug Design (eds Klyosov, A. A., Witczak, Z. J. & Platt, D.) 3–49 ACS Symposium series vol. 932 (American Chemical Society, 2006).
Zomer, E., Klyosov, A. A. & Platt, D. in Glycobiology and Drug Design (ed. Klyosov, A. A.) 3–69 ACS Symposium series 1102 (American Chemical Society, 2013).
Miller, M. C. et al. Binding of polysaccharides to human galectin-3 at a noncanonical site in its carbohydrate recognition domain. Glycobiology 26, 88–99 (2016).
Traber, P. G. et al. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS ONE 8, e75361 (2013). This article evaluates GR-MD-02 (belapectin) and GM-CT-01 (Davanat) in an experimental model of liver fibrosis.
Traber, P. G. & Zomer, E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE 8, e83481 (2013).
Harrison, S. A. et al. Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment. Pharmacol. Ther. 44, 1183–1198 (2016).
Chalasani, N. et al. Effects of Belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology 158, 1334–1345 (2020).
Zhang, L. et al. RN1, a novel galectin-3 inhibitor, inhibits pancreatic cancer cell growth in vitro and in vivo via blocking galectin-3 associated signaling pathways. Oncogene 36, 1297–1308 (2017).
Xue, H. et al. Selective effects of ginseng pectins on galectin-3-mediated T cell activation and apoptosis. Carbohydr. Polym. 219, 121–129 (2019).
Dings, R. P. M. & Mayo, K. H. A journey in structure-based drug discovery: from designed peptides to protein surface topomimetics as antibiotic and antiangiogenic agents. Acc. Chem. Res. 40, 1057–1065 (2007).
Mayo, K. H. in Galectins and Disease Implications for Targeted Therapeutics 3–61 ACS Symposium series 1115 (American Chemical Society, 2013).
Griffioen, A. W. et al. Anginex, a designed peptide that inhibits angiogenesis. Biochem. J. 354, 233–242 (2001).
van der Schaft, D. W. J. et al. The designer antiangiogenic peptide anginex targets tumor endothelial cells and inhibits tumor growth in animal models. FASEB J. 16, 1991–1993 (2002).
Dings, R. P. M., Yokoyama, Y., Ramakrishnan, S., Griffioen, A. W. & Mayo, K. H. The designed angiostatic peptide anginex synergistically improves chemotherapy and antiangiogenesis therapy with angiostatin. Cancer Res. 63, 382–385 (2003). This study reports the synergistic antitumour effects of Anginex, an angiostatic peptide that binds GAL1, with chemotherapy and anti-angiogenic therapy.
Thijssen, V. L. J. L. et al. Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc. Natl Acad. Sci. USA 103, 15975–15980 (2006). This pioneering study reports the role of GAL1 as a target of the anti-angiogenic peptide Anginex.
Brandwijk, R. J. M. G. E. et al. Cloning an artificial gene encoding angiostatic anginex: From designed peptide to functional recombinant protein. Biochem. Biophys. Res. Commun. 333, 1261–1268 (2005).
Ma, K. et al. Recombinant adeno-associated virus-delivered anginex inhibits angiogenesis and growth of HUVECs by regulating the Akt, JNK and NF-κB signaling pathways. Oncol. Rep. 35, 3505–3513 (2016).
Mayo, K. H. et al. Design of a partial peptide mimetic of anginex with antiangiogenic and anticancer activity. J. Biol. Chem. 278, 45746–45752 (2003).
Dings, R. P. M. et al. Structure-based optimization of angiostatic agent 6DBF7, an allosteric antagonist of galectin-1. J. Pharmacol. Exp. Ther. 344, 589–599 (2013).
Dings, R. P. M. et al. Antitumor agent calixarene 0118 targets human galectin-1 as an allosteric inhibitor of carbohydrate binding. J. Med. Chem. 55, 5121–5129 (2012).
Koonce, N. A., Griffin, R. J. & Dings, R. P. M. Galectin-1 inhibitor OTX008 induces tumor vessel normalization and tumor growth inhibition in human head and neck squamous cell carcinoma models. Int. J. Mol. Sci. 18, 2671–2680 (2017).
Zucchetti, M. et al. Pharmacokinetics and antineoplastic activity of galectin-1-targeting OTX008 in combination with sunitinib. Cancer Chemother. Pharmacol. 72, 879–887 (2013).
Rezai, K. et al. Abstract 33: OTX008 pharmacokinetics (PK) during the first-in-man phase I study in patients with advanced solid tumors. Cancer Res. 73 (Suppl. 8), 33 (2013).
Leung, Z. et al. Galectin-1 promotes hepatocellular carcinoma and the combined therapeutic effect of OTX008 galectin-1 inhibitor and sorafenib in tumor cells. J. Exp. Clin. Cancer Res. 38, 423 (2019).
Gheysen, L. et al. New treatment strategy targeting galectin-1 against thyroid cancer. Cells 10, 1112–1120 (2021).
Dings, R. P. M. et al. Polycationic calixarene PTX013, a potent cytotoxic agent against tumors and drug resistant cancer. Invest. New Drugs 31, 1142–1150 (2013).
Läppchen, T. et al. Novel analogs of antitumor agent calixarene 0118: synthesis, cytotoxicity, click labeling with 2-[18F]fluoroethylazide, and in vivo evaluation. Eur. J. Med. Chem. 89, 279–295 (2015).
Blanchard, H., Bum-Erdene, K., Bohari, M. H. & Yu, X. Galectin-1 inhibitors and their potential therapeutic applications: a patent review. Expert. Opin. Ther. Pat. 26, 537–554 (2016).
Miller, M. C. et al. Targeting the CRD F-face of human galectin-3 and allosterically modulating glycan binding by angiostatic PTX008 and a structurally optimized derivative. ChemMedChem 16, 713–723 (2021).
Zou, J., Glinsky, V. V., Landon, L. A., Matthews, L. & Deutscher, S. L. Peptides specific to the galectin-3 carbohydrate recognition domain inhibit metastasis-associated cancer cell adhesion. Carcinogenesis 26, 309–318 (2005).
Geng, Q., Sun, X., Gong, T. & Zhang, Z.-R. Peptide–drug conjugate linked via a disulfide bond for kidney targeted drug delivery. Bioconjug. Chem. 23, 1200–1210 (2012).
Kopansky, E., Shamay, Y. & David, A. Peptide-directed HPMA copolymer-doxorubicin conjugates as targeted therapeutics for colorectal cancer. J. Drug Target. 19, 933–943 (2011).
Yang, Y. et al. Targeting prostate carcinoma by G3-C12 peptide conjugated N-(2-hydroxypropyl)methacrylamide copolymers. Mol. Pharm. 11, 3251–3260 (2014).
Yang, Y. et al. Treatment of prostate carcinoma with (galectin-3)-targeted HPMA copolymer-(G3-C12)-5-fluorouracil conjugates. Biomaterials 33, 2260–2271 (2012).
Liu, T. & Huang, Q. Biodegradable brush-type copolymer modified with targeting peptide as a nanoscopic platform for targeting drug delivery to treat castration-resistant prostate cancer. Int. J. Pharm. 511, 1002–1011 (2016).
Kanda, A., Noda, K., Saito, W. & Ishida, S. Aflibercept traps galectin-1, an angiogenic factor associated with diabetic retinopathy. Sci. Rep. 5, 17946 (2015).
Danhier, F., Messaoudi, K., Lemaire, L., Benoit, J.-P. & Lagarce, F. Combined anti-Galectin-1 and anti-EGFR siRNA-loaded chitosan-lipid nanocapsules decrease temozolomide resistance in glioblastoma: In vivo evaluation. Int. J. Pharm. 481, 154–161 (2015). This study highlights the impact of combining GAL1 and EGFR siRNA in the treatment of patients with glioblastoma resistant to temozolomide.
Storti, P. et al. Galectin-1 suppression delineates a new strategy to inhibit myeloma-induced angiogenesis and tumoral growth in vivo. Leukemia 30, 2351–2363 (2016).
Tsai, Y.-T. et al. A DNA Aptamer targeting galectin-1 as a novel immunotherapeutic strategy for lung cancer. Mol. Ther. Nucleic Acids 18, 991–998 (2019).
Zuckerman, J. E. & Davis, M. E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 14, 843–856 (2015).
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
Nelson, A. L., Dhimolea, E. & Reichert, J. M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 9, 767–774 (2010).
Ouyang, J. et al. Viral induction and targeted inhibition of galectin-1 in EBV+ posttransplant lymphoproliferative disorders. Blood 117, 4315–4322 (2011).
Pérez Sáez, J. M. et al. Characterization of a neutralizing anti-human galectin-1 monoclonal antibody with angioregulatory and immunomodulatory activities. Angiogenesis 24, 1–5 (2021). This study highlights the dual immunomodulatory and angioregulatory activities of a GAL1-neutralizing mAb.
Femel, J. et al. Vaccination against galectin-1 promotes cytotoxic T-cell infiltration in melanoma and reduces tumor burden. Cancer Immunol. Immunother. 71, 2029–2040 (2022).
Rao, T. D. et al. Expression of the carboxy-terminal portion of MUC16/CA125 induces transformation and tumor invasion. PLoS ONE 10, e0126633 (2015).
Stasenko, M. et al. Targeting galectin-3 with a high-affinity antibody for inhibition of high-grade serous ovarian cancer and other MUC16/CA-125-expressing malignancies. Sci. Rep. 11, 3718 (2021). This study reports the therapeutic relevance of a GAL3 blocking mAb in high-grade serous ovarian cancer.
Sun, D. et al. Anti-Gal-3 antibodies and methods of use. International Patent Application. Patent no. WO/2021/146218 A1 (2021).
Lee, M., Filipovic, A. & Henry, C. J. Combinatorial inhibition of galectin-9 and CHK1 represent a novel treatment strategy for T-cell acute lymphoblastic leukemia. Blood 138, 4400 (2021).
Mirandola, L. et al. Galectin-3 inhibition suppresses drug resistance, motility, invasion and angiogenic potential in ovarian cancer. Gynecol. Oncol. 135, 573–579 (2014).
John, C. M., Leffler, H., Kahl-Knutsson, B., Svensson, I. & Jarvis, G. A. Truncated galectin-3 inhibits tumor growth and metastasis in orthotopic nude mouse model of human breast cancer. Clin. Cancer Res. 9, 2374 (2003).
Mirandola, L. et al. Galectin-3C inhibits tumor growth and increases the anticancer activity of bortezomib in a murine model of human multiple myeloma. PLoS ONE 6, e21811 (2011).
St-Gelais, J., Denavit, V. & Giguère, D. Efficient synthesis of a galectin inhibitor clinical candidate (TD139) using a Payne rearrangement/azidation reaction cascade. Org. Biomol. Chem. 18, 3903–3907 (2020).
Stowell, S. R. et al. Ligand reduces galectin-1 sensitivity to oxidative inactivation by enhancing dimer formation. J. Biol. Chem. 284, 4989–4999 (2009).
Guardia, C. M. et al. Structural basis of redox-dependent modulation of galectin-1 dynamics and function. Glycobiology 24, 428–441 (2014).
Di Lella, S. et al. When galectins recognize glycans: from biochemistry to physiology and back again. Biochemistry 50, 7842–7857 (2011).
Salomonsson, E., Thijssen, V. L., Griffioen, A. W., Nilsson, U. J. & Leffler, H. The anti-angiogenic peptide anginex greatly enhances galectin-1 binding affinity for glycoproteins. J. Biol. Chem. 286, 13801–13804 (2011).
Gohil, S. H., Iorgulescu, J. B., Braun, D. A., Keskin, D. B. & Livak, K. J. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat. Rev. Clin. Oncol. 18, 244–256 (2021).
Kearney, C. J. et al. SUGAR-seq enables simultaneous detection of glycans, epitopes, and the transcriptome in single cells. Sci. Adv. 7, eabe3610 (2021).
Peixoto, A., Miranda, A., Santos, L. L. & Ferreira, J. A. A roadmap for translational cancer glycoimmunology at single cell resolution. J. Exp. Clin. Cancer Res. 41, 143 (2022).
Matthews, S. E. in Calixarenes and Beyond (eds Neri, P., Sessler, J. L. & Wang, M.-X.) 559–600 (Springer International Publishing, 2016).
Glinsky, V. V. & Raz, A. Modified citrus pectin anti-metastatic properties: one bullet, multiple targets. Carbohydr. Res. 344, 1788–1791 (2009).
Zhang, T. et al. Macromolecular assemblies of complex polysaccharides with galectin-3 and their synergistic effects on function. Biochem. J. 474, 3849–3868 (2017).
Nanthakumar, C. B. et al. Dissecting fibrosis: therapeutic insights from the small-molecule toolbox. Nat. Rev. Drug Discov. 14, 693–720 (2015).
Miller, M. C., Klyosov, A. & Mayo, K. H. The α-galactomannan Davanat binds galectin-1 at a site different from the conventional galectin carbohydrate binding domain. Glycobiology 19, 1034–1045 (2009).
Miller, M. C., Klyosov, A. A. & Mayo, K. H. Structural features for α-galactomannan binding to galectin-1. Glycobiology 22, 543–551 (2012).
Leclere, L. et al. Heat-modified citrus pectin induces apoptosis-like cell death and autophagy in HepG2 and A549 cancer cells. PLoS ONE 10, e0115831 (2015).
Li, L. et al. The effect of the size of fluorescent dextran on its endocytic pathway. Cell Biol. Int. 39, 531–539 (2015).
Pilch, J. et al. The anti-angiogenic peptide anginex disrupts the cell membrane. J. Mol. Biol. 356, 876–885 (2006).
RodrÍguez, E., Schetters, S. T. T. & van Kooyk, Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 18, 204–211 (2018).
Farrants, H. et al. Chemogenetic control of nanobodies. Nat. Methods 17, 279–282 (2020).
Hockl, P. F. et al. Glyco-nano-oncology: novel therapeutic opportunities by combining small and sweet. Pharmacol. Res. 109, 45–54 (2016).
Zhong, X., Qian, X., Chen, G. & Song, X. The role of galectin-3 in heart failure and cardiovascular disease. Clin. Exp. Pharmacol. Physiol. 46, 197–203 (2019).
Lau, E. S. et al. Galectin-3 inhibition with modified citrus pectin in hypertension. JACC Basic Transl. Sci. 6, 12–21 (2021).
Chen, W.-S., Cao, Z., Leffler, H., Nilsson, U. J. & Panjwani, N. Galectin-3 inhibition by a small-molecule inhibitor reduces both pathological corneal neovascularization and fibrosis. Invest. Ophthalmol. Vis. Sci. 58, 9–20 (2017).
Al-Obaidi, N. et al. Galectin-1 is a new fibrosis protein in type 1 and type 2 diabetes. FASEB J. 33, 373–387 (2019).
Neelamegham, S. et al. Updates to the symbol nomenclature for glycans guidelines. Glycobiology 29, 620–624 (2019).
Stowell, S. R. et al. Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J. Biol. Chem. 283, 10109–10123 (2008).
Kopitz, J., von Reitzenstein, C., Burchert, M., Cantz, M. & Gabius, H.-J. Galectin-1 is a major receptor for ganglioside GM1, a product of the growth-controlling activity of a cell surface ganglioside sialidase, on human neuroblastoma cells in culture*. J. Biol. Chem. 273, 11205–11211 (1998).
Boscher, C. et al. Galectin-3 protein regulates mobility of N-cadherin and GM1 ganglioside at cell-cell junctions of mammary carcinoma cells. J. Biol. Chem. 287, 32940–32952 (2012).
Acknowledgements
Work in the authors’ laboratories is supported by grants from the Agency for Promotion of Science, Technology and Innovation (PICT 2015-0564 and 2018-2766 to K.V.M., PICT 2018-0955 to A.J.C., PICT 2019-0532 to D.O.C. and PICT 2017-0494 and PICT 2020-01552 to G.A.R.) as well as NIH grant CA221208 to G.A.R. The authors are also thankful for generous support from Sales Foundation, Bunge & Born Foundation, Williams Foundation and Richard Lounsbery Foundation, as well as donations from Ferioli-Ostry and Caraballo families to G.A.R. They thank H. Rosenberg for critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed data for the article, made substantial contributions to discussions of the content, wrote the article, and reviewed and edited the article before its submission.
Corresponding author
Ethics declarations
Competing interests
G.A.R. and D.O.C. are co-inventors in the US Patent 10294295B2: ‘Methods for modulating angiogenesis of cancers refractory to anti-VEGF treatment’.
Peer review
Peer review information
Nature Reviews Drug Discovery thanks Gerardo Vasta and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- ADME(T)
-
Acronym for the study of absorption, distribution, metabolism, excretion and (toxicity), key processes in drug pharmacokinetics.
- Antennae
-
Branches of the core N-glycan structure (Manα1–3(Manα1–6)Manβ1–4GlcNAcβ1–4GlcNAcβ1–Asn-X-Ser/Thr) initiated by the action of N-acetylglucosaminyltransferases, resulting in mono-, di-, tri- and tetra-antennary complex N-glycans.
- Cluster glycoside effect
-
Increased binding activity observed in multivalent versus monovalent interactions on a per mole or valence-corrected basis, previously known as ‘avidity’. Multivalent interactions are typically stronger than the sum of individual monovalent interactions.
- Child–Pugh
-
Classification that assesses liver function and prognosis in patients with cirrhosis. Based on a score that includes serum albumin and serum bilirubin levels, ascites and encephalopathy, it includes three classes (A, B, C). Classes B and C are the most severe forms of hepatic dysfunction, with life expectancy of 4–14 years (B) and 1–3 years (C).
- Cyclodextrins
-
Cyclic oligosaccharides composed of α(1–4)-linked d-glucopyranose units that bear a hydrophilic outer surface and a hydrophobic central cavity.
- Doxorubicin
-
Anthracycline antibiotic with broad use as a chemotherapeutic agent.
- F-face of the GAL3 β-sandwich
-
All members of the galectin family, including GAL3, present a conserved carbohydrate recognition domain with an 11-stranded, β-sandwich fold that presents two faces: the six β-stranded S-face (key for interaction with β/α-galactosides), and an opposing F-face (five β-strands) where some larger polysaccharides can interact.
- Glycoconjugates
-
Macromolecules that contain carbohydrates in their structure. Carbohydrates can be covalently linked to proteins or lipids.
- Glycodendrimers
-
Radially symmetrical, carbohydrate-based repetitively branched molecules with well-defined, homogeneous and monodisperse structure.
- Idiopathic pulmonary fibrosis
-
(IPF). A chronic, progressive inflammatory lung disease that causes fibrosis.
- Mucin 1
-
Glycoconjugate that belongs to the family of large, highly glycosylated proteins that contain tandem repeats of amino acids rich in serine and threonine (mucins). These Ser/Thr residues can be O-glycosylated with the monosaccharide N-acetylgalactosamine (GalNAc) and further elongated (O-GalNAc glycans). This modification is characteristic of mucins and is therefore termed mucin-type O-glycosylation. Mucins can be secreted or be transmembrane proteins.
- N-acetyllactosamine
-
(LacNAc). A Galβ(1–3)GlcNAc (type 1) or Galβ(1–4)GlcNAc (type 2) disaccharide.
- Nonalcoholic steatohepatitis
-
(NASH). An inflammatory condition in which patients build up an excess of fat in the liver, leading to damage of this organ. This pathology is not related to alcohol consumption and results in progressive fibrosis that can lead to cirrhosis and hepatocellular carcinoma.
- Neoglycoproteins
-
Synthetic or modified glycoproteins. This term typically refers to natural proteins that have been modified by the addition of structurally well-defined glycans.
- Small interfering RNA
-
(siRNA). A class of double-stranded RNA molecules, usually about 20–25 nucleotides in length, designed to target a specific mRNA for degradation. Thus, siRNA prevents the production of specific proteins based on the nucleotide sequences of their corresponding mRNA. The process is called RNA interference (RNAi).
- Sorafenib
-
An anticancer agent that inhibits multiple kinases and affects the vascular endothelial growth factor (VEGF), Raf–Ras and FMS-like tyrosine kinase 3 (FLT3) pathways.
- Thomsen–Friedenreich antigen
-
(Tf antigen). The core 1 O-GalNAc glycan Galβ(1–3)GalNAc, also known as TF(a), T antigen, Thomsen–Friedenreich disaccharide and/or CD176. In some publications the definition of Tf antigen includes the covalently bound Ser/Thr (α-linkage).
- Topomimetic
-
Agent with stereostructural elements that imitate the topographical features of proteins or peptides.
- Tumour-associated carbohydrate antigens
-
(TACAs). Glycan structures that are frequently found in tumour cells as a consequence of malignancy. They include under- or overexpression of naturally occurring glycans, but also neo-expression of specific carbohydrate structures.
- Valency
-
Number of individual structural units connected to a core structure in a multivalent ligand.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mariño, K.V., Cagnoni, A.J., Croci, D.O. et al. Targeting galectin-driven regulatory circuits in cancer and fibrosis. Nat Rev Drug Discov 22, 295–316 (2023). https://doi.org/10.1038/s41573-023-00636-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-023-00636-2
This article is cited by
-
Galectins in epithelial-mesenchymal transition: roles and mechanisms contributing to tissue repair, fibrosis and cancer metastasis
Biological Research (2024)
-
Engagement of sialylated glycans with Siglec receptors on suppressive myeloid cells inhibits anticancer immunity via CCL2
Cellular & Molecular Immunology (2024)
-
Membrane organization by tetraspanins and galectins shapes lymphocyte function
Nature Reviews Immunology (2024)
-
Pectin: Health-promoting properties as a natural galectin-3 inhibitor
Glycoconjugate Journal (2024)
-
Anchoring immunosuppression to inflamed tissue
Nature Biomedical Engineering (2023)
