
Abstract
The Hippo pathway is an evolutionarily conserved signalling pathway with key roles in organ development, epithelial homeostasis, tissue regeneration, wound healing and immune modulation. Many of these roles are mediated by the transcriptional effectors YAP and TAZ, which direct gene expression via control of the TEAD family of transcription factors. Dysregulated Hippo pathway and YAP/TAZ–TEAD activity is associated with various diseases, most notably cancer, making this pathway an attractive target for therapeutic intervention. This Review highlights the key findings from studies of Hippo pathway signalling across biological processes and diseases, and discusses new strategies and therapeutic implications of targeting this pathway.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
References
Jia, J., Zhang, W., Wang, B., Trinko, R. & Jiang, J. The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev. 17, 2514–2519 (2003).
Justice, R. W., Zilian, O., Woods, D. F., Noll, M. & Bryant, P. J. The Drosophila tumor suppressor gene Warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 9, 534–546 (1995).
Kango-Singh, M. et al. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development 129, 5719–5730 (2002).
Pantalacci, S., Tapon, N. & Leopold, P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 5, 921–927 (2003).
Tapon, N. et al. Salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 110, 467–478 (2002).
Udan, R. S., Kango-Singh, M., Nolo, R., Tao, C. & Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 5, 914–920 (2003).
Wu, S., Huang, J., Dong, J. & Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with Salvador and Warts. Cell 114, 445–456 (2003).
Xu, T., Wang, W., Zhang, S., Stewart, R. A. & Yu, W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 121, 1053–1063 (1995).
Meng, Z., Moroishi, T. & Guan, K. L. Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17 (2016).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Zanconato, F., Cordenonsi, M. & Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 29, 783–803 (2016).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10 (2018).
Fan, R., Kim, N. G. & Gumbiner, B. M. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl Acad. Sci. USA 110, 2569–2574 (2013).
Garcia-Escudero, R. et al. Overexpression of PIK3CA in head and neck squamous cell carcinoma is associated with poor outcome and activation of the YAP pathway. Oral. Oncol. 79, 55–63 (2018).
Lamar, J. M. et al. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl Acad. Sci. USA 109, E2441–2450 (2012).
Lee, C. K. et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 363, 644–649 (2019).
Yang, C. S. et al. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. 19, e43577 (2018).
Bianchi, A. B. et al. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc. Natl Acad. Sci. USA 92, 10854–10858 (1995).
Murakami, H. et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res. 71, 873–883 (2011).
Feng, X. et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 25, 831–845 (2014).
Yu, F. X. et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 25, 822–830 (2014).
Tanas, M. R. et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci. Transl. Med. 3, 98ra82 (2011).
Tanas, M. R. et al. Mechanism of action of a WWTR1(TAZ)–CAMTA1 fusion oncoprotein. Oncogene 35, 929–938 (2016).
Antonescu, C. R. et al. Novel YAP1–TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 52, 775–784 (2013).
Dhanasekaran, S. M. et al. Transcriptome meta-analysis of lung cancer reveals recurrent aberrations in NRG1 and Hippo pathway genes. Nat. Commun. 5, 5893 (2014).
Gujral, T. S. & Kirschner, M. W. Hippo pathway mediates resistance to cytotoxic drugs. Proc. Natl Acad. Sci. USA 114, E3729–E3738 (2017).
Ghiso, E. et al. YAP-dependent AXL overexpression mediates resistance to EGFR inhibitors in NSCLC. Neoplasia 19, 1012–1021 (2017).
Lee, J. E. et al. Hippo pathway effector YAP inhibition restores the sensitivity of EGFR-TKI in lung adenocarcinoma having primary or acquired EGFR-TKI resistance. Biochem. Biophys. Res. Commun. 474, 154–160 (2016).
Kapoor, A. et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 179, 1239 (2019).
Shao, D. D. et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184 (2014).
Lin, L. et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 47, 250–256 (2015).
Wilson, F. H. et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell 27, 397–408 (2015).
Nguyen, C. D. K. & Yi, C. YAP/TAZ signaling and resistance to cancer therapy. Trends Cancer 5, 283–296 (2019).
Calses, P. C., Crawford, J. J., Lill, J. R. & Dey, A. Hippo pathway in cancer: aberrant regulation and therapeutic opportunities. Trends Cancer 5, 297–307 (2019).
Nehme, N. T. et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 119, 3458–3468 (2012).
Abdollahpour, H. et al. The phenotype of human STK4 deficiency. Blood 119, 3450–3457 (2012).
Du, X. et al. Mst1/Mst2 regulate development and function of regulatory T cells through modulation of Foxo1/Foxo3 stability in autoimmune disease. J. Immunol. 192, 1525–1535 (2014).
Tang, F. et al. The kinases NDR1/2 act downstream of the Hippo homolog MST1 to mediate both egress of thymocytes from the thymus and lymphocyte motility. Sci. Signal. 8, ra100 (2015).
Dang, T. S. et al. Defective leukocyte adhesion and chemotaxis contributes to combined immunodeficiency in humans with autosomal recessive MST1 deficiency. J. Clin. Immunol. 36, 117–122 (2016).
Xu, X. et al. Mst1 kinase regulates the actin-bundling protein L-plastin to promote T cell migration. J. Immunol. 197, 1683–1691 (2016).
Du, X. et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α+ dendritic cells. Nature 558, 141–145 (2018).
Li, C. et al. Dendritic cell MST1 inhibits TH17 differentiation. Nat. Commun. 8, 14275 (2017).
Katagiri, K., Imamura, M. & Kinashi, T. Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nat. Immunol. 7, 919–928 (2006).
Zhou, D. et al. The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naive T cells. Proc. Natl Acad. Sci. USA 105, 20321–20326 (2008).
Mou, F. et al. The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. J. Exp. Med. 209, 741–759 (2012).
Geng, J. et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat. Immunol. 18, 800–812 (2017).
Stampouloglou, E. et al. Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol. 18, e3000591 (2020).
Ni, X. et al. YAP is essential for Treg-mediated suppression of antitumor immunity. Cancer Discov. 8, 1026–1043 (2018).
Thaventhiran, J. E. et al. Activation of the Hippo pathway by CTLA-4 regulates the expression of Blimp-1 in the CD8+ T cell. Proc. Natl Acad. Sci. USA 109, E2223–E2229 (2012).
Wang, S. et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKε-mediated phosphorylation. Nat. Immunol. 18, 733–743 (2017).
Zhang, Q. et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol. 19, 362–374 (2017).
Guo, X. et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 31, 247–259 (2017).
Wang, G. et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 6, 80–95 (2016).
Lee, B. S. et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem. Biophys. Res. Commun. 491, 493–499 (2017).
Janse van Rensburg, H. J. et al. The Hippo pathway component TAZ promotes immune evasion in human cancer through PD-L1. Cancer Res. 78, 1457–1470 (2018).
Kim, M. H. et al. YAP-induced PD-L1 expression drives immune evasion in BRAFi-resistant melanoma. Cancer Immunol. Res. 6, 255–266 (2018).
Feng, J. et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 36, 5829–5839 (2017).
Miao, J. et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 8, 114576–114587 (2017).
Ramjee, V. et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J. Clin. Invest. 127, 899–911 (2017).
Moroishi, T. et al. The Hippo pathway kinases LATS1/2 suppress cancer immunity. Cell 167, 1525–1539.e17 (2016).
Karpowicz, P., Perez, J. & Perrimon, N. The Hippo tumor suppressor pathway regulates intestinal stem cell regeneration. Development 137, 4135–4145 (2010).
Shaw, R. L. et al. The Hippo pathway regulates intestinal stem cell proliferation during Drosophila adult midgut regeneration. Development 137, 4147–4158 (2010).
Staley, B. K. & Irvine, K. D. Warts and Yorkie mediate intestinal regeneration by influencing stem cell proliferation. Curr. Biol. 20, 1580–1587 (2010).
Cai, J. et al. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 24, 2383–2388 (2010).
Barry, E. R. et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 493, 106–110 (2013).
Gregorieff, A., Liu, Y., Inanlou, M. R., Khomchuk, Y. & Wrana, J. L. Yap-dependent reprogramming of Lgr5+ stem cells drives intestinal regeneration and cancer. Nature 526, 715–718 (2015).
Yui, S. et al. YAP/TAZ-dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell 22, 35–49.e7 (2018).
Azzolin, L. et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170 (2014).
Serra, D. et al. Self-organization and symmetry breaking in intestinal organoid development. Nature 569, 66–72 (2019).
Ayyaz, A. et al. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569, 121–125 (2019).
Grijalva, J. L. et al. Dynamic alterations in Hippo signaling pathway and YAP activation during liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 307, G196–G204 (2014).
Wang, C. et al. Differences in Yes-associated protein and mRNA levels in regenerating liver and hepatocellular carcinoma. Mol. Med. Rep. 5, 410–414 (2012).
Lu, L., Finegold, M. J. & Johnson, R. L. Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp. Mol. Med. 50, e423 (2018).
Bai, H. et al. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology 56, 1097–1107 (2012).
Camargo, F. D. et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17, 2054–2060 (2007).
Lu, L. et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl Acad. Sci. USA 107, 1437–1442 (2010).
Varelas, X. et al. The Hippo pathway regulates Wnt/β-catenin signaling. Dev. Cell 18, 579–591 (2010).
Zhou, D. et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16, 425–438 (2009).
Benhamouche, S. et al. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev. 24, 1718–1730 (2010).
Zhang, N. et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev. Cell 19, 27–38 (2010).
Yimlamai, D. et al. Hippo pathway activity influences liver cell fate. Cell 157, 1324–1338 (2014).
Yovchev, M. et al. Experimental model for successful liver cell therapy by Lenti TTR-YapERT2 transduced hepatocytes with tamoxifen control of Yap subcellular location. Sci. Rep. 6, 19275 (2016).
Loforese, G. et al. Impaired liver regeneration in aged mice can be rescued by silencing Hippo core kinases MST1 and MST2. EMBO Mol. Med. 9, 46–60 (2017).
Lodge, E. J. et al. Homeostatic and tumourigenic activity of SOX2+ pituitary stem cells is controlled by the LATS/YAP/TAZ cascade. eLife https://doi.org/10.7554/eLife.43996 (2019).
Hogan, B. L. et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15, 123–138 (2014).
Liu, Z. et al. MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep. 16, 1810–1819 (2016).
Sun, T. et al. TAZ is required for lung alveolar epithelial cell differentiation after injury. JCI Insight https://doi.org/10.1172/jci.insight.128674 (2019).
LaCanna, R. et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Invest. 130, 2107–2122 (2019).
Szymaniak, A. D., Mahoney, J. E., Cardoso, W. V. & Varelas, X. Crumbs3-mediated polarity directs airway epithelial cell fate through the Hippo pathway effector Yap. Dev. Cell 34, 283–296 (2015).
Mahoney, J. E., Mori, M., Szymaniak, A. D., Varelas, X. & Cardoso, W. V. The Hippo pathway effector Yap controls patterning and differentiation of airway epithelial progenitors. Dev. Cell 30, 137–150 (2014).
Zhao, R. et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self-renewal of stem cells. Dev. Cell 30, 151–165 (2014).
Elbediwy, A. et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 143, 1674–1687 (2016).
Schlegelmilch, K. et al. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 144, 782–795 (2011).
Silvis, M. R. et al. α-Catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci. Signal. 4, ra33 (2011).
Beverdam, A. et al. Yap controls stem/progenitor cell proliferation in the mouse postnatal epidermis. J. Invest. Dermatol. 133, 1497–1505 (2013).
Lee, M. J., Byun, M. R., Furutani-Seiki, M., Hong, J. H. & Jung, H. S. YAP and TAZ regulate skin wound healing. J. Invest. Dermatol. 134, 518–525 (2014).
Zhang, H., Pasolli, H. A. & Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl Acad. Sci. USA 108, 2270–2275 (2011).
von Gise, A. et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl Acad. Sci. USA 109, 2394–2399 (2012).
Xin, M. et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl Acad. Sci. USA 110, 13839–13844 (2013).
Lin, Z. et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res. 115, 354–363 (2014).
Ito, M. et al. Characterization of a small molecule that promotes cell cycle activation of human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell Cardiol. 128, 90–95 (2019).
Hara, H. et al. Discovery of a small molecule to increase cardiomyocytes and protect the heart after ischemic injury. JACC Basic. Transl. Sci. 3, 639–653 (2018).
Bassat, E. et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 547, 179–184 (2017).
Morikawa, Y. et al. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in Hippo-deficient mice. Sci. Signal. 8, ra41 (2015).
Vite, A., Zhang, C., Yi, R., Emms, S. & Radice, G. L. α-Catenin-dependent cytoskeletal tension controls Yap activity in the heart. Development 145, dev149823 (2018).
Nowell, C. S. et al. Chronic inflammation imposes aberrant cell fate in regenerating epithelia through mechanotransduction. Nat. Cell Biol. 18, 168–180 (2016).
Mindos, T. et al. Merlin controls the repair capacity of Schwann cells after injury by regulating Hippo/YAP activity. J. Cell Biol. 216, 495–510 (2017).
Zhao, K. et al. Muscle Yap is a regulator of neuromuscular junction formation and regeneration. J. Neurosci. 37, 3465–3477 (2017).
Deng, Y. et al. Yap1 regulates multiple steps of chondrocyte differentiation during skeletal development and bone repair. Cell Rep. 14, 2224–2237 (2016).
Hu, J. K. et al. An FAK–YAP–mTOR signaling axis regulates stem cell-based tissue renewal in mice. Cell Stem Cell 21, 91–106.e6 (2017).
Hayashi, S., Tamura, K. & Yokoyama, H. Yap1, transcription regulator in the Hippo signaling pathway, is required for Xenopus limb bud regeneration. Dev. Biol. 388, 57–67 (2014).
Hayashi, S. et al. Transcriptional regulators in the Hippo signaling pathway control organ growth in Xenopus tadpole tail regeneration. Dev. Biol. 396, 31–41 (2014).
Mateus, R. et al. Control of tissue growth by Yap relies on cell density and F-actin in zebrafish fin regeneration. Development 142, 2752–2763 (2015).
Lin, A. Y. & Pearson, B. J. Planarian yorkie/YAP functions to integrate adult stem cell proliferation, organ homeostasis and maintenance of axial patterning. Development 141, 1197–1208 (2014).
Caliari, S. R. et al. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Sci. Rep. 6, 21387 (2016).
Mannaerts, I. et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 63, 679–688 (2015).
Szeto, S. G. et al. YAP/TAZ are mechanoregulators of TGF-β–Smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. 27, 3117–3128 (2016).
Liu, F. et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 308, L344–L357 (2015).
Piersma, B. et al. YAP1 Is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am. J. Pathol. 185, 3326–3337 (2015).
Zhao, B. et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971 (2008).
Aikawa, T., Gunn, J., Spong, S. M., Klaus, S. J. & Korc, M. Connective tissue growth factor-specific antibody attenuates tumor growth, metastasis, and angiogenesis in an orthotopic mouse model of pancreatic cancer. Mol. Cancer Ther. 5, 1108–1116 (2006).
Huang, W. T., Vayalil, P. K., Miyata, T., Hagood, J. & Liu, R. M. Therapeutic value of small molecule inhibitor to plasminogen activator inhibitor-1 for lung fibrosis. Am. J. Respir. Cell Mol. Biol. 46, 87–95 (2012).
Ledwozyw, A. The effect of β-aminopropionitrile on bleomycin-induced lung injury in rats. Acta Physiol. Hung. 83, 91–99 (1995).
Barry-Hamilton, V. et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 16, 1009–1017 (2010).
Gokey, J. J. et al. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight https://doi.org/10.1172/jci.insight.98738 (2018).
Machado, M. V. et al. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J. Hepatol. 63, 962–970 (2015).
Chen, P. et al. Pathogenesis of non-alcoholic fatty liver disease mediated by YAP. Hepatol. Int. 12, 26–36 (2018).
Wang, X. et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 24, 848–862 (2016).
Liang, M. et al. Yap/Taz deletion in Gli+ cell-derived myofibroblasts attenuates fibrosis. J. Am. Soc. Nephrol. 28, 3278–3290 (2017).
Kramann, R. et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16, 51–66 (2015).
Mitani, A. et al. Transcriptional coactivator with PDZ-binding motif is essential for normal alveolarization in mice. Am. J. Respir. Crit. Care Med. 180, 326–338 (2009).
Toyama, T. et al. Therapeutic targeting of TAZ and YAP by dimethyl fumarate in systemic sclerosis fibrosis. J. Invest. Dermatol. 138, 78–88 (2018).
Varelas, X. et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 10, 837–848 (2008).
Hiemer, S. E., Szymaniak, A. D. & Varelas, X. The transcriptional regulators TAZ and YAP direct transforming growth factor β-induced tumorigenic phenotypes in breast cancer cells. J. Biol. Chem. 289, 13461–13474 (2014).
Chan, M. W., Hinz, B. & McCulloch, C. A. Mechanical induction of gene expression in connective tissue cells. Methods Cell Biol. 98, 178–205 (2010).
Haak, A. J. et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 11, eaau6296 (2019).
Gill, M. K. et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat. Commun. 9, 3510 (2018).
Lagares, D. et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci. Transl. Med. 9, eaal3765 (2017).
Liu-Chittenden, Y. et al. Genetic and pharmacological disruption of the TEAD–YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305 (2012).
Dasari, V. R. et al. Verteporfin exhibits YAP-independent anti-proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget 8, 28628–28640 (2017).
Wang, C. et al. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am. J. Cancer Res. 6, 27–37 (2016).
Zhang, H. et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci. Signal. 8, ra98 (2015).
Song, S. et al. A novel YAP1 inhibitor targets CSC-enriched radiation-resistant cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol. Cancer Ther. 17, 443–454 (2018).
Basu, D. et al. Identification, mechanism of action, and antitumor activity of a small molecule inhibitor of Hippo, TGF-β, and Wnt signaling pathways. Mol. Cancer Ther. 13, 1457–1467 (2014).
Smith, S. A. et al. Antiproliferative and antimigratory effects of a novel YAP–TEAD interaction inhibitor identified using in silico molecular docking. J. Med. Chem. 62, 1291–1305 (2019).
Kurppa, K. J. et al. Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell 37, 104–122.e12 (2020).
Santucci, M. et al. The Hippo pathway and YAP/TAZ–TEAD protein–protein interaction as targets for regenerative medicine and cancer treatment. J. Med. Chem. 58, 4857–4873 (2015).
Gibault, F., Sturbaut, M., Bailly, F., Melnyk, P. & Cotelle, P. Targeting transcriptional enhanced associate domains (TEADs). J. Med. Chem. 61, 5057–5072 (2018).
Noland, C. L. et al. Palmitoylation of TEAD transcription factors is required for their stability and function in Hippo pathway signaling. Structure 24, 179–186 (2016).
Chan, P. et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 12, 282–289 (2016).
Pobbati, A. V. et al. Targeting the central pocket in human transcription factor TEAD as a potential cancer therapeutic strategy. Structure 23, 2076–2086 (2015).
Bum-Erdene, K. et al. Small-molecule covalent modification of conserved cysteine leads to allosteric inhibition of the TEAD·Yap protein–protein interaction. Cell Chem. Biol. 26, 378–389.e13 (2019).
Maeda, T., Chapman, D. L. & Stewart, A. F. Mammalian vestigial-like 2, a cofactor of TEF-1 and MEF2 transcription factors that promotes skeletal muscle differentiation. J. Biol. Chem. 277, 48889–48898 (2002).
Pobbati, A. V., Chan, S. W., Lee, I., Song, H. & Hong, W. Structural and functional similarity between the VGLL1–TEAD and the YAP–TEAD complexes. Structure 20, 1135–1140 (2012).
Vaudin, P., Delanoue, R., Davidson, I., Silber, J. & Zider, A. TONDU (TDU), a novel human protein related to the product of vestigial (Vg) gene of Drosophila melanogaster interacts with vertebrate TEF factors and substitutes for Vg function in wing formation. Development 126, 4807–4816 (1999).
Jiao, S. et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 25, 166–180 (2014).
Zhao, B. et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761 (2007).
Noto, A. et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 36, 4573–4584 (2017).
Lin, K. C. et al. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat. Cell Biol. 19, 996–1002 (2017).
Magico, A. C. & Bell, J. B. Identification of a classical bipartite nuclear localization signal in the Drosophila TEA/ATTS protein scalloped. PLoS ONE 6, e21431 (2011).
Qi, Y. et al. A splicing isoform of TEAD4 attenuates the Hippo–YAP signalling to inhibit tumour proliferation. Nat. Commun. 7, ncomms11840 (2016).
Fu, V., Plouffe, S. W. & Guan, K. L. The Hippo pathway in organ development, homeostasis, and regeneration. Curr. Opin. Cell Biol. 49, 99–107 (2017).
Fan, F. et al. Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Sci. Transl. Med. 8, 352ra108 (2016).
Zhou, D. et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl Acad. Sci. USA 108, E1312–E1320 (2011).
Lin, C., Yao, E. & Chuang, P. T. A conserved MST1/2–YAP axis mediates Hippo signaling during lung growth. Dev. Biol. 403, 101–113 (2015).
Takahashi, Y. et al. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin. Cancer Res. 11, 1380–1385 (2005).
Cordenonsi, M. et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772 (2011).
Diaz-Martin, J. et al. Nuclear TAZ expression associates with the triple-negative phenotype in breast cancer. Endocr. Relat. Cancer 22, 443–454 (2015).
Chan, S. W. et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 68, 2592–2598 (2008).
Cancer Genome Atlas Research Network. et al. Integrated genomic and molecular characterization of cervical cancer. Nature 543, 378–384 (2017).
Liu, T. et al. Clinical significance of Yes-associated protein overexpression in cervical carcinoma: the differential effects based on histotypes. Int. J. Gynecol. Cancer 23, 735–742 (2013).
Deng, J. et al. LATS1 suppresses proliferation and invasion of cervical cancer. Mol. Med. Rep. 15, 1654–1660 (2017).
He, C. et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 7, 1426–1449 (2015).
Wang, Y. et al. Comprehensive molecular characterization of the Hippo signaling pathway in cancer. Cell Rep. 25, 1304–1317.e5 (2018).
Wang, Y., Xie, C., Li, Q., Xu, K. & Wang, E. Clinical and prognostic significance of Yes-associated protein in colorectal cancer. Tumour Biol. 34, 2169–2174 (2013).
Lee, K. W. et al. Significant association of oncogene YAP1 with poor prognosis and cetuximab resistance in colorectal cancer patients. Clin. Cancer Res. 21, 357–364 (2015).
Imajo, M., Ebisuya, M. & Nishida, E. Dual role of YAP and TAZ in renewal of the intestinal epithelium. Nat. Cell Biol. 17, 7–19 (2015).
Cancer Genome Atlas Research Network. et al. Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175 (2017).
Muramatsu, T. et al. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis 32, 389–398 (2011).
Song, S. et al. The Hippo coactivator YAP1 mediates EGFR overexpression and confers chemoresistance in esophageal cancer. Clin. Cancer Res. 21, 2580–2590 (2015).
Song, S. et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 74, 4170–4182 (2014).
Ge, L. et al. Yes-associated protein expression in head and neck squamous cell carcinoma nodal metastasis. PLoS ONE 6, e27529 (2011).
Eun, Y. G. et al. Clinical significance of YAP1 activation in head and neck squamous cell carcinoma. Oncotarget 8, 111130–111143 (2017).
Hiemer, S. E. et al. A YAP/TAZ-regulated molecular signature is associated with oral squamous cell carcinoma. Mol. Cancer Res. 13, 959–968 (2015).
Zender, L. et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125, 1253–1267 (2006).
Han, S. X. et al. Expression and clinical significance of YAP, TAZ, and AREG in hepatocellular carcinoma. J. Immunol. Res. 2014, 261365 (2014).
Song, H. et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl Acad. Sci. USA 107, 1431–1436 (2010).
Lee, K. P. et al. The Hippo–Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc. Natl Acad. Sci. USA 107, 8248–8253 (2010).
Fitamant, J. et al. YAP inhibition restores hepatocyte differentiation in advanced HCC, leading to tumor regression. Cell Rep. 10, 1692–1707 (2015).
Bueno, R. et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 48, 407–416 (2016).
Hmeljak, J. et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 8, 1548–1565 (2018).
Sekido, Y. Inactivation of Merlin in malignant mesothelioma cells and the Hippo signaling cascade dysregulation. Pathol. Int. 61, 331–344 (2011).
Mizuno, T. et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 31, 5117–5122 (2012).
Tanahashi, K. et al. Activation of Yes-associated protein in low-grade meningiomas is regulated by merlin, cell density, and extracellular matrix stiffness. J. Neuropathol. Exp. Neurol. 74, 704–709 (2015).
Baia, G. S. et al. Yes-associated protein 1 is activated and functions as an oncogene in meningiomas. Mol. Cancer Res. 10, 904–913 (2012).
Schramm, A. et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 47, 872–877 (2015).
Wang, M. et al. Transcriptional co-activator TAZ sustains proliferation and tumorigenicity of neuroblastoma by targeting CTGF and PDGF-β. Oncotarget 6, 9517–9530 (2015).
Seong, B. K. et al. A metastatic mouse model identifies genes that regulate neuroblastoma metastasis. Cancer Res. 77, 696–706 (2017).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Malik, S. A., Khan, M. S., Dar, M., Hussain, M. U. & Mudassar, S. TAZ is an independent prognostic factor in non-small cell lung carcinoma: elucidation at protein level. Cancer Biomark. 18, 389–395 (2017).
Xie, M. et al. Prognostic significance of TAZ expression in resected non-small cell lung cancer. J. Thorac. Oncol. 7, 799–807 (2012).
Wang, H. et al. Tankyrase inhibitor sensitizes lung cancer cells to endothelial growth factor receptor (EGFR) inhibition via stabilizing angiomotins and inhibiting YAP signaling. J. Biol. Chem. 291, 15256–15266 (2016).
Berger, A. C. et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 33, 690–705.e9 (2018).
Jeong, W. et al. Activation of YAP1 is associated with poor prognosis and response to taxanes in ovarian cancer. Anticancer Res. 34, 811–817 (2014).
Xia, Y. et al. YAP promotes ovarian cancer cell tumorigenesis and is indicative of a poor prognosis for ovarian cancer patients. PLoS ONE 9, e91770 (2014).
Zhang, X. et al. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene 30, 2810–2822 (2011).
Hua, G. et al. YAP induces high-grade serous carcinoma in fallopian tube secretory epithelial cells. Oncogene 35, 2247–2265 (2016).
Merritt, N. M. et al. A comprehensive evaluation of Hippo pathway silencing in sarcomas. Oncotarget 9, 31620–31636 (2018).
Eisinger-Mathason, T. S. et al. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc. Natl Acad. Sci. USA 112, E3402–E3411 (2015).
Zhang, Z. et al. Structure-based design and synthesis of potent cyclic peptides inhibiting the YAP–TEAD protein–protein interaction. ACS Med. Chem. Lett. 5, 993–998 (2014).
Konradi, A. W., Lin, T. T.-L. T. Benzosulfonyl compounds https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019040380 (2018).
Acknowledgements
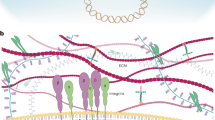
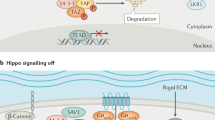
K.-L.G. is supported by grants from NIH (CA196878, CA217642, GM51586). X.V. is supported by grants from NIH (R01HL124392 and R21HD094012) and the American Cancer Society (130257-RSG-17-138-01-CSM). The authors acknowledge P. Cotelle for Fig. 3c,d, C. Noland for Fig. 3a and P. Calses for Fig. 4.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
K.-L.G. is a co-founder and has an equity interest in Vivace Therapeutics, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. A.D. is an employee of Genentech and shareholder at Roche.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Mechanotransduction
-
The process through which cells sense and respond to mechanical stimuli by converting them into biochemical signals for cellular signalling and responses.
- Cholestatic liver injury
-
A condition with reduced or blocked flow of bile from the liver.
- Pneumonectomy
-
The surgical removal of the lung or a lobe of the lung.
- Proteotoxic
-
The impairment of cellular function caused by protein misfolding.
Rights and permissions
About this article

Cite this article
Dey, A., Varelas, X. & Guan, KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov 19, 480–494 (2020). https://doi.org/10.1038/s41573-020-0070-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-020-0070-z
This article is cited by
-
Biomaterial-based mechanical regulation facilitates scarless wound healing with functional skin appendage regeneration
Military Medical Research (2024)
-
Transcriptional regulation of cancer stem cell: regulatory factors elucidation and cancer treatment strategies
Journal of Experimental & Clinical Cancer Research (2024)
-
Characterization of the proteome of stable and unstable carotid atherosclerotic plaques using data-independent acquisition mass spectrometry
Journal of Translational Medicine (2024)
-
RGS20 promotes non-small cell lung carcinoma proliferation via autophagy activation and inhibition of the PKA-Hippo signaling pathway
Cancer Cell International (2024)
-
STAT3–mediated up-regulation of DAB2 via SRC-YAP1 signaling axis promotes Helicobacter pylori-driven gastric tumorigenesis
Biomarker Research (2024)
