
Abstract
Therapeutic targeting of noncoding RNAs (ncRNAs), such as microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), represents an attractive approach for the treatment of cancers, as well as many other diseases. Over the past decade, substantial effort has been made towards the clinical application of RNA-based therapeutics, employing mostly antisense oligonucleotides and small interfering RNAs, with several gaining FDA approval. However, trial results have so far been ambivalent, with some studies reporting potent effects whereas others demonstrated limited efficacy or toxicity. Alternative entities such as antimiRNAs are undergoing clinical testing, and lncRNA-based therapeutics are gaining interest. In this Perspective, we discuss key challenges facing ncRNA therapeutics — including issues associated with specificity, delivery and tolerability — and focus on promising emerging approaches that aim to boost their success.
Similar content being viewed by others

Introduction
Noncoding RNAs (ncRNAs) are generated from the larger part of the genome that does not encode proteins but produces noncoding transcripts that regulate gene expression and protein function. The two major classes of ncRNA are the well-studied short microRNAs (miRNAs) and the more recently identified long ncRNAs (lncRNAs) (Box 1). Deregulation of both types of transcript has been linked to every cancer investigated to date and affects all major cancer hallmarks1,2,3,4,5. In addition, they have been linked to complex biological processes such as immune cell development and function, immune disorders6, neural development and neurological diseases7,8,9. Therapeutic targeting of such naturally occurring ncRNAs thus represents a very promising approach for the treatment of various diseases.
Various RNA-based therapies, including antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), short hairpin RNAs (shRNAs), ASO anti-microRNAs (antimiRs), miRNA mimics, miRNA sponges, therapeutic circular RNAs (circRNAs) and CRISPR–Cas9-based gene editing have been developed and several excellent reviews describe these agents10,11,12 (Box 2). Currently, 11 RNA-based therapeutics are approved by the FDA and/or the European Medicines Agency (EMA) (Table 1), aiming at gene modifications in liver, muscle or the central nervous system. All these therapeutics are either siRNAs or ASOs that cause specific gene downregulation, or ASOs that target pre-mRNA splicing (that is, inducing exon skipping or inclusion). In addition, a plethora of RNA therapeutics are in phase II or III clinical development, including newer entities such as miRNA mimics and antimiRs (Table 2), but no lncRNA-based therapeutics have entered the clinic.
The use of miRNA-based therapeutics has dual advantages10,13,14. First, miRNAs are naturally occurring molecules in human cells, unlike man-made chemotherapy compounds or ASOs, and therefore have all the mechanisms in place for their processing and downstream target selection (Fig. 1). Second, miRNAs act by targeting multiple genes within one pathway, thus causing a broader yet specific response. The miR-15–miR-16 cluster, downregulating multiple anti-apoptotic factors including BCL-2 and MCL1, represents an excellent example of a miRNA acting at multiple levels to affect the same cancer hallmark15,16. The use or targeting of naturally occurring miRNAs could therefore represent a promising alternative to existing RNA-based therapies and may potentially boost therapeutic effects compared with synthetic siRNAs or ASOs that influence only a single target gene.

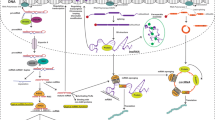
MicroRNA (miRNA) biogenesis is a multistep process (see blue boxes) consisting of transcription of a pri-miRNA by RNA polymerase II or III, its nuclear processing into a pre-miRNA by Drosha and DGCR8, nuclear export of the pre-miRNA by exportin 5, cytoplasmic processing by Dicer and TRBP into a mature miRNA duplex and its helicase-mediated unwinding. The passenger strand is degraded, and the mature miRNA strand is integrated into the RNA-induced silencing complex (RISC) to mediate either translational repression or mRNA degradation depending on the extent of complementarity to the mRNA target. Translational repression is mediated through effects on translation initiation, elongation and termination as well as co-translational degradation. mRNA degradation is mediated through mechanisms resulting first in mRNA deadenylation (step 1), followed by de-capping (step 2) and concluded by exonuclease-mediated 5′ to 3′ degradation (step 3). Ways to interfere with the endogenous miRNA pathway (see red boxes) include inhibition of biogenesis at the nuclear or cytoplasmic level, miRNA replacement therapy and functional inhibition of the mature miRNA or the interaction with its target mRNA. As oligonucleotides are not readily taken up into cells, commonly used delivery methods are shown and include conjugation to various chemical or biological entities as well as delivery within lipid particles, polymers and viral or bacterial vector systems.
The diverse functional repertoire of lncRNAs reveals various opportunities for their therapeutic targeting, the means of which need to be adjusted to the mode of action of the lncRNA. LncRNA targeting may include transcriptional inhibition, post-transcriptional inhibition, steric hindrance of secondary structure formation or protein interactions, introduction of synthetic (for example, circular) lncRNAs, and modulation of lncRNA genomic loci or expression patterns via CRISPR–Cas9 and CRISPR–Cas13, respectively (reviewed in ref.17). An interesting development is the exploration of natural antisense transcripts (NATs): lncRNAs that are transcribed in the antisense direction to coding genes, and negatively regulate them in cis. ASOs that target NATs, termed ‘antagoNATs’ have shown very promising preclinical results for gene reactivation in the central nervous system. AntagoNATs successfully upregulated brain-derived neurotrophic factor (BDNF), a protein highly involved in memory formation18, as well as the healthy allele of SCN1A, the haploinsufficiency of which causes the brain disorder Dravet syndrome19. Notably, BDNF-AS-targeting antagoNATs were successfully delivered across the blood–brain barrier using a minimally invasive nasaldepot (MIND) in a mouse model. MIND is aimed at directing drug delivery to the olfactory submucosal space and achieved approximately 40% efficacy compared with riskier invasive delivery techniques20. Such promising developments suggest that the entrance of lncRNA-based therapeutics into clinical testing is imminent.
The translation of all RNA-based therapeutics into the clinic has been hampered by issues associated with specificity, delivery and tolerability. Specificity issues include undesired on-target effects due to uptake in cells other than the cells of interest, or off-target effects caused by either sequence similarities or overdosing to levels much higher than expected endogenously. Delivery issues are related to three major points: the instability of ‘naked’, chemically unmodified RNA structures; their inefficient intracellular delivery, which requires the exploitation of endosomal escape mechanisms; and the lack of delivery vehicles suitable for targeting the organ and cell type of interest. In line with this, clinical trials have been terminated most often owing to a lack of efficacy (Table 3), one example being the nuclease-resistant ASO Genasense (G3139) that targets BCL2 mRNA, which was discontinued owing to limited therapeutic effect21; this stands in sharp contrast to the very successful use of venetoclax, a small-molecule inhibitor of the BCL-2 protein22. Tolerability issues are caused by the recognition of RNA structures by pathogen-associated molecular pattern (PAMP) receptors, such as Toll-like receptors (TLRs), leading to adverse immune effects. A prime example is the miR-34 mimic MRX34, which caused significant adverse reactions in five patients, including a case of cytokine release syndrome, in a multicentre phase I clinical trial in patients with advanced malignancies23,24. Conversely, miR-16 restitution therapy in patients with mesothelioma (MesomiR-1)25, treatment of keloid scars via intradermal injection of miR-29 mimic remlarsen26, as well as the first studies with cobomarsen (anti-miR-155) in cutaneous T cell lymphoma did not identify life-threatening toxicities10,11. These findings suggest that miRNAs are suitable for therapeutic development if toxicities are carefully assessed and delivery methods are improved.
In this Perspective, we discuss potential approaches to address the drawbacks that have been experienced regarding the clinical translation of RNA-based therapeutics so far, including the hurdles of immune responses, low specificity and aspecific delivery, with a particular focus on miRNA- and lncRNA-based therapeutics. By giving an overview of new and promising preclinical and clinical advancements, we hope to further facilitate the development of RNA-based life-saving interventions for the treatment of any type of disease in which RNAs are pathogenic, including RNAs produced from the human genome and RNAs from xeno-genomes such as RNA viruses (for example, SARS-CoV-2)27 (Box 3).

The hurdle of immunogenicity
As a viral defence mechanism, our immune system recognizes both single-stranded (ss) and double-stranded (ds) RNA via diverse extra- and intracellular PAMP receptors28. Extracellular recognition is mediated by TLRs 3, 7 and 8 in endosomes, and intracellular recognition is mediated by receptors in the cytoplasm such as EIF2AK2, RIG1, IFIH1 and NOD1/2. Immune activation by RNA interference (RNAi) was first described in vitro, with seven individual siRNAs causing a IFNγ-mediated response through cytoplasmic recognition of the 21 nt dsRNA molecules by the tyrosine kinase EIF2AK2 (ref.29). Although delivery of naked siRNA was first shown not to trigger an interferon response in animals30, intraocular injections of anti-vascular endothelial growth factor receptor 1 (VEGFR1) siRNAs bevasiranib (NCT00499590) and AGN 211745 (NCT00395057) were later shown to owe their anti-angiogenic effects to direct TLR3 stimulation instead of to the desired silencing effect31, resulting in termination of clinical development (Table 3). The predominant pathway recognizing RNA therapeutics is TLR signalling, which is mediated through myeloid differentiation factor 88 (MyD88) and activates various pathways, resulting in nuclear factor-κB (NF-κB) activation and the production of pro-inflammatory cytokines (IL-6, IL-8, IL-12 and TNF), or a type I interferon response, which ultimately leads to the activation of diverse downstream immune responses32. TLR7 and TLR8 stimulation by miRNA was shown to be dependent on the presence of GU-rich sequences (for example, 5′-UGUGU-3′ or 5′-GUCCUUCAA-3′) and caused a IFNα-mediated inflammatory response, activation of NF-κB and production of inflammatory cytokines (for example, IL-6 and TNF) in dendritic cells and macrophages. This effect observed in vitro after transfection and in vivo after delivery using liposomes was triggered by both ssRNA and dsRNA and occurred in the endosomal compartment33,34,35,36,37. It was furthermore shown that ssRNA is more prone to cause immune stimulation than dsRNA38; consequently, all siRNAs currently in clinical use or development are double-stranded (Tables 1,2).
Endogenous miRNAs may also act as TLR agonists, as shown for multiple tumour-secreted miRNAs. Lung tumour cell-derived, exosome-secreted miR-21 and miR-29a could reach the endosomal compartment of surrounding macrophages to activate TLR8 (and TLR7 in mice), causing NF-κB activation and production of inflammatory cytokines (for example, IL-6 and TNF)39. Of note, the miRNA-mediated activation of TLR8 in tumour-associated macrophages has been shown to be involved in the acquisition of resistance to chemotherapy40. The let-7 miRNA family caused TLR7 stimulation in macrophages, microglia and neurons, which was also dependent on the presence of GU-rich sequences (5′-GUUGUGU-3′ and variants thereof)41. Moreover, not only cellular miRNAs, but also viral miRNAs directly activate TLRs and their downstream signalling, showing that this is a more widespread response to miRNAs than initially reported42,43. Single-stranded small GU-rich RNAs putatively transcribed from the SARS-CoV genome also induced inflammatory cytokine secretion (TNF, IL-6, IL-12) via TLR7 and TLR8 stimulation in vitro44.
Second-generation chemical modifications now commonly applied to RNA-based therapeutics were developed to reduce the immunostimulatory potential of synthetic RNA therapeutics. 2′-ribose modifications on siRNAs (for example, 2′-F, 2′-O-Me and 2′-H), especially when applied to uridines within GU-rich sequences, can abrogate TLR stimulation38,45,46. The presence of 2 nt 3′-overhangs, as present in endogenous miRNAs, assists evasion from RNA recognition protein RIG1, which only responds to blunt-ended short RNAs47. However, 5′-triphosphate-modified siRNAs caused RIG1 recognition48, showing that chemical modifications may also be immunogenic. Despite significant advancements, there are still reports of adverse immune responses in clinical trials employing such modified reagents. For example, systemic injection of a liposome-delivered siRNA targeting apolipoprotein B (TKM-ApoB, PRO-040201) for treatment of hypercholesterolaemia was terminated after phase I owing to the occurrence of flu-like symptoms in a patient receiving the highest dose (see Related links). In addition, while the 23 nt ds miR-34a mimic MRX34 had shown promising preclinical results regarding efficacy and safety, with liposome-delivered MRX34 demonstrating efficient antitumour effects in preclinical studies49,50, the first-in-human MRX34 clinical trial was terminated owing to immune-related adverse effects in five patients, which included enterocolitis, hypoxia, systemic inflammatory response syndrome, cytokine release syndrome, hepatic failure and respiratory failure24. This potent immune effect was surprising, as there had been no reports of immunogenicity of miR-34a in preclinical studies as measured by IL-1β, IL-6 and TNF secretion in mice51. Notably, a study using the same delivery vehicle to deliver ssDNA molecules (PNT2258) also showed no evidence of immune stimulation52. In non-human primates, MRX34 most strongly located to liver, bone marrow and spleen53. Pharmacodynamic analysis in phase I trial patients showed downregulation of miR-34a target genes in white blood cells and increased levels of miR-34a in tumour tissue. Despite this, it remains unclear whether the three patients who responded to MRX34 therapy (4% response rate) experienced gene silencing-mediated effects or immune-mediated antitumour activity24. Of note, the prominent immunotherapy target programmed cell death 1 ligand 1 (PDL1) is a known target of miR-34a54 and could be involved in the responses observed. As the natural sequence of miR-34a contains GU-rich sequences (5′-UGGCAGUGUCUUAGCUGGUUGU-3′) and the precise chemical formulation of MRX34 is elusive, TLR stimulation by this therapeutic cannot be excluded and requires further investigation. On the other hand, MesomiR-1 (miR-16 mimic) administration showed a favourable safety profile in a phase I clinical trial, with minor immune reactions attributed to the carrier25.
Third-generation modifications such as phosphoramidate morpholino oligomers (PMOs) can further reduce immunogenicity by neutralizing the charge of small RNA therapeutics, thereby prohibiting their interaction with proteins, including TLRs. This technology is used in eteplirsen, a 30 nt ASO approved for treatment of Duchenne muscular dystrophy, which induces the skipping of exon 51 of dystrophin pre-mRNA in muscle cells, thereby removing a premature stop codon in a subset of eligible patients. Despite systemic (intravenous) injection, there are no reports of adverse immune reactions in response to eteplirsen in clinical trials55,56,57.
In summary, the discussion above indicates that although there have been some advancements addressing the issue of immunogenicity of RNA therapeutics, the hurdle is not yet overcome and there is still much to explore. Potential solutions to help address this issue will be discussed below.
Potential solutions
Expanding immune-related adverse reaction screening with TLR interaction methods
Not all miRNAs and miRNA therapeutics similarly induce immunogenicity, and the apparent difficulty of predicting such responses prompts the use of broader screening methods in preclinical studies. As immune responses may differ between animal models and humans, such screening methods should employ various cell types of human origin. Primary cells should be preferred as cell lines may have impaired response pathways. The use of co-culture and organoid systems58 as well as patient-derived xenograft models59 could furthermore grant a better assessment of systemic responses. In the clinical assessment of kinase inhibitors, screenings for cytokine production or kinase activity are already standard practice60. Such practice should also be adapted for RNA therapeutics and include in vivo immune screening, the assessment of large panels of immune cell markers and testing of direct TLR stimulation. A comprehensive database can be developed for the systematic collection of examples of miRNAs that target TLRs (out of more than 3,000 human miRNAs already known61), or of exact immune adverse reactions and gravity of symptoms for each tested RNA-based therapeutic. This will support large data analyses and allow selection of the RNA therapeutics with the smallest potential immunogenicity before the initiation of clinical studies.
Using ‘tiny’ antisense RNAs
As efficient activation of TLRs requires a length of at least 21 nucleotides for ssRNA31,62, the design of smaller RNA therapeutics could circumvent the issue. Locked nucleic acid (LNA)-modified antimiRs with a short sequence of 7–8 nucleotides, termed ‘tiny’ LNAs, target the 5′-seed region of miRNAs and can enable antagonism and inhibition of an entire miRNA family sharing that seed sequence. A shorter sequence obviously increases the potential for off-target effects, although targeting of two miR-122 family members using tiny LNAs showed few off-target effects in vitro with no measurable effects on mRNAs that contained the complementary sequence63. Tiny LNAs targeting the miR-17/miR-18/miR-19 (ref.64), miR-122 and let-7 (ref.63) families successfully downregulated multiple family members simultaneously. Intravenous injection of tiny LNAs in murine cancer models showed antitumour effects after treatment with anti-miR-21 in breast cancer63 and anti-miR-17 and anti-miR-19 in medulloblastoma64. In addition, miR-34-targeting tiny LNA injection in a murine cardiac stress model showed promising results, improving cardiac parameters, an effect that could not be achieved by inhibition of a single miR-34 family member65. Despite these successes in preclinical studies, tiny LNAs have not yet been clinically assessed. The successful use of shorter RNA therapeutics will depend on the detailed analyses of the correlation between the lengths of the RNA therapeutic, the in vivo efficacy and off-target binding to other RNAs or DNA by perfect or imperfect complementarity to exclude any unexpected and/or undesired effects. LNA-antimiRs that have been in clinical development (miravirsen, MRG-110 and cobomarsen (terminated, see Table 3)) are small sequences with a length of 14–16 nt, which may assist in escaping TLR recognition, but are not considered ‘tiny’ LNAs.
Applying metronomic miRNA therapy
By analogy with metronomic chemotherapy66, we define metronomic RNA therapy as regular and frequent administration of limited drug doses over a prolonged period, to achieve a low, but active, dose range without inducing excessive toxicity or immunogenicity. The addition of metronomic cyclophosphamide to the combination trastuzumab plus pertuzumab showed markedly longer progression-free survival for older patients with metastatic HER2-positive breast cancer67,68. Continuous administration of RNA therapeutics over months, instead of daily administration in cycles of weeks interrupted by periods of no administration, could achieve similar results.
To safely perform this, extensive pharmacodynamic and pharmacokinetic studies are necessary to grant a precise understanding of delivery systems as well as the circulation half-life of the chemically diverse subsets of RNA therapeutics. siG12D-LODER (local drug elutor) is a biodegradable polymeric matrix implanted into the pancreas to deliver a siRNA targeting G12D-mutated KRAS mRNA over a period of 12 weeks69. A currently ongoing phase II clinical trial (NCT01676259) will show whether siG12D-LODER together with chemotherapy improves the survival of patients with unresectable advanced pancreatic cancer. For the clinical application of metronomic RNA therapy, such locally implanted delivery devices that release their therapeutics over a long period of time could be appealing if drug release from the device is stable. Furthermore, such low-dose RNA therapy may grant ‘second chances’ for therapeutics that showed immune stimulation such as the miRNA mimic MRX34. Indeed, the miR-16 mimic MesomiR-1 was reported to be dosed approximately 1,000-fold lower compared with MRX34 with no adverse immune effects observed in its phase I clinical trial25. Metronomic RNA therapy is furthermore well suited as a combinatorial therapy, for instance, the combination of chemotherapy and miRNA metronomic therapeutics for elderly and/or frail patients.
Combinatorial RNA therapeutics
Combination therapies can also be used to reduce the required dose of RNA therapeutics and thus their immunogenicity, for example, if used in synergy with chemotherapy, radiotherapy, small-molecule-based or immunotherapies70,71. Moreover, combinatorial RNA therapeutics could induce a dual effect, as ncRNAs are involved in resistance to commonly used therapeutics72. A large-scale screening revealed that individual siRNAs, for instance, those that target genes involved in mitotic spindle assembly, could increase the sensitivity of lung cancer cell lines to paclitaxel, allowing a 1,000-fold reduction in the required dosage of the drug73. Natural miRNAs can play a similar role, as shown for miR-155 in lung cancer, which caused significant resistance to chemotherapy via a negative feedback mechanism with TP53. The application of anti-miR-155 therapy resensitized lung cancer cells to chemotherapy both in vitro and in vivo74. Similarly, miR-34a mimics sensitized lung cancer cells to erlotinib, an EGFR-specific tyrosine kinase inhibitor75 and radiation76. In addition, many lncRNAs have also been linked to drug resistance in cancer77,78, thus broadly enlarging the pool of ncRNA targets that may be exploited in the future. Furthermore, combinatorial therapeutics are being investigated for non-cancerous diseases. A recent phase II clinical trial reported the use of a single injection of the anti-miR-122 therapeutic RG-101 in combination with viral protein inhibitor GSK2878175, administered orally for 12 weeks as a single-visit curative regimen for chronic hepatitis C (EudraCT 2015-004702-42). This study reported high cure rates with an oral GSK2878175 administration scheme of longer than 9 weeks79; however, the development of RG-101 was halted owing to adverse effects observed in a different clinical trial (discussed below)80.
The development of nanoparticles (NPs) that can co-deliver multiple therapeutics in parallel is particularly interesting for combinatorial therapeutics and many advancements have been made in this field. Polymeric micelles carrying methotrexate (that is, a chemotherapeutic and immunosuppressive agent) and anti-Notch1 siRNA for the treatment of rheumatoid arthritis showed promising results in mouse models81. Long circulating NPs with a pH-regulated drug release mechanism have been developed to deliver siRNA–mitoxantrone drug complexes directly into the tumour tissue. The delivery of siRNA targeting PLK1 (Polo-like kinase, a mitotic cell cycle regulator) together with a miR-200c mimic (miR-200c acts as a tumour suppressor by blocking epithelial-to-mesenchymal transition) caused tumour regression in a murine breast cancer model82. Furthermore, the combination of miR-520d-3p and anti-EphA2 siRNA therapeutics in liposomal NPs was tested in preclinical models. EphA2 is an ovarian cancer oncoprotein, the downregulation of which suppresses tumour growth and tumour cell migration and invasion83. The synergistic effect of anti-Eph2A siRNA and miR-520d-3p therapy was likely caused by the concordant targeting of related receptor EphB2 by miR-520-3p84. Another interesting approach is the combination of multiple RNA therapeutics to target not only multiple genes but also multiple cell types. Proof of this concept has been provided by use of siRNAs that target vascular endothelial growth factor (VEGF) and placental growth factor (PIGF) delivered by multi-functionalized NPs to both breast cancer cells and tumour-associated macrophages. This approach caused a synergistic effect, addressing the role of VEGF and PIGF in promoting tumour proliferation and immunosuppression by creating a pro-oncogenic nature in the tumour microenvironment85.
Another interesting approach for combinatorial RNA therapy is the development of bioengineered RNA molecules capable of delivering multiple small RNAs at once. Such a combinatorial bioengineered RNA agent (CO-BERA) has been developed based on a transfer RNA (tRNA)–pre-miR-34a carrier to deliver combinations of NRF2–siRNA, anti-miR-21-5p, let-7c-5p, miR-124-3p and miR-34a-3p. The employed combinations of siRNAs, antimiRs and miRNA mimics successfully inhibited the growth of multiple lung cancer cell lines in vitro86. A similar approach also integrated RNA aptamers within bioengineered RNA therapeutics87,88.
Small-molecule inhibitors of miRNAs
Small molecules have important advantages over the use of oligonucleotides, that is, good solubility, bioavailability, and metabolic stability89. They may act on miRNAs at the transcriptional or post-transcriptional level or may influence miRNA processing (Fig. 1) to regulate a specific miRNA90. The main methods to screen for small-molecule inhibitors of miRNAs (SMIRs) are either computer-assisted design through sequence screening91 or high-throughput screening of (existing) compounds for interaction with miRNAs or miRNA precursors92,93 or effects on miRNA pathways94.
Computational models to predict the miRNA–SMIR association95 may employ miRNA structure-based models (that mainly work on the miRNA precursor 3D structure to predict the appropriate small-molecule interactors)96, and gene expression profile-based models (developed on the basis of the expression profile of genes targeted by small molecules and/or miRNAs or on the basis of previously known miRNA–SMIR interactions), which are sometimes combined with the structural similarity of small molecules and miRNAs97. Moreover, databases that record the relationships between small molecules and miRNAs, such as SM2miR98, Inforna 2.0 (ref.99), mTD100, NRDTD101 and ChemiRs102 offer a reasonably comprehensive repository of data regarding the influence of small molecules on miRNA expression.
High-throughput screening of large compound libraries use either the interactions of specific miRNAs with their biogenesis machinery or effects on miRNA expression levels as efficacy measures. For example, >100,000 compounds were assessed for interactors with let-7 binding motifs in RNA binding protein LIN28, which identified TPEN and LI71 as inhibitors of LIN28-mediated let-7 biogenesis92. Screening for miRNA–Dicer interactions revealed that 4 of 14 tested aminoglycosides (streptomycin, neomycin, tobramycin and amikacin) specifically inhibited pre-miR-27a processing94, and 3 polyamine derivatives from a 640-compound library interfered with pre-miR-372 processing103. Of the latter, the most active inhibitor, a spermine–amidine conjugate, showed anti-proliferative activity in gastric cancer cells that overexpressed miR-372 (ref.103). Last, testing of more than 40,000 compounds for their ability to inhibit oncogenic miR-29a led to the discovery of five inhibitors of pre-miR-29a93.
Functionally, SMIRs may interfere with the transcription or the nuclear or cytoplasmic processing of a specific miRNA (Fig. 2). The first identified SMIRs interfered with miRNA transcription and include miR-21-inhibiting azobenzene-2 (ref.104) and miR-122-reducing ‘miR-122 inhibitor 2’. The latter efficiently reduced the expression of liver-specific miR-122, resulting in a reduction in hepatitis C virus replication105. Aza-flavanones specifically inhibited transcription of miR-4644, an inhibitor of cell death, and induced cell death in human carcinoma cells106. Other SMIRs act by inhibiting the biogenesis of specific miRNAs, blocking either nuclear pri-miRNA processing by Drosha or cytoplasmic pre-miRNA processing by Dicer (Fig. 1). Targaprimir-96 is a small dimeric molecule that binds to the internal loop of miR-96 to interfere with Drosha binding. Targaprimir-96 treatment in breast cancer cells triggered apoptosis without affecting healthy cells and this effect was reversed by overexpression of pri-miR-96 (ref.107). Owing to its prevalence as an oncomiR in many types of cancer108, multiple compounds were identified that interfere with Dicer processing of pre-miR-21, including the aminoglycoside streptomycin (a tuberculosis therapeutic)109, a peptide possessing a binding pocket for pre-miR-21 (ref.110) and the compound AC1MMYR2, which showed potent antitumour effects in murine glioma and breast cancer models111. BzDANP binds to pre-miRNAs possessing a C bulge near the Dicer processing site, that is, miR-29a112 and miR-136 (ref.113). The binding of BzDANP to miR-136 caused formation of a ternary complex together with Dicer, slowing the kinetics of pre-miR-136 processing and consequently lowering mature miR-136 levels113. Targapremir-210 binds close to the Dicer processing site on pre-miR-210. MiR-210 is a major regulator of the hypoxic response (via a positive feedback loop with hypoxia-inducible factor 1α) and thereby influences breast cancer metastasis114,115, and accordingly, treatment of triple-negative breast cancer cells with targapremir-210 triggered apoptosis under hypoxic conditions and inhibited tumorigenesis in a murine xenograft model116. Other examples include the multi-tyrosine kinase inhibitor linifanib, which inhibits cytoplasmic miR-10b maturation through an unknown mechanism117, and an aminoglycoside–nucleobase conjugate that inhibited Dicer processing of multiple oncogenic miRNAs including miR-372, miR-373, miR-17 and miR-21, but possibly also other miRNAs90.

a | Small-molecule inhibitors of miRNA (SMIRs) may act at the transcriptional level or may affect the nuclear or cytoplasmic maturation steps of the microRNA (miRNA). The exact mechanism by which azobenzene-2, miR-122 inhibitor 2 and aza-flavanones inhibit the transcription of specific miRNA host genes to primary RNA transcripts (pri-miRNAs) is unknown. Targaprimir-96 binds to the internal loop of pri-miR-96 to prevent its processing by Drosha. Multiple small molecules interfere with Dicer processing, including targapremir-210, which binds to the Dicer cleavage site, and targapremir-18a, which binds to a 1 nt bulge present in three of the six miRNAs of the miR-17–92 cluster. BzDANP similarly binds to a C bulge present in miR-29a and miR-136, causing complex formation and slowing of Dicer processing. Linifanib inhibits the processing of pre-miR-10 via an unknown mechanism. Proximity-enabled Dicer inactivation makes use of two small molecules, a miRNA binder and a weak Dicer inhibitor that is active upon proximation. Use of a photocleavable linker can grant specific Dicer inactivation that can be terminated using light. b | The first small molecules applied to modify long noncoding RNA (lncRNA) expression levels can be classified as interaction element blockers (IEBs) and structural element lockers (SELs). NP-C86 works as an IEB for GAS5, blocking its interaction with UPF1, which normally results in nonsense-mediated decay of GAS5, thus increasing the stability and half-life of GAS5. Multiple SELs are being developed for MALAT1, which carries a stabilizing triple helix structure at its 3′ end. The SELs are aimed at disrupting the stabilizing triple helix, consequently resulting in MALAT1 destabilization and downregulation.
A more tailorable approach can be developed using a miRNA binding molecule linked to an effector domain. One such approach links a short 2′-5′ poly(A) oligonucleotide to a miRNA binding molecule, causing selective recruitment of RNase L to degrade the RNA structure. This principle has been shown to work in vitro, where selective degradation of oncogenic pri-miR-96 induced apoptosis in triple-negative breast cancer cells118, and in vivo, where selective degradation of pre-miR-21 reduced the occurrence of metastases in a murine breast cancer model119. A similar approach consists of linkage of a miRNA binding molecule to a weak Dicer inhibitor, a method termed ‘proximity-enabled Dicer inhibition’. This system was used to successfully inhibit miR-21 biogenesis in vitro, and the use of a photocleavable linker enabled inactivation of the reaction by light120,121.
Other small-molecule inhibitors can be envisioned that target the hairpin structure, which can be very similar between two or more oncogenic miRNAs such as miR-10b and miR-21, the concomitant downregulation of which reduces the proliferation of glioma cells122. Such an approach could synergize the killing of malignant cells and in addition reduce potential resistance, as the two miRNAs induce cell death through independent targets. Although there are so far no reports of small molecules targeting miRNA hairpin structures, the hairpin structure of a promoter G-quadruplex structure in hTERT has recently been targeted successfully using the small molecule RG1603 (ref.123), implying the feasibility of this approach. Targeting of the mature miRNA or its seed sequence could represent another tactic, whereby the latter would enable inhibition of an entire miRNA family, akin to what was achieved using tiny LNA antisense technology.
Although SMIRs have been reported as efficient miRNA inhibitors, several challenges remain regarding specificity, optimal potency, delivery and lack of toxicity that still hinder their clinical application, and there are currently no clinical trials involving SMIRs.
Targeting ncRNAs with interaction element blockers and structural element lockers
A new type of RNA therapy can be envisaged based on the targeting of interactor elements (IEs), that is, docking sites on ncRNAs for multiple types of molecule (DNA, RNA, proteins and lipids) and/or targeting structural elements (SEs), which directly affect the conformation of a ncRNA and indirectly the functional interactions with interactor molecules (Fig. 2b). Consequently, such new therapeutics will influence signalling pathways and disease phenotypes. Small molecules are obvious candidates for such developments owing to their stability, lower toxicity and pharmacokinetic and dynamic properties. IE blockers (IEBs) can also be envisioned as antisense oligonucleotides able to hide the IE from the interaction with its effector, while an oligonucleotide SE locker (SEL) could disrupt the SE sequence to affect secondary and tertiary structures of ncRNAs.
The small molecule NP-C86 has been shown to inhibit the interaction of the GAS5 lncRNA with UPF1, a protein involved in nonsense-mediated decay and degradation of GAS5. Blocking this interaction resulted in increased stability of GAS5, which in turn increased its interaction with the insulin receptor and caused an upregulation of glucose intake in adipocytes124. NP-C86 is thus an IEB and may find future applications in the treatment of patients with diabetes. MALAT1 is strongly associated with cancer metastasis and contains a triple helix structure at its 3′-end that protects the lncRNA from exonuclease degradation and assists its nuclear accumulation. Multiple studies have attempted to identify SELs that target this stabilizing triplex structure. Fluorescence-based screening of a diphenylfuran (DPF)-scaffold-based library identified a DPF-based small molecule that specifically targets the triple helix structural element in MALAT1 (ref.125). However, it remains to be elucidated whether the binding of this molecule results in efficient MALAT1 destabilization. Similarly, another high-throughput screening identified two small molecules that target the MALAT1 triple helix, but not other similar structures present for instance in NEAT1. The compounds significantly reduced levels of MALAT1 as well as downstream genes and attenuated the branching of mammary gland organoids126. However, the development of MALAT1-targeting small molecules should be addressed with caution as a dual function has been described for this lncRNA127. Accordingly, these molecules have not yet entered clinical testing.
The hurdle of specificity
The quality of an RNA therapeutic is determined by the strength of its on-target specificity as well as the absence of off-target and undesired on-target effects. Many developments in RNAi therapeutic design have already improved on-target specificity and decreased unwanted off-target effects. Second-generation chemical modifications have significantly improved the potency of antisense therapeutics128, and third-generation LNA modifications show the highest potency129. Studies on siRNA specificity have shown that off-target genes with three or four base mismatches and additional G-U mismatches can be efficiently downregulated through translational repression130, similar to the effect of miRNA binding with mismatches (Fig. 1). Sequence-mediated off-targeting was mainly caused by complementarity to the first eight nucleotides at the 5′-end of the siRNA, again mirroring the endogenous function of miRNA seed sequences131,132. Placement of a 2′-O-methyl ribosyl substitution at position 2 of the guide strand could abrogate this seed effect131. Other off-target effects are caused by loading of the passenger instead of the guide strand into the RNA-induced silencing complex (RISC). Multiple approaches have been suggested to improve guide-strand RISC loading, including the incorporation of LNAs at the 5′-end of the passenger strand133,134, 2′-F-O-Me-phosphorodithioate modifications135, trimming136 or fragmentation137 of the passenger strand, its 5′-biotinylation138, and creation of so called Dicer-substrate siRNAs (longer siRNAs with asymmetrical termini and containing DNA nucleotides)139. Yet other off-target effects may stem from interactions of RNA therapeutics with proteins. Oblimersen (Genasense), a first-generation 18 nt phosphothiorate-modified ASO that targets BCL2 mRNA, was tested in several clinical trials but showed limited efficacy21,140,141. Molecular studies comparing oblimersen with BCL2-targeting siRNAs later showed several off-target effects, including upregulation of stress-response genes and downregulation of proliferation-associated genes142, as well as downregulation of several apoptosis and glycolysis-related proteins143. These effects were in part defined to be a class effect of phosphothiorate-modified ASOs, as the identical sequence formulated with a phosphodiester backbone did not show the same effect142,143. Detailed molecular studies showed that phosphothiorate oligonucleotides such as oblimersen could directly bind and inhibit the mitochondrial channel protein VDAC, resulting in apoptosis144,145. In accordance with an off-target effect, oblimersen also induced apoptosis in BCL-2-negative cell lines146. An unexpected side effect was also noted in the clinical trial phase IB of the anti-miR-122 therapeutic RG-101, which was terminated owing to high levels of bilirubin in the blood (see Related links). Although the precise molecular mechanism causing impaired bilirubin transport is not yet clear, hepatocyte-specific loss of miR-122 in mice affected lipid, but not bilirubin, protein or glucose metabolism147, indicating the absence of an undesired on-target effect. Liver-specific deletion of Dicer to abrogate global miRNA production also did not affect bilirubin metabolism in mice148, arguing against a global impact on miRNA processing. The anti-miR-122 therapeutic miravirsen showed a favourable safety profile with no reports of adverse immune effects or liver toxicity149.
Undesired on-targeting is also common, especially when the RNA therapeutic is administered systemically. For example, the advanced tumour therapeutic MRX34 was shown to be taken up by white blood cells24, and miR-34a plays major roles in T cells150 and macrophages151. In vitro analysis of macrophages and T cells upon incubation with a miR-34a mimic showed changes in chemokine profiles that would be detrimental to tumour lysis152. Whether this caused the low response rate (4%) or was involved in the adverse immune effects observed in the clinical trial of MRX34 (ref.24) remains to be elucidated. Similarly, the increase in chemotherapy-induced neuropathy in patients who received the XIAP-targeting ASO AEG35156 could be due to downregulation of XIAP in oligodendrocytes, glial or neuronal cells instead of tumour cells153.
Another important consideration is the dosing of the RNA therapeutic. For example, it has been shown that off-targeting is more sensitive to siRNA concentration than on-target silencing132. In addition, as many RNA therapeutics make use of the endogenous RNAi machinery, coarse overdosing can cause saturation of the system and prevent the function of endogenous miRNAs. The first report of this phenomenon indicated that strong shRNA overexpression in hepatocytes caused global downregulation of miRNAs, resulting in liver toxicity and mortality in mice154. A large-scale screening study confirmed that global upregulation of miRNA target genes is a general phenomenon caused by the introduction of exogenous small RNAs155.
When using miRNA mimics and antimiR approaches, dosing has additional implications. On the one hand, miRNAs are known to target multiple different mRNAs156. It has for example been demonstrated that one cluster of miRs (the miR-15a–16-1 cluster) alone can affect (by direct and indirect targeting) about 14% of the entire transcriptome in leukaemic cells15. This is both concerning, as it could lead to unwanted silencing of genes, and advantageous, as miRNAs can affect entire signalling pathways by synchronous silencing of different genes involved in that pathway. On the other hand, each mRNA target is regulated by multiple miRNAs and may possess different miRNA binding sites in close vicinity157, resulting in binding competition. Therefore, the question that arises is how much the concentration of an overexpressed or downregulated miRNA affects the targetome (that is, the full spectrum of target genes silenced by a specific miRNA). Experimental approaches designed to identify the plethora of targets of a specific miRNA introduce an arbitrary amount of miRNA, not considering how the quality and quantity of targeted mRNAs could be affected. Let us consider the example of the oncogenic miR-17–92 cluster, in which miRNAs may be upregulated 2- to 36-fold in a pathological setting (that is, in lymphoma cell lines or Burkitt lymphoma tissues)158. Transient transduction of miR-17–92 cluster mimics at concentrations commonly used in experimental settings caused not only a supraphysiological upregulation of up to 400-fold, but also nonspecific gene expression changes and accumulation of mutated and tailed mimic variants, whereas lower concentrations conversely did not result in specific gene knock-down158. If miRNAs interact with their targets in a competitive manner, perturbation of the level of a single miRNA will affect the targeting of the mRNA by other miRNAs. In a mathematical model of such combinatorial regulation, miRNA overexpression or depletion could also lead to positive regulation of target mRNAs through changes in competitive interactions159. Studies to address the effect of different miRNA concentrations on the targetome are thus warranted and seem the most logical basis for any future translational study with miRNA therapeutics. Unfortunately, dosing represents an important limitation in the field of miRNA therapeutics, and clinical studies are rarely able to determine the precise dose of an RNA therapeutic successfully delivered to the cell type of interest.
In summary, several issues persist with regard to specificity, including unexpected off-target effects and undesired on-target effects, and the relationship between dosing and specificity remains largely unexplored in the clinical setting. The following considerations may help in overcoming these issues in the future.
Potential solutions
Cell-specific miRNA modulation
One possible approach to limit the detrimental overexpression of a miRNA or antimiR in undesirable cells, is to express the RNA therapeutic using a suitable vector under the control of a specific promoter, which is overexpressed in the cells of interest. For instance, MYCN is a well-known driving oncogene in neuroblastoma160. Therefore, more selective expression of a therapeutic miRNA in neuroblastoma cells could be achieved by cloning the miRNA into a vector under the promoter for MYCN. If this vector enters a non-neuroblastoma cell with little to no expression of MYCN, the expression of the miRNA will be minimal. In contrast, high expression will be achieved in MYCN-overexpressing cancer cells. In such a way, anti-miR-155 could be a suitable therapeutic in neuroblastoma, as it was proved to contribute to resistance to chemotherapy through exosomal crosstalk between neuroblastoma cells and human monocytes in vitro40. Such cell-specific miRNA modulation represents an alternative approach that has not been applied clinically. Dosing should be monitored closely when using this approach, as, for instance, shRNA expression from strong promoters caused neurotoxicity owing to saturation of the RNAi machinery161,162. Conversely, expression of miRNA mimics from pri-miR-encoding plasmids resulted in miRNA expression at physiological levels and caused specific gene targeting158.
Pseudo cell-specific targeting
In cases where a miRNA or lncRNA is specifically overexpressed in a diseased cell type but shows low expression in normal cell types, pseudo cell-specific targeting could be applied. This principle assumes that the downregulation of a lowly expressed transcript in normal cells will not produce strong adverse effects. This is the case for a large proportion of lncRNAs that are expressed at very low levels in normal cells, and therefore antisense therapy will have little to no effect (and consequently no toxicity) in normal cells. One such example is targeting of the human-specific lncRNAs from the category of pyknon transcripts163. These are noncoding transcripts that contain one or more very short distinct sequences (about 16 nt long) that are human and/or primate specific and work as sponges of miRNAs. Having at least 40 copies in the genome, each pyknon is quite poorly expressed in normal cells. Therefore, in the case of overexpressed pyknon transcripts in malignant cells, a nanodelivery system containing siRNA specifically targeting the transcript was used and achieved therapeutic efficacy in mouse models of colon cancer164,165. The clinical application of this approach requires careful preclinical investigation of the exact tissue expression patterns of the ncRNA, especially for such transcripts that are non-conserved and primate and/or human specific, as are many lncRNAs and some miRNAs166.
Targeting precursor miRNAs
Molecules that are able to interfere with miRNA genesis and function are a potent therapeutic strategy. Peptide nucleic acid (PNA) oligomers that target pre-miRNA are exploited to inhibit miRNA maturation. PNAs bind to their RNA targets with high specificity as even a single mismatch strongly influences the association constant of PNA–RNA interactions167. The targeting of pre-miRNAs was first attempted using PNAs with perfect complementarity to the sense strand of the mature miRNA168. As a pre-miRNA naturally has mismatches with its antisense strand, a synthetic nucleic acid with perfect complementarity disrupts the stem–loop structure of the pre-miRNA, thereby disrupting its recognition by Dicer, which depends on the integrity of the stem–loop structure. Using PNAs, miR-210 maturation was successfully inhibited in K562 cells, blocking mithramycin-induced erythroid differentiation, and PNA-backbone modifications significantly improved cellular uptake and resistance to enzymatic degradation168,169,170. Interestingly, pre-miRNAs and their processing machinery including Dicer and Ago2 are also present in tumour-derived microvesicles171, indicating that therapeutics targeting pre-miRNAs could also disrupt exosomal signalling between tumour cells and the microenvironment. SMIRs, as described above, may similarly be used to target precursor miRNAs.
Using circular transcripts
RNAs in circular form are much more stable than their linear counterparts. Multiple circular small RNAs have been synthesized, including dumbbell shaped (dbRNA)172,173,174, covalently closed175,176 and linker-circularized177,178 siRNAs. Interestingly, many of these circularized siRNAs were not only more stable but also mediated more efficient and prolonged RNAi effects172,173,176. dbRNA, for example, mediated stronger and longer RNAi effects at lower concentrations173. A miR-34a-3p- and miR-34a-5p-containing dbRNA (db34a) inhibited angiogenesis in an in vitro model as well as in a zebrafish model174. Covalent closure of only the passenger strand to prevent its RISC loading could significantly reduce off-target effects in vitro175. In fact, circularization seems to overcome multiple hurdles experienced by linear siRNAs, showing less immune stimulation176,178, higher stability and bioavailability172,174,175,177, high efficacy172,173,176 and increased cellular uptake176.
The discovery of circular lncRNAs (circRNAs) as miRNA sponges further broadens the possibilities for blocking the function of oncogenic RNAs. The first characterized circRNA was the ciRS-7 transcript, which is highly expressed in the human brain and contains more than 70 conserved miR-7 binding sites179. It has been established that circRNAs are especially abundant in brain tissue180, whereas they are mostly downregulated in cancer tissue181. The latter may be attributed to the dilution of these generally low transcribed but highly stable transcripts in quickly proliferating tissues182. CircRNA deregulation has been linked to several diseases, including cancer, diabetes and atherosclerosis (reviewed in181). Endogenous or synthetic circRNAs thus have the potential to be exploited as potent and highly stable miRNA sponge therapeutics. Foreign circRNAs were shown to cause an intron-mediated immune response through PAMP receptor RIG1 (ref.183). N6-methyladenosine (m6A) modifications, as occur in endogenous circRNA, were able to alleviate immune stimulation184. After further characterization of their immunogenic properties, synthetic circRNAs thus have great potential as miRNA sponge therapeutics. Synthetic circular mRNAs are further being explored to reconstitute protein expression and were shown to be less immunogenic (no RIG1 or TLR stimulation in vitro) and potently translated185.
Ex vivo manipulations
Although certainly not new, ex vivo manipulations may be more appreciated after successes such as the use of chimeric antigen receptor (CAR) T cells186 and with new possibilities offered by the CRISPR–Cas9 system187,188. Most importantly, off-target effects can be monitored before re-implantation. The autologous tumour cell immunotherapeutic Vigil has shown promising results in advanced malignancies in phase I189 and II190 clinical trials. Vigil is generated from the patient’s tumour cells modified to express recombinant granulocyte–macrophage colony-stimulating factor (GM-CSF) and a bifunctional shRNA that targets furin. Furin inhibition results in downregulation of immunosuppressive cytokines TGFβ1 and TGFβ2, thereby supporting T cell responses in the tumour. Bifunctional shRNAs cause both mRNA degradation and inhibition of translation, causing stronger and prolonged RNAi effects and allowing lower dosing to reduce off-targeting191. TGFβ1 and TGFβ2 were inhibited 90–100% in bi-shRNA furin-treated cells189, and treatment of patients with advanced cancer with Vigil caused an increase in circulating activated T cells and significantly improved survival189,190. No long-term toxicity was reported in a 3-year follow up in patients with Ewing sarcoma who had received Vigil192.
The CRISPR–Cas9 system187,188 offers new opportunities for therapeutic targeting of coding and noncoding genes. The system is capable of genome editing by active CRISPR–Cas9 or Cas12a (Cpf1)193, gene interference or activation by catalytically dead (d)Cas9 fused to a repressive or activating effector domain194, or RNA editing by the Cas13 variant195. Clinical trials applying CRISPR–Cas for genome editing ex vivo are actively ongoing. For example, the disruption of the CCR5 gene, the predominant co-receptor for entry of the HIV virus into human T cells, in CD34+ haematopoietic stem cells (HSCs) of HIV-infected subjects with haematological malignancies is being attempted (NCT03164135). The dCas9 system has so far been employed only in vitro, for example, to reactivate HIV long terminal repeat (LTR) transcription in latently infected cells to trigger their suicide death, presenting a potential curative treatment194. Importantly, these HIV LTR-specific MS2-mediated single guide RNAs showed very few genome-wide off-target effects196. Multiple lncRNAs have also been shown to activate (for example, MALAT1, HEAL, SAF) or repress (lincRNA-p21, NEAT1, GAS5, 7 SK, NRON, TAR-gag) HIV transcription, while lncRNAs NRON and uc002yug.2 contribute to HIV latency (reviewed in197). Furthermore, CRISPR–Cas9 is used to target the BCL11A gene enhancer, the disruption of which induced the expression of fetal haemoglobin ex vivo198 in CD34+ cells for treatment of β-thalassaemia (NCT03655678). The lncRNA UCA1 was recently shown to be crucially involved in human haem metabolism and erythrocyte differentiation of CD34+ HSCs199. This therefore shows that lncRNAs can expand the amount of therapeutic CRISPR–Cas9 ex vivo editing targets. For the CRISPR–Cas9 system to be applied in vivo, delivery issues especially regarding the large Cas proteins must be overcome and immunological responses (reviewed in200) must be carefully assessed.
The hurdle of delivery
Efficient delivery of RNA therapeutics not only to the organ and cell type of interest but also across the cell membrane to perform their intracellular functions is one of the greatest challenges in the field. Accordingly, the first and foremost reason for clinical trial termination regarding RNA therapeutics is lack of efficacy (Table 3). Efficient delivery of oligonucleotides is challenging owing to their instability, negative charge and hydrophilic nature preventing diffusion through cell membranes201. First- and second-generation chemical modifications improve stability and uptake by inducing resistance to nuclease degradation and increasing interaction with proteins129. Third-generation antisense technologies such as neutrally charged PMOs are highly stable but face difficulties with cellular uptake and require administration in high doses202. Different delivery systems are also being used, including lipid- and polymer-based vectors and ligand–oligonucleotide conjugate delivery systems. These delivery systems are taken up via different endocytosis mechanisms and consequently, endosomal escape of the RNA therapeutic must be facilitated to prevent its lysosomal degradation. Numerous strategies can be employed, such as the use of cationic lipids, viral or bacterial agents, or cell-penetrating peptides (CPPs), which are reviewed in detail elsewhere203,204.
Lipid nanoparticles (LNPs) can be easily modified, coupled to targeting moieties and are characterized by high biodegradability and biocompatibility with low immunogenicity. Surface modification with for example, hyaluronic acid (HA) or polyethylene glycol (PEG) reduces nonspecific uptake, cellular toxicity and aggregation, and improves characteristics such as tumour targeting and stability. Cellular uptake is mediated by endocytosis, and interactions between fusogenic cationic LNPs and anionic membrane lipids mediate the release of their cargo into the cytoplasm205. The FDA-approved siRNA therapeutic patisiran uses LNPs as a delivery method206, as did the miRNA mimic MRX34 (ref.23), and many more lipid-based RNA therapeutic delivery systems are being tested in clinical trials.
Polymers (synthetic or naturally based) are successful alternatives as miRNA delivery vectors. Polymer-based carriers are characterized over lipid carriers by their versatility in size, molecular composition and structure. Polymers such as polyethylene imine (PEI), polylactic-co-glycolic acid (PLGA), poly-amidoamine (PAMAM) and chitosan are being actively investigated in vitro and in vivo for the delivery of miRNA mimics or antimiRs, either alone or in combination with chemotherapies for enhanced therapeutics. Many miRNAs have been delivered either systemically or locally through polymers, including miR-221/miR-222 (ref.207), miR-21 (ref.208,209), miR-150 (ref.210,211), miR-34a212, miR-145 and miR-33a213. Other studies address polymer combinations, such as PLGA–PEI NPs modified with HA (for specific internalization by tumour cells) to deliver miR-145. After local administration of this construct to xenograft tumour-bearing mice, intact miRNA plasmid vectors were delivered, and stable expression of miR-145 was achieved followed by a significant antitumour effect214. PEI–PLGA–HA NPs were furthermore used to encapsulate miR-542-3p and doxorubicin for targeted delivery to triple-negative breast cancer cells215. An interesting development is also the LODER system, a miniature biodegradable polymeric matrix designed to slowly release siRNA against G12D-mutated KRAS upon local injection for the treatment of pancreatic cancer216,217. siG12D-LODER is currently being tested in a phase II clinical trial (NCT01676259).
The conjugation of oligonucleotides to various entities is being further explored for their delivery. Ligand conjugation is a major clinical strategy for delivery of RNA therapeutics to hepatocytes, offering selective delivery via receptor-mediated mechanisms218. N-acetylgalactosamine (GalNAc) is a high-affinity ligand for the hepatocyte-specific asialoglycoprotein receptor (ASGR), which mediates clathrin-mediated endocytosis. The endosomal release mechanism of GalNAc-conjugated RNAs is thought to be relatively inefficient, and successful delivery is owed to the fast cycling of the ASGR204. Owing to its advanced liver-targeted delivery, various RNA therapeutics employ GalNAc conjugation (Tables 1,2), including the anti-miRNA RG-125 (AZD4076), which targets miR-103/miR-107 for treatment of nonalcoholic steatohepatitis in individuals with type 2 diabetes or pre-diabetes219, and is currently being tested in a phase I/II clinical trial (NCT02612662). The conjugation of oligonucleotides to antibodies represents another strategy with promising preclinical developments. Conjugation of RNAs to antibodies may for instance be achieved via electrostatic interactions, affinity conjugation via biotin or avidin, direct conjugation or double strand hybridization220. In addition, various other strategies have been explored, such as the use of an azide-functionalized linker peptide on the antibody and conjugation to dibenzylcyclooctyne-bearing RNAs221, or antibodies with a reactive lysine residue combined with β-lactam linker-functionalized RNAs222. Although antibody conjugation is promising regarding target-organ delivery, for instance, a siRNA that targets myostatin was successfully delivered to skeletal muscle of mice by conjugation to an anti-CD71 antibody223, a major limitation is their entrapment in the endocytic compartment, which often results in limited efficacy224. The most promising developments regarding improved intracellular escape were seen with CPP conjugation. The efficacy of the splice-modifying PMO ASOs currently approved for treatment of Duchenne muscular dystrophy could be greatly enhanced upon conjugation to CPPs225. The CPP Pip8b2, for example, increased the uptake of DMD-targeting ASOs in vitro226, and HA2-ApoE(131–150) increased endosomal escape of nusinersen in mice227.
Other notable developments regarding delivery methods include, for instance, metal-based NPs, particularly gold NPs (AuNPs). Gold nanostructures show low toxicity profiles, may be conjugated to various compounds including carbohydrates, amino acids, peptides, proteins and oligonucleotides, and, most importantly, passively accumulate in tumour tissues228. The successful delivery of ss and ds oligonucleotides of up to 50 nucleotides or base pairs using AuNPs was first shown in vitro in a murine melanoma cell line229. Spherical nucleic acids (SNAs), that is, gold NPs densely coated with oligonucleotides230, have shown notable results as delivery vehicles to brain tumours. Systemic administration of miR-182 mimic-coated SNAs showed successful delivery to tumour in a murine glioblastoma model231. SNAs may also be combined with liposomal delivery, for instance, anti-miR-92b-loaded SNAs in apolipoprotein E (ApoE)-coated liposomes successfully crossed the blood–brain barrier in a murine glioblastoma model and reduced tumour cell viability232. Their clinical safety was proved in a phase 0 clinical trial in which SNAs coated with siRNAs targeting the Bcl2Like12 oncogene were systemically administered to patients with glioblastoma (NCT03020017)233. The safety and proof of concept of adeno-associated virus 5 (AAV5)-mediated delivery is currently being tested for the potential treatment of Huntington disease (rAAV5-miHTT; NCT04120493). AAV5 has shown effective delivery to the brain in non-human primates234. With a huntingtin-targeting miRNA in a miR-451 backbone that is flanked by inverted terminal repeats to mediate genome integration, rAAV5-miHTT is intended as a one-time gene therapy235. Additional developments are being made with respect to the route of administration, such as inhalation (for example, pressurized dry powder inhalers, nebulizers, soft mist inhalers) that allow delivery of RNA-based therapeutics to specific regions in the pulmonary tract236,237.
In summary, LNPs, polymers, RNA conjugations, NP and virus-based approaches for delivery are already either used in clinically approved therapeutics or have entered clinical testing. Below, we highlight two alternative approaches that we think are promising candidates for clinical translation.
Potential solutions
Exosome-mediated miRNA delivery
Cell-derived membrane vesicles (exosomes and microvesicles) have been reported to be effective delivery vectors. They are small sized (nanoscale) vesicles, secreted by various cells, and they contribute to intercellular communication through the delivery of different types of cargo, including proteins (cytoplasmic proteins, membrane proteins), lipids and nucleic acids (DNA, mRNAs and ncRNAs) involved in cell-to-cell communication238,239. Exosomes are thus considered natural carriers of miRNAs and may present an ideal delivery system owing to their negligible antigenicity, minimal cytotoxicity and their ability to bypass the endocytic pathway and circumvent phagocytosis240. The precise uptake mechanism for exosomes may differ depending on targeted cell type241. The exosomal transmembrane protein CD47 was shown to inhibit the phagocytosis of exosome delivery vehicles by monocytes, which increased their delivery to pancreatic tumours and the improved targeting of oncogenic KRASG12D (ref.242). The use of iExosomes (that is, engineered exosomes) loaded with anti-KRAS-G12D siRNA for the treatment of pancreatic cancer is currently being tested in a phase I clinical trial (NCT03608631).
Exosome biodistribution studies in animal models indicated accumulation in lung, liver and spleen upon intravenous injection, and in draining lymph nodes upon lymphatic injection243. However, a very recent study showed that genetic modification of the membrane proteins can vastly change the distribution patterns of exosomes244. Other work similarly suggests differences in biodistribution based on exosome subtypes245 and the preferential uptake of self-produced exosomes in parental cells246. These findings imply that exosome biodistribution can be tailored to specific needs based on their specific composition as well as the cells used for their production. Indeed, neuron-specific targeting of exosomes was achieved in mice through expression of exosomal membrane protein Lamp2B fused to the RVG peptide that specifically localizes to neurons247.
Exosomes separated from bone marrow-derived mesenchymal stem cells were induced to deliver LNA–anti-miR-142-3p to suppress the expression level of miR-142-3p to reduce the tumorigenicity of breast cancer cells. MiR-142-3p repression reduced the levels of miR-150 via derepression of miR-142-3p target gene adenomatous polyposis coli (APC) and consequent repression of Wnt–β-catenin signalling, which in turn induces miR-150 (ref.248). Furthermore, exosomes can be engineered to introduce multiple compounds, for example, an oligonucleotide and a chemotherapeutic agent, for a more efficient effect. For example, purified exosomes loaded with miR-21 and 5-fluorouracil (5-FU) were efficiently taken up by 5-FU-resistant HCT-1165FR cell lines. The combined delivery of miR-21 and 5-FU effectively reversed drug resistance of the cells and significantly elevated the cytotoxicity of cells compared with single treatment with 5-FU or miR-21 (ref.249).
Although using exosomes as a delivery platform is a promising strategy, large-scale production of exosomes is not easily available or commercially viable owing to the high cost. Production through chemically induced membrane blebbing has been suggested, which was shown to produce large numbers of highly homogeneous vesicles termed ‘extracellular blebs’250. In addition, food-derived exosomes have shown promising results in preclinical studies, including bovine milk-derived251 and grape-derived exosomes252. Grapefruit-derived exosomes coated with plasma membrane of activated leukocytes were successfully delivered to tumour inflammatory sites to attenuate tumour growth and colitis in mouse models. The homing of drug-loaded exosomes to inflammatory sites was mediated by chemokine receptors CXCR2 and LFA1 (ref.253). Furthermore, exosome production from delicate systems such as mesenchymal stem cells is being developed, employing, for example, 3D scaffolds, large-scale cell expansion methods and molecular modifications to increase exosome yield or genetic packaging (reviewed in254).
Bacteriophage and bacterial minicell delivery vehicles
Several studies have reported bacteriophages as a safe and effective delivery vehicle that can be loaded with RNAs or other drugs. For example, modification of MS2 bacteriophages to be used as virus-like particles (VLPs) could protect miRNAs against degradation. Self-assembly of MS2 by interaction between a specific MS2 cistron, a 19 nt sequence known as a pac site, and the bacteriophage coat protein allows the bacteriophage particles to be loaded with RNAs or other molecules. The covalent linkage of pre-miR-146a to modified MS2 bacteriophage particles significantly induced the expression of miR-146a in vitro and in vivo and resulted in target gene repression255. PP7 bacteriophages were similarly used to package pre-miR-23b. Insertion of a cell-penetrating peptide (CPP) into the AB loop of the PP7 bacteriophages facilitated the delivery of the pre-miR-23b into SK-HEP-1 hepatoma cells. Pre-miR-23b delivery using PP7 VLPs was followed by pre-miRNA processing to produce mature miR-23b, which consequently repressed its target gene cadherin and reduced cell migration256. Bacteriophages, like many other delivery agents and drugs, accumulate preferentially in liver and spleen as well as in solid tumours owing to the enhanced permeation and retention effect257,258. The modification of bacteriophage biodistribution is so far grossly understudied, but for example, splenic accumulation could be inhibited by PEG conjugation257 and their encapsulation by liposomes has improved oral administration259. Bacteriophages are currently being extensively explored in clinical trials as antibacterial agents, but not yet as delivery vehicles for RNA therapeutics. An important limitation could be the lack of naturally developed internalization and endosomal escape mechanisms.
Achromosomal bacterial minicells are small size vesicles that are assembled by bacteria. Minicells with recombinant-plasmid DNA molecules are produced by transforming the parental bacterial strains with a plasmid of choice, before inducing production of minicells260. The ongoing MesomiR-1 phase I study (NCT02369198) is the first human study in which EnGeneIC Dream Vectors (EDV), that is, non-living bacterial minicells targeted with anti-EGFR antibody that encapsulate a tumour-suppressive miR-16 mimic (termed ‘TargomiRs’), have been applied to patients with non-small cell lung cancer or mesothelioma261,262. This approach may be especially beneficial for granting optimal intracellular delivery, as bacteria have a variety of natural endosomal escape mechanisms in place263. Minicells have shown promising results for specific delivery, including for example, EGFR-mediated delivery of doxorubicin-loaded minicells to the core of late-stage brain tumours in dogs264, and the folic acid-mediated delivery of anti-VEGFA shRNA to murine xenograft tumours265.
Outlook
The successful application of RNA-based therapeutics requires an unprecedented interdisciplinary approach, including technical advancements in molecular biology, immunology, pharmacology, chemistry and nanotechnology. An optimal RNA therapeutic should be extensively tested for immunogenicity, chemically modified to improve pharmacokinetics and pharmacodynamics, delivered under consideration of biodistribution patterns and intracellular escape mechanisms, specifically and potently interact with the intended target and be dosed at an optimal level to trigger the desired effect. Significant advancements in each of these areas has been made for RNA therapeutics individually, but their successful translation depends on further interdisciplinary development to improve tolerance, specificity and delivery. An example of such an interdisciplinary approach is the delivery of a miRNA inhibitor to heart tissue using an ultrasound and microbubble-targeted delivery system that suppressed cardiac hypertrophy in a mouse model266. Although challenging, with the many new, promising and creative developments that are emerging through valuable preclinical work, the challenges faced in the field of RNA therapeutics will ultimately be overcome.
References
Calin, G. A. & Croce, C. M. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 66, 7390–7394 (2006).
Lenkala, D. et al. The impact of microRNA expression on cellular proliferation. Hum. Genet. 133, 931–938 (2014).
Ivey, K. N. & Srivastava, D. microRNAs as developmental regulators. Cold Spring Harb. Perspect. Biol. 7, a008144 (2015).
Gutschner, T. & Diederichs, S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 9, 703–719 (2012).
Schmitt, A. M. & Chang, H. Y. Long noncoding RNAs in cancer pathways. Cancer Cell 29, 452–463 (2016).
Mehta, A. & Baltimore, D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 16, 279–294 (2016).
Amit, M. et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 578, 449–454 (2020).
Andersen, R. E. & Lim, D. A. Forging our understanding of lncRNAs in the brain. Cell Tissue Res. 371, 55–71 (2018).
Constantin, L. Circular RNAs and neuronal development. Adv. Exp. Med. Biol. 1087, 205–213 (2018).
Ling, H., Fabbri, M. & Calin, G. A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 12, 847–865 (2013).
Rupaimoole, R. & Slack, F. J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 16, 203–222 (2017).
van Rooij, E. & Olson, E. N. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat. Rev. Drug Discov. 11, 860–872 (2012).
Slack, F. J. & Chinnaiyan, A. M. The role of non-coding RNAs in oncology. Cell 179, 1033–1055 (2019).
Shah, M. Y., Ferrajoli, A., Sood, A. K., Lopez-Berestein, G. & Calin, G. A. MicroRNA therapeutics in cancer - an emerging concept. EBioMedicine 12, 34–42 (2016).
Calin, G. A. et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc. Natl Acad. Sci. USA 105, 5166–5171 (2008).
Cimmino, A. et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl Acad. Sci. USA 102, 13944–13949 (2005).
Arun, G., Diermeier, S. D. & Spector, D. L. Therapeutic targeting of long non-coding RNAs in cancer. Trends Mol. Med. 24, 257–277 (2018).
Modarresi, F. et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 30, 453–459 (2012).
Hsiao, J. et al. Upregulation of haploinsufficient gene expression in the brain by targeting a long non-coding RNA improves seizure phenotype in a model of dravet syndrome. EBioMedicine 9, 257–277 (2016).
Padmakumar, S. et al. Minimally invasive nasal depot (MIND) technique for direct BDNF AntagoNAT delivery to the brain. J. Control. Rel. 331, 176–186 (2021).
Moulder, S. L. et al. Phase I/II study of G3139 (Bcl-2 antisense oligonucleotide) in combination with doxorubicin and docetaxel in breast cancer. Clin. Cancer Res. 14, 7909–7916 (2008).
Croce, C. M. & Reed, J. C. Finally, an apoptosis-targeting therapeutic for cancer. Cancer Res. 76, 5914–5920 (2016).
Beg, M. S. et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. New Drugs 35, 180–188 (2017).
Hong, D. S. et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 122, 1630–1637 (2020).
van Zandwijk, N. et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 18, 1386–1396 (2017).
Gallant-Behm, C. L. et al. A microRNA-29 mimic (Remlarsen) represses extracellular matrix expression and fibroplasia in the skin. J. Invest. Dermatol. 139, 1073–1081 (2019).
Chakraborty, C., Sharma, A. R., Sharma, G., Bhattacharya, M. & Lee, S. S. SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): diagnostic and proposed therapeutic options. Eur. Rev. Med. Pharmacol. Sci. 24, 4016–4026 (2020).
Kumar, H., Kawai, T. & Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30, 16–34 (2011).
Sledz, C. A., Holko, M., de Veer, M. J., Silverman, R. H. & Williams, B. R. Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 5, 834–839 (2003).
Heidel, J. D., Hu, S., Liu, X. F., Triche, T. J. & Davis, M. E. Lack of interferon response in animalsto naked siRNAs. Nat. Biotechnol. 22, 1579–1582 (2004).
Kleinman, M. E. et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 452, 591–597 (2008).
Barton, G. M. & Medzhitov, R. Toll-like receptor signaling pathways. Science 300, 1524–1525 (2003).
Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529 (2004).
Sioud, M. Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J. Mol. Biol. 348, 1079–1090 (2005).
Hornung, V. et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 11, 263–270 (2005).
Judge, A. D. et al. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 23, 457–462 (2005).
Forsbach, A. et al. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol. 180, 3729–3738 (2008).
Sioud, M. Single-stranded small interfering RNA are more immunostimulatory than their double-stranded counterparts: a central role for 2′-hydroxyl uridines in immune responses. Eur. J. Immunol. 36, 1222–1230 (2006).
Fabbri, M. et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl Acad. Sci. USA 109, E2110–E2116 (2012).
Challagundla, K. B. et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J. Natl Cancer Inst. 107, djv135 (2015).
Lehmann, S. M. et al. An unconventional role for miRNA: let-7 activates toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 15, 827–835 (2012).
Dragomir, M. P., Knutsen, E. & Calin, G. A. SnapShot: unconventional miRNA functions. Cell 174, 1038–1038.e1 (2018).
Tudor, S. et al. Cellular and Kaposi’s sarcoma-associated herpes virus microRNAs in sepsis and surgical trauma. Cell. Death Dis. 5, e1559 (2014).
Li, Y. et al. Extraordinary GU-rich single-strand RNA identified from SARS coronavirus contributes an excessive innate immune response. Microbes Infect. 15, 88–95 (2013).
Morrissey, D. V. et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 23, 1002–1007 (2005).
Judge, A. D., Bola, G., Lee, A. C. & MacLachlan, I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol. Ther. 13, 494–505 (2006).
Marques, J. T. et al. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat. Biotechnol. 24, 559–565 (2006).
Chen, X. et al. 5′-triphosphate-siRNA activates RIG-I-dependent type I interferon production and enhances inhibition of hepatitis B virus replication in HepG2.2.15 cells. Eur. J. Pharmacol. 721, 86–95 (2013).
Liu, C. et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 17, 211–215 (2011).
Bader, A. G. miR-34 - a microRNA replacement therapy is headed to the clinic. Front. Genet. 3, 120 (2012).
Daige, C. L. et al. Systemic delivery of a miR34a mimic as a potential therapeutic for liver cancer. Mol. Cancer Ther. 13, 2352–2360 (2014).
Tolcher, A. W. et al. A phase 1 study of the BCL2-targeted deoxyribonucleic acid inhibitor (DNAi) PNT2258 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 73, 363–371 (2014).
Kelnar, K. & Bader, A. G. A qRT-PCR method for determining the biodistribution profile of a miR-34a mimic. Methods Mol. Biol. 1317, 125–133 (2015).
Wang, X. et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 27, 443–452 (2015).
Cirak, S. et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378, 595–605 (2011).
Mendell, J. R. et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 74, 637–647 (2013).
Charleston, J. S. et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology 90, e2146–e2154 (2018).
Artegiani, B. & Clevers, H. Use and application of 3D-organoid technology. Hum. Mol. Genet. 27, R99–R107 (2018).
Aparicio, S., Hidalgo, M. & Kung, A. L. Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer 15, 311–316 (2015).
He, J. et al. FRET biosensor-based kinase inhibitor screen for ERK and AKT activity reveals differential kinase dependencies for proliferation in TNBC cells. Biochem. Pharmacol. 169, 113640 (2019).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73 (2013).
Dalpke, A. & Helm, M. RNA mediated Toll-like receptor stimulation in health and disease. RNA Biol. 9, 828–842 (2012).
Obad, S. et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet. 43, 371–378 (2011).
Murphy, B. L. et al. Silencing of the miR-17~92 cluster family inhibits medulloblastoma progression. Cancer Res. 73, 7068–7078 (2013).
Bernardo, B. C. et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl Acad. Sci. USA 109, 17615–17620 (2012).
Simsek, C., Esin, E. & Yalcin, S. Metronomic chemotherapy: a systematic review of the literature and clinical experience. J. Oncol. 2019, 5483791 (2019).
Wildiers, H. et al. Pertuzumab and trastuzumab with or without metronomic chemotherapy for older patients with HER2-positive metastatic breast cancer (EORTC 75111-10114): an open-label, randomised, phase 2 trial from the Elderly Task Force/Breast Cancer Group. Lancet Oncol. 19, 323–336 (2018).
Romero, D. Breast cancer: metronomic chemotherapy for elderly and/or frail patients. Nat. Rev. Clin. Oncol. 15, 266 (2018).
Golan, T. et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 6, 24560–24570 (2015).
Cortez, M. A. et al. Role of miRNAs in immune responses and immunotherapy in cancer. Genes Chromosomes Cancer 58, 244–253 (2019).
Smolle, M. A., Calin, H. N., Pichler, M. & Calin, G. A. Noncoding RNAs and immune checkpoints-clinical implications as cancer therapeutics. FEBS J. 284, 1952–1966 (2017).
De Los Santos, M. C., Dragomir, M. P. & Calin, G. A. The role of exosomal long non-coding RNAs in cancer drug resistance. Cancer Drug Resist. 2, 1178–1192 (2019).
Whitehurst, A. W. et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 446, 815–819 (2007).
Van Roosbroeck, K. et al. Combining anti-Mir-155 with chemotherapy for the treatment of lung cancers. Clin. Cancer Res. 23, 2891–2904 (2017).
Zhao, J., Kelnar, K. & Bader, A. G. In-depth analysis shows synergy between erlotinib and miR-34a. PLoS ONE 9, e89105 (2014).
Cortez, M. A. et al. In vivo delivery of miR-34a sensitizes lung tumors to radiation through RAD51 regulation. Mol. Ther. Nucleic Acids 4, e270 (2015).
Leucci, E. Cancer development and therapy resistance: spotlights on the dark side of the genome. Pharmacol. Ther. 189, 22–30 (2018).
Jiang, W. et al. Long non-coding RNAs as a determinant of cancer drug resistance: towards the overcoming of chemoresistance via modulation of lncRNAs. Drug Resist. Updat. 50, 100683 (2020).
Deng, Y. et al. Randomized clinical trials towards a single-visit cure for chronic hepatitis C: oral GSK2878175 and injectable RG-101 in chronic hepatitis C patients and long-acting injectable GSK2878175 in healthy participants. J. Viral Hepat. 27, 699–708 (2020).
van der Ree, M. H. et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. Lancet 389, 709–717 (2017).
Zhao, G. & Zhang, H. Notch-1 siRNA and methotrexate towards a multifunctional approach in rhematoid arthritis management: a nanomedicine approach. Pharm. Res. 35, 123 (2018).
Wang, Y. et al. Endosomolytic and tumor-penetrating mesoporous silica nanoparticles for siRNA/miRNA combination cancer therapy. ACS Appl. Mater. Interfaces 12, 4308–4322 (2020).
Lu, C. et al. EphA2 overexpression promotes ovarian cancer growth. Cancer Biol. Ther. 7, 1098–1103 (2008).
Nishimura, M. et al. Therapeutic synergy between microRNA and siRNA in ovarian cancer treatment. Cancer Discov. 3, 1302–1315 (2013).
Song, Y., Tang, C. & Yin, C. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi-functionalized nanoparticles to tumor-associated macrophages and breast cancer cells. Biomaterials 185, 117–132 (2018).
Petrek, H., Batra, N., Ho, P. Y., Tu, M. J. & Yu, A. M. Bioengineering of a single long noncoding RNA molecule that carries multiple small RNAs. Appl. Microbiol. Biotechnol. 103, 6107–6117 (2019).
Chen, Q. X., Wang, W. P., Zeng, S., Urayama, S. & Yu, A. M. A general approach to high-yield biosynthesis of chimeric RNAs bearing various types of functional small RNAs for broad applications. Nucleic Acids Res. 43, 3857–3869 (2015).
Ho, P. Y. et al. Bioengineered noncoding RNAs selectively change cellular miRNome profiles for cancer therapy. J. Pharmacol. Exp. Ther. 365, 494–506 (2018).
Zhang, S., Chen, L., Jung, E. J. & Calin, G. A. Targeting microRNAs with small molecules: from dream to reality. Clin. Pharmacol. Ther. 87, 754–758 (2010).
Vo, D. D. et al. Targeting the production of oncogenic microRNAs with multimodal synthetic small molecules. ACS Chem. Biol. 9, 711–721 (2014).
Periwal, V. & Scaria, V. Machine learning approaches toward building predictive models for small molecule modulators of miRNA and its utility in virtual screening of molecular databases. Methods Mol. Biol. 1517, 155–168 (2017).
Wang, L. et al. Small-molecule inhibitors disrupt let-7 oligouridylation and release the selective blockade of let-7 processing by LIN28. Cell. Rep. 23, 3091–3101 (2018).
Fukuzumi, T., Murata, A., Aikawa, H., Harada, Y. & Nakatani, K. Exploratory study on the RNA-binding structural motifs by library screening targeting pre-miRNA-29a. Chemistry 21, 16859–16867 (2015).
Bose, D., Jayaraj, G. G., Kumar, S. & Maiti, S. A molecular-beacon-based screen for small molecule inhibitors of miRNA maturation. ACS Chem. Biol. 8, 930–938 (2013).
Chen, X., Guan, N. N., Sun, Y. Z., Li, J. Q. & Qu, J. MicroRNA-small molecule association identification: from experimental results to computational models. Brief. Bioinform. 21, 47–61 (2020).
Jiang, W. et al. Identification of links between small molecules and miRNAs in human cancers based on transcriptional responses. Sci. Rep. 2, 282 (2012).
Li, J. et al. Network-based identification of microRNAs as potential pharmacogenomic biomarkers for anticancer drugs. Oncotarget 7, 45584–45596 (2016).
Liu, X. et al. SM2miR: a database of the experimentally validated small molecules’ effects on microRNA expression. Bioinformatics 29, 409–411 (2013).
Disney, M. D. et al. Inforna 2.0: a platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol. 11, 1720–1728 (2016).
Chen, X., Xie, W. B., Xiao, P. P., Zhao, X. M. & Yan, H. mTD: a database of microRNAs affecting therapeutic effects of drugs. J. Genet. Genomics 44, 269–271 (2017).
Chen, X. et al. NRDTD: a database for clinically or experimentally supported non-coding RNAs and drug targets associations. Database https://doi.org/10.1093/database/bax057 (2017).
Su, E. C. et al. ChemiRs: a web application for microRNAs and chemicals. BMC Bioinform. 17, 167 (2016).
Staedel, C. et al. Modulation of oncogenic miRNA biogenesis using functionalized polyamines. Sci. Rep. 8, 1667 (2018).
Gumireddy, K. et al. Small-molecule inhibitors of microrna miR-21 function. Angew. Chem. Int. Ed. Engl. 47, 7482–7484 (2008).
Young, D. D., Connelly, C. M., Grohmann, C. & Deiters, A. Small molecule modifiers of microRNA miR-122 function for the treatment of hepatitis C virus infection and hepatocellular carcinoma. J. Am. Chem. Soc. 132, 7976–7981 (2010).
Chandrasekhar, S. et al. Aza-flavanones as potent cross-species microRNA inhibitors that arrest cell cycle. Bioorg. Med. Chem. Lett. 22, 645–648 (2012).
Velagapudi, S. P. et al. Design of a small molecule against an oncogenic noncoding RNA. Proc. Natl Acad. Sci. USA 113, 5898–5903 (2016).
Bautista-Sánchez, D. et al. The promising role of miR-21 as a cancer biomarker and its importance in RNA-based therapeutics. Mol. Ther. Nucleic Acids 20, 409–420 (2020).
Bose, D. et al. The tuberculosis drug streptomycin as a potential cancer therapeutic: inhibition of miR-21 function by directly targeting its precursor. Angew. Chem. Int. Ed. Engl. 51, 1019–1023 (2012).
Bose, D. et al. Selective inhibition of miR-21 by phage display screened peptide. Nucleic Acids Res. 43, 4342–4352 (2015).
Shi, Z. et al. AC1MMYR2, an inhibitor of dicer-mediated biogenesis of Oncomir miR-21, reverses epithelial-mesenchymal transition and suppresses tumor growth and progression. Cancer Res. 73, 5519–5531 (2013).
Murata, A., Otabe, T., Zhang, J. & Nakatani, K. BzDANP, a small-molecule modulator of pre-miR-29a maturation by dicer. ACS Chem. Biol. 11, 2790–2796 (2016).
Otabe, T., Nagano, K., Kawai, G., Murata, A. & Nakatani, K. Inhibition of pre-miRNA-136 processing by Dicer with small molecule BzDANP suggested the formation of ternary complex of pre-miR-136-BzDANP-Dicer. Bioorg. Med. Chem. 27, 2140–2148 (2019).
Kelly, T. J., Souza, A. L., Clish, C. B., & Puigserver, P. A Hypoxia-Induced Positive Feedback Loop Promotes Hypoxia-Inducible Factor 1α Stability through miR-210 Suppression of Glycerol-3-Phosphate Dehydrogenase 1-Like. Mol. Cell. Biol. 31, 2696–2706 (2011).
Kulshreshtha, R. et al. A MicroRNA Signature of Hypoxia. Mol. Cell. Biol. 27, 1859–1867 (2007).
Costales, M. G. et al. Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J. Am. Chem. Soc. 139, 3446–3455 (2017).
Monroig-Bosque, P. D. C. et al. OncomiR-10b hijacks the small molecule inhibitor linifanib in human cancers. Sci. Rep. 8, 13106 (2018).
Costales, M. G., Matsumoto, Y., Velagapudi, S. P. & Disney, M. D. Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc. 140, 6741–6744 (2018).
Costales, M. G. et al. Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl Acad. Sci. USA 117, 2406–2411 (2020).
Yan, H. & Liang, F. S. miRNA inhibition by proximity-enabled Dicer inactivation. Methods 167, 117–123 (2019).
Yan, H., Bhattarai, U., Song, Y. & Liang, F. S. Design, synthesis and activity of light deactivatable microRNA inhibitor. Bioorg. Chem. 80, 492–497 (2018).
Dong, C. G. et al. Co-inhibition of microRNA-10b and microRNA-21 exerts synergistic inhibition on the proliferation and invasion of human glioma cells. Int. J. Oncol. 41, 1005–1012 (2012).
Song, J. H. et al. Small-molecule-targeting hairpin loop of hTERT promoter G-quadruplex induces cancer cell death. Cell. Chem. Biol. 26, 1110–1121.e4 (2019).
Shi, Y. et al. Stabilization of lncRNA GAS5 by a small molecule and its implications in diabetic adipocytes. Cell. Chem. Biol. 26, 319–330.e6 (2019).
Donlic, A. et al. Discovery of small molecule ligands for MALAT1 by tuning an RNA-binding scaffold. Angew. Chem. Int. Ed. Engl. 57, 13242–13247 (2018).
Abulwerdi, F. A. et al. Selective small-molecule targeting of a triple helix encoded by the long noncoding RNA, MALAT1. ACS Chem. Biol. 14, 223–235 (2019).
Chen, Q., Zhu, C. & Jin, Y. The oncogenic and tumor suppressive functions of the long noncoding RNA MALAT1: an emerging controversy. Front. Genet. 11, 93 (2020).
Egli, M. & Manoharan, M. Re-engineering RNA molecules into therapeutic agents. Acc. Chem. Res. 52, 1036–1047 (2019).
Crooke, S. T., Seth, P. P., Vickers, T. A. & Liang, X. H. The interaction of phosphorothioate-containing RNA targeted drugs with proteins is a critical determinant of the therapeutic effects of these agents. J. Am. Chem. Soc. 142, 14754–14771 (2020).
Saxena, S., Jónsson, Z. O. & Dutta, A. Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J. Biol. Chem. 278, 44312–44319 (2003).
Jackson, A. L. et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12, 1179–1187 (2006).
Ui-Tei, K., Naito, Y., Nishi, K., Juni, A. & Saigo, K. Thermodynamic stability and Watson-Crick base pairing in the seed duplex are major determinants of the efficiency of the siRNA-based off-target effect. Nucleic Acids Res. 36, 7100–7109 (2008).
Elmén, J. et al. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 33, 439–447 (2005).
Mook, O. R., Baas, F., de Wissel, M. B. & Fluiter, K. Evaluation of locked nucleic acid-modified small interfering RNA in vitro and in vivo. Mol. Cancer Ther. 6, 833–843 (2007).
Wu, S. Y. et al. 2’-OMe-phosphorodithioate-modified siRNAs show increased loading into the RISC complex and enhanced anti-tumour activity. Nat. Commun. 5, 3459 (2014).
Chang, C. I. et al. Asymmetric shorter-duplex siRNA structures trigger efficient gene silencing with reduced nonspecific effects. Mol. Ther. 17, 725–732 (2009).
Bramsen, J. B. et al. Improved silencing properties using small internally segmented interfering RNAs. Nucleic Acids Res. 35, 5886–5897 (2007).
Mobergslien, A. & Sioud, M. Exploring 5′-biotinylation of the sense strand to improve siRNA specificity and potency. Methods Mol. Biol. 2115, 163–170 (2020).
Snead, N. M. et al. Molecular basis for improved gene silencing by Dicer substrate interfering RNA compared with other siRNA variants. Nucleic Acids Res. 41, 6209–6221 (2013).
Rudin, C. M. et al. Randomized phase II study of carboplatin and etoposide with or without the bcl-2 antisense oligonucleotide oblimersen for extensive-stage small-cell lung cancer: CALGB 30103. J. Clin. Oncol. 26, 870–876 (2008).
Sternberg, C. N. et al. Docetaxel plus oblimersen sodium (Bcl-2 antisense oligonucleotide): an EORTC multicenter, randomized phase II study in patients with castration-resistant prostate cancer. Ann. Oncol. 20, 1264–1269 (2009).
Anderson, E. M. et al. Gene profiling study of G3139- and Bcl-2-targeting siRNAs identifies a unique G3139 molecular signature. Cancer Gene Ther. 13, 406–414 (2006).
Winkler, J., Stessl, M., Amartey, J. & Noe, C. R. Off-target effects related to the phosphorothioate modification of nucleic acids. ChemMedChem 5, 1344–1352 (2010).
Lai, J. C. et al. A pharmacologic target of G3139 in melanoma cells may be the mitochondrial VDAC. Proc. Natl Acad. Sci. USA 103, 7494–7499 (2006).
Tan, W., Loke, Y. H., Stein, C. A., Miller, P. & Colombini, M. Phosphorothioate oligonucleotides block the VDAC channel. Biophys. J. 93, 1184–1191 (2007).
Pisano, M. et al. Targeting Bcl-2 protein in treatment of melanoma still requires further clarifications. Ann. Oncol. 19, 2092–2093 (2008).
Wen, J. & Friedman, J. R. miR-122 regulates hepatic lipid metabolism and tumor suppression. J. Clin. Invest. 122, 2773–2776 (2012).
Hand, N. J., Master, Z. R., Le Lay, J. & Friedman, J. R. Hepatic function is preserved in the absence of mature microRNAs. Hepatology 49, 618–626 (2009).
van der Ree, M. H. et al. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antivir. Res. 111, 53–59 (2014).
Hart, M. et al. miR-34a: a new player in the regulation of T cell function by modulation of NF-κB signaling. Cell Death Dis. 10, 46 (2019).
Xu, Y. et al. Macrophage miR-34a is a key regulator of cholesterol efflux and atherosclerosis. Mol. Ther. 28, 202–216 (2020).
Hart, M. et al. Wrinkle in the plan: miR-34a-5p impacts chemokine signaling by modulating CXCL10/CXCL11/CXCR3-axis in CD4(+), CD8(+) T cells, and M1 macrophages. J. Immunother. Cancer 8, e001617 (2020).
LaCasse, E. C. Pulling the plug on a cancer cell by eliminating XIAP with AEG35156. Cancer Lett. 332, 215–224 (2013).
Grimm, D. et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441, 537–541 (2006).
Khan, A. A. et al. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 27, 549–555 (2009).
Brennecke, J., Stark, A., Russell, R. B. & Cohen, S. M. Principles of microRNA-target recognition. PLoS Biol. 3, e85 (2005).
Krek, A. et al. Combinatorial microRNA target predictions. Nat. Genet. 37, 495–500 (2005).
Jin, H. Y. et al. Transfection of microRNA mimics should be used with caution. Front. Genet. 6, 340 (2015).
Nyayanit, D. & Gadgil, C. J. Mathematical modeling of combinatorial regulation suggests that apparent positive regulation of targets by miRNA could be an artifact resulting from competition for mRNA. RNA 21, 307–319 (2015).
Matthay, K. K. et al. Neuroblastoma. Nat. Rev. Dis. Prim. 2, 16078 (2016).
McBride, J. L. et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl Acad. Sci. USA 105, 5868–5873 (2008).
Martin, J. N. et al. Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther. 18, 666–673 (2011).
Rigoutsos, I. et al. N-BLR, a primate-specific non-coding transcript leads to colorectal cancer invasion and migration. Genome Biol. 18, 98 (2017).
Pichler, M. et al. Therapeutic potential of FLANC, a novel primate-specific long non-coding RNA in colorectal cancer. Gut 69, 1818–1831 (2020).
Dragomir, M. P., Kopetz, S., Ajani, J. A. & Calin, G. A. Non-coding RNAs in GI cancers: from cancer hallmarks to clinical utility. Gut 69, 748–763 (2020).
Awan, H. M., Shah, A., Rashid, F. & Shan, G. Primate-specific long non-coding RNAs and microRNAs. Genomics Proteom. Bioinform. 15, 187–195 (2017).
Jensen, K. K., Orum, H., Nielsen, P. E. & Nordén, B. Kinetics for hybridization of peptide nucleic acids (PNA) with DNA and RNA studied with the BIAcore technique. Biochemistry 36, 5072–5077 (1997).
Avitabile, C. et al. Targeting pre-miRNA by peptide nucleic acids: a new strategy to interfere in the miRNA maturation. Artif. DNA PNA XNA 3, 88–96 (2012).
Manicardi, A. et al. Cellular uptakes, biostabilities and anti-miR-210 activities of chiral arginine-PNAs in leukaemic K562 cells. Chembiochem 13, 1327–1337 (2012).
Avitabile, C., Fabbri, E., Bianchi, N., Gambari, R. & Romanelli, A. Inhibition of miRNA maturation by peptide nucleic acids. Methods Mol. Biol. 1095, 157–164 (2014).
Clancy, J. W., Zhang, Y., Sheehan, C. & D’Souza-Schorey, C. An ARF6-Exportin-5 axis delivers pre-miRNA cargo to tumour microvesicles. Nat. Cell Biol. 21, 856–866 (2019).
Abe, N., Abe, H. & Ito, Y. Dumbbell-shaped nanocircular RNAs for RNA interference. J. Am. Chem. Soc. 129, 15108–15109 (2007).
Abe, N. et al. Synthesis, structure, and biological activity of dumbbell-shaped nanocircular RNAs for RNA interference. Bioconjug. Chem. 22, 2082–2092 (2011).
Gnanamony, M. et al. Circular dumbbell miR-34a-3p and -5p suppresses pancreatic tumor cell-induced angiogenesis and activates macrophages. Oncol. Lett. 21, 75 (2021).
Zhang, L. et al. Circular siRNAs for reducing off-target effects and enhancing long-term gene silencing in cells and mice. Mol. Ther. Nucleic Acids 10, 237–244 (2018).
Hagiwara, K. et al. Development of prodrug type circular siRNA for in vivo knockdown by systemic administration. Nucleic Acid. Ther. 30, 346–364 (2020).
Griepenburg, J. C., Ruble, B. K. & Dmochowski, I. J. Caged oligonucleotides for bidirectional photomodulation of let-7 miRNA in zebrafish embryos. Bioorg. Med. Chem. 21, 6198–6204 (2013).
Kimura, Y. et al. Intracellular build-up RNAi with single-strand circular RNAs as siRNA precursors. Chem. Commun. 56, 466–469 (2020).
Hansen, T. B. et al. Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388 (2013).
Rybak-Wolf, A. et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 58, 870–885 (2015).
Kristensen, L. S. et al. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 20, 675–691 (2019).
Bachmayr-Heyda, A. et al. Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci. Rep. 5, 8057 (2015).
Chen, Y. G. et al. Sensing self and foreign circular RNAs by intron identity. Mol. Cell 67, 228–238.e5 (2017).
Chen, Y. G. et al. N6-methyladenosine modification controls circular RNA immunity. Mol. Cell 76, 96–109.e9 (2019).
Wesselhoeft, R. A. et al. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol. Cell 74, 508–520.e4 (2019).
Hollyman, D. et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J. Immunother. 32, 169–180 (2009).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Senzer, N. et al. Phase I trial of “bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol. Ther. 20, 679–686 (2012).
Oh, J. et al. Phase II study of Vigil® DNA engineered immunotherapy as maintenance in advanced stage ovarian cancer. Gynecol. Oncol. 143, 504–510 (2016).
Rao, D. D. et al. Bifunctional short hairpin RNA (bi-shRNA): design and pathway to clinical application. Methods Mol. Biol. 942, 259–278 (2013).
Ghisoli, M. et al. Three-year follow up of GMCSF/bi-shRNA(furin) DNA-transfected autologous tumor immunotherapy (Vigil) in metastatic advanced Ewing’s sarcoma. Mol. Ther. 24, 1478–1483 (2016).
Swarts, D. C. & Jinek, M. Cas9 versus Cas12a/Cpf1: Structure-function comparisons and implications for genome editing. Wiley Interdiscip. Rev. RNA https://doi.org/10.1002/wrna.1481 (2018).
Zhang, Y. et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 5, 16277 (2015).
Cox, D. B. T. et al. RNA editing with CRISPR-Cas13. Science 358, 1019–1027 (2017).
Zhang, Y. et al. Comprehensive off-target analysis of dCas9-SAM-mediated HIV reactivation via long noncoding RNA and mRNA profiling. BMC Med. Genomics 11, 78 (2018).
Shen, L. et al. Roles and potential applications of lncRNAs in HIV infection. Int. J. Infect. Dis. 92, 97–104 (2020).
Khosravi, M. A. et al. Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: a promising approach for gene therapy of beta thalassemia disease. Eur. J. Pharmacol. 854, 398–405 (2019).
Liu, J. et al. Long non-coding RNA-dependent mechanism to regulate heme biosynthesis and erythrocyte development. Nat. Commun. 9, 4386 (2018).
Chew, W. L. Immunity to CRISPR Cas9 and Cas12a therapeutics. Wiley Interdiscip. Rev. Syst. Biol. Med. https://doi.org/10.1002/wsbm.1408. (2018).
Baumann, V. & Winkler, J. miRNA-based therapies: strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med. Chem. 6, 1967–1984 (2014).
Dzierlega, K. & Yokota, T. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 27, 407–416 (2020).
Johannes, L. & Lucchino, M. Current challenges in delivery and cytosolic translocation of therapeutic RNAs. Nucleic Acid. Ther. 28, 178–193 (2018).
Dowdy, S. F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 35, 222–229 (2017).
Zelphati, O. & Szoka, F. C. Jr. Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl Acad. Sci. USA 93, 11493–11498 (1996).
Suhr, O. B. et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J. Rare Dis. 10, 109 (2015).
Zhou, Z., Kennell, C., Lee, J. Y., Leung, Y. K. & Tarapore, P. Calcium phosphate-polymer hybrid nanoparticles for enhanced triple negative breast cancer treatment via co-delivery of paclitaxel and miR-221/222 inhibitors. Nanomedicine 13, 403–410 (2017).
Gao, S. et al. miRNA oligonucleotide and sponge for miRNA-21 inhibition mediated by PEI-PLL in breast cancer therapy. Acta Biomater. 25, 184–193 (2015).
Chen, C. Y. et al. MicroRNA delivery mediated by PEGylated polyethylenimine for prostate cancer therapy. Open Chem. 16, 1257–1267 (2018).
Biray Avcı, Ç., Özcan, İ., Balcı, T., Özer, Ö. & Gündüz, C. Design of polyethylene glycol-polyethylenimine nanocomplexes as non-viral carriers: mir-150 delivery to chronic myeloid leukemia cells. Cell Biol. Int. 37, 1205–1214 (2013).
Arora, S. et al. Synthesis, characterization, and evaluation of poly (D,L-lactide-co-glycolide)-based nanoformulation of miRNA-150: potential implications for pancreatic cancer therapy. Int. J. Nanomed. 9, 2933–2942 (2014).
Cosco, D. et al. Delivery of miR-34a by chitosan/PLGA nanoplexes for the anticancer treatment of multiple myeloma. Sci. Rep. 5, 17579 (2015).
Ibrahim, A. F. et al. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 71, 5214–5224 (2011).
Liang, G. et al. Cationic microRNA-delivering nanocarriers for efficient treatment of colon carcinoma in xenograft model. Gene Ther. 23, 829–838 (2016).
Wang, S., Zhang, J., Wang, Y. & Chen, M. Hyaluronic acid-coated PEI-PLGA nanoparticles mediated co-delivery of doxorubicin and miR-542-3p for triple negative breast cancer therapy. Nanomedicine 12, 411–420 (2016).
Ramot, Y. et al. Preclinical safety evaluation in rats of a polymeric matrix containing an siRNA drug used as a local and prolonged delivery system for pancreatic cancer therapy. Toxicol. Pathol. 44, 856–865 (2016).
Zorde Khvalevsky, E. et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl Acad. Sci. USA 110, 20723–20728 (2013).
Østergaard, M. E. et al. Efficient synthesis and biological evaluation of 5’-GalNAc conjugated antisense oligonucleotides. Bioconjug. Chem. 26, 1451–1455 (2015).
Sundqvist, M. et al. Preclinical pharmacokinetics-pharmacodynamics modelling to guide first-time-in-human studies with the anti-miR-103/107, RG-125 (AZD4076). 51st EASD Annual Meeting. Diabetologia 58, 1–607 (2015).
Dugal-Tessier, J., Thirumalairajan, S. & Jain, N. Antibody-oligonucleotide conjugates: a twist to antibody-drug conjugates. J. Clin. Med. 10, 838 (2021).
Huggins, I. J. et al. Site selective antibody-oligonucleotide conjugation via microbial transglutaminase. Molecules 24, 3287 (2019).
Nanna, A. R. et al. Generation and validation of structurally defined antibody-siRNA conjugates. Nucleic Acids Res. 48, 5281–5293 (2020).
Sugo, T. et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Rel. 237, 1–13 (2016).
Cuellar, T. L. et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 43, 1189–1203 (2015).
Tsoumpra, M. K. et al. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine 45, 630–645 (2019).
Tone, Y. et al. Immortalized canine dystrophic myoblast cell lines for development of peptide-conjugated splice-switching oligonucleotides. Nucleic Acid Ther. 31, 172–181 (2021).
Dastpeyman, M. et al. Endosomal escape cell-penetrating peptides significantly enhance pharmacological effectiveness and CNS activity of systemically administered antisense oligonucleotides. Int. J. Pharm. 599, 120398 (2021).
Khoshnevisan, K. et al. The promising potentials of capped gold nanoparticles for drug delivery systems. J. Drug Target. 26, 525–532 (2018).
Jewell, C. M. et al. Oligonucleotide delivery by cell-penetrating “striped” nanoparticles. Angew. Chem. Int. Ed. Engl. 50, 12312–12315 (2011).
Jensen, S. A. et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci. Transl. Med. 5, 209ra152 (2013).
Kouri, F. M. et al. miR-182 integrates apoptosis, growth, and differentiation programs in glioblastoma. Genes Dev. 29, 732–745 (2015).
Grafals-Ruiz, N. et al. Brain targeted gold liposomes improve RNAi delivery for Glioblastoma. Int. J. Nanomed. 15, 2809–2828 (2020).
Kumthekar, P. et al. A first-in-human phase 0 clinical study of RNA interference-based spherical nucleic acids in patients with recurrent glioblastoma. Sci. Transl. Med. 13, eabb3945 (2021).
Samaranch, L. et al. MR-guided parenchymal delivery of adeno-associated viral vector serotype 5 in non-human primate brain. Gene Ther. 24, 253–261 (2017).
Keskin, S. et al. AAV5-miHTT lowers huntingtin mRNA and protein without off-target effects in patient-derived neuronal cultures and astrocytes. Mol. Ther. Methods Clin. Dev. 15, 275–284 (2019).
Qiu, Y., Lam, J. K., Leung, S. W. & Liang, W. Delivery of RNAi therapeutics to the airways-from bench to bedside. Molecules 21, 1249 (2016).
Moschos, S. A., Usher, L. & Lindsay, M. A. Clinical potential of oligonucleotide-based therapeutics in the respiratory system. Pharmacol. Ther. 169, 83–103 (2017).
Bayraktar, R., Van Roosbroeck, K. & Calin, G. A. Cell-to-cell communication: microRNAs as hormones. Mol. Oncol. 11, 1673–1686 (2017).
El-Daly, S. M., Bayraktar, R., Anfossi, S. & Calin, G. A. The interplay between microRNAs and the components of the tumor microenvironment in B-cell malignancies. Int. J. Mol. Sci. 21, 3387 (2020).
Zhou, Y. et al. Exosome-mediated small RNA delivery for gene therapy. Wiley Interdiscip. Rev. RNA 7, 758–771 (2016).
McKelvey, K. J., Powell, K. L., Ashton, A. W., Morris, J. M. & McCracken, S. A. Exosomes: mechanisms of uptake. J. Circ. Biomark. 4, 7 (2015).
Kamerkar, S. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546, 498–503 (2017).
Jung, K. O. et al. Identification of lymphatic and hematogenous routes of rapidly labeled radioactive and fluorescent exosomes through highly sensitive multimodal imaging. Int. J. Mol. Sci. 21, 7850 (2020).
Wu, A. Y. et al. Multiresolution imaging using bioluminescence resonance energy transfer identifies distinct biodistribution profiles of extracellular vesicles and exomeres with redirected tropism. Adv. Sci. 7, 2001467 (2020).
Gupta, D. et al. Quantification of extracellular vesicles in vitro and in vivo using sensitive bioluminescence imaging. J. Extracell. Vesicles 9, 1800222 (2020).
Xu, L. et al. Design of experiment (DoE)-driven in vitro and in vivo uptake studies of exosomes for pancreatic cancer delivery enabled by copper-free click chemistry-based labelling. J. Extracell. Vesicles 9, 1779458 (2020).
Alvarez-Erviti, L. et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345 (2011).
Naseri, Z., Oskuee, R. K., Forouzandeh-Moghadam, M. & Jaafari, M. R. Delivery of LNA-antimiR-142-3p by mesenchymal stem cells-derived exosomes to breast cancer stem cells reduces tumorigenicity. Stem Cell. Rev. Rep. 16, 541–556 (2020).
Liang, G. et al. Engineered exosomes for targeted co-delivery of miR-21 inhibitor and chemotherapeutics to reverse drug resistance in colon cancer. J. Nanobiotechnol. 18, 10 (2020).
Thone, M. N. & Kwon, Y. J. Extracellular blebs: artificially-induced extracellular vesicles for facile production and clinical translation. Methods 177, 135–145 (2020).
Munagala, R., Aqil, F., Jeyabalan, J. & Gupta, R. C. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 371, 48–61 (2016).
Ju, S. et al. Grape exosome-like nanoparticles induce intestinal stem cells and protect mice from DSS-induced colitis. Mol. Ther. 21, 1345–1357 (2013).
Wang, Q. et al. Grapefruit-derived nanovectors use an activated leukocyte trafficking pathway to deliver therapeutic agents to inflammatory tumor sites. Cancer Res. 75, 2520–2529 (2015).
Phan, J. et al. Engineering mesenchymal stem cells to improve their exosome efficacy and yield for cell-free therapy. J. Extracell. Vesicles 7, 1522236 (2018).
Pan, Y. et al. Development of a microRNA delivery system based on bacteriophage MS2 virus-like particles. FEBS J. 279, 1198–1208 (2012).
Sun, Y., Sun, Y. & Zhao, R. Establishment of microRNA delivery system by PP7 bacteriophage-like particles carrying cell-penetrating peptide. J. Biosci. Bioeng. 124, 242–249 (2017).
Farkas, M. E. et al. PET Imaging and biodistribution of chemically modified bacteriophage MS2. Mol. Pharm. 10, 69–76 (2013).
Lin, Y. W. et al. Pharmacokinetics/pharmacodynamics of antipseudomonal bacteriophage therapy in rats: a proof-of-concept study. Clin. Microbiol. Infect. 26, 1229–1235 (2020).
Otero, J. et al. Biodistribution of liposome-encapsulated bacteriophages and their transcytosis during oral phage therapy. Front. Microbiol. 10, 689 (2019).
Giacalone, M. J. et al. The use of bacterial minicells to transfer plasmid DNA to eukaryotic cells. Cell. Microbiol. 8, 1624–1633 (2006).
Reid, G. et al. Restoring expression of miR-16: a novel approach to therapy for malignant pleural mesothelioma. Ann. Oncol. 24, 3128–3135 (2013).
van Zandwijk, N. et al. P1.02 - MesomiR 1: a phase I study of TargomiRs in patients with refractory malignant pleural mesothelioma (MPM) and lung cancer (NSCLC). Ann. Oncol. 26, ii16 (2015).
Varkouhi, A. K., Scholte, M., Storm, G. & Haisma, H. J. Endosomal escape pathways for delivery of biologicals. J. Control. Rel. 151, 220–228 (2011).
MacDiarmid, J. A. et al. Targeted doxorubicin delivery to brain tumors via minicells: proof of principle using dogs with spontaneously occurring tumors as a model. PLoS ONE 11, e0151832 (2016).
Jivrajani, M. & Nivsarkar, M. Ligand-targeted bacterial minicells: Futuristic nano-sized drug delivery system for the efficient and cost effective delivery of shRNA to cancer cells. Nanomedicine 12, 2485–2498 (2016).
Kopechek, J. A. et al. Ultrasound and microbubble-targeted delivery of a microRNA inhibitor to the heart suppresses cardiac hypertrophy and preserves cardiac function. Theranostics 9, 7088–7098 (2019).
Burnett, J. C. & Rossi, J. J. RNA-based therapeutics: current progress and future prospects. Chem. Biol. 19, 60–71 (2012).
Fizazi, K. et al. Final overall survival (OS) from the AFFINITY phase 3 trial of custirsen and cabazitaxel/prednisone in men with previously treated metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol. 27, 1–36 (2016).
Warren, M. S., Hughes, S. G., Singleton, W., Yamashita, M. & Genovese, M. C. Results of a proof of concept, double-blind, randomized trial of a second generation antisense oligonucleotide targeting high-sensitivity C-reactive protein (hs-CRP) in rheumatoid arthritis. Arthritis Res. Ther. 17, 80 (2015).
Nguyen, Q. D. et al. Dose-ranging evaluation of intravitreal siRNA PF-04523655 for diabetic macular edema (the DEGAS study). Invest. Ophthalmol. Vis. Sci. 53, 7666–7674 (2012).
ISIS Pharmaceuticals. ISIS 104838 discontinuation. 10-K (2005).
Oza, A. M. et al. Phase II study of CGP 69846A (ISIS 5132) in recurrent epithelial ovarian cancer: an NCIC clinical trials group study (NCIC IND.116). Gynecol. Oncol. 89, 129–133 (2003).
Paz-Ares, L. et al. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 24, 1428–1434 (2006).
Hutvágner, G. et al. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293, 834–838 (2001).
Lee, Y. et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415–419 (2003).
Yi, R., Qin, Y., Macara, I. G. & Cullen, B. R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 17, 3011–3016 (2003).
Gregory, R. I. et al. The microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240 (2004).
Forman, J. J., Legesse-Miller, A. & Coller, H. A. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc. Natl Acad. Sci. USA 105, 14879–14884 (2008).
Tay, Y., Zhang, J., Thomson, A. M., Lim, B. & Rigoutsos, I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455, 1124–1128 (2008).
Cabili, M. N. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927 (2011).
Fabbri, M., Girnita, L., Varani, G. & Calin, G. A. Decrypting noncoding RNA interactions, structures, and functional networks. Genome Res. 29, 1377–1388 (2019).
Rinn, J. L. & Chang, H. Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166 (2012).
Guttman, M. & Rinn, J. L. Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346 (2012).
Dragomir, M. & Calin, G. A. Circular RNAs in cancer- lessons learned from microRNAs. Front. Oncol. 8, 179 (2018).
Khvorova, A. & Watts, J. K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 35, 238–248 (2017).
Ochoa, S. & Milam, V. T. Modified nucleic acids: expanding the capabilities of functional oligonucleotides. Molecules 25, 4659 (2020).
Crooke, S. T. Molecular mechanisms of antisense oligonucleotides. Nucleic Acid. Ther. 27, 70–77 (2017).
Singh, N. N., Luo, D. & Singh, R. N. Pre-mRNA splicing modulation by antisense oligonucleotides. Methods Mol. Biol. 1828, 415–437 (2018).
Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001).
Rao, D. D. et al. Preclinical justification of pbi-shRNA EWS/FLI1 Lipoplex (LPX) treatment for Ewing’s sarcoma. Mol. Ther. 24, 1412–1422 (2016).
Wang, Z. et al. Preclinical biodistribution and safety evaluation of a pbi-shRNA STMN1 Lipoplex after subcutaneous delivery. Toxicol. Sci. 155, 400–408 (2017).
van Rooij, E. & Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 6, 851–864 (2014).
Krützfeldt, J. et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438, 685–689 (2005).
Gebert, L. F. et al. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 42, 609–621 (2014).
Ebert, M. S., Neilson, J. R. & Sharp, P. A. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 4, 721–726 (2007).
Lima, J. F., Cerqueira, L., Figueiredo, C., Oliveira, C. & Azevedo, N. F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 15, 338–352 (2018).
Kluiver, J. et al. Generation of miRNA sponge constructs. Methods 58, 113–117 (2012).
Chang, S. Construction of multi-potent microRNA sponge and its functional evaluation. Methods Mol. Biol. 1699, 201–209 (2018).
Jung, J. et al. Simultaneous inhibition of multiple oncogenic miRNAs by a multi-potent microRNA sponge. Oncotarget 6, 20370–20387 (2015).
Das, S. et al. Divergent effects of miR-181 family members on myocardial function through protective cytosolic and detrimental mitochondrial microRNA targets. J. Am. Heart Assoc. 6, e004694 (2017).
Bernardo, B. C., Gregorevic, P., Ritchie, R. H. & McMullen, J. R. Generation of microRNA-34 sponges and tough decoys for the heart: developments and challenges. Front. Pharmacol. 9, 1090 (2018).
Wang, Z. The principles of MiRNA-masking antisense oligonucleotides technology. Methods Mol. Biol. 676, 43–49 (2011).
Murakami, K. & Miyagishi, M. Tiny masking locked nucleic acids effectively bind to mRNA and inhibit binding of microRNAs in relation to thermodynamic stability. Biomed. Rep. 2, 509–512 (2014).
Gilot, D. et al. A non-coding function of TYRP1 mRNA promotes melanoma growth. Nat. Cell Biol. 19, 1348–1357 (2017).
Girardi, E., López, P. & Pfeffer, S. On the importance of host microRNAs during viral infection. Front. Genet. 9, 439 (2018).
Trobaugh, D. W. & Klimstra, W. B. MicroRNA regulation of RNA virus replication and pathogenesis. Trends Mol. Med. 23, 80–93 (2017).
Esau, C. et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell. Metab. 3, 87–98 (2006).
Roberts, A. P., Lewis, A. P. & Jopling, C. L. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 39, 7716–7729 (2011).
Sedano, C. D. & Sarnow, P. Hepatitis C virus subverts liver-specific miR-122 to protect the viral genome from exoribonuclease Xrn2. Cell. Host Microbe 16, 257–264 (2014).
Chahal, J. et al. miR-122 and Ago interactions with the HCV genome alter the structure of the viral 5’ terminus. Nucleic Acids Res. 47, 5307–5324 (2019).
Amador-Cañizares, Y., Bernier, A., Wilson, J. A. & Sagan, S. M. miR-122 does not impact recognition of the HCV genome by innate sensors of RNA but rather protects the 5′ end from the cellular pyrophosphatases, DOM3Z and DUSP11. Nucleic Acids Res. 46, 5139–5158 (2018).
Scheel, T. K. et al. A broad RNA virus survey reveals both miRNA dependence and functional sequestration. Cell. Host Microbe 19, 409–423 (2016).
Trobaugh, D. W. et al. Cooperativity between the 3’ untranslated region microRNA binding sites is critical for the virulence of eastern equine encephalitis virus. PLoS Pathog. 15, e1007867 (2019).
Zheng, H. et al. MicroRNA-221-5p inhibits porcine epidemic diarrhea virus replication by targeting genomic viral RNA and activating the NF-κB pathway. Int. J. Mol. Sci. 19, 3381 (2018).
Lee, Y. R. et al. Honeysuckle aqueous extracts induced let-7a suppress EV71 replication and pathogenesis in vitro and in vivo and is predicted to inhibit SARS-CoV-2. Viruses 13, 308 (2021).
Lai, F. W., Stephenson, K. B., Mahony, J. & Lichty, B. D. Human coronavirus OC43 nucleocapsid protein binds microRNA 9 and potentiates NF-κB activation. J. Virol. 88, 54–65 (2014).
Vitravene Study Group. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am. J. Ophthalmol. 133, 467–474 (2002).
Stein, C. A. & Castanotto, D. FDA-approved oligonucleotide therapies in 2017. Mol. Ther. 25, 1069–1075 (2017).
Stelma, F. et al. Immune phenotype and function of natural killer and T cells in chronic hepatitis C patients who received a single dose of anti-MicroRNA-122, RG-101. Hepatology 66, 57–68 (2017).
Janssen, H. L. et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 368, 1685–1694 (2013).
Li, Y. P., Gottwein, J. M., Scheel, T. K., Jensen, T. B. & Bukh, J. MicroRNA-122 antagonism against hepatitis C virus genotypes 1-6 and reduced efficacy by host RNA insertion or mutations in the HCV 5’ UTR. Proc. Natl Acad. Sci. USA 108, 4991–4996 (2011).
Li, Y. P., Van Pham, L., Uzcategui, N. & Bukh, J. Functional analysis of microRNA-122 binding sequences of hepatitis C virus and identification of variants with high resistance against a specific antagomir. J. Gen. Virol. 97, 1381–1394 (2016).
Ottosen, S. et al. In vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob. Agents Chemother. 59, 599–608 (2015).
Hum, C. et al. MicroRNA mimics or inhibitors as antiviral therapeutic approaches against COVID-19. Drugs 81, 517–531 (2021).
Kheirkhah, A. H. et al. Mesenchymal stem cell derived-exosomes as effective factors in reducing cytokine storm symptoms of COVID-19. Protein Pept. Lett. https://doi.org/10.2174/0929866528666210222150347 (2021).
Schultz, I. C., Bertoni, A. P. S. & Wink, M. R. Mesenchymal stem cell-derived extracellular vesicles carrying miRNA as a potential multi target therapy to COVID-19: an in silico analysis. Stem Cell. Rev. Rep. 17, 341–356 (2021).
Guterres, A., de Azeredo Lima, C. H., Miranda, R. L. & Gadelha, M. R. What is the potential function of microRNAs as biomarkers and therapeutic targets in COVID-19? Infect. Genet. Evol. 85, 104417 (2020).
Saçar Demirci, M. D. & Adan, A. Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. PeerJ 8, e9369 (2020).
Khan, M. A., Sany, M. R. U., Islam, M. S. & Islam, A. B. M. M. K. Epigenetic regulator miRNA pattern differences among SARS-CoV, SARS-CoV-2, and SARS-CoV-2 world-wide isolates delineated the mystery behind the epic pathogenicity and distinct clinical characteristics of pandemic COVID-19. Front. Genet. 11, 765 (2020).
Rohani, N., Ahmadi Moughari, F. & Eslahchi, C. DisCoVering potential candidates of RNAi-based therapy for COVID-19 using computational methods. PeerJ 9, e10505 (2021).
Natarelli, L. et al. MicroRNAs and long non-coding RNAs as potential candidates to target specific motifs of SARS-CoV-2. Noncoding RNA 7, 14 (2021).
Bartoszewski, R. et al. SARS-CoV-2 may regulate cellular responses through depletion of specific host miRNAs. Am. J. Physiol. Lung Cell. Mol. Physiol. 319, L444–L455 (2020).
Mohammadi-Dehcheshmeh, M. et al. A transcription regulatory sequence in the 5’ untranslated region of SARS-CoV-2 is vital for virus replication with an altered evolutionary pattern against human inhibitory microRNAs. Cells 10, 319 (2021).
Satyam, R. et al. miRNAs in SARS-CoV 2: a spoke in the wheel of pathogenesis. Curr. Pharm. Des. https://doi.org/10.2174/1381612826999201001200529 (2020).
Merino, G. A. et al. Novel SARS-CoV-2 encoded small RNAs in the passage to humans. Bioinformatics 36, 5571–5581 (2020).
Çetin, Z., Bayrak, T., Oğul, H., Saygılı, E. İ. & Akkol, E. K. Predicted SARS-CoV-2 miRNAs associated with epigenetic viral pathogenesis and the detection of new possible drugs for Covid-19. Curr. Drug Deliv. https://doi.org/10.2174/1567201818666210301102 (2021).
Lu, D. et al. MicroRNAs targeting the SARS-CoV-2 entry receptor ACE2 in cardiomyocytes. J. Mol. Cell. Cardiol. 148, 46–49 (2020).
Mone, P. et al. miR-24 targets SARS-CoV-2 co-factor Neuropilin-1 in human brain microvascular endothelial cells: insights for COVID-19 neurological manifestations. Res. Sq. https://doi.org/10.21203/rs.3.rs-192099/v1 (2021).
Centa, A. et al. Deregulated miRNA expression is associated with endothelial dysfunction in post-mortem lung biopsies of COVID-19 patients. Am. J. Physiol. Lung Cell. Mol. Physiol. 320, L405–L412 (2020).
Vishnubalaji, R., Shaath, H. & Alajez, N. M. Protein coding and long noncoding RNA (lncRNA) transcriptional landscape in SARS-CoV-2 infected bronchial epithelial cells highlight a role for interferon and inflammatory response. Genes 11, 760 (2020).
Laha, S. et al. In silico analysis of altered expression of long non-coding RNA in SARS-CoV-2 infected cells and their possible regulation by STAT1, STAT3 and interferon regulatory factors. Heliyon 7, e06395 (2021).
Mallick, B., Ghosh, Z. & Chakrabarti, J. MicroRNome analysis unravels the molecular basis of SARS infection in bronchoalveolar stem cells. PLoS ONE 4, e7837 (2009).
Wei, L., Li, J., Han, Z., Chen, Z. & Zhang, Q. Silencing of lncRNA MALAT1 prevents inflammatory injury after lung transplant ischemia-reperfusion by downregulation of IL-8 via p300. Mol. Ther. Nucleic Acids 18, 285–297 (2019).
Arora, S., Singh, P., Dohare, R., Jha, R. & Ali Syed, M. Unravelling host-pathogen interactions: ceRNA network in SARS-CoV-2 infection (COVID-19). Gene 762, 145057 (2020).
Turjya, R. R., Khan, M. A. & Islam, A. B. M. M. K. Perversely expressed long noncoding RNAs can alter host response and viral proliferation in SARS-CoV-2 infection. Future Virol. 15, 577–593 (2020).
Hadjicharalambous, M. R. et al. Long non-coding RNAs are central regulators of the IL-1β-induced inflammatory response in normal and idiopathic pulmonary lung fibroblasts. Front. Immunol. 9, 2906 (2018).
Acknowledgements
G.A.C. is the Felix L. Haas Endowed Professor in Basic Science. Work in G.A.C.’s laboratory is supported by National Institutes of Health (NIH/NCATS) grant UH3TR00943-01 through the NIH Common Fund, Office of Strategic Coordination (OSC), the NCI grants 1R01 CA182905-01 and 1R01CA222007-01A1, an NIGMS 1R01GM122775-01 grant, a Team DOD (CA160445P1) grant, a Chronic Lymphocytic Leukaemia Moonshot Flagship project, a CLL Global Research Foundation 2019 grant, a CLL Global Research Foundation 2020 grant, donor support through J. Ajani and the Estate of C.G. Johnson, Jr. S.M.E. was supported by a Fulbright foundation fellowship and is currently supported by the Science and Technology Development Fund (STDF, grant #26388). Work in M.F.’s laboratory is supported by the National Institutes of Health (NIH/NCI) grant 5P30CA071789-20 and by the NIH/NCI grants R01CA215753 and R01CA219024.
Author information
Authors and Affiliations
Contributions
All authors contributed to the development of presented concepts and writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
G.A.C. is one of the scientific founders of Ithax Pharmaceuticals. The other authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related Links
Dicerna Prioritizes Resources to Advance GalXC Product Candidates: https://www.itnonline.com/content/dicerna-prioritizes-resources-advance-galxc-product-candidates
miRagen decides to discontinue further internal development of Cobomarsen: http://investors.miragen.com/press-releases/press-release/2020/miRagen-Announces-Internal-Review-of-Preliminary-Topline-Data-for-the-Phase-2-SOLAR-Clinical-Trial-of-Cobomarsen-in-Patients-with-Cutaneous-T-Cell-Lymphoma-CTCL/default.aspx
PRO-040201: https://www.creative-biolabs.com/gene-therapy/pro040201.htm
Regulus to discontinue clinical development of HCV candidate RG-101: https://www.pharmaceutical-business-review.com/clinical-trials/news/regulus-to-terminate-development-of-hcv-candidate-rg-101-130617-5841251
Trial Termination Aganirsen: http://strong-nvg.com/trial-termination/
Wave Life Sciences Discontinues Development of Suvodirsen for DMD: https://musculardystrophynews.com/2019/12/17/wave-life-sciences-discontinues-suvodirsen-development-for-dmd/
Rights and permissions
About this article

Cite this article
Winkle, M., El-Daly, S.M., Fabbri, M. et al. Noncoding RNA therapeutics — challenges and potential solutions. Nat Rev Drug Discov 20, 629–651 (2021). https://doi.org/10.1038/s41573-021-00219-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-021-00219-z
This article is cited by
-
Tumorigenesis of basal muscle invasive bladder cancer was mediated by PTEN protein degradation resulting from SNHG1 upregulation
Journal of Experimental & Clinical Cancer Research (2024)
-
KIT mutations and expression: current knowledge and new insights for overcoming IM resistance in GIST
Cell Communication and Signaling (2024)
-
Non-coding RNA in the gut of the blood-feeding parasitic worm, Haemonchus contortus
Veterinary Research (2024)
-
EIF4A3-mediated biogenesis of circSTX6 promotes bladder cancer metastasis and cisplatin resistance
Journal of Experimental & Clinical Cancer Research (2024)
-
4D label-free proteomics analysis of oxygen-induced retinopathy with or without anti-VEGF treatment
BMC Genomics (2024)
