
Abstract
Uncontrolled haemorrhage is a major preventable cause of death in patients with traumatic injury. Trauma-induced coagulopathy (TIC) describes abnormal coagulation processes that are attributable to trauma. In the early hours of TIC development, hypocoagulability is typically present, resulting in bleeding, whereas later TIC is characterized by a hypercoagulable state associated with venous thromboembolism and multiple organ failure. Several pathophysiological mechanisms underlie TIC; tissue injury and shock synergistically provoke endothelial, immune system, platelet and clotting activation, which are accentuated by the ‘lethal triad’ (coagulopathy, hypothermia and acidosis). Traumatic brain injury also has a distinct role in TIC. Haemostatic abnormalities include fibrinogen depletion, inadequate thrombin generation, impaired platelet function and dysregulated fibrinolysis. Laboratory diagnosis is based on coagulation abnormalities detected by conventional or viscoelastic haemostatic assays; however, it does not always match the clinical condition. Management priorities are stopping blood loss and reversing shock by restoring circulating blood volume, to prevent or reduce the risk of worsening TIC. Various blood products can be used in resuscitation; however, there is no international agreement on the optimal composition of transfusion components. Tranexamic acid is used in pre-hospital settings selectively in the USA and more widely in Europe and other locations. Survivors of TIC experience high rates of morbidity, which affects short-term and long-term quality of life and functional outcome.
Similar content being viewed by others

Introduction
Injury is the fourth leading cause of mortality worldwide, accounting for 9% of deaths globally (4.9 million people) in 2016 (ref.1). Moreover, the burden is highest in individuals <50 years of age, among whom injury as a cause of death is second only to infectious diseases. Early preventable deaths after injury in civilian2 and military3 settings are primarily attributable to uncontrolled haemorrhage2,3,4,5,6,7,8, whereas later preventable deaths are typically due to hypercoagulability9. Consequently, there is intense interest worldwide in the pathogenesis of trauma-induced coagulopathy (TIC) to attenuate its adverse effects on the outcomes of seriously injured patients.
Impaired coagulation following sudden death from injury has been observed for centuries10 and, in the 1960s, the first clinical laboratory documentation of the temporal changes in coagulation following severe injury were documented11. However, early endogenous drivers of coagulopathy were not specifically investigated until 1982, when a case series of patients with major abdominal vascular injuries highlighted TIC as a common direct cause of early post-injury mortality: 89% of the deaths were bleeding-related, yet half occurred after mechanical control of bleeding sites — in other words, they were due to coagulopathy12. The remaining ongoing quagmire is the inability to distinguish between patients with exsanguinating injuries whose TIC is the result of metabolic failure (that is, who are bleeding because they are dying) from patients whose TIC is the cause of the ongoing blood loss (that is, who are dying because they are bleeding)13. Furthermore, not all patients with abnormalities in laboratory coagulation tests are bleeding14.
Despite the long-term fascination with changes in coagulation resulting from shock and tissue injury15, there is no standard definition of TIC, which refers to abnormal coagulation capacity attributable to trauma. The term TIC was established during the Trans-Agency Consortium for Trauma Induced Coagulopathy Workshop conducted by the National Institutes of Health in April 2010 to describe the variety of phenomena that characterize this condition. TIC can manifest as a spectrum of phenotypes from hypocoagulation to hypercoagulation (Fig. 1), as a function of several interactive factors, including (but not limited to) tissue injury, presence of shock and, in particular, time from injury (Fig. 2). Coagulopathy, metabolic acidosis and hypothermia were initially emphasized as the three pillars of life-threatening post-injury bleeding (known as the ‘lethal triad’).

Physiological clot formation and degradation represent a delicate balance of prothrombotic or antithrombotic and fibrinolytic or antifibrinolytic processes. Early and late phenotypes of trauma-induced coagulation (TIC) result from the collective insults of tissue injury, shock and traumatic brain injury (TBI), as well as individual responses to these insults. Furthermore, the mechanisms underlying the various phenotypes can occur at different times after injury. Consequently, there are a myriad of TIC phenotypes that change over time. Adapted with permission from Gonzalez, E. et al. Goal-directed hemostatic resuscitation of trauma-induced coagulopathy: a pragmatic randomized clinical trial comparing a viscoelastic assay to conventional coagulation assays. Ann. Surg. 263, 1051–1059 (https://journals.lww.com/annalsofsurgery/)39.

Progress in understanding the pathogenesis of trauma-induced coagulation (TIC) has been moved forward by the concept of the cell-based model of coagulation, which emphasizes the fundamental role of platelets as a platform for clotting factor assembly and their interaction with endothelium that culminates in thrombin generation and incorporation of fibrin to form a haemostatic plug. Although there are several hypotheses on the driving mechanisms, tissue injury and shock (1) synergistically activate the endothelium, platelets and the immune system (2) to generate an array of mediators that reduce fibrinogen, impair platelet function and compromise thrombin generation (3), ultimately resulting in inadequate clot formation for haemostasis (4). Increased fibrinolysis via plasmin generation further compromises haemostatic capacity. These defects are accentuated by ongoing blood loss, haemodilution, metabolic acidosis and hypothermia. A colour gradient indicates that the mechanism can result in both hypocoagulation and hypercoagulation. DAMPs, damage-associated molecular patterns; HMGB1, high mobility group protein B1; PAI-1, plasminogen activator inhibitor-1; tPA, tissue plasminogen factor.
For discussion purposes, we suggest the terms ‘early TIC’ and ‘late TIC’ but acknowledge that the phenotypes can vary substantially within these time periods. Early TIC (generally within 6 hours of injury) is characterized by the inability to achieve haemostasis, which may lead to uncontrolled haemorrhage and protracted shock; whereas late TIC (usually >24 hours after injury) is represented by a hypercoagulable state, which may result in excessive macro-clotting and micro-clotting leading to thromboembolic events (for example, deep venous thrombosis (DVT) and pulmonary embolism) or to acute respiratory distress syndrome (ARDS) and multiple organ failure. Of note, early and late TIC are not mutually exclusive, that is, patients may develop early TIC due to massive blood loss but die of extensive microvascular occlusion recognized as irreversible shock. Furthermore, the transition from hypocoagulability to hypercoagulability may occur within minutes or hours or be delayed for days.
Notably, disseminated intravascular coagulation (DIC) is a syndrome related to but distinct from TIC. DIC is defined as “an acquired syndrome characterized by the intravascular activation of coagulation with a loss of localization arising from different causes”16. A consensus statement from the International Society on Thrombosis and Haemostasis (ISTH) clarified the common as well as distinct mechanisms of DIC versus TIC17. Early TIC is dominated by acute blood loss with associated shock (and ischaemia–reperfusion damage), impaired clot formation and, in severe cases, hyperfibrinolysis (Fig. 1). In TIC, tissue factor (TF; a procoagulant factor) facilitates clot formation at sites of endothelial injury, whereas in DIC there is unbridled systemic clotting often promoted by TF expression on several cell surfaces. Ultimately, the late systemic prothrombotic–antifibrinolytic TIC phenotype mirrors certain DIC phenotypes18.
In this Primer, we describe what is known of TIC, but perhaps more importantly we acknowledge what remains to be defined. Our primary objective is to provide a broad picture of the entity TIC to inspire investigators from diverse disciplines to pursue answers to the substantial gaps in knowledge.
Epidemiology
Uncontrolled bleeding has been reported to cause 25% of all injury-related deaths19,20,21,22,23,24,25,26,27, and 40–80% of potentially preventable deaths28, both in military and in civilian settings (Supplementary Table 1). At least a quarter of the haemorrhagic deaths probably have a TIC component29. Uncontrolled bleeding as a cause of death following injury is observed globally; for example, Australian25 and Canadian30 studies implicated haemorrhage in 15–33% of injury-related deaths. In Stavanger, Norway, 25% of trauma-related deaths between 1996 and 2004 were due to exsanguination26. In a Turkish hospital, from 2010 to 2013, circulatory collapse accounted for 33% of injury-related deaths31. In Brazil, haemorrhage caused 18% of trauma-related deaths in an urban hospital32. Two European studies33,34 found lower proportions of haemorrhagic deaths; however, these studies underestimated death attributable to bleeding because they classified polytrauma, chest injury and cardiac arrest as separate, non-haemorrhagic causes of death. Differences in populations, injury mechanisms and health-care resources explain the disparities in statistics. Since the 1990s, when bleeding caused over one third of trauma fatalities20,, we have made little progress, as currently haemorrhage accounts for 20–34% of trauma-related mortality24,35. Although a reduction in bleeding-related deaths was observed in a US urban trauma centre after implementing a bleeding control bundle-of-care (from 36% to 25%)27, haemorrhage remained frequent among potentially preventable deaths despite the bundle (decreasing from 48% to 43%)36.
Understanding the timing of haemorrhagic deaths is crucial to determine when haemostatic therapies are most effective, and which outcomes (such as the need for massive transfusion, all-cause or haemorrhagic deaths, and early or late mortality) they may affect4,37. Trauma-related deaths immediately after injury are often due to irreparable injuries; thus, haemostatic interventions are more likely to affect haemorrhagic deaths over the ensuing hours. Randomized controlled trials (RCTs)2,5,6,38,39,40,41 and observational studies30,31,42 unequivocally show that haemorrhagic deaths occur within 24 hours of injury, mostly within 3–6 hours. Traumatic brain injury (TBI) is also a prevalent cause of death in the 6–24-hour period, and multiple organ failure becomes prevalent after the first week2. In the CRASH-2 trial, representing mainly developing countries, 34% of all deaths were attributed to bleeding, 50% of which were due to haemorrhage occurring within 10 hours40. Analyses of three recent US RCTs focusing on post-injury haemorrhage control, with comparable populations, methods and health-care resources, showed that most haemorrhagic deaths occurred in the first 6 hours7,43. Half of all deaths in the first 3–6 hours in these three RCTs were due to haemorrhage.
The incidence of TIC diagnosed via laboratory tests varies (Supplementary Table 2), but most studies converge around a TIC incidence of 25% of severely injured patients, with an associated 35–50% mortality. Children in general develop TIC later and less frequently than adults44, and TIC in children is typically associated with TBI. Older individuals are more vulnerable to TIC than younger adults45,46. Severe tissue injury and shock or hypoperfusion are the major risk factors for TIC (Supplementary Table 2). Studies in civilian47 and military48 populations have indicated that TIC is more severe when both severe tissue injury and shock are present. Metabolic acidosis and penetrating injury are commonly reported risk factors for TIC (Table S2). Long pre-hospital times49 and pre-hospital treatment with crystalloid solutions49,50 worsen TIC. The severity of TIC correlates with the severity of TBI (Table S2), but studies51,52 have suggested that hypoperfusion is an important cofactor. An often-neglected factor is hypocalcaemia, caused by both shock and blood products containing citrate (especially plasma and platelets), which has an anticoagulant effect by chelating calcium ions, and it has been suggested that the ‘lethal triad’ should include hypocalcaemia and become the ‘lethal diamond’53,54. Of note, it is important to recognize that although TIC is common in severely injured individuals, many patients with laboratory-based TIC do not have substantial bleeding14.
Mechanisms/pathophysiology
The biochemical reactions of physiological haemostasis are subject to control at several levels. Some control mechanisms act on the various factors and steps of the coagulation cascade; additional regulation levels involve the anticoagulants and protease inhibitors, as well as the cellular and tissue localization of coagulation55. These control mechanisms are barriers to the activation and spread of coagulation and thereby prevent clot formation at inappropriate times and places. Physiological haemostasis is terminated when the area of injury is surrounded by a platelet–fibrin clot that stops bleeding, forms a physical barrier to the diffusion of activated factors and provides a provisional scaffold for healing processes. Coagulopathy occurs not only when procoagulants are consumed or diluted, but also when one or more of the control mechanisms are disrupted. Thus, not only can the amount of thrombin generation be abnormal, but thrombin localization can also be abnormal. Because trauma is such a heterogeneous event, it is difficult to define a dominant mechanism of TIC. Furthermore, haemostatic function changes over time as bleeding continues, compensatory mechanisms are engaged and inflammation progresses.
Cell-based model of haemostasis
The cell-based model of haemostasis proposes that cells have active roles in regulating and localizing the coagulation reactions56. Receptors, lipids and other structures of cell surfaces are crucial to defining the roles of specific cell types in haemostasis. Platelets and endothelial cells are the two key players. Platelets adhere at a site of injury and provide the surface on which procoagulant reactions occur, and they control the rate and localization of thrombin production57. Endothelial cells are physiologically actively antithrombotic and prevent propagation of clotting from a site of injury throughout the vasculature55.
Impaired cell-mediated regulation of haemostasis can lead to haemostasis failure, even when the levels of the protein components are within normal ranges. This concept is particularly relevant to understanding the mechanisms of bleeding and thrombosis induced by trauma. In the cell-based model of haemostasis, the overlapping events of initiation (via the extrinsic pathway on TF-bearing cells), amplification (positive feedback of thrombin on platelets) and propagation of large-scale thrombin generation (via the intrinsic pathway on activated platelets) are regulated by cell surfaces rather than by the protein components alone (Fig. 3).

In the cell-based model of coagulation, initiation occurs on tissue factor (TF)-bearing cells, via the extrinsic pathway, and results in the activation of small amounts of thrombin. Thrombin generated on the TF-bearing cell amplifies the procoagulant response by activating additional coagulation factors and platelets. The large burst of thrombin required for formation of a fibrin clot is generated on platelet surfaces during the propagation phase. Adapted with permission from ref.318, Wiley.
Haemorrhagic shock
The pathophysiology of haemorrhagic shock is fundamentally blood volume depletion with diminished oxygen delivery to the microcirculation, ultimately resulting in metabolic acidosis. Although isolated transient haemorrhagic shock may be tolerated, when it is compounded by tissue injury, haemodilution and coagulation factor abnormalities, it is a major driver of TIC. It is important to distinguish early, hypocoagulable TIC (Fig. 2) from iatrogenic coagulopathy due to resuscitation with large volumes of cold fluids and blood products, which leads to dilution of enzymes required for clot formation, and hypothermia, which impairs clotting factor activity and platelet function12,58. The hypocoagulable TIC phenotype can be attributed partially to metabolic acidosis as a result of reduced oxygen delivery to tissue beds and organs49,59,60,61,62,63. In animal studies and in vitro experiments, acidosis has been shown to retard fibrin polymerization and clot strengthening in viscoelastic haemostatic assays (VHAs)60; to decrease factors V and IX activity and platelet aggregation64; to increase fibrinogen consumption61; to reduce platelet count, thrombin generation and maximum clot strength; and to induce abnormal conventional coagulation test results59. A pH drop from 7.4 to 7.2 reduces the activity of each of the coagulation proteases by more than half63,65. Hypothermia is now less frequent with modern haemostatic, goal-directed resuscitation with warm fluids66,67, but it should not be overlooked. An in vitro study of blood from healthy volunteers68 found a substantial reduction in both platelet function and coagulation enzyme activity at temperatures <33 °C. Hypothermia remains a marker for poor a prognosis after haemorrhage, probably representing metabolic dysfunction49,69,70,71. Shock also leads to auto-dilution72.
Activation of protein C is suggested to contribute to TIC29,51,52,73. Trauma-induced hypoperfusion has been associated with protein C activation and reduced levels of plasminogen activator inhibitor-1 (PAI-1)54. Elevated activated protein C (aPC) predicts adverse outcomes after injury; however, the mechanistic role of aPC in TIC has been disputed. Specifically, platelets and plasma factor Va are resistant to aPC cleavage at concentrations of aPC seen in TIC72. Although it has been hypothesized that aPC inactivates PAI-1, therefore PA1-1 is unavailable to bind t-PA, it seems more likely that the enormous release of t-PA from the endothelium is due to adrenaline, vasopressin and thrombin signalling as well as hypoperfusion, which drives the fibrinolytic phenotype of TIC74.
In addition, metabolic by-products, such as succinate, have been associated with early TIC75, and oxidative stress has been shown to modify fibrinogen polymerization resulting in weakened clots76. Finally, hypocalcaemia is another mechanism by which haemorrhagic shock can impair coagulation. Calcium has an important role in the formation and stabilization of fibrin polymerization sites and, consequently, it affects all platelet-dependent functions77. Of note, laboratory coagulation tests may not detect the negative effect of hypocalcaemia on coagulation, as blood samples are re-calcified prior to being assayed. Hypocalcaemia is prevalent after haemorrhage, owing to resuscitation with citrated blood products, low hepatic clearance of citrate due to defective hepatic perfusion78, and other still poorly understood shock-related mechanisms53,67.
As haemorrhagic shock progresses, hypercoagulability ensues, owing to prothrombotic changes and fibrinolysis shutdown (see below) that promote organ damage by generating thrombi and occluding the microvascular circulation, leading ultimately to organ failure9,79. Hypocoagulability and increased fibrinolysis during shock may well represent intrinsic mechanisms to prevent these events from occurring; it remains debatable whether these are adaptive or pathological responses80.
Tissue injury
Tissue injury promotes both early hypocoagulability and contributes to later hypercoagulability. Tissue damage with endothelial disruption activates the coagulation system at the injury site via TF, a transmembrane protein expressed within the sub-endothelium that becomes exposed. TF complexes to factor VIIa and activates the coagulation system, resulting in thrombin generation and fibrin formation81. Moreover, tissue trauma provokes the release of damage-associated molecular patterns (DAMPs), which stimulate inflammatory pathways by the release of several mediators. Inflammation and coagulation are interrelated processes that robustly influence one another.
The development of TIC is typically associated with the severity and extent of tissue injury48,82. Tissue damage and shock-related hypoperfusion frequently occur together; however, their synergistic contribution to TIC remains unclear. Multiple potential pathways have been suggested, including an early effect of DAMPs on platelet function, with DAMPS rendering platelets hyporesponsive80,83. Furthermore, an initial thrombin surge activates endogenous anticoagulation pathways, and some clotting factor consumption may occur in TIC, although with mechanisms distinct from those in DIC17. It is also possible that the specific organ(s) affected by tissue damage contribute to TIC. For example, TBI creates a hypocoagulable state that has been suggested to be partially attributable to the cerebral tissue releasing high concentrations of potent procoagulant molecules such as phospholipids and also brain-derived cellular microvesicles, which stimulate clotting that depletes clotting factors systemically and provokes platelet inhibitors84,85. Damage to organs with high contents of tissue plasminogen factor (tPA; which is profibrinolytic), such as the pancreas, lung and urogenital system, may also compromise haemostasis via fibrinolytic activation. However, the exact contribution of these organ injuries is unknown. It is similarly unclear whether any pre-existing chronic conditions in these tPA-rich organs may modulate TIC dynamics. In addition, tissue injury has also been directly correlated with fibrinolysis shutdown through release of cellular by-products of injury, as well as mechanical trauma to red blood cells (RBCs) and platelets, leading to the release of their contents86. A recent study suggested that myosin can bind factors Xa and Va, thereby increasing their ability to create prothrombinase and generate thrombin87. In both preclinical models and patient studies, tissue injury results in the production of extracellular vesicles from multiple cellular sources, which are strongly prothrombotic and may result in coagulation factor depletion after injury88,89.
Endothelial dysfunction
The endothelial cell surface network governs coagulation, inflammation, microcirculation, and barrier function crucial to vascular homeostasis and oxygen delivery (Fig. 4). Trauma-associated damage to this network, termed the endotheliopathy of trauma (EOT), is characterized by loss of barrier function, leukocyte adhesion, endothelial activation, clinical expression of coagulopathy, micro-thrombosis and macro-thrombosis, and organ dysfunction. Mechanistically, it is probable that TIC contributes to EOT as well as vice versa90. The role of the intrinsic (also known as contact) pathway activation of the coagulation system as a result of exposure to disrupted endothelium remains unclear91. In the intrinsic pathway, a negatively charged surface activates factor XII, and factor XIIa cleavage of prekallikrein results in the serine protease kallikrein, which can cleave high molecular weight kininogen to generate bradykinin. Bradykinin can both induce the expression of TF and generate tPA.

Projecting beyond the cell membrane of healthy endothelial cells is a glycocalyx of polysaccharides linked to membrane and trans-membrane proteoglycans, which is fortified with soluble glycoproteins that coordinate coagulation and immune functions. The glycocalyx provides cytoprotection, membrane integrity and anti-apoptotic antithrombotic signalling. Clot formation relies on platelet plug construction (primary haemostasis), which begins with platelet tethering and adhesion to exposed extravascular matrices including tissue factor and collagen via von-Willebrand factor (vWF). Extravascular adhesion and thrombin stimulation activate platelets, resulting in procoagulant calcium mobilization, structural changes, soluble factor degranulation, phosphatidylserine exposure and glycoprotein (GP) IIb/IIIa receptor conformational change to accept fibrin binding. Additionally, platelets control local fibrinolysis via degranulation of soluble factors from alpha granules including plasminogen activator inhibitor-1 (PAI-1) and α2 antiplasmin to maintain prothrombotic, antifibrinolytic clot architecture. Secondarily, activated platelets recruit leukocytes to local environments. Further, via reciprocal release of trophogens, platelets promote endothelial stability and angiogenesis in return for endothelial control of platelet-dependent haemostasis and release of cytokines that signal megakaryopoiesis. However, in trauma-induced coagulopathy, platelet activation pathways are maladaptive, that is, they result in primary and secondary platelet function failures. This is characterized by altered and shed glycoprotein VI and Ibα, impaired extracellular and intracellular calcium, circulating soluble platelet inhibitors, altered granule content and loss of endothelial protection and trophogenesis. Further, a procoagulant and pro-inflammatory milieu is promoted by circulating platelet–leukocyte aggregates (PLAs) and platelet ballooning, sustained exocytosis and impaired clearance of vWF by ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin motifs 13), and metalloproteinase (MMP) cleavage of the protective ectodomains of glycocalyx components exposing neutrophil adhesion receptors for neutrophil binding and release of chemoattractant molecules and cytokines. In this setting, the endothelium becomes denuded and leaky. These trauma-induced coagulopathy (TIC)-associated procoagulant and pro-inflammatory platelet and endothelial biologies are associated with micro-thrombosis and macro-thrombosis. EV, extravcellular vesicle; PAF, platelet activating factor; PAR, protease-activating receptor, PGI2, prostaglandin I2; RBC, red blood cell; TP, TXA2/PG endoperoxidases; TXA, tranexamic acid; TXA2, thromboxane A2.
EOT is mediated by hypoperfusion and is characterized by circulating markers of shed endothelial glycocalyx associated with coagulopathy, inflammatory complications, vascular thrombosis, organ failure and death92,93,94 (Fig. 4). The glycosaminoglycan syndecan-1 is the most well-characterized circulating biomarker induced by sheddases90 (membrane-bound enzymes that cleave and release ectodomains) in TIC, as its heparan sulfate ectodomain is shed with hypoperfusion, catecholamine surges and oxidative stress. It remains controversial whether auto-heparinization due to the heparan sulfate domain contributes to impaired clot formation, as endogenous anticoagulation due to heparan is variably identifiable by viscoelastic assays95. Pathological cleavage of the syndecan-1 ectodomain may be mediated by matrix metalloproteinases (MMPs) of the ADAM (a disintegrin and metalloproteinase) family. However, it is unclear whether poor outcomes associated with shed proteoglycans are due to direct or downstream effects of altered protective glycoproteins. Experimental work suggests that the tissue injury-driven and shock-driven activation of the thrombin–thrombomodulin system, and ultimate depletion of protein C, diminish endogenous cytoprotective effects on the endothelium51,73,96. Additionally, altered platelet–endothelium regulation in TIC may disrupt an important symbiosis, as soluble CD40, a platelet ligand for endothelial inflammatory cascades, is associated with TIC97. Further, both sustained exocytosis of structurally ultra-large von Willebrand factor (vWF; an adhesive protein) and impaired clearance of vWF by ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin motifs 13) have been identified in injured patients with TIC98, and are associated with prothrombotic and pro-inflammatory biology98,99, highlighting the importance of endothelial biology in mediating micro-thrombosis and macro-thrombosis (Fig. 4). Cerebral endothelial release of vWF has been suggested to be important in TIC provoked by TBI100.
Animal studies have shown that endothelial barrier function is restored with plasma90,101,102,103,104, and early plasma transfusion of injured patients is associated with reduced levels of circulating shed syndecan-1 ectodomain105, providing mechanistic insights for improved outcomes5,6. Plasma transfusion might reduce syndecan-1 shedding via tissue inhibitor of metalloproteinase (TIMP) or decreased activation of ADAM MMPs106. Additionally, new hypotheses on the mechanisms of tranexamic acid (TXA) in injured patients centre on abrogation of the EOT through serine protease inhibition, suppression of the release of DAMP mitochondrial DNA, stimulation of mitochondrial respiration and increase in oxidative phosphorylation107,108. It remains unknown whether the EOT is cause or effect in TIC, but investigations to identify therapeutic targets for recovery of endothelial cell surface networks, including characterization of soluble reparative molecules in plasma, are continuing.
Platelet dysfunction
Despite being subcellular in size and anucleate in structure, platelets are biologically dynamic in coordinating haemostasis, endothelial health and immune function109,110,111 (Fig. 4). Interest in the role of platelets in TIC intensified following the description of the cell-based model of haemostasis in 2001 (ref.56). Subsequent accumulating evidence has supported the presence of quantitative and qualitative112 deficits in all platelet functions113,114 in human and animal TIC models, and it has implicated platelets in the pathogenesis of post-injury venous thromboembolism (VTE) and multiple organ failure.
Failures of both primary (haemostatic) and secondary (immunoregulatory) platelet functions are characteristic of TIC and can be identified in up to 50% of injured patients, regardless of injury severity or presence of shock112 (Fig. 4). Secondary platelet function should not be confused with the term secondary haemostasis. Quantitative consumptive and dilutional thrombocytopenia are independently associated with bleeding115,116. However, most patients with TIC have preserved platelet counts and evidence of circulating populations of activated platelets, yet paradoxically impaired ex vivo aggregation responses117,118. This phenomenon is described as ‘platelet exhaustion’, due to injury and shock119 and is driven by endothelial release of TF, platelet activating factor and vWF99,120 that activates platelets beyond what is needed for primary haemostasis at the local sites of injury, thereby creating a pool of activated circulating platelets that are ‘spent’ or exhausted following the release of their procoagulant and anticoagulant factors. These circulating exhausted platelets cannot contribute to primary haemostasis or ex vivo aggregation assays that require platelets to respond to stimulation112,119. Injured patients with impaired platelet aggregation responses also exhibit increased sensitivity to tPA-mediated fibrinolysis, perhaps due to impaired platelet PAI-1 release121. Importantly, these acquired platelet dysfunctions of TIC may not be reversed by transfusion of platelets stored at room temperature122,123, perhaps owing to injury-induced and shock-induced circulating platelet inhibitors124. Recent work suggests cold-stored platelets may be more effective in restoring platelet contribution to haemostasis125,126.
Efforts for deeper molecular phenotyping89,127,128,129,130 have uncovered multiple molecular phenotypes of platelet dysfunction characteristic of TIC, both adaptive and maladaptive in nature (Fig. 4). Whereas the primary effects of platelets contribute to early TIC and haemorrhage, TIC-associated immunoregulation of platelets probably contributes to later TIC hypercoagulability114,131. Specifically, injury induced platelet activation stimulates platelet and leukocyte binding, creating circulating platelet–leukocyte aggregates, which are associated with promoting a procoagulant milieu through the release of platelet factor 4 and increased expression of TF, fibrinogen and factor Xa in animal models118.
Further, platelet-mediated Toll-like receptor 4 (TLR4) signalling, attachment of histone H4 to platelets, platelet ballooning (a shape change that has procoagulant effects), recruitment of monocytes by platelet-derived high mobility group protein B1 (HMGB1), and neutrophil extracellular trap formation89,132,133 are all pro-inflammatory mechanisms identified in association with early failures in platelet haemostasis and later hypercoagulability. Whether the diverse qualitative changes in platelet behaviour characteristic of TIC are favourable remains unclear80, and investigations beyond platelet biomarkers including microfluidics, cell culture, mitochondrial respiration, ultrastructure microscopy and genomic methods, are necessary to uncover platelet targets for alternative TIC therapies beyond human-donated blood products89,99,127,134,135.
Inappropriate thrombin generation
In initial phases of bleeding, thrombin generation may be insufficient, whereas later excessive thrombin generation may contribute to adverse thrombotic events. Insufficient thrombin concentration results in clots composed of thick fibrin fibres with diminished stability, which are prone to fibrinolysis. Thus, the balance between thrombin generation and inhibition is crucial to haemostatic capacity. Depletion of endogenous inhibitors after injury can offset a decrease in procoagulants and increase the risk of thromboembolic complications136,137.
Thrombin generation can be altered by dilution of coagulation factors following fluid therapy, rapid coagulation factor consumption immediately after injury, shock-related systemic acidosis and hypothermia65,138,139. Severely injured patients are prone to having reduced levels of factor V5,140,141, factor VII141, factor X141 and fibrinogen early after injury139,141. However, the reports of decreases in the activity of coagulation factors following severe injury are inconsistent. Concentrations of coagulation factors >30% of physiological levels are generally accepted as sufficient for effective haemostasis142, although this threshold is based on studies on single factor deficiencies. Data from the pre-hospital COMBAT study revealed that coagulation factor activities in severely wounded patients were all >64% of physiological values upon hospital arrival, suggesting adequate factor activity for clot formation. Consequently, we do not know the optimal threshold for clotting factor activity levels after injury, when there are multiple deficiencies coexisting5.
Importantly, a reduction in procoagulants is not necessarily accompanied by impaired thrombin generation141,143. Even though the levels of multiple procoagulants were reduced in patients with traumatic injury, circulating markers of thrombin generation (including prothrombin fragment 1.2 and thrombin–antithrombin complexes) were higher than in uninjured individuals or patients without evidence of TIC141. Elevation of these markers reflects formation of thrombi in sites where they are needed and may constitute a physiological response to injury, with increased thrombin generation in vivo leading to depletion of both procoagulant and anticoagulant factors. Importantly, standard coagulation assays do not reflect the activity of the anticoagulant systems. Thus, a slightly prolonged prothrombin time (PT), international normalized ratio (INR) or activated partial thromboplastin time (aPTT) could reflect a modest depletion of procoagulants, which is not necessarily accompanied by diminished thrombin generation and a bleeding tendency in vivo, as it is offset by depletion of anticoagulants143,144. Blood samples from patients with traumatic injury displayed a higher peak ‘native’ (no activator added) plasma thrombin concentration than samples from healthy individuals despite prolonged standard coagulation tests143. Recent data indicated that upon hospital admission, patients with traumatic injury exhibited 2.5-fold higher average plasma thrombin generation capacity than uninjured individuals145. However, low thrombin generation capacity was evident in 17% of severely injured patients, and a low peak concentration was linked to a fourfold increased odds of requiring a massive transfusion and a threefold greater odds of 30-day mortality145. Furthermore, there may be substantial differences between plasma and whole-blood thrombin assays146. Recent data from whole-blood assays indicate that patients who required a massive transfusion had thrombin generation levels below that in healthy controls18. With respect to late TIC, thrombin is at the cross-road of coagulation and inflammation (Fig. 5), and excessive thrombin generation may have an important role in delayed hypercoagulability in injured patients137.

Once it is activated by the coagulation cascade, the serin protease thrombin can function in procoagulant, anticoagulant, antifibrinolytic and pro-inflammatory or anti-inflammatory pathways. PAR1, protease-activated receptor 1; TAFI, thrombin-activatable fibrinolysis inhibitor. Adapted with permission from ref.319, Wiley.
Fibrinogen depletion
Fibrinogen is the most abundant coagulation factor in blood, with circulating levels in the range of 2–4 g/l in a healthy adult and a circulating half-life of ~4 days147. Conversion of fibrinogen to fibrin occurs via thrombin-mediated cleavage at two sites, exposing binding sites for other fibrin molecules, thereby giving rise to spontaneous polymerization. Each fibrin fibre comprises several hundred to several thousand protofibrils aligned side by side, which provide extraordinary strength and resilience to the scaffold protein77. Fibrin fibres are crosslinked by the transglutaminase enzyme, activated factor XIII, that provides additional mechanical strength and resistance to fibrinolytic degradation148. In addition, fibrinogen binds with high affinity to integrin αIIbβ3 (also termed glycoprotein IIb/IIIa) on platelets, thereby facilitating further platelet aggregation and generating force to contract the fibrin matrix and stabilize the forming clot149.
Fibrinogen is synthesized by hepatocytes, with ~98% of circulating human fibrinogen being derived from the liver150. Circulating fibrinogen levels increase up to 20-fold in the acute phase response, mediated by IL-6 release following tissue injury, infection and inflammation151. Despite its high circulating concentrations, fibrinogen is the first coagulation factor to reach critically low levels in severe bleeding events152,153. In major trauma, key contributors to hypofibrinogenaemia include haemodilution (due to fluid resuscitation), blood loss, consumption in clot formation at the wound sites, hypothermia (which impairs fibrinogen synthesis), fibrinogenolysis and increased degradation due to acidosis138,153. Trauma and haemorrhagic shock are associated with a hyperfibrinolytic state, that occurs in the first few minutes and sometimes persists for hours after injury154. These observations are linked to excessive release of tPA from the endothelium, which overwhelms the availability of its natural inhibitor PAI-1 (ref.155), thereby driving activation of circulating plasminogen to plasmin. Increased plasmin generation shifts the balance of the fibrinolytic system, promoting premature breakdown of fibrin in clots and also fibrinogen degradation.
Low fibrinogen levels upon admission are independently associated with an increase in injury severity and shock156. Moreover, the fibrinogen level upon admission is an independent predictor of the need for transfusion, and 24-hour and 28-day mortality156,157,158. Fibrinogen level has been identified as the most important independent predictor of mortality, but whether this value represents a biomarker (as opposed to a mediator) in patients with traumatic injury remains to be determined.
Dysregulated fibrinolysis
Fibrinolysis activation following severe injury has been documented for over half a century11. Although the exact pathophysiology remains unclear, haemorrhagic shock is common in patients who present to the hospital with elevated fibrinolytic activity9,159,160,161,162. Hyperfibrinolysis is associated with elevated levels of tPA155,163. The source of tPA release during haemorrhagic shock is presumed to be Weibel–Palade vesicles in the endothelium, which are released in response to multiple stimuli164. Weibel–Palade vesicles also store vWF165, and circulating levels of these factors are increased following trauma166.
Hyperfibrinolysis is exacerbated by loss of fibrinolytic inhibitors155,163,166, including α2 antiplasmin167, and platelet dysfunction168 (Fig. 2). Elevated tPA activity with PAI-1 depletion is the hallmark of patients with traumatic injury with hyperfibrinolysis52,155,163,169. In addition, depletion of secondary tPA inhibitors (plasma protease C1 inhibitor and α1 antitrypsin) or factors that modulate inhibitor function (such as vitronectin, the cofactor for PAI-1) also occurs162. Platelet alpha granules are the primary circulating source of PAI-1, which is secreted following stimulation and retained on the surface of activated platelets170. PAI-1 can also be generated in several cells, including endothelial cells. Additional factors that govern clot dissolution, including thrombin-activatable fibrinolysis inhibitor (TAFI; alternatively named carboxypeptidase U, encoded by CPB2) and factor XIII168, are depleted in hyperfibrinolytic patients with traumatic injury171. The antifibrinolytic function of factor XIII is conferred by crosslinking of the plasmin inhibitor α2 antiplasmin into the forming fibrin matrix172. Depletion of factor XIII levels to ~50% has a negative effect on clot stability173. This observation is important, as factor XIII circulates in complex with fibrinogen, which is also depleted in trauma156,158.
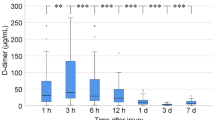
Hyperfibrinolysis is suppressed in most patients with traumatic injury by a surge of PAI-1 that initiates at 2 hours from injury and results in shutdown of fibrinolytic activity in the majority of patients by 12 hours174. This concept, termed fibrinolysis shutdown175, is evident in a broad range of diseases, including viral infections such as COVID-19 (ref.176). Although PAI-1 upregulation occurring hours after injury seems to be a physiological event, fibrinolysis shutdown that occurs within an hour of severe injury is associated with twofold to sixfold increased mortality177. These patients exhibit hallmarks of prior fibrinolysis activation, including elevated levels of D-dimer (a degradation product of crosslinked fibrin) and depletion of fibrinolytic inhibitors, and have low systemic fibrinolytic activity on presentation to the emergency department162. The precise mechanism of acute fibrinolysis shutdown remains unclear. There is some evidence that the plasminogen-binding protein, S100-A10, is shed into the circulation and may associate with tPA, thereby impeding fibrinolysis178. Resuscitation promotes PAI-1 elevation in most injured patients, and the increase is pathological if sustained beyond 24 hours174.
Prior fibrinolytic activation with subsequent shutdown is associated with ongoing coagulation abnormalities, including platelet dysfunction and prolonged PT178,179. It remains controversial whether these patients may have fibrinolysis shutdown at the systemic level while having ongoing bleeding at the local tissue level, a phenomenon termed ‘occult’ hyperfibrinolysis178. Regardless of terms, patients with low fibrinolytic activity, measured by viscoelastic activity tests and elevated D-dimer or plasmin–antiplasmin levels, have increased mortality compared with patients with balanced fibrinolytic activity, with significantly less blood product utilization than patients with hyperfibrinolysis162,178,179. Patients in fibrinolysis shutdown tend to have delayed mortality from brain injury and organ failure, whereas patients with hyperfibrinolysis die early from haemorrhage9. To add complexity, a subset of patients with traumatic injury do not generate a robust fibrinolytic response and present to the hospital in a low fibrinolytic state, which is also associated with increased mortality166. Hypofibrinolysis, defined as lack of fibrinolysis activation with low fibrinolytic activity, remains poorly described in trauma but may contribute to thrombotic complications.
Ongoing work on fibrinolysis in trauma has focused on the temporal changes of fibrinolysis following injury. Most patients with traumatic injury transition to a depressed fibrinolytic state following severe injury180. Patients with traumatic injury who retain low fibrinolytic activity beyond 24 hours (both adults174,180,181 and children182) exhibit increased mortality. This phenomenon could be attributed to elevated PAI-1, which is associated with poor outcomes in sepsis, but requires further investigation in trauma. Alternative mechanisms to inhibit fibrinolysis include activation of a persistent inflammatory state, in which neutrophil elastase has been demonstrated to reduce fibrinolytic activity183.
Sex dimorphism
Sex dimorphisms in coagulation have been described in humans, with women manifesting a more hypercoagulable profile than men184. As women often have less severe and less penetrating trauma (both important TIC risk factors) than men, isolating the independent role of sex in TIC is difficult (Supplementary Tables 1,2). The effect of sex on post-injury morbidity and mortality has been somewhat controversial185,186,187,188. In one study, men up to 50 years of age with blunt injuries had a higher risk of death than women189; among those of ≥50 years of age, no difference in survival was noted following blunt trauma, but women with penetrating injuries had a higher mortality than men. Other studies across the world have shown that following trauma, premenopausal women have a survival advantage over men185,190,191. The presence of TIC may change this picture, as a multicentre trauma study187 found increased mortality among women presenting with TIC, independent of age.
With regard to TIC-associated hypercoagulability, we also observe disparities between women and men. Although men have higher VTE rates than women in their lifetime192,193, women are at increased risk of VTE during pregnancy, when using sex hormones and after ovarian stimulation. In trauma, there is controversy, with some studies showing no sex differences194,195 in VTE rates, and others showing an increased risk in men193. Interestingly, the study showing an increased risk in men included post-discharge VTEs, which represented 62% of the events. In studies using native thromboelastography (TEG), healthy women showed faster clot initiation and stronger clots than men184,196. These differences were more pronounced in pregnant women than in their non-pregnant counterparts184, further suggesting that female sex hormones are involved in coagulation. Oestrogen increases the levels of many clotting factors via gene transcription197. Lower oestradiol levels were associated with higher levels of PAI-1 in a large prospective study of women in the age range 42–52 years198. There were no significant differences in haemostatic factors from before to after menopause, but hormone therapy was associated with lower PAI-1 concentrations. Most studies have found no cyclic variation in coagulation and fibrinolytic factors199. In one study200 using rapid TEG (with TF activation), injured women had faster clot formation, increased clot strength and less fibrinolysis than men, after adjustment for risk factors. Moreover, women were less likely than men to die when presenting with abnormal clot strength or hyperfibrinolysis, despite being older, having longer time from injury to admission, and presenting with lower systolic blood pressure. This sex-specific relative hypercoagulability did not seem to increase the risk of thrombotic morbidity, and it was not dependent on age. It is conceivable that epigenetic or post-translational processes due to lifetime exposure to female sex hormones could alter platelet progenitor function or cellular clotting biology, leading to a persistent hypercoagulable state during menopause201. The same group202 described that healthy women of 18–55 years of age had shorter time to clot formation, higher rate of clot propagation, and increased clot strength than their men counterpart. The study showed higher levels of total and functional fibrinogen in women than men, but no difference in fibrinolysis. Collectively, these findings suggest that more circulating functional fibrinogen and faster coagulation activation may be involved in women’s resilience to TIC. Other studies have found that men have lower fibrinogen levels as well as platelet count and function then women203. Platelets express receptors for oestrogens, which might affect their function and haemostatic ability204, and testosterone reduces agonist-induced platelet aggregation205; however, there are conflicting results regarding platelet aggregation over the menstrual cycle206,207. Platelets from healthy men who were pretreated with oestradiol approximated the activation response to platelet-activating factor of platelets from women208, suggesting that donor sex should be considered in platelet transfusions and encouraging investigation of the therapeutic potential of oestradiol in TIC. Timing is also a potential factor, as serial viscoelastic tests suggest that women convert to a hypercoagulable profile after injury earlier than their male counterparts209.
Diagnosis, screening and prevention
Clinical trials have demonstrated challenges in identifying patients at risk of major bleeding, and, therefore clinically relevant TIC. First, there is controversy over the definition of massive transfusion. Early definitions included the military description of 10 units of RBCs in a 24-hour period210. These definitions have matured to focus on shorter intervals207,208, for example, >4 units of RBCs in the first hour211,212,213, on the basis that the median time to death from bleeding is <3 hours6,9,51, and that longer time frames may lead to survivor bias (that is, patients may die of haemorrhage early, before having the opportunity to receive more blood). Second, although several scores have been proposed, the positive predictive value remains low. Consequently, the lack of scoring systems with good predictive performance represents a major challenge in identifying patients who will develop TIC, and consequently in designing clinical studies. For example, in the large CRASH-2 international trial of TXA for traumatic haemorrhage214, which included >20,000 patients, only half of those who were clinically assessed as being at risk of major bleeding received a blood transfusion.
Diagnosis
Early TIC
While the rates of patients receiving a massive transfusion requiring trauma team intervention are low (3–17%), massively bleeding patients are at great risk of TIC215,216,217. Identification of TIC within a cohort of massively bleeding patients can be augmented by laboratory testing. The conventional tests include a platelet count, Clauss assay to measure fibrinogen level, PT and aPTT. Major limiting factors with these assays are the time to obtaining results from multiple tests and the inability to identify hyperfibrinolysis. The alternative currently is VHAs, which provide several measurements in a single readout (Fig. 6). These individual VHA measurements correspond to the requirement for a specific blood component better than conventional tests202. Conventional coagulation assays can take up to 40 minutes before actionable data are available, whereas VHAs provide real-time data with results in half the time215, with some newer VHAs providing actionable results in 5 minutes, enabling the identification of patients at risk of massive bleeding167,218. Additionally, a clinical scoring system for assessing TIC, which includes subclassifications for the anatomic location of injury and interventions required for bleeding control, has been proposed219. This scoring system correlates well with laboratory-detected coagulopathy and blood transfusions but requires assessment in the operating room166. The rapid availability of the comprehensive information provided by VHAs has led to the recommendation that VHAs should replace conventional coagulation testing in TIC assessment215, although their additional costs limit their accessibility in under-resourced settings. VHAs use to guide resuscitation in trauma has been associated with reduced mortality in a US-based RCT39. The recent ITACTIC study220, however, found no difference in clinical outcomes between resuscitations guided by VHAs and those guided by conventional coagulation tests. However, the VHA transfusion thresholds were based on the same thresholds as for conventional testing used in the control group, thereby creating a circular logic that resulted in the two groups being treated similarly. The conclusion from the ITACTIC study is that resuscitation based on such VHA thresholds did not offer benefit over conventional test guidance, but the study did not provide evidence for different, outcome-based VHA resuscitation thresholds. Although the evidence in trauma is limited at this time, substantial evidence from elective cardiac and liver transplant surgery studies provides further support for the use of VHAs221.

Thromboelastography (TEG) and rotational thromboelastometry (ROTEM) are currently the most widely used viscoelastic assays to assess and manage trauma-induced coagulation320,321. In a typical TEG device, whole blood is transferred to a cylindrical cup, a stationary pin is inserted into the blood, and an oscillating rotational movement is applied to the cup. In typical ROTEM devices, the cup is stationary and the pin oscillates. Both instruments are point-of-care devices and provide similar measurements to reflect the phases of clot formation and clot degradation. Both assays measure speed of thrombin generation, that is, time to until clot firmness of 2 mm (reaction time (R) in TEG and clotting time (CT) in ROTEM); speed of clot formation that includes the contribution of fibrinogen (α angle in both assays); maximum clot strength (maximal amplitude (MA) in TEG and maximal clot firmness (MCF) in ROTEM); and magnitude of fibrinolysis (LY30; that is, the percentage reduction in the area under the curve at 30 min after MA in TEG, and LI30, the residual clot firmness at 30 min after CT in ROTEM). Additional measurements include the contribution of fibrinogen to clot formation independent of platelets (FF, functional fibrinogen, in TEG, and FIBTEM in ROTEM), and a variety of other measurements to reflect platelet function, presence of heparin, presence of thrombin inhibitors and tranexamic acid (TXA) reversibility of fibrinolysis. For TEG, K time is the time interval between reaching a clot amplitude of 2 mm and amplitude of 20 mm, mainly dependent on fibrinogen cleavage and fibrin polymerization; and EPL is estimated percent lysis determined prior to LY30. For ROTEM, A10 is estimated clot firmness at 10 min; CFT is firmness time, the interval between reaching a clot amplitude 2 mm and an amplitude of 20 mm; and ML is maximal lysis between MCF and the lowest amplitude. Adapted from ref.322, Springer Nature Limited.
TIC has been historically defined by PT or the more commonly used INR, with prolongation detected in one in four severely injured patients requiring the intervention of the trauma team82. Mortality in this cohort can be up to fourfold higher than in similarly injured patients without a prolonged PT, and the need for blood product resuscitation is significantly higher. Thus, prolonged PT, INR or aPTT were proposed as clinical tests to identify TIC. However, several studies have subsequently shown that PT or INR may be abnormal after injury despite normal clotting factor activity levels222,223. Furthermore, the exact definition of TIC based on conventional coagulation assays remains a topic of debate as investigators argue over the PT or INR and aPTT thresholds82. Of note, PT or INR reflect only the contribution of plasma proteins to clot formation, without regard for the central role of platelets. Consequently, VHAs have been adopted for the diagnosis of TIC in many countries224,225,226,227, owing to their assessment of whole-blood clot formation and degradation in real time, although there has been criticism regarding assay reproducibility of older versions of VHA devices228. Viscoelastic evidence of decreased clot strength has repeatedly been associated with massive transfusion and increased mortality in trauma229,230,231, although there is disagreement on specific thresholds defining hyperfibrinolysis. A recent study indicates that cut-off values should not be fixed but combined with clinical signs of injury severity and shock232.
Given the vast array of coagulation changes in patients with traumatic injury, defining TIC with a single laboratory measurement is imprecise. TIC is a complex process that involves the endothelium, platelets, circulating coagulation factors and the immune system17,233, and no single assay or set of assays available to date can effectively integrate the measurements of the crucial coordinated events involved in vascular homeostasis to provide a comprehensive evaluation. Ex vivo laboratory assays to assess coagulation fail to account for at least some of these important events. Hyperfibrinolytic phenotypes identified by VHAs are frequently associated with early mortality rates exceeding 50%9,155,159, whereas fibrinolysis shutdown is associated with delayed mortality161. Importantly, the findings of coagulation tests are similar in both paediatric patients and adults, with both populations showing similar phenotypes of TIC, and VHAs predict outcomes in a similar manner in children234 as in adults. Even in the setting of an abnormal laboratory test result, the clinical status of the patient ultimately drives decision making; abnormal laboratory results should not be corrected with blood products in a patient with no clinical signs of coagulopathy and requiring no surgical or interventional haemostasis.
Late TIC
Shock is the dominant risk factor in early TIC, whereas tissue injury is more influential in late TIC. Interestingly, the patients at greatest risk of late TIC or hypercoagulability are those who developed severe early TIC or hypocoagulability. Patients with spine, pelvic and femur fractures, which limit post-injury mobilization, are particularly at high risk of late thrombotic complications. Early TIC is mostly an endogenous coagulation response to tissue injury and haemorrhagic shock. However, with resuscitation, the administration of fluids, blood products and haemostatic agents results in potential secondary coagulopathies as well as haemodilution, acidosis and hypothermia17. Further, in the event of ongoing blood loss due to lack of mechanical control, this coagulopathy is exacerbated. Following resolution of haemorrhage and hypoperfusion, most patients transition from a hypocoagulable to a hypercoagulable state within 24 hours235. Late hypercoagulable TIC presents a new set of challenges in clinical management focused on prevention of thrombotic complications17. Thus, we need to initiate thrombotic pharmacological prophylaxis early in the hospital course. However, the specific laboratory definition of late hypercoagulable TIC remains elusive, but several studies have identified increased clot strength and fibrinolysis shutdown, as measured by VHAs, following resuscitation as risk factors for VTE236,237,238 and stroke239. Repeated VHAs during hospital admission182 identified changes in TIC phenotype associated with thrombotic complications. However, the measures to mitigate late TIC are presently focused on best practice of haemostatic resuscitation with blood products and subsequent thromboprophylaxis. Effective prevention of thromboembolic complications requires a better understanding of the underlying mechanisms, which seem likely to be at the crossroads of inflammation and coagulation extending beyond Virchov’s triad (hypercoagulability, stasis, endothelial damage). Although we have evidence that early thromboembolism prophylaxis is beneficial, its precise timing after injury to prevent thrombotic complications without increasing bleeding risk remains to be determined.
Management
The priorities in the management of the patient at risk for life-threatening bleeding are: mechanically stopping the bleeding, which is fundamentally limited to tourniquets and direct pressure on bleeding sites in the field and rapid access to a capable trauma surgeon; reversing hypovolaemic shock, largely through restoration of circulating blood volume; and restoring clotting homeostasis through administering the right blood products to the right patient at the right time. Damage control resuscitation39,240 and damage control surgery241,242 are further measures in patients at high risk of TIC. Importantly, global variations exist with respect to resuscitation strategies (Fig. 7). For example, the European guidelines243 on the management of major bleeding and coagulopathy following trauma strongly emphasize fibrinogen replacement to overcome rapid depletion of fibrinogen in patients with traumatic injury. This approach varies from a largely ‘plasma and platelets first’ approach to haemostatic resuscitation in the United States. Further complicating this issue is the lack of stored blood in many under-resourced settings, making recommendations for management regionally dependent.

There is substantial variation in management algorithms for early, hypocoagulable trauma-induced coagulopathy (TIC) throughout the world. These algorithms are designed for the general trauma patient. Isolated traumatic brain injury (TBI) provokes a unique TIC phenotype and, therefore, may warrant TBI-specific blood product thresholds for thromboelastography (TEG) and rotational thromboelastometry (ROTEM). a | An example of a US goal-driven approach for a patient at risk of a massive transfusion. Resuscitation is initiated with a balanced blood product strategy with a focus on reversing haemorrhagic shock. In many institutions this consists of a plasma, platelet and RBC formula at a ratio of 1:1:1 with some centres using a plasma to RBC ratio of 1:2, and more recently some have employed low-titre type O-positive whole blood. Regardless of the initial ratio, crystalloids are not administered until shock is reversed, and calcium chloride is given empirically. RBC are continued to maintain haemoglobin >10 g/dl. A rapid TEG measurement is obtained as soon as possible to guide subsequent blood product and tranexamic acid (TXA) administration, according to thresholds derived from clinical studies. If the patient requires >4 RBC during the first hour and TEG results are unavailable, cryoprecipitate and platelets are delivered. b | Examples of algorithms used in some European countries. Isotonic crystalloids and vasopressors are begun initially, and supplemented with fibrinogen and RBC or plasma and RBC at a ratio of 1:2 in the patient at risk of a massive transfusion. TXA is given to all patients, and calcium is corrected according to laboratory testing. RBC are continued to maintain haemoglobin >7 g/dl. Blood products are subsequently provided according to a goal-directed platform based on either ROTEM or conventional coagulation assays. ACT, activated clotting time; aPTT, activated partial thromboplastin time; CCA, conventional coagulation assay; LY30, percentage reduction in the area under the curve at 30 min after MA in TEG; MA, maximum amplitude; PT, prothrombin time; RBC, red blood cell; SBP, systolic blood pressure; VHA, viscoelastic haemostatic assay.
Current post-injury resuscitation protocols are not sex-specific, theoretically exposing women to unnecessary transfusions. Given the low representation of women in cohorts of patients with traumatic injury, a type 2 error cannot be excluded as an explanation for the lack of sex differences in transfusion requirements187,222. Carefully sized, inevitably large, RCTs testing sex-specific thresholds for haemostatic resuscitation, considering menstrual cycle, pregnancy and menopause, will ultimately be required.
Pre-hospital care
The initial management of TIC focuses on preventing progression to haemorrhagic shock, by arresting the bleeding and restoring circulating blood volume. Efforts have been made to raise public awareness of strategies such as tourniquets and direct compression of bleeding wounds to slow haemorrhage (for example, the Stop the Bleed campaign and the STOP the Bleeding Campaign)244,245. Pre-hospital health-care providers also initiate resuscitation of the critically ill patient with traumatic injury to increase intravascular volume to preserve organ perfusion. The fluids administered in the pre-hospital setting can help but also potentially harm the patient. Resuscitation using large volumes of crystalloids can increase blood pressure but may also exacerbate coagulopathy and burst the forming clots246, if blood pressure is raised too rapidly. High-volume crystalloid resuscitation has been associated with hyperfibrinolysis upon presentation to the hospital159, partially through dilution of circulating antifibrinolytic proteins247, and is independently associated with morbidity248. Although permissive hypotension with low-volume crystalloid administration was demonstrated to be effective for the management of patients with traumatic injury in a landmark study in the pre-hospital setting249, a subsequent subgroup analysis indicated the benefit was limited to patients with pericardial tamponade. While a permissive hypotensive strategy in actively bleeding patients is advocated in patients with traumatic injury until definitive bleeding control can be achieved, the optimal level of hypotension remains to be established, particularly in patients with associated TBI.
Additional therapies in the pre-hospital setting include the transfusion of blood products. Pre-hospital plasma resuscitation reduces mortality in patients who require helicopter transportation with prolonged transport times6. However, pre-hospital plasma resuscitation in an urban setting with short transportation times did not reduce mortality and was associated with a prolonged INR5. In a post hoc analysis of two clinical trials of pre-hospital plasma transfusion, the benefit of this measure seemed to be limited to those with a blunt injury and transport times >20 minutes250. Ongoing work is evaluating the potential role of lyophilized plasma in the pre-hospital setting (as it overcomes the logistical challenges of thawing plasma in the mobile setting) and of whole blood as a pre-hospital resuscitation strategy (which has been proven to be feasible251, but its effect on coagulopathy remains to be determined252).
Tranexamic acid
TXA has been shown to reduce mortality in a large international trauma study214, and, therefore, it has been implemented in some pre-hospital systems and included in the WHO list of essential medicines for the treatment of trauma253. However, the patient group that would benefit the most from TXA remains unclear. The Study of Tranexamic Acid During Air and Ground Medical Prehospital Transport (STAAMP)254 trial was a phase III, multicentre RCT of TXA versus placebo, given within an estimated 2 hours of injury in the pre-hospital setting to patients with hypotension or tachycardia. There was no significant difference in the primary outcome of 30-day mortality. However, in a pre-planned subgroup analysis, patients with severe shock (systolic blood pressure <70 mmHg) who received TXA within 1 hour of injury had a significant reduction in 30-day mortality. The Prehospital TXA for TBI trial255 was a randomized, double-blind, placebo-controlled multicentre phase II trial designed to assess the safety and efficacy of TXA given within 2 hours of injury in the pre-hospital setting to patients with moderate to severe TBI but without haemorrhagic shock. Similarly, no significant differences in the primary outcome of favourable neurological function at 6 months or in 28-day mortality were observed. A secondary analysis of this study determined that the highest TXA dosage tested was associated with a trend towards reduced mortality254, although at the expense of an increase in the rate of seizures from 2% (placebo) to 6%. However, at this time, no RCTs investigating TXA have randomized patients on the basis of their fibrinolytic status.
The potential risk of VTE with TXA treatment has been a topic of debate. Studies in both civilian256 and military medicine257 suggest an association with increased VTE rates, although these studies were limited by their retrospective design. The use of TXA as an antifibrinolytic drug could theoretically increase the incidence of post-injury fibrinolysis shutdown, and the observation that late (>3 hours after injury) TXA administration is associated with death21 indicates that TXA should be administered early and probably not to patients with evidence of fibrinolysis shutdown258,259. A small single-centre randomized trial in patients with severe traumatic injury suggested a small but significant dose-dependent increase in a composite outcome of thrombotic events in patients receiving TXA260. The recent HALT-IT trial261 of TXA in patients with gastrointestinal haemorrhage indicated a significant increase in VTE in those receiving TXA given over 24 hours. By contrast, the CRASH-2 (ref.214) and CRASH-3 (ref.262) trials randomized tens of thousands of patients at risk of haemorrhage after injury to TXA or placebo, and neither study demonstrated an increase in– VTE. This finding aligns with the observation of safety and low VTE rates in randomized trials investigating the use of TXA in patients with other bleeding conditions such as post-partum haemorrhage263. The CRASH-2 and CRASH-3 trials have been criticized for reporting VTE rates substantially lower than those found in most studies, probably due to the low rate of patients who actually required transfusion and lack of VTE screening. As expected, the STAAMP and Prehospital TXA for TBI trials had substantially higher VTE rates than those reported in the CRASH studies264. At this moment, the use of TXA varies widely between Europe and North America. In Europe, most emergency medical services (EMS) deliver TXA in the field, and most hospitals administer TXA to patients at risk for TIC. In the USA, TXA is given more selectively, with many EMS providing TXA for severely injured patients in the field, whereas in-hospital TXA is limited to selected groups, often guided by VHA.
Hospital care
There is wide variation around the globe in protocols for the treatment of severe traumatic haemorrhage243,265,266,267,268,269, which, to some extent, may reflect availability of resources and the most frequent types of injuries in the areas served by the hospitals. However, when resources are available, most guidelines emphasize high ratios of plasma to RBCs, restricted use of crystalloids (with some emphasizing the role of isotonic crystalloid solutions243), permissive hypotension until active bleeding is controlled (except in the setting of TBI, when it has been contraindicated270,271), and goal-directed haemostatic resuscitation with blood components and antifibrinolytics once laboratory test results are available. While European guidelines recommend restricted, balanced crystalloids with inotropic agents to restore volume, and fibrinogen and prothrombin complex concentrate (PCC) to provide clotting factors243, US guidelines emphasize plasma with the goal of volume resuscitation as well as restoration of endothelial function267.
Empirical blood product resuscitation is warranted in patients who arrive at the hospital in overt haemorrhagic shock, to restore circulating blood volume and thereby attenuate the development of worsening coagulopathy. This approach includes a high ratio of plasma to RBCs210; the exact ratio of plasma to RBCs remains debated but should be at minimum 1:2. The only RCT evaluating 1:1 and 1:2 ratios demonstrated no benefit in survival with the 1:1 ratio38, but suggested a shorter time to haemostasis in this group. This study also included early platelet transfusions in the 1:1 arm (because apheresis platelets (~6 units) could not be split-up in the first round of transfusion), which has been associated with improved outcomes in trauma in a retrospective study272. Of note, these improved outcomes are limited to patients with traumatic injury undergoing a massive transfusion, and there is evidence that patients with traumatic injury who do not receive a massive transfusion may be harmed by these protocols. High ratios of fibrinogen or cryoprecipitate to RBCs have also been advocated to decrease trauma mortality273, but the results of ongoing RCTs are awaited before this approach can be recommended. If empirical ratios of plasma to RBCs are used to resuscitate massively bleeding patients, cryoprecipitate or fibrinogen and platelets should be administered if the patient requires >4 units of RBCs before laboratory results are available213. An alternative to high ratio resuscitation (and a historical practice) is whole blood274. Low anti-A and anti-B titre, group O whole blood (LTOWB) was the standard for trauma resuscitation until blood component separation in the early 1980s, and it has been shown to be feasible and safe as initial fluid in some US centres275,276. In a single-centre study in injured adults, the use of LTOWB has been associated with reduced transfusion volumes and increased survival compared with individual blood component therapy277. Of note, bicarbonate should be given only if severe acidosis persists despite resuscitation, as bicarbonate therapy has been shown to be ineffective or harmful, owing to excess HCO3-derived CO2, which is very soluble across cell membranes, causing a cellular respiratory acidosis not reflected in arterial pH or PaCO2 measurements278,279.
Coagulation monitoring
All patients with traumatic injury and major bleeding need ongoing coagulation assessment regardless of their transfusion strategy, as demonstrated in a RCT of VHA-guided goal-directed resuscitation, in which mortality was reduced by almost 50%39. Goal-directed resuscitation targets the patient’s specific coagulation phenotype, with the objective of achieving a balanced coagulation profile without excessive blood component use, an approach that has been associated with the best outcome280. The choice of specific haemostatic adjuncts (including plasma, cryoprecipitate or fibrinogen, platelets and TXA) can be directed with algorithms based on VHAs or equivalent haemostatic assays281,282,283 (Fig. 6). A recent meta-analysis supports this concept, as pooled data from randomized control studies has shown that patients undergoing VHA-based emergency resuscitation received less plasma and platelets than those treated on the basis of conventional coagulation testing, with an associated reduction in postoperative mechanical ventilation days221. However, a reliable test to ascertain when to administer platelets in trauma patients is lacking. Impairment in platelet-dependent clotting does not predict the need for platelet transfusion, nor do platelet transfusions reverse these impairments122,123,284. Ultimately, the clinical status of the patient should dictate empirical platelet transfusion. Although no specific studies have evaluated the timing of coagulation assessment in trauma, the dynamic nature of resuscitation warrants early and repeated monitoring until haemostasis is achieved, and a return to balanced resuscitation is warranted if haemostasis is lost.
Special considerations
Trauma patients who present to the hospital with occult bleeding but who are physiologically compensated pose a different challenge in resuscitation. A common source for occult bleeding is injury to solid abdominal organs, which can be managed predominantly with non-operative interventions. Targeted resuscitation of these patients is an important approach, as most transition to a hypercoagulable state by 24–48 hours236,237.
TBI poses unique challenges in the management of coagulation. Unlike the abdominal and thoracic cavities that can accommodate moderate amounts of bleeding, the fixed space in the skull warrants a more aggressive approach to obtain timely haemostasis to avoid intracranial hypertension. The CRASH-3 study identified a survival benefit of early TXA administration in patients with mild-to-moderate but not severe TBI262. However, the protective effect of TXA in TBI remains unclear and may be more related to countering inflammation than improving haemostasis. One major challenge in TBI-associated TIC is the use of pharmacological prophylaxis against VTE. The transition to hypercoagulability after injury poses a major clinical threat, with high VTE rates noted in patients who have TBI. The decision to initiate anticoagulation to prevent post-injury clotting and thrombotic complications needs to be weighed against the risk of exacerbating bleeding, particularly in patients with substantial brain injury. Early VTE prophylaxis in patients with TBI has been associated with a reduction in VTE incidence without worsening of intracranial haemorrhage when started after a head CT scan indicates stabilization285. Low molecular weight heparin (LMWH) has been shown to be superior to unfractionated heparin (UFH) in this cohort, with improved survival and fewer thromboembolic complications286. Owing to patient immobility and the hypercoagulable state, thromboprophylaxis is administered as soon as possible after the bleeding risk has subsided, and individual patient factors must guide the choice of UFH or LMWH287,288.
Quality of life
Data from the WHO Global Burden of Disease and Injury study showed that, despite sizeable improvements since the 1990s, injury remains a substantial cause of morbidity worldwide289. Few studies have provided information on the long-term outcomes in patients with TIC with the hypocoagulable phenotype. The Trauma Recovery Project (TRP) is a large prospective study in which the quality of life (QOL) as well as functional and psychological (depression and post-traumatic stress disorder (PTSD)) outcomes in injured patients without serious head injury are assessed290,291. Post-injury functional limitation was present in >75% of adults at the 12-month and 18-month follow-up. Depression was present in 60% of patients at discharge and 31% at the 6-month follow-up. In subsequent TRP studies, women were more likely than men to have poor QOL at 6, 12 and 18 months228,290. The TRP in Adolescents 1999–2002 study292, which focused on injured adolescents without serious brain trauma, showed acute stress disorder in 40% of individuals upon discharge, which was associated with large QOL deficits at 3, 6, 12 and 24 months. The long-term PTSD rate was 27%293. In a longitudinal study of 156 children with blunt trauma (but no head or spinal cord injuries), physical scores remained lower than in age-matched healthy controls at 6 months294.
In the CONTROL international RCT of activated factor VII295, which included severely injured adults with refractory bleeding but no severe brain injuries, survivors reported very poor QOL 3 months after injury, with over 70% reporting moderate or extreme difficulties in usual activities, pain or discomfort and mobility limitations. Over half of the patients reported self-care problems and anxiety or depression. In an Australian trauma centre, Glasgow Outcome Score–Extended (GOSE) was assessed at 6 and 12 months in adults who required massive blood transfusion after injury (patients with TBI were not excluded), and massive transfusion was independently associated with unfavourable outcomes among survivors at 6 months after injury296.
The hypercoagulable extreme of TIC, prevalent in patients who survive the initial resuscitation, is linked to multiple organ failure and ARDS, as well as macro-thrombotic complications such as VTE9,166, all of which are associated with prolonged hospitalization. Patients requiring prolonged intensive care develop a state of chronic critical illness (CCI; defined as ≥14 days in ICU with persistent organ dysfunction)297,298, which was associated with dismal long-term outcomes in a series of trauma patients without TBI297,299. Individuals with CCI but not TBI had significantly lower physical function and QOL than their counterparts who rapidly recovered, at 3, 6 and 12 months299. Of 135 adult patients with blunt trauma and haemorrhagic shock who survived beyond 48 hours after injury, 19% developed CCI; these patients were more likely to require long-term care and, at 4 months, scored lower in general health measures297.
The incidence of VTE after injury depends on whether routine surveillance is implemented (as opposed to symptom-driven diagnosis) and the use of thromboprophylaxis. A comparison of two US trauma centres showed that serial VTE surveillance via ultrasonography detected DVT in 9% of patients admitted for >48 hours, whereas surveillance of only symptomatic patients showed a 2% DVT incidence, despite similar thromboprophylaxis protocols300. Pulmonary embolism was diagnosed in 0.4% of these patients, a result similar to the finding of another US multicentre investigation301. Analyses of the US National Trauma Data Bank showed in-hospital VTE rates of around 1%194,302,303. In a Swiss trauma centre, VTE rates were in the range 7–10%304, and in Germany a rate of 2% was reported305. A higher VTE incidence has been reported in a military population than in civilians (22% and 2%, respectively)306,307,308. A review of long-term functional outcomes after an acute pulmonary embolism showed that more than half of the patients reported dyspnoea and poor physical performance309, with rates of chronic thromboembolic pulmonary hypertension as high as 3.8% at 2 years310. The incidence of post-thrombotic syndrome, a chronic debilitating consequence of acute DVT, has been estimated at 20–50% even when appropriately treated311.
Outlook
Perhaps the most conspicuous gaps to be addressed in the management of TIC are a clinical definition and the ability to further distinguish between the dynamic early hypocoagulable state and the late hypercoagulable state on the bases of mechanistic foundations. The initial proposal to use PT or INR to define early TIC was subsequently questioned, and these measurements are rather considered biomarkers of injury. Clinical coagulation scoring systems have been developed but are relatively insensitive unless based on body cavity exposure in the operating room. Definitions of massive transfusion do not capture the effect of TIC on TBI. Additionally, multiple TIC phenotypes exist, and these need to be defined to optimize goal-directed therapy. Finally, in patients with refractory early TIC, we are unable to distinguish those who are dying because they are bleeding from those bleeding because they are dying.
Mechanisms
The investigation of the fundamental mechanisms of TIC has been pursued since the work of W.B. Cannon in World War I312. Although there has been remarkable progress, M. Stefanini’s words in his address to the New York Academy of Medicine in 1954 remain applicable: “The ponderous literature on the subject of hemostasis could perhaps be considered a classical example of the infinite ability of the human mind for abstract speculation. For several years, the number of working theories of the hemostatic mechanisms greatly exceeded and not always respected the confirmed experimental facts. In recent years, however, the revived interest in this field has led to an accumulation of new findings, which has been almost too rapid for their orderly incorporation into a logical working pattern”313. Stefanini’s words still apply to today, but we have made substantial progress since the 1990s, stimulated by the introduction of the cell-based model of haemostasis and the confirmation that uncontrolled cavitary bleeding was the leading cause of death in the war in Iraq3. Nevertheless, there are many gaps to be addressed.
Several hypotheses have been proposed on the mechanisms driving early TIC, but definitive evidence remains elusive for many. Notably among the controversial proposals are the roles of thrombin-induced activation of protein C and heparan sulfate shed from the disrupted endothelial glycocalyx. Shock and tissue ischaemia seem to dominate in early TIC, but the mediators remain to be identified, including metabolites. The contributing role of tissue injury is also unclear and may be crucial in determining early fibrinolytic phenotypes. A study of military casualties showed that blast injuries (due to explosions) caused significantly more plasmin activation than gunshot wounds141. Interestingly, isolated shock or TBI are not associated with a pronounced TIC but, in combination with tissue injury, provoke a severe early TIC. In fact, the mechanisms driving TIC from TBI seem to be unique and need to be elucidated. There is clear evidence of crosstalk between inflammation and coagulation, but the direct links in TIC remain to be elucidated.
Early TIC has been suggested to result from a combination of inadequate thrombin generation, platelet dysfunction, fibrinogen depletion and hyperfibrinolysis. However, the relative contribution of these abnormalities remains unclear, and some of these coagulation aberrations may be biomarkers rather than critical mechanistic drivers. Although thrombin generation has been described as accelerated following trauma, recent evidence indicates it is depressed in patients requiring a massive transfusion18. By contrast, excessive thrombin generation has been associated with VTE137. What constitutes appropriate thrombin concentration in the evolution of TIC is unknown. Platelet dysfunction has been well documented following severe injury, but its aetiology, mechanism and role in early and late TIC remain unclear. Dysfunctional circulating platelets may represent platelets that have already functioned, rather than the haemostatic capacity of the platelet reserves in the local injury environment. Fibrinogen is the first circulating coagulation protein to decrease following severe injury, but what critical level warrants replacement remains to be established. The overlapping roles of platelets and fibrinogen in clot formation compound the issue; that is, one of these components may compensate for a deficiency in the other. In addition, the effect of fibrinolysis phenotypes in early and late TIC remains debated. Whereas hyperfibrinolysis may compromise haemostasis, fibrinolysis shutdown may contribute to later ARDS and multiple organ failure. These observations continue to raise the question as to whether TXA should be used selectively following injury based on the degree of shock, particularly in the USA. Finally, the endothelium is the ‘black box’ in TIC. Endothelial activation has been invoked as a dominant feature of early TIC, but this suggestion is based on the presence of shed biomarkers, rather than real-time assessment of endothelial behaviour. EOT, and more specifically barrier breakdown, has been documented in vitro and in vivo, but the relationship with TIC remains speculative.
Diagnosis
The optimal coagulation test to assess early and late TIC has yet to be developed. The shortcomings of traditional plasma-based tests (PT or INR and aPTT) have been recognized, and these assays have been replaced to some degree by VHAs (TEG and rotational thromboelastometry (ROTEM)). Although TEG and ROTEM whole-blood assays reflect the various phases of clot formation, they are not performed on activated endothelium with physiological shear stress. Furthermore, these devices cannot reliably ascertain platelet functional capability, and their precision in measuring fibrinolysis is debated314. Microfluidic devices have been employed for research and will probably emerge for clinical use soon. Consequently, at this time, we are unable to accurately determine the specific blood products and their optimal administration timing required to avert a massive transfusion or progressive TBI. We are currently unable to clearly discern the transition from hypocoagulability to hypercoagulability, and this state may differ between the arterial (high shear stress) and venous (low shear stress) systems. Consequently, we continue to debate when to initiate VTE preventive therapy and the optimal preventive agents.
Management
Ultimately, the goal of personalized medicine for injured patients at risk of TIC is to deliver the right product(s) at the right time to the right patient. However, our current understanding of the pathophysiology of TIC remains incomplete despite intense research focus, and compounded by limitations in diagnostic testing, rendering current clinical decisions imprecise; these decisions should be continually refined by a thorough appraisal of the literature on TIC (Box 1). Unanswered questions include whether cryoprecipitate is superior to fibrinogen concentrates, whether PCCs are equivalent to fresh frozen plasma, and how to identify the patients who can benefit the most from TXA. Although initial administration of LTOWB may seem attractive, returning to stored whole blood throughout resuscitation is not consistent with our knowledge of the varied phenotypes of TIC. Furthermore, type O blood has lower vWF activity levels, and has been shown to be associated with post-injury massive transfusion and adverse outcomes315. Although other studies were unable to reproduce this association316,317, a type II error was a concern in these other investigations. Protocols to address severe bleeding vary widely around the globe; while some of the variations are necessary and appropriate to match the local available resources and predominant injury patterns, others reflect the lack of solid scientific evidence.
Change history
22 April 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41572-022-00360-y
References
World Health Organization. Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2016 (WHO, 2018).
Tisherman, S. A. et al. Detailed description of all deaths in both the shock and traumatic brain injury hypertonic saline trials of the Resuscitation Outcomes Consortium. Ann. Surg. 261, 586–590 (2015).
Eastridge, B. J. et al. Death on the battlefield (2001-2011): implications for the future of combat casualty care. J. Trauma Acute Care Surg. 73, S431–S437 (2012).
Fox, E. E. et al. Earlier endpoints are required for hemorrhagic shock trials among severely injured patients. Shock. 47, 567–573 (2017).
Moore, H. B. et al. Plasma-first resuscitation to treat haemorrhagic shock during emergency ground transportation in an urban area: a randomised trial. Lancet 392, 283–291 (2018). This randomized controlled trial shows that pre-hospital plasma in a ground ambulance system does not improve survival in seriously injured patients.
Sperry, J. L. et al. Prehospital plasma during air medical transport in trauma patients at risk for hemorrhagic shock. N. Engl. J. Med. 379, 315–326 (2018). This randomized controlled trial shows that pre-hospital plasma during helicopter transport improves survival in seriously injured patients.
Sauaia A., Moore E. E., Wade C., Holcomb J. B. in Trauma Induced Coagulopathy 2nd edn (eds Moore H. B., Neal M. D. & Moore E. E.) 13-27 (Springer, 2021).
Kalkwarf, K. J. et al. Bleeding to death in a big city: an analysis of all trauma deaths from hemorrhage in a metropolitan area over one year. J. Trauma Acute Care Surg. 89, 716–722 (2020).
Moore, H. B. et al. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J. Trauma Acute Care Surg. 77, 811–817 (2014). Distinct phenotypes of fibrinolysis were identified by thromboelastography, indicating increased mortality for both hyperfibrinolysis and fibrinolysis shutdown.
Macfarlane, R. G. & Biggs, R. Fibrinolysis; its mechanism and significance. Blood 3, 1167–1187 (1948).
Innes, D. & Sevitt, S. Coagulation and fibrinolysis in injured patients. J. Clin. Pathol. 17, 1–13 (1964).
Kashuk, J. L., Moore, E. E., Millikan, J. S. & Moore, J. B. Major abdominal vascular trauma–a unified approach. J. Trauma 22, 672–679 (1982). The first clinical study to propose the ‘lethal triad’ as a unifying mechanism for hypocoagulable TIC.
Morton, A. P. et al. Revisiting early postinjury mortality: are they bleeding because they are dying or dying because they are bleeding? J. Surg. Res. 179, 5–9 (2013). This clinical review highlights the challenge of determining whether a critically injured patient is dying because the patient has acquired TIC or TIC reflects irreversible metabolic failure.
Chang, R. et al. Abnormalities of laboratory coagulation tests versus clinically evident coagulopathic bleeding: results from the prehospital resuscitation on helicopters study (PROHS). Surgery. 163, 819–826 (2018).
Moore E. & Moore H. in Trauma Induced Coagulopathy 2nd edn (ed. Moore H. B. Moore, E. E. & Neal M. D.) 3-11(Springer 2020).
Gando, S., Levi, M. & Toh, C. H. Disseminated intravascular coagulation. Nat. Rev. Dis. Prim. 2, 16037 (2016).
Moore, H. B. et al. Defining trauma-induced coagulopathy with respect to future implications for patient management: communication from the SSC of the ISTH. J. Thromb. Haemost. 18, 740–747 (2020). The ISTH developed a consensus statement that outlines the key differences between TIC and DIC; DIC is well described in association with sepsis.
Coleman J. J., et al. Whole blood thrombin generation is impaired in injured patients requiring a massive transfusion. J. Am. Coll. Surg. https://doi.org/10.1016/j.jamcollsurg.2020.12.058 (2021).Together with Cardenas et al. (J. Trauma Acute Care Surg., 2014), this study shows that thrombin generation, measured in plasma or whole blood, is impaired in injured patients requiring a massive transfusion.
Sobrino, J. & Shafi, S. Timing and causes of death after injuries. Proceedings 26, 120–123 (2013).
Sauaia, A. et al. Epidemiology of trauma deaths: a reassessment. J. Trauma. 38, 185–193 (1995).
CRASH-2 Collaborators. The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 377, 1096–1101 (2011).
Kahl, J. E. et al. The changing nature of death on the trauma service. J. Trauma Acute Care Surg. 75, 195–201 (2013).
Shackford, S. R. et al. The epidemiology of traumatic death. A population-based analysis. Arch. Surg. 128, 571–575 (1993).
Callcut, R. A. et al. The why and how our trauma patients die: a prospective multicenter Western Trauma Association study. J. Trauma Acute Care Surg. 86, 864–870 (2019).
Evans, J. A. et al. Epidemiology of traumatic deaths: comprehensive population-based assessment. World J. Surg. 34, 158–163 (2010).
Soreide, K. et al. Epidemiology and contemporary patterns of trauma deaths: changing place, similar pace, older face. World J. Surg. 31, 2092–2103 (2007).
Oyeniyi, B. T. et al. Trends in 1029 trauma deaths at a level 1 trauma center: impact of a bleeding control bundle of care. Injury 48, 5–12 (2017).
Teixeira, P. G. R. et al. Preventable or potentially preventable mortality at a mature trauma center. J. Trauma Acute Care Surg. 63, 1338–1347 (2007).
Brohi, K., Singh, J., Heron, M. & Coats, T. Acute traumatic coagulopathy. J. Trauma Acute Care Surg. 54, 1127–1130 (2003). Together with MacLeod et al. (2003), this article documents evidence of TIC in 25% of injured patients arriving at the hospital prior to resuscitation.
Roberts, D. J. et al. One thousand consecutive in-hospital deaths following severe injury: has the etiology of traumatic inpatient death changed in Canada? Can. J. Surg. 61, 150–152 (2018).
Arslan, E. D. et al. Assessment of traumatic deaths in a level one trauma center in Ankara, Turkey. Eur. J. Trauma Emerg. Surg. 41, 319–323 (2015).
Trajano, A. D., Pereira, B. M. & Fraga, G. P. Epidemiology of in-hospital trauma deaths in a Brazilian university hospital. BMC Emerg. Med. 14, 22 (2014).
Kleber, C. et al. Overall distribution of trauma-related deaths in Berlin 2010: advancement or stagnation of German trauma management? World J. Surg. 36, 2125–2130 (2012).
Jochems, D., Leenen, L. P. H., Hietbrink, F., Houwert, R. M. & van Wessem, K. J. P. Increased reduction in exsanguination rates leaves brain injury as the only major cause of death in blunt trauma. Injury 49, 1661–1667 (2018).
Drake, S. A. et al. Establishing a regional trauma preventable/potentially preventable death rate. Ann. Surg. 271, 375–382 (2018).
Koh, E. Y. et al. Trends in potentially preventable trauma deaths between 2005-2006 and 2012-2013. Am. J. Surg. 218, 501–506 (2019).
Brenner, A. et al. Outcome measures in clinical trials of treatments for acute severe haemorrhage. Trials 19, 533 (2018).
Holcomb, J. B. et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 313, 471–482 (2015). This randomized controlled trial did not detect a difference in survival employing a transfusion strategy of 1:1:1 ratio versus a 1:1:2 ratio of plasma to platelets to RBCs, but more patients in the 1:1:1 group achieved haemostasis and fewer experienced death due to exsanguination by 24 hours.
Gonzalez, E. et al. Goal-directed hemostatic resuscitation of trauma-induced coagulopathy: a pragmatic randomized clinical trial comparing a viscoelastic assay to conventional coagulation assays. Ann. Surg. 263, 1051–1059 (2016). This randomized controlled trial indicates improved survival when blood products are delivered to seriously injured patients on the basis of thromboelastography compared with conventional coagulation testing.
Roberts, I. et al. The CRASH-2 trial: a randomised controlled trial and economic evaluation of the effects of tranexamic acid on death, vascular occlusive events and transfusion requirement in bleeding trauma patients. Health Technol. Assess. 17, 1–79 (2013).
Moore, E. E. et al. Human polymerized hemoglobin for the treatment of hemorrhagic shock when blood is unavailable: the USA multicenter trial. J. Am. Coll. Surg. 208, 1–13 (2009).
Holcomb, J. B. et al. The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study: comparative effectiveness of a time-varying treatment with competing risks. JAMA Surg. 148, 127–136 (2013).
Holcomb, J. B. et al. Evidence based and clinically relevant outcomes for hemorrhage control trauma trials. Ann. Surg. 273, 395–401 (2021).
Strumwasser, A. et al. The impact of acute coagulopathy on mortality in pediatric trauma patients. J. Trauma Acute Care Surg. 81, 312–318 (2016).
Wafaisade, A. et al. Acute coagulopathy in isolated blunt traumatic brain injury. Neurocriti. Care 12, 211–219 (2010).
Peltan, I. D., Vande Vusse, L. K., Maier, R. V. & Watkins, T. R. An international normalized ratio-based definition of acute traumatic coagulopathy is associated with mortality, venous thromboembolism, and multiple organ failure after injury. Crit. Care Med. 43, 1429–1438 (2015).
Frith, D. et al. Definition and drivers of acute traumatic coagulopathy: clinical and experimental investigations. J. Thromb. Haemost. 8, 1919–1925 (2010).
Niles, S. E. et al. Increased mortality associated with the early coagulopathy of trauma in combat casualties. J. Trauma 64, 1459–1463; discussion 1463–1465 (2008).
Kutcher, M. E. et al. Evolving beyond the vicious triad: differential mediation of traumatic coagulopathy by injury, shock, and resuscitation. J. Trauma Acute Care Surg. 78, 516–523 (2015).
Maegele, M. et al. Early coagulopathy in multiple injury: an analysis from the German Trauma Registry on 8724 patients. Injury. 38, 298–304 (2007).
Brohi, K. et al. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann. Surg. 245, 812–818 (2007). This provocative report describes the potential role of activated protein C in the pathogenesis of TIC.
Cohen, M. J. et al. Early coagulopathy after traumatic brain injury: the role of hypoperfusion and the protein C pathway. J. trauma. 63, 1254–1261; discussion 1261–1262 (2007).
Moore, H. B. et al. Forgot calcium? Admission ionized-calcium in two civilian randomized controlled trials of pre-hospital plasma for traumatic hemorrhagic shock. J. Trauma Acute Care Surg. 88, 588–596 (2020).
Ditzel, R. M. Jr. et al. A review of transfusion- and trauma-induced hypocalcemia: is it time to change the lethal triad to the lethal diamond? J. Trauma Acute Care Surg. 88, 434–439 (2020).
van ‘t Veer, C. & Mann, K. G. Regulation of tissue factor initiated thrombin generation by the stoichiometric inhibitors tissue factor pathway inhibitor, antithrombin-III, and heparin cofactor-II. J. Biol. Chem. 272, 4367–4377 (1997).
Hoffman, M. & Monroe, D. M. III A cell-based model of hemostasis. Thromb. Haemost. 85, 958–965 (2001). The seminal work that introduced the novel concept of the essential role of cellular elements in clot formation.
Li, Z., Delaney, M. K., O’Brien, K. A. & Du, X. Signaling during platelet adhesion and activation. Arterioscler. Thromb. Vasc. Biol. 30, 2341–2349 (2010).
Rossaint, R. et al. Key issues in advanced bleeding care in trauma. Shock 26, 322–331 (2006).
Martini, W. Z., Pusateri, A. E., Uscilowicz, J. M., Delgado, A. V. & Holcomb, J. B. Independent contributions of hypothermia and acidosis to coagulopathy in swine. J. Trauma 58, 1002–1009; discussion 1009–1010 (2005).
Engström, M., Schött, U., Romner, B. & Reinstrup, P. Acidosis impairs the coagulation: a thromboelastographic study. J. Trauma 61, 624–628 (2006).
Martini, W. Z. & Holcomb, J. B. Acidosis and coagulopathy: the differential effects on fibrinogen synthesis and breakdown in pigs. Ann. Surg. 246, 831–835 (2007).
Lier, H., Krep, H., Schroeder, S. & Stuber, F. Preconditions of hemostasis in trauma: a review. The influence of acidosis, hypocalcemia, anemia, and hypothermia on functional hemostasis in trauma. J. Trauma. 65, 951–960 (2008).
Mitrophanov, A. Y., Szlam, F., Sniecinski, R. M., Levy, J. H. & Reifman, J. Controlled multifactorial coagulopathy: effects of dilution, hypothermia, and acidosis on thrombin generation in vitro. Anesth. Analg. 130, 1063–1076 (2020).
Marumo, M., Suehiro, A., Kakishita, E., Groschner, K. & Wakabayashi, I. Extracellular pH affects platelet aggregation associated with modulation of store-operated Ca2+ entry. Thromb. Res. 104, 353–360 (2001).
Meng, Z. H., Wolberg, A. S., Monroe, D. M. III & Hoffman, M. The effect of temperature and pH on the activity of factor VIIa: implications for the efficacy of high-dose factor VIIa in hypothermic and acidotic patients. J. Trauma. 55, 886–891 (2003).
Butler F. K., Jr., et al. Advanced resuscitative care in tactical combat casualty care: TCCC guidelines change 18-01:14 October 2018. J. Spec. Oper. Med.18:37-55.
Magnotti, L. J. et al. Admission ionized calcium levels predict the need for multiple transfusions: a prospective study of 591 critically ill trauma patients. J. Trauma. 70, 391–395; discussion 395–397 (2011).
Wolberg, A. S., Meng, Z. H., Monroe, D. M. III & Hoffman, M. A systematic evaluation of the effect of temperature on coagulation enzyme activity and platelet function. J. Trauma Acute Care Surg. 56, 1221–1228 (2004).
Kashuk, J. L. et al. Postinjury life threatening coagulopathy: is 1:1 fresh frozen plasma:packed red blood cells the answer? J. Trauma. 65, 261–270; discussion 270–271 (2008).
Tauber, H. et al. Prevalence and impact of abnormal ROTEM® assays in severe blunt trauma: results of the ‘Diagnosis and Treatment of Trauma-Induced Coagulopathy (DIA-TRE-TIC) study’. Br. J. Anaesth. 107, 378–387 (2011).
Thorn, S., Lefering, R., Maegele, M., Gruen, R. L. & Mitra, B. Early prediction of acute traumatic coagulopathy: a validation of the COAST score using the German Trauma Registry. Eur. J. Trauma Emerg. Surg. https://doi.org/10.1007/s00068-019-01142-0 (2019).
Cap, A. & Hunt, B. Acute traumatic coagulopathy. Curr. Opin. Crit. Care 20, 638–645 (2014).
Cohen, M. J. et al. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann. Surg. 255, 379–385 (2012).
Cap, A. & Hunt, B. J. The pathogenesis of traumatic coagulopathy. Anaesthesia. 70, 96–101 (2015).
Peltz, E. D. et al. Pathologic metabolism: an exploratory study of the plasma metabolome of critical injury. J. Trauma Acute Care Surg. 78, 742–751 (2015).
White, N. J. et al. Post-translational oxidative modification of fibrinogen is associated with coagulopathy after traumatic injury. Free Radic. Biol. Med. 96, 181–189 (2016).
Weisel, J. W. & Litvinov, R. I. Mechanisms of fibrin polymerization and clinical implications. Blood. 121, 1712–1719 (2013).
Steele, T., Kolamunnage-Dona, R., Downey, C., Toh, C. H. & Welters, I. Assessment and clinical course of hypocalcemia in critical illness. Crit. Care 17, R106 (2013).
Hardaway, R. M. & Mc, K. D. Intravascular thrombi and the intestinal factor of irreversible shock. Ann. Surg. 150, 261–265 (1959).
Vulliamy, P. et al. Alterations in platelet behavior after major trauma: adaptive or maladaptive? Platelets 32, 295–304 (2020). This review reflects our current understanding of adaptive and maladaptive alterations in platelet biology induced by severe trauma, and identifies existing knowledge gaps as well as their therapeutic implications.
Gando, S. Tissue factor in trauma and organ dysfunction. Semin. Thromb. Hemost. 32, 48–53 (2006).
MacLeod, J. B., Lynn, M., McKenney, M. G., Cohn, S. M. & Murtha, M. Early coagulopathy predicts mortality in trauma. J. trauma. 55, 39–44 (2003).
Neal, M. D. The great platelet paradox: evolution of platelet contribution to hemostasis, inflammation, and thrombosis after injury. Blood Adv. 4, 2556 (2020).
Maegele, M. et al. Coagulopathy and haemorrhagic progression in traumatic brain injury: advances in mechanisms, diagnosis, and management. Lancet Neurol. 16, 630–647 (2017).
Zhang, J., Zhang, F. & Dong, J. F. Coagulopathy induced by traumatic brain injury: systemic manifestation of a localized injury. Blood 131, 2001–2006 (2018).
Moore, H. B. et al. Hemolysis exacerbates hyperfibrinolysis, whereas platelolysis shuts down fibrinolysis: evolving concepts of the spectrum of fibrinolysis in response to severe injury. Shock 43, 39–46 (2015).
Deguchi, H. et al. Prothrombotic skeletal muscle myosin directly enhances prothrombin activation by binding factors Xa and Va. Blood 128, 1870–1878 (2016).
Dyer, M. R. et al. Platelet-derived extracellular vesicles released after trauma promote hemostasis and contribute to DVT in mice. J. Thromb. Haemost. 17, 1733–1745 (2019).
Vulliamy, P. et al. Histone H4 induces platelet ballooning and microparticle release during trauma hemorrhage. Proc. Natl Acad. Sci. USA 116, 17444–17449 (2019).
Kozar, R. A. & Pati, S. Syndecan-1 restitution by plasma after hemorrhagic shock. J. Trauma Acute Care Surg. 78, S83–S86 (2015).
Simão, F. & Feener, E. P. The effects of the contact activation system on hemorrhage. Front. Med. 4, 121 (2017).
Johansson, P. I., Stensballe, J., Rasmussen, L. S. & Ostrowski, S. R. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann. Surg. 254, 194–200 (2011).
Gonzalez Rodriguez, E. et al. Syndecan-1: a quantitative marker for the endotheliopathy of trauma. J. Am. Coll. Surg. 225, 419–427 (2017).
Ban, K. et al. Plasma-Mediated gut protection after hemorrhagic shock is lessened in syndecan-1−/− mice. Shock 44, 452–457 (2015).
Ostrowski, S. R. & Johansson, P. I. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J. Trauma Acute Care Surg. 73, 60–66 (2012).
Brohi, K. et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J. Trauma 64, 1211–1217; discussion 1217 (2008).
Johansson, P. I. et al. High sCD40L levels early after trauma are associated with enhanced shock, sympathoadrenal activation, tissue and endothelial damage, coagulopathy and mortality. J. Thromb. Haemost. 10, 207–216 (2012).
Dyer, M. R. et al. Traumatic injury results in prolonged circulation of ultralarge von Willebrand factor and a reduction in ADAMTS13 activity. Transfusion 60, 1308–1318 (2020).
Kornblith, L. Z. et al. Perhaps it’s not the platelet: ristocetin uncovers the potential role of von Willebrand factor in impaired platelet aggregation following traumatic brain injury. J. Trauma Acute Care Surg. 85, 873–880 (2018).
Xu, X., Kozar, R., Zhang, J. & Dong, J.-F. Diverse activities of von Willebrand factor in traumatic brain injury and associated coagulopathy. J. Thromb. Haemost. 18, 3154–3162 (2020).
Pati, S. et al. Protective effects of fresh frozen plasma on vascular endothelial permeability, coagulation, and resuscitation after hemorrhagic shock are time dependent and diminish between days 0 and 5 after thaw. J. Trauma 69, S55–S63 (2010).
Kozar, R. A. et al. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth. Analg. 112, 1289–1295 (2011). This study shows that plasma resuscitation of haemorrhagic shock restores the endothelial glycocalyx and alleviates trauma endotheliopathy.
Peng, Z. et al. Fresh frozen plasma lessens pulmonary endothelial inflammation and hyperpermeability after hemorrhagic shock and is associated with loss of syndecan 1. Shock 40, 195–202 (2013).
Haywood-Watson, R. J. et al. Modulation of syndecan-1 shedding after hemorrhagic shock and resuscitation. PLoS ONE 6, e23530 (2011).
Gruen, D. S. et al. Prehospital plasma is associated with distinct biomarker expression following injury. JCI Insight 5, e135350 (2020).
Fitzgerald, M. L., Wang, Z., Park, P. W., Murphy, G. & Bernfield, M. Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell Biol. 148, 811–824 (2000).
Prudovsky, I. et al. Tranexamic acid suppresses the release of mitochondrial DNA, protects the endothelial monolayer and enhances oxidative phosphorylation. J. Cell Physiol. 234, 19121–19129 (2019).
Diebel, M. E., Martin, J. V., Liberati, D. M. & Diebel, L. N. The temporal response and mechanism of action of tranexamic acid in endothelial glycocalyx degradation. J. Trauma Acute Care Surg. 84, 75–80 (2018).
Weyrich, A. S. & Zimmerman, G. A. Platelets: signaling cells in the immune continuum. Trends Immunol. 25, 489–495 (2004).
Rondina, M. T., Weyrich, A. S. & Zimmerman, G. A. Platelets as cellular effectors of inflammation in vascular diseases. Circ. Res. 112, 1506–1519 (2013).
Nachman, R. L. & Rafii, S. Platelets, petechiae, and preservation of the vascular wall. N. Engl. J. Med. 359, 1261–1270 (2008).
Kutcher, M. E. et al. Characterization of platelet dysfunction after trauma. J. Trauma Acute Care Surg. 73, 13–19 (2012). This seminal report characterizes platelet dysfunction in the majority of injured patients evident at the time of hospital presentation.
Tweardy, D. J. et al. Essential role for platelets in organ injury and inflammation in resuscitated hemorrhagic shock. Shock 26, 386–390 (2006).
Ding, N. et al. Toll-like receptor 4 regulates platelet function and contributes to coagulation abnormality and organ injury in hemorrhagic shock and resuscitation. Circ. Cardiovasc. Genet. 7, 615–624 (2014).
Brown, L. M., Call, M. S., Knudson, M. M. & Cohen, M. J., Trauma Outcomes Group. A normal platelet count may not be enough: the impact of admission platelet count on mortality and transfusion in severely injured trauma patients. J. Trauma 71, S337–S342 (2011).
Stansbury, L. G. et al. The clinical significance of platelet counts in the first 24 hours after severe injury. Transfusion 53, 783–789 (2013).
Kornblith, L. Z. et al. Fibrinogen and platelet contributions to clot formation: implications for trauma resuscitation and thromboprophylaxis. J. Trauma Acute Care Surg. 76, 255–256; discussion 262–263 (2014).
Zipperle, J. et al. Potential role of platelet-leukocyte aggregation in trauma-induced coagulopathy: ex vivo findings. J. Trauma Acute Care Surg. 82, 921–926 (2017).
Starr, N. E. et al. Identification of injury and shock driven effects on ex-vivo platelet aggregometry: a cautionary tale of phenotyping. J. Trauma Acute Care Surg. 89, 20–28 (2020).
Plautz, W. E. et al. Von Willebrand factor as a thrombotic and inflammatory mediator in critical illness. Transfusion 60, S158–S166 (2020).
Moore, H. B. et al. Shock induced systemic hyperfibrinolysis is attenuated by plasma first resuscitation. J. Trauma Acute Care Surg. 79, 897–903; discussion 903–904 (2015).
Kornblith, L. Z. et al. It’s about time: transfusion effects on postinjury platelet aggregation over time. J. Trauma Acute Care Surg. 87, 1042–1051 (2019).
Vulliamy, P. et al. Platelet transfusions reduce fibrinolysis but do not restore platelet function during trauma hemorrhage. J. Trauma Acute Care Surg. 83, 388–397 (2017).
Fields, A. T. et al. Good platelets gone bad: the effects of trauma patient plasma on healthy platelet aggregation. Shock 55, 189–197 (2021).
Nair, P. M., Pidcoke, H. F., Cap, A. P. & Ramasubramanian, A. K. Effect of cold storage on shear-induced platelet aggregation and clot strength. J. Trauma Acute Care Surg. 77, S88–S93 (2014).
Reddoch, K. M. et al. Hemostatic function of apheresis platelets stored at 4°C and 22°C. Shock 41, 54–61 (2014).
Kornblith, L. Z. et al. A journey upstream: fluctuating platelet-specific genes in cell free plasma as proof-of-concept for using RNA sequencing to improve understanding of post-injury platelet biology. J. Trauma Acute Care Surg. 88, 742–751 (2020).
Li, R., Elmongy, H., Sims, C. & Diamond, S. L. Ex vivo recapitulation of trauma-induced coagulopathy and preliminary assessment of trauma patient platelet function under flow using microfluidic technology. J. Trauma Acute Care Surg. 80, 440–449 (2016).
Colace, T. V., Jobson, J. & Diamond, S. L. Relipidated tissue factor linked to collagen surfaces potentiates platelet adhesion and fibrin formation in a microfluidic model of vessel injury. Bioconjug Chem. 22, 2104–2109 (2011).
Neeves, K. B. et al. Microfluidic focal thrombosis model for measuring murine platelet deposition and stability: PAR4 signaling enhances shear-resistance of platelet aggregates. J. Thromb. Haemost. 6, 2193–2201 (2008).
Vogel, S. et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Invest. 125, 4638–4654 (2015).
Dyer, M. R. et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci. Rep. 8, 2068 (2018).
Stark, K. et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 128, 2435–2449 (2016).
Verni, C. C., Davila, A. Jr., Balian, S., Sims, C. A. & Diamond, S. L. Platelet dysfunction during trauma involves diverse signaling pathways and an inhibitory activity in patient-derived plasma. J. Trauma Acute Care Surg. 86, 250–259 (2019).
Lee, M. Y., Verni, C. C., Herbig, B. A. & Diamond, S. L. Soluble fibrin causes an acquired platelet glycoprotein VI signaling defect: implications for coagulopathy. J. Thromb. Haemost. 15, 2396–2407 (2017).
Butenas, S., van’t Veer, C. & Mann, K. G. “Normal” thrombin generation. Blood. 94, 2169–2178 (1999).
Park, M. S. et al. Thrombin generation profiles as predictors of symptomatic venous thromboembolism after trauma: a prospective cohort study. J. Trauma Acute Care Surg. 83, 381–387 (2017). This study shows that postinjury enhanced thrombin generation is a risk factor for venous thromboembolism.
Martini, W. Z. Coagulopathy by hypothermia and acidosis: mechanisms of thrombin generation and fibrinogen availability. J. Trauma 67, 202–208; discussion 208–209 (2009).
Floccard, B. et al. Early coagulopathy in trauma patients: an on-scene and hospital admission study. Injury 43, 26–32 (2012).
Rizoli, S. B. et al. Clotting factor deficiency in early trauma-associated coagulopathy. J. Trauma 71, S427–S434 (2011).
Woolley, T. et al. A prospective observational study of acute traumatic coagulopathy in traumatic bleeding from the battlefield. Transfusion 60, S52–S61 (2020).
Dzik, W. H. The James Blundell Award Lecture 2006: transfusion and the treatment of haemorrhage: past, present and future. Transfus. Med. 17, 367–374 (2007).
Dunbar, N. M. & Chandler, W. L. Thrombin generation in trauma patients. Transfusion 49, 2652–2660 (2009).
Negrier, C., Shima, M. & Hoffman, M. The central role of thrombin in bleeding disorders. Blood Rev. 38, 100582 (2019).
Cardenas, J. C. et al. Measuring thrombin generation as a tool for predicting hemostatic potential and transfusion requirements following trauma. J. Trauma Acute Care Surg. 77, 839–845 (2014).
Coleman, J. R. et al. Whole blood thrombin generation is distinct from plasma thrombin generation in healthy volunteers and after severe injury. Surgery 166, 1122–1127 (2019).
Mosesson, M. W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 3, 1894–1904 (2005).
Muszbek, L., Bereczky, Z., Bagoly, Z., Komaromi, I. & Katona, E. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol. Rev. 91, 931–972 (2011).
Kononova, O. et al. Mechanistic basis for the binding of RGD- and AGDV-peptides to the platelet integrin αIIbβ3. Biochemistry 56, 1932–1942 (2017).
Tennent, G. A. et al. Human plasma fibrinogen is synthesized in the liver. Blood. 109, 1971–1974 (2007).
Levy, J. H., Szlam, F., Tanaka, K. A. & Sniecienski, R. M. Fibrinogen and hemostasis: a primary hemostatic target for the management of acquired bleeding. Anesth. Analg. 114, 261–274 (2012).
Hiippala, S. T., Myllyla, G. J. & Vahtera, E. M. Hemostatic factors and replacement of major blood loss with plasma-poor red cell concentrates. Anesth. Analg. 81, 360–365 (1995).
Schlimp, C. J. & Schochl, H. The role of fibrinogen in trauma-induced coagulopathy. Hamostaseologie 34, 29–39 (2014). Fibrinogen is the first clotting factor to become depleted in the bleeding injured patient; based on this study, fibrinogen levels should be maintained at >150 mg/dl in bleeding patients.
Raza, I. et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J. Thromb. Haemost. 11, 307–314 (2013).
Chapman, M. P. et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J. Trauma Acute Care Surg. 80, 16–23; discussion 23–25 (2016). This study shows that hyperfibrinolysis seems to be due to extensive tPA release that overwhelms circulating PAI-1 levels.
Rourke, C. et al. Fibrinogen levels during trauma hemorrhage, response to replacement therapy, and association with patient outcomes. J. Thromb. Haemost. 10, 1342–1351 (2012).
McQuilten, Z. K., Wood, E. M., Bailey, M., Cameron, P. A. & Cooper, D. J. Fibrinogen is an independent predictor of mortality in major trauma patients: a five-year statewide cohort study. Injury 48, 1074–1081 (2017).
Inaba, K. et al. Impact of fibrinogen levels on outcomes after acute injury in patients requiring a massive transfusion. J. Am. Coll. Surg. 216, 290–297 (2013).
Cotton, B. A. et al. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J. Trauma Acute Care Surg. 73, 365–370; discussion 370 (2012).
Schochl, H., Frietsch, T., Pavelka, M. & Jambor, C. Hyperfibrinolysis after major trauma: differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J. Trauma. 67, 125–131 (2009).
Moore, H. B. et al. Acute fibrinolysis shutdown after injury occurs frequently and increases mortality: a multicenter evaluation of 2,540 severely injured patients. J. Am. Coll. Surg. 222, 347–355 (2016).
Moore, H. B. et al. Does tranexamic acid improve clot strength in severely injured patients who have elevated fibrin degradation products and low fibrinolytic activity, measured by thrombelastography? J. Am. Coll. Surg. 229, 92–101 (2019).
Cardenas, J. C. et al. Elevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patients. Shock 41, 514–521 (2014).
Schillemans, M., Karampini, E., Kat, M. & Bierings, R. Exocytosis of Weibel-Palade bodies: how to unpack a vascular emergency kit. J. Thromb. Haemost. 17, 6–18 (2019).
Huber, D. et al. Tissue-type plasminogen activator (t-PA) is stored in Weibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood 99, 3637–3645 (2002).
Moore, H. B. et al. Fibrinolysis shutdown is associated with a fivefold increase in mortality in trauma patients lacking hypersensitivity to tissue plasminogen activator. J. Trauma acute care Surg. 83, 1014–1022 (2017).
Barrett, C. D. et al. Plasmin TEG rapidly identifies trauma patients at risk for massive transfusion, mortality and hyperfibrinolysis: a diagnostic tool to resolve an international debate on TXA? J. Trauma Acute Care Surg. 89, 991–998 (2020).
Moore, H. B. et al. Viscoelastic measurements of platelet function, not fibrinogen function, predicts sensitivity to tissue-type plasminogen activator in trauma patients. J. Thromb. Haemost. 13, 1878–1887 (2015).
Davenport, R. A. et al. Activated protein C drives the hyperfibrinolysis of acute traumatic coagulopathy. Anesthesiology. 126, 115–127 (2017).
Morrow, G. B., Whyte, C. S. & Mutch, N. J. Functional plasminogen activator inhibitor 1 is retained on the activated platelet membrane following platelet activation. Haematologica. 105, 2824–2833 (2019).
Bouma, B. N. & Mosnier, L. O. Thrombin activatable fibrinolysis inhibitor (TAFI) at the interface between coagulation and fibrinolysis. Pathophysiol. Haemost. Thromb. 33, 375–381 (2003).
Mutch, N. J. et al. Model thrombi formed under flow reveal the role of factor XIII-mediated cross-linking in resistance to fibrinolysis. J. Thromb. Haemost. 8, 2017–2024 (2010).
Fraser, S. R., Booth, N. A. & Mutch, N. J. The antifibrinolytic function of factor XIII is exclusively expressed through α2-antiplasmin cross-linking. Blood 117, 6371–6374 (2011).
Moore, H. B. et al. Reperfusion shutdown: delayed onset of fibrinolysis resistance after resuscitation from hemorrhagic shock is associated with increased circulating levels of plasminogen activator inhibitor-1 and postinjury complications. Blood 128, 206 (2016).
Chakrabarti, R., Hocking, E. D. & Fearnley, G. R. Reaction pattern to three stresses–electroplexy, surgery, and myocardial infarction–of fibrinolysis and plasma fibrinogen. J. Clin. Pathol. 22, 659–662 (1969).
Wright, F. L. et al. Fibrinolysis shutdown correlates to thromboembolic events in severe COVID-19 infection. JACS. 231, 193–203.e1 (2020).
Moore, H. B. et al. Fibrinolysis shutdown in trauma: historical review and clinical implications. Anesth Analg. 129, 762–773 (2019). Postinjury fibrinolysis shutdown has been documented recently with the advent of viscoelastic haemostatic assays, but has been recognized in experimental work for the past 50 years.
Gall, L. S. et al. The S100A10 pathway mediates an occult hyperfibrinolytic subtype in trauma patients. Ann. Surg. 269, 1184–1191 (2019).
Cardenas, J. C. et al. TEG lysis shutdown represents coagulopathy in bleeding trauma patients: analysis of the PROPPR cohort. Shock 51, 273–283 (2019).
Roberts, D. J. et al. Time course and outcomes associated with transient versus persistent fibrinolytic phenotypes after injury: a nested, prospective, multicenter cohort study. J. Trauma Acute Care Surg. 86, 206–213 (2019).
Meizoso, J. P. et al. Persistent fibrinolysis shutdown is associated with increased mortality in severely injured trauma patients. J. Am. Coll. Surg. 224, 575–582 (2017).
Leeper, C. M., Neal, M. D., McKenna, C. J. & Gaines, B. A. Trending fibrinolytic dysregulation: fibrinolysis shutdown in the days after injury is associated with poor outcome in severely injured children. Ann. Surg. 266, 508–515 (2017).
Barrett, C. D. et al. Human neutrophil elastase mediates fibrinolysis shutdown through competitive degradation of plasminogen and generation of angiostatin. J. Trauma. Acute Care Surg. 83, 1053–1061 (2017).
Gorton, H. J., Warren, E. R., Simpson, N. A., Lyons, G. R. & Columb, M. O. Thromboelastography identifies sex-related differences in coagulation. Anesth. Analg. 91, 1279–1281 (2000).
Haider, A. H. et al. Evidence of hormonal basis for improved survival among females with trauma-associated shock: an analysis of the National Trauma Data Bank. J. Trauma 69, 537–540 (2010).
Napolitano, L. M. et al. Gender differences in adverse outcomes after blunt trauma. J. Trauma 50, 274–280 (2001).
Brown, J. B. et al. Characterization of acute coagulopathy and sexual dimorphism after injury: females and coagulopathy just do not mix. J. Trauma Acute Care Surg. 73, 1395–1400 (2012).
Magnotti, L. J., Fischer, P. E., Zarzaur, B. L., Fabian, T. C. & Croce, M. A. Impact of gender on outcomes after blunt injury: a definitive analysis of more than 36,000 trauma patients. J. Am. Coll. Surg. 206, 984–991; discussion 991–992 (2008).
George, R. L. et al. Age-related gender differential in outcome after blunt or penetrating trauma. Shock 19, 28–32 (2003).
Wohltmann, C. D. et al. A multicenter evaluation of whether gender dimorphism affects survival after trauma. Am. J. Surg. 181, 297–300 (2001).
Croce, M. A., Fabian, T. C., Malhotra, A. K., Bee, T. K. & Miller, P. R. Does gender difference influence outcome? J. Trauma 53, 889–894 (2002).
Roach, R. E. J., Cannegieter, S. C. & Lijfering, W. M. Differential risks in men and women for first and recurrent venous thrombosis: the role of genes and environment. J. Thromb. Haemost. 12, 1593–1600 (2014).
Park, M. S. et al. Risk factors for venous thromboembolism after acute trauma: a population-based case-cohort study. Thromb. Res. 144, 40–45 (2016).
Knudson, M. M., Gomez, D., Haas, B., Cohen, M. J. & Nathens, A. B. Three thousand seven hundred thirty-eight posttraumatic pulmonary emboli: a new look at an old disease. Ann. Surg. 254, 625–632 (2011).
Berndtson, A. E., Costantini, T. W., Smith, A. M., Kobayashi, L. & Coimbra, R. Does sex matter? Effects on venous thromboembolism risk in screened trauma patients. J. Trauma Acute Care Surg. 81, 493–499 (2016).
Francis, J. L., Francis, D. A. & Gunathilagan, G. J. Assessment of hypercoagulability in patients with cancer using the Sonoclot analyzer and thromboelastography. Thromb. Res. 74, 335–346 (1994).
Farsetti, A. et al. Molecular basis of estrogen regulation of Hageman factor XII gene expression. Endocrinology 136, 5076–5083 (1995).
Sowers, M. R. et al. Hemostatic factors and estrogen during the menopausal transition. J. Clin. Endocrinol. Metab. 90, 5942–5948 (2005).
Knol, H. M., Kemperman, R. F., Kluin-Nelemans, H. C., Mulder, A. B. & Meijer, K. Haemostatic variables during normal menstrual cycle. A systematic review. Thromb. Haemost. 107, 22–29 (2012).
Coleman, J. R. et al. Trauma resuscitation consideration: sex matters. J. Am. Coll. Surg. 228, 760–768.e1 (2019).
Heldring, N. et al. Estrogen receptors: how do they signal and what are their targets. Physiol. Rev. 87, 905–931 (2007).
Coleman, J. R. et al. Untangling sex dimorphisms in coagulation: initial steps towards precision medicine for trauma resuscitation. Ann. Surg. 271, e128–e130 (2020).
Gee, A. C. et al. The influence of sex hormones on coagulation and inflammation in the trauma patient. Shock 29, 334–341 (2008).
Dupuis, M. et al. Effects of estrogens on platelets and megakaryocytes. Int. J. Mol. Sci. 20, 3111 (2019).
Haque, S. F. et al. Sex difference in platelet aggregation detected by new aggregometry using light scattering. Endocr. J. 48, 33–41 (2001).
Berlin, G., Hammar, M., Tapper, L. & Tynngård, N. Effects of age, gender and menstrual cycle on platelet function assessed by impedance aggregometry. Platelets 30, 473–479 (2019).
Teran, E., Escudero, C. & Vivero, S. Physiological changes in platelet aggregation and nitric oxide levels during menstrual cycle in healthy women. Nitric Oxide 7, 217–220 (2002).
Coleman, J. R. et al. Female platelets have distinct functional activity compared with male platelets: implications in transfusion practice and treatment of trauma-induced coagulopathy. J. Trauma Acute Care Surg. 87, 1052–1060 (2019).
Schreiber, M. A., Differding, J., Thorborg, P., Mayberry, J. C. & Mullins, R. J. Hypercoagulability is most prevalent early after injury and in female patients. J. Trauma 58, 475–480; discussion 480–481 (2005).
Borgman, M. A. et al. The ratio of blood products transfused affects mortality in patients receiving massive transfusions at a combat support hospital. J. Trauma 63, 805–813 (2007). This military study introduces the concept of a high ratio of plasma to RBCs for haemostatic resuscitation.
Meyer, D. E. et al. A comparison of resuscitation intensity and critical administration threshold in predicting early mortality among bleeding patients: a multicenter validation in 680 major transfusion patients. J. Trauma Acute Care Surg. 85, 691–696 (2018).
Savage, S. A. et al. The new metric to define large-volume hemorrhage: results of a prospective study of the critical administration threshold. J. Trauma Acute Care Surg. 78, 224–229; discussion 229–230 (2015).
Nunns, G. R. et al. Empiric transfusion strategies during life-threatening hemorrhage. Surgery 164, 306–311 (2018).
CRASH-2 Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 376, 23–32 (2010). This randomized controlled trial of 20,211 injured patients conducted in 40 countries indicates a survival benefit of TXA when given <3 hours from injury.
Holcomb, J. B. et al. Admission rapid thrombelastography can replace conventional coagulation tests in the emergency department: experience with 1974 consecutive trauma patients. Ann. Surg. 256, 476–486 (2012).
Moore, H. B. et al. Viscoelastic tissue plasminogen activator challenge predicts massive transfusion in 15 minutes. J. Am. Coll. Surg. 225, 138–147 (2017).
Ives, C. et al. Hyperfibrinolysis elicited via thromboelastography predicts mortality in trauma. J. Am. Coll. Surg. 215, 496–502 (2012).
Kelly, J. M., Rizoli, S., Veigas, P., Hollands, S. & Min, A. Using rotational thromboelastometry clot firmness at 5 minutes (ROTEM((R)) EXTEM A5) to predict massive transfusion and in-hospital mortality in trauma: a retrospective analysis of 1146 patients. Anaesthesia 73, 1103–1109 (2018).
Neal, M. D. et al. Clinical assessment of trauma-induced coagulopathy and its contribution to postinjury mortality: a TACTIC proposal. J. Trauma Acute Care Surg. 79, 490–492 (2015).
Baksaas-Aasen, K. et al. Viscoelastic haemostatic assay augmented protocols for major trauma haemorrhage (ITACTIC): a randomized, controlled trial. Intensive Care Med. 47, 49–59 (2020).
Dias, J. D., Sauaia, A., Achneck, H. E., Hartmann, J. & Moore, E. E. Thromboelastography-guided therapy improves patient blood management and certain clinical outcomes in elective cardiac and liver surgery and emergency resuscitation: a systematic review and analysis. J. Thromb. Haemost. 17, 984–994 (2019).
McCully, S. P. et al. The international normalized ratio overestimates coagulopathy in stable trauma and surgical patients. J. Trauma Acute Care Surg. 75, 947–953 (2013).
Stettler, G. R. et al. Variability in international normalized ratio and activated partial thromboplastin time after injury are not explained by coagulation factor deficits. J. Trauma Acute Care Surg. 87, 582–589 (2019).
Prat, N. J. et al. Rotational thromboelastometry significantly optimizes transfusion practices for damage control resuscitation in combat casualties. J. Trauma Acute Care Surg. 83, 373–380 (2017). Implementation of ROTEM at a military base improved blood product use compared with an approach involving a fixed ratio of plasma to RBCs.
Kashuk, J. L. et al. Postinjury coagulopathy management: goal directed resuscitation via POC thrombelastography. Ann. Surg. 251, 604–614 (2010).
Cotton, B. A. et al. Rapid thrombelastography delivers real-time results that predict transfusion within 1 hour of admission. J. Trauma 71, 71:407–414; discussion 414–417 (2011).
Cotton, B. A. et al. Admission rapid thrombelastography predicts development of pulmonary embolism in trauma patients. J. Trauma Acute Care Surg. 72, 1470–1475; discussion 1475–1477 (2012).
Holbrook, T. L., Hoyt, D. B. & Anderson, J. P. The importance of gender on outcome after major trauma: functional and psychologic outcomes in women versus men. J. Trauma 50, 270–273 (2001).
Pezold, M. et al. Viscoelastic clot strength predicts coagulation-related mortality within 15 minutes. Surgery 151, 48–54 (2012).
Nystrup, K. B., Windelov, N. A., Thomsen, A. B. & Johansson, P. I. Reduced clot strength upon admission, evaluated by thrombelastography (TEG), in trauma patients is independently associated with increased 30-day mortality. Scand. J. Trauma. Resusc. Emerg. Med. 19, 52 (2011).
Plotkin, A. J. et al. A reduction in clot formation rate and strength assessed by thrombelastography is indicative of transfusion requirements in patients with penetrating injuries. J. Trauma 64, S64–S68 (2008).
Vigneshwar, N. et al. Precision medicine or one size does not fit all: clinical tolerance to hyperfibrinolysis differs by shock and injury severity. Ann. Surg. https://doi.org/10.1097/SLA.0000000000004548 (2020).
Kornblith, L. Z., Moore, H. B. & Cohen, M. J. Trauma-induced coagulopathy: the past, present, and future. J. Thromb. Haemost. 17, 852–862 (2019).
Leeper, C. M., Neal, M. D., McKenna, C., Sperry, J. L. & Gaines, B. A. Abnormalities in fibrinolysis at the time of admission are associated with deep vein thrombosis, mortality, and disability in a pediatric trauma population. J. Trauma Acute Care Surg. 82, 27–34 (2017).
Moore, H. B. & Moore, E. E. Temporal changes in fibrinolysis following injury. Semin. Thromb. Hemost. 46, 189–198 (2020).
Coleman, J. R. et al. It’s sooner than you think: blunt solid organ injury patients are already hypercoagulable upon hospital admission – results of a bi-institutional, prospective study. Am. J. Surg. 218, 1065–1073 (2019).
Chapman, B. C. et al. Hypercoagulability following blunt solid abdominal organ injury: when to initiate anticoagulation. Am. J. Surg. 206, 917–922; discussion 922–923 (2013).
Gary, J. L. et al. Can thrombelastography predict venous thromboembolic events in patients with severe extremity trauma? J. Orthop. Trauma 30, 294–298 (2016).
Sumislawski, J. J. et al. Not all in your head (and neck): stroke after blunt cerebrovascular injury is associated with systemic hypercoagulability. J. Trauma Acute Care Surg. 87, 1082–1087 (2019).
Holcomb, J. B. et al. Damage control resuscitation: directly addressing the early coagulopathy of trauma. J. Trauma 62, 307–310 (2007).
Moore, E. E. Staged laparotomy for the hypothermia, acidosis, and coagulopathy syndrome. Am. J. Surg. 172, 405–410 (1996).
Stone, H. H., Strom, P. R. & Mullins, R. J. Management of the major coagulopathy with onset during laparotomy. Ann. Surg. 197, 532–535 (1983).
Spahn, D. R. et al. The European guideline on management of major bleeding and coagulopathy following trauma: fifth edition. Crit. Care 23, 98 (2019). These European guidelines reflect the currently most accepted protocol for the management of TIC, although there is some variation among western European countries. It brings a wealth of curated references on TIC mechanisms, diagnosis and treatment.
Goolsby, C. et al. Stop the Bleed Education Consortium: education program content and delivery recommendations. J. Trauma Acute Care Surg. 84, 205–210 (2018).
Rossaint, R. et al. The STOP the bleeding campaign. Crit. Care 17, 136 (2013).
Shaftan, G. W., Chiu, C. J., Dennis, C. & Harris, B. Fundamentals of physiologic control of arterial hemorrhage. Surgery 58, 851–856 (1965).
Moore, H. B. et al. Plasma is the physiologic buffer of tissue plasminogen activator-mediated fibrinolysis: rationale for plasma-first resuscitation after life-threatening hemorrhage. J. Am. Coll. Surg. 220, 872–879 (2015).
Neal, M. D. et al. Crystalloid to packed red blood cell transfusion ratio in the massively transfused patient: when a little goes a long way. J. Trauma Acute Care Surg. 72, 892–898 (2012).
Bickell, W. H. et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. N. Engl. J. Med. 331, 1105–1109 (1994).
Pusateri, A. E. et al. Association of prehospital plasma transfusion with survival in trauma patients with hemorrhagic shock when transport times are longer than 20 minutes: a post hoc analysis of the PAMPer and COMBAT clinical trials. JAMA Surg. 155, e195085 (2019).
Nadler, R. et al. Early experience with transfusing low titer group O whole blood in the pre-hospital setting in Israel. Transfusion 60, S10–S16 (2020).
Leeper, C. M., Yazer, M. H. & Neal, M. D. Whole-blood resuscitation of injured patients: innovating from the past. JAMA Surg. 155, 771–772 (2020).
World Health Organization. Summary of the Report of the 18th Meeting of the WHO Expert Committee on the Selection and Use of Essential Medicines. https://www.who.int/selection_medicines/committees/TRS_web_summary.pdf (2021).
Guyette, F. X. et al. Tranexamic acid during prehospital transport in patients at risk for hemorrhage after injury: a double-blind, placebo-controlled, randomized clinical trial. JAMA Surg. 156, 11–20 (2020). This randomized controlled trial of pre-hospital TXA administered during air medical or ground transport of severely injured patients without TBI does not indicate improved survival, but shows that it is safe and improves survival in patients in severe shock.
Rowell, S. E. et al. Effect of out-of-hospital tranexamic acid vs placebo on 6-month functional neurologic outcomes in patients with moderate or severe traumatic brain injury. JAMA 324, 961–974 (2020). This randomized controlled trial of pre-hospital TXA for TBI does not indicate improved neurological recovery, although a subgroup receiving a dose of 2 g appeared to have improved survival.
Myers, S. P. et al. Tranexamic acid administration is associated with an increased risk of posttraumatic venous thromboembolism. J. Trauma Acute Care Surg. 86, 20–27 (2019).
Morrison, J. J., Dubose, J. J., Rasmussen, T. E. & Midwinter, M. J. Military Application of Tranexamic Acid in Trauma Emergency Resuscitation (MATTERs) study. Arch. Surg. 147, 113–119 (2012).
Moore, E. E. et al. Rationale for the selective administration of tranexamic acid to inhibit fibrinolysis in the severely injured patient. Transfusion 56, S110–S114 (2016).
Moore, H. B. et al. Tranexamic acid is associated with increased mortality in patients with physiological fibrinolysis. J. Surg. Res. 220, 438–443 (2017).
Spinella, P. C. et al. The immunologic effect of early intravenous two and four gram bolus dosing of tranexamic acid compared to placebo in patients with severe traumatic bleeding (TAMPITI): a randomized, double-blind, placebo-controlled, single-center trial. Front. Immunol. 11, 2085 (2020).
Roberts, I. et al. Effects of a high-dose 24-h infusion of tranexamic acid on death and thromboembolic events in patients with acute gastrointestinal bleeding (HALT-IT): an international randomised, double-blind, placebo-controlled trial. Lancet 395, 1927–1936 (2020).
CRASH-3 Trial Collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet 394, 1713–1723 (2019).
Shakur, H. et al. Antifibrinolytic drugs for treating primary postpartum haemorrhage. Cochrane Database Syst. Rev. 2, CD012964 (2018).
Myers, S. P. & Neal, M. D. Venous thromboembolism after tranexamic acid administration: legitimate risk or statistical confounder? ANZ. J. Surg. 90, 425–426 (2020).
Etchill, E. et al. The confusion continues: results from an American Association for the Surgery of Trauma survey on massive transfusion practices among United States trauma centers. Transfusion 56, 2478–2486 (2016).
National Blood Authority. Patient blood management guidelines: module 1. Critical bleeding massive transfusion. https://www.blood.gov.au/system/files/documents/pbm-module1_0.pdf (2011).
American College of Surgeons. ACS TQIP massive transfusion in trauma guidelines. https://www.facs.org/-/media/files/quality-programs/trauma/tqip/transfusion_guildelines.ashx (2014).
Norfolk D. (ed.) Handbook of Transfusion Medicine 5th edn 82–87 (TSO, 2014).
Miyata, S. et al. Transfusion guidelines for patients with massive bleeding. Jpn. J. Transfus. Cell Ther. 65, 21–92 (2019).
Carney, N. et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery 80, 6–15 (2017).
Nevin, D. G. & Brohi, K. Permissive hypotension for active haemorrhage in trauma. Anaesthesia. 72, 1443–1448 (2017).
Holcomb, J. B. et al. Increased platelet:RBC ratios are associated with improved survival after massive transfusion. J. Trauma 71, S318–S328 (2011).
Itagaki, Y. et al. Early administration of fibrinogen concentrate is associated with improved survival among severe trauma patients: a single-centre propensity score-matched analysis. World J. Emerg. Surg. 15, 7 (2020).
Black, J. A., Pierce, V. S., Kerby, J. D. & Holcomb, J. B. The evolution of blood transfusion in the trauma patient: whole blood has come full circle. Semin. Thromb. Hemost. 46, 215–220 (2020).
Yazer, M. H. et al. Initial safety and feasibility of cold-stored uncrossmatched whole blood transfusion in civilian trauma patients. J. Trauma Acute Care Surg. 81, 21–26 (2016).
Shea, S. M. et al. The use of low-titer group O whole blood is independently associated with improved survival compared to component therapy in adults with severe traumatic hemorrhage. Transfusion 60 (Suppl 3), S2–S9 (2020).
Williams, J. et al. Safety profile and impact of low-titer group O whole blood for emergency use in trauma. J. Trauma Acute Care Surg. 88, 87–93 (2020).
Wilson, R. F., Spencer, A. R., Tyburski, J. G., Dolman, H. & Zimmerman, L. H. Bicarbonate therapy in severely acidotic trauma patients increases mortality. J. Trauma Acute Care Surg. 74, 45–50; discussion 50 (2013).
Corwin, G. S. et al. Characterization of acidosis in trauma patient. J. Emerg. Trauma. Shock 13, 213–218 (2020).
Moore, H. B. et al. Targeting resuscitation to normalization of coagulating status: hyper and hypocoagulability after severe injury are both associated with increased mortality. Am. J. Surg. 214, 1041–1045 (2017).
Einersen, P. M. et al. Rapid thrombelastography thresholds for goal-directed resuscitation of patients at risk for massive transfusion. J. Trauma Acute Care Surg. 82, 114–119 (2017).
Baksaas-Aasen, K. et al. Data-driven development of ROTEM and TEG algorithms for the management of trauma hemorrhage: a prospective observational multicenter study. Ann. Surg. 270, 1178–1185 (2019).
Stettler, G. R. et al. Rotational thromboelastometry thresholds for patients at risk for massive transfusion. J. Surg. Res. 228, 154–159 (2018).
Henriksen, H. H. et al. Impact of blood products on platelet function in patients with traumatic injuries: a translational study. J. Surg. Res. 214, 154–161 (2017).
Spano, P. J. II et al. Anticoagulant chemoprophylaxis in patients with traumatic brain injuries: a systematic review. J. Trauma Acute Care Surg. 88, 454–460 (2020).
Benjamin, E., Recinos, G., Aiolfi, A., Inaba, K. & Demetriades, D. Pharmacological thromboembolic prophylaxis in traumatic brain injuries: low molecular weight heparin is superior to unfractionated heparin. Ann. Surg. 266, 463–469 (2017).
Hecht, J. P. et al. Association of timing of initiation of pharmacologic venous thromboembolism prophylaxis with outcomes in trauma patients. J. Trauma Acute Care Surg. 90, 54–63 (2021).
Spyropoulos, A. C. et al. Scientific and Standardization Committee communication: clinical guidance on the diagnosis, prevention, and treatment of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 18, 1859–1865 (2020).
Haagsma, J. A. et al. The global burden of injury: incidence, mortality, disability-adjusted life years and time trends from the Global Burden of Disease study 2013. Injury Prev. 22, 3–18 (2016).
Holbrook, T. L. & Hoyt, D. B. The impact of major trauma: quality-of-life outcomes are worse in women than in men, independent of mechanism and injury severity. J. Trauma Acute Care Surg. 56, 284–290 (2004).
Holbrook, T. L., Anderson, J. P., Sieber, W. J., Browner, D. & Hoyt, D. B. Outcome after major trauma: 12-month and 18-month follow-up results from the Trauma Recovery Project. J. Trauma 46, 765–771; discussion 771–773 (1999).
Holbrook, T. L. et al. High rates of acute stress disorder impact quality-of-life outcomes in injured adolescents: mechanism and gender predict acute stress disorder risk. J. Trauma 59, 1126–1130 (2005).
Holbrook, T. L. et al. Long-term posttraumatic stress disorder persists after major trauma in adolescents: new data on risk factors and functional outcome. J. Trauma 58, 764–769; discussion 769–771 (2005).
Winthrop, A. L. et al. Quality of life and functional outcome after pediatric trauma. J. Trauma. Acute Care Surg. 58, 468–474 (2005).
Christensen, M. C., Banner, C., Lefering, R., Vallejo-Torres, L. & Morris, S. Quality of life after severe trauma: results from the global trauma trial with recombinant factor VII. J. Trauma 70, 1524–1531 (2011).
Mitra, B. et al. Long-term outcomes of patients receiving a massive transfusion after trauma. Shock 42, 307–312 (2014).
Mira, J. C. et al. The epidemiology of chronic critical illness after severe traumatic injury at two level one trauma centers. Crit. Care Med. 45, 1989–1996 (2017).
Stortz, J. A. et al. Evidence for persistent immune suppression in patients who develop chronic critical illness after sepsis. Shock 49, 249–258 (2018).
Gardner, A. K. et al. The development of chronic critical illness determines physical function, quality of life, and long-term survival among early survivors of sepsis in surgical ICUs. Crit. Care Med. 47, 566–573 (2019).
Shackford, S. R. et al. Determining the magnitude of surveillance bias in the assessment of lower extremity deep venous thrombosis: a prospective observational study of two centers. J. Trauma Acute Care Surg. 80, 734–739; discussion 740–741 (2016).
Coleman, J. J. et al. Factors associated with pulmonary embolism within 72 hours of admission after trauma: a multicenter study. J. Am. Coll. Surg. 220, 731–736 (2015).
Nastasi, A. J. et al. Characterizing the relationship between age and venous thromboembolism in adult trauma patients: findings from the National Trauma Data Bank and the National Inpatient Sample. J. Surg. Res. 216, 115–122 (2017).
Dietch, Z. C. et al. Rate of lower-extremity ultrasonography in trauma patients is associated with rate of deep venous thrombosis but not pulmonary embolism. Surgery 158, 379–385 (2015).
Stein, A. L. et al. Impact of a goal-directed factor-based coagulation management on thromboembolic events following major trauma. Scand. J. Trauma Resusc. Emerg. Med. 27, 117 (2019).
Paffrath, T. et al. Venous thromboembolism after severe trauma: incidence, risk factors and outcome. Injury. 41, 97–101 (2010).
Hutchison, T. N. et al. Venous thromboembolism during combat operations: a 10-y review. J. Surg. Res. 187, 625–630 (2014).
Holley, A. B., Petteys, S., Mitchell, J. D., Holley, P. R. & Collen, J. F. Thromboprophylaxis and VTE rates in soldiers wounded in Operation Enduring Freedom and Operation Iraqi Freedom. Chest 144, 966–973 (2013).
Lundy, J. B. et al. Frequency and relevance of acute peritraumatic pulmonary thrombus diagnosed by computed tomographic imaging in combat casualties. J. Trauma Acute Care Surg. 75, S215–S220 (2013).
Klok, F. A. et al. The post-PE syndrome: a new concept for chronic complications of pulmonary embolism. Blood Rev. 28, 221–226 (2014).
Pengo, V. et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N. Engl. J. Med. 350, 2257–2264 (2004).
Anderson, D. R. et al. American Society of Hematology 2019 guidelines for management of venous thromboembolism: prevention of venous thromboembolism in surgical hospitalized patients. Blood Adv. 3, 3898–3944 (2019).
Cannon, W. B. & Grabfield, G. P. Factors affecting the coagulation time of blood. Am. J. Physiol. 34, 232–242 (1914).
Stefanini, M. Basic mechanisms of hemostasis. Bull. N. Y. Acad. Med. 30, 239–277 (1954).
Kitchen, D. P., Kitchen, S., Jennings, I., Woods, T. & Walker, I. Quality assurance and quality control of thrombelastography and rotational thromboelastometry: the UK NEQAS for blood coagulation experience. Semin. Thromb. Hemost. 36, 757–763 (2010).
Takayama, W. et al. The impact of blood type O on mortality of severe trauma patients: a retrospective observational study. Crit. Care 22, 100 (2018).
Griffin, R. L., Jansen, J. O., Bosarge, P. L., Marques, M. B. & Kerby, J. D. The association between ABO blood type and mortality among severely injured trauma patients. Shock 54, 205–208 (2020).
Hamsen, U. et al. The influence of ABO blood group on mortality in major trauma. Orthop. Rev. 11, 8214 (2019).
Hoffman, M. & Cichon, L. J. H. Practical coagulation for the blood banker. Transfusion 53, 1594–1602 (2013).
Crawley, J. T., Zanardelli, S., Chion, C. K. & Lane, D. A. The central role of thrombin in hemostasis. J. Thromb. Haemost. 5, 95–101 (2007).
Gonzalez, E., Moore, E. E. & Moore, H. B. Management of trauma-induced coagulopathy with thrombelastography. Crit. Care Clin. 33, 119–134 (2017).
Veigas, P. V., Callum, J., Rizoli, S., Nascimento, B. & da Luz, L. T. A systematic review on the rotational thrombelastometry (ROTEM®) values for the diagnosis of coagulopathy, prediction and guidance of blood transfusion and prediction of mortality in trauma patients. Scand. J. Trauma. Resusc. Emerg. Med. 24, 114 (2016).
Harr, J. N. et al. Viscoelastic hemostatic fibrinogen assays detect fibrinolysis early. Eur. J. Trauma. Emerg. Surg. 41, 49–56 (2015).
Acknowledgements
E.E.M. and A.S. appreciate the generous support from the National Institutes of Health for their inflammation and coagulation research over the past 35 years (NIGMS: 1-6 P50 GM49222, 1-6 T32 GM08315, 1-2 U54 GM 62119, RM1 GM 131968 and NHLBI: UM1 HL120877). H.B.M. acknowledges the generous support from the National Institutes of Health (NHBLI: K99HL1518870) L.Z.K. acknowledges the generous support from the National Institutes of Health for her platelet biology research (NIGMS: K23GM130892-01). M.D.N. acknowledges the generous support by the National Institutes of Health (NIGMS: R35 GM119526 and NHLBI R01 HL141080).
Author information
Authors and Affiliations
Contributions
Introduction (all authors); Epidemiology (all authors); Mechanisms/pathophysiology (all authors); Diagnosis, screening and prevention (all authors); Management (all authors); Quality of life (all authors); Outlook (all authors); Overview of Primer (E.E.M.).
Corresponding author
Ethics declarations
Competing interests
E.E.M. currently receives research support from Haemonetics, Instrumentation Laboratory, Stago, Hemosonics, Diapharma, Prytime, Humacyte and Genentech; he is a co-founder of ThromboTherapeutics; he is listed as inventor on the following patents relating to blood coagulation or fibrinolysis (including assays): WO-2016073668-A1 (assignee: The Regents Of The University Of Colorado; status: published); US-9354243-B2 (assignee: Haemonetics Corporation, The Regents Of The University Of Colorado, A Body Corporate; status: granted); WO-2019014595-A1 (assignee: Thrombo Therapeutics, Inc.; status: published); EP-3215634-A1 (assignee: The Regents of the University of Colorado; status: published); EP-3303943-A1 (assignee: The Regents of The University of Colorado, A Body Corporate; status: published); WO-2021158799-A1 (assignee: The Regents Of The University Of Colorado, A Body Corporate; status: published); US-2020208194-A1 (assignee: Massachusetts Institute Of Technology, University Of Colorado; status: published). H.B.M. receives research support from Haemonetics and Instrumentation Laboratory; he is a co-founder of ThromboTherapeutics; he is listed as inventor on the following patents relating to blood coagulation or fibrinolysis (including assays): WO-2016073668-A1 (assignee: The Regents Of The University Of Colorado; status: published); WO-2019014595-A1 (assignee: Thrombo Therapeutics, Inc.; status: published); EP-3215634-A1 (assignee: The Regents of the University of Colorado; status: published); US-2020208194-A1 (assignee: Massachusetts Institute Of Technology, University Of Colorado; status: published). M.D.N. receives research support from Haemonetics, Janssen Pharmaceuticals, Accriva Diagnostics and Instrumentation Laboratory; he serves on the Scientific Advisory Board with equity stake in Haima Therapeutics. All other authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Disease Primers thanks P. Cabrales, M. Levi, I. Marzi, S. Ostrowski, P. Spinella and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Potentially preventable deaths
-
The three criteria that must be present in a trauma-related death to qualify as potentially preventable are: the injury must have been survivable, the delivery of care was suboptimal, and the error in care must have been directly or indirectly implicated in the death of the patient.
- Bleeding control bundle-of-care
-
A series of measures to optimize bleeding control, including: accurate identification of the bleeding patient; damage control resuscitation; haemostatic techniques with tourniquets, pelvic binders or haemostatic dressings; resuscitative endovascular balloon occlusion of the aorta; thromboelastography coagulation monitoring; tranexamic acid administration for substantial hyperfibrinolysis; decreased time to operating room and interventional radiology; and goal-directed resuscitation with blood products.
- Massive transfusion
-
Several definitions exist. The most frequently used is >10 units of red blood cells (RBCs) per 24 hours, although this definition is liable to substantial survivor bias. Other definitions include: the critical administration threshold (CAT, ≥3 RBC units per hour in the first hour or in any of the first 4 hours from arrival); >4 RBC units or death in the first hour after injury, a definition that has the advantage of minimizing survivor bias; and >4 RBC units within the first hour, which is also known as the resuscitation intensity definition.
- Crystalloid solutions
-
Isotonic plasma volume expanders that contain electrolytes.
- Viscoelastic haemostatic assays
-
These assays measure change in viscoelastic properties of the whole blood during clot formation, strengthening and dissolution. The most commonly used devices are thromboelastography (TEG) and rotational thromboelastometry (ROTEM).
- Goal-directed resuscitation
-
A procedure consisting of haemostatic resuscitation with blood components guided by viscoelastic haemostatic assays that is directed at normalizing coagulation.
- Auto-dilution
-
A shift of interstitial fluid into the vascular compartment in response to haemorrhagic shock, which may impair haemostatic capacity.
- Secondary haemostasis
-
Secondary haemostasis refers to the deposition of insoluble fibrin, generated by the proteolytic coagulation cascade, into the platelet plug, which forms a mesh that is incorporated into and around the platelet plug.
- Primary haemostasis
-
Primary haemostasis refers to platelet aggregation and plug formation on an injury site.
- Prothrombin time
-
(PT). A conventional coagulation assay that evaluates the extrinsic and the common pathways of the coagulation cascade. The PT result (measured in seconds) in a healthy individual varies between different types and batches of the tissue factor used by the manufacturer.
- International normalized ratio
-
(INR). The INR was devised to standardize the PT results. Manufacturers assign an International Sensitivity Index (IST) for their tissue factor and the INR is calculated as (PT test/PT normal)
- Activated partial thromboplastin time
-
(aPTT). PTT is a conventional coagulation assay that measures the clotting activity of the intrinsic pathway cascade. It tests the function of all clotting factors except factor VII and factor XIII (fibrin stabilizing factor). aPTT is often used to monitor patients’ responses to unfractionated heparin infusion, to target therapeutic anticoagulation. Activation occurs via exposure to a negatively charged substrate, which replicates contact activation and enhances the speed of the test.
- Damage control resuscitation
-
A procedure consisting of limited (to avoid haemodilution) crystalloid fluid, permissive hypotension and administration of balanced blood components in severely injured patients to attenuate TIC.
- Damage control surgery
-
A procedure consisting of essential operative manoeuvres: control of mechanical bleeding, shunting critical arteries, controlling gastrointestinal spillage and temporary packing of bleeding sites in patients manifesting TIC due to ongoing shock. Patients will need to return to the operating room to complete definitive reconstruction after they have stabilized.
- Cryoprecipitate
-
A plasma-derived blood product for transfusion that contains fibrinogen (factor I), factor VIII, factor XIII, von Willebrand factor and fibronectin.
- Glasgow Outcome Score–Extended
-
(GOSE). Global scale for functional outcome that rates patient status into eight categories (death, vegetative state, lower severe disability, upper severe disability, lower moderate disability, upper moderate disability, lower good recovery and upper good recovery).
Rights and permissions
About this article

Cite this article
Moore, E.E., Moore, H.B., Kornblith, L.Z. et al. Trauma-induced coagulopathy. Nat Rev Dis Primers 7, 30 (2021). https://doi.org/10.1038/s41572-021-00264-3
Accepted:
Published:
DOI: https://doi.org/10.1038/s41572-021-00264-3
This article is cited by
-
Transforming research to improve therapies for trauma in the twenty-first century
Critical Care (2024)
-
Association between tranexamic acid administration and mortality based on the trauma phenotype: a retrospective analysis of a nationwide trauma registry in Japan
Critical Care (2024)
-
Trends in pre-hospital volume resuscitation of blunt trauma patients: a 15-year analysis of the British (TARN) and German (TraumaRegister DGU®) National Registries
Critical Care (2024)
-
Derivation of Coagulation Phenotypes and the Association with Prognosis in Traumatic Brain Injury: A Cluster Analysis of Nationwide Multicenter Study
Neurocritical Care (2024)
-
Effect of low fibrinogen level on in-hospital mortality and 6-month functional outcome of TBI patients, a single center experience
Neurosurgical Review (2024)
