
Abstract
Cellular senescence is a state of stable, terminal cell cycle arrest associated with various macromolecular changes and a hypersecretory, pro-inflammatory phenotype. Entry of cells into senescence can act as a barrier to tumorigenesis and, thus, could in principle constitute a desired outcome for any anticancer therapy. Paradoxically, studies published in the past decade have demonstrated that, in certain conditions and contexts, malignant and non-malignant cells with lastingly persistent senescence can acquire pro-tumorigenic properties. In this Review, we first discuss the major mechanisms involved in the antitumorigenic functions of senescent cells and then consider the cell-intrinsic and cell-extrinsic factors that participate in their switch towards a tumour-promoting role, providing an overview of major translational and emerging clinical findings. Finally, we comprehensively describe various senolytic and senomorphic therapies and their potential to benefit patients with cancer.
Key points
-
Cellular senescence is a natural barrier to tumorigenesis; senescent cells are widely detected in premalignant lesions from patients with cancer.
-
Cellular senescence is induced by anticancer therapy and can contribute to some treatment-related adverse events (TRAEs).
-
Senescent cells exert both protumorigenic and antitumorigenic effects via cell-autonomous and paracrine mechanisms.
-
Pharmacological modulation of senescence-associated phenotypes has the potential to improve therapy efficacy and reduce the incidence of TRAEs.
Similar content being viewed by others
Introduction
Cellular senescence is a stress-inducible state of terminal proliferative arrest accompanied by a hypersecretory phenotype referred to as senescence-associated secretory phenotype (SASP). Senescent cells can have context-dependent beneficial or detrimental roles in various physiological and pathological settings. The initial physiological evidence of senescence included the finding of senescent cells in the limb, hindbrain, neural tube and several organs of developing mouse embryos as early as day 9.5 (ref.1), in a study that also showed that defective senescence programmes or removal of senescent cells during various developmental stages results in patterning abnormalities1.
The contribution of senescent cells to tissue regeneration and remodelling can also be observed in adult tissues. In a mouse model that enables the identification of senescent cells, removal of such cells during wound healing delayed tissue repair2, whereas in a mouse model of oncogene-induced senescence (OIS), local transplantation of keratinocytes transiently exposed to SASP factors accelerated tissue repair3. Interestingly, the senescence programme is associated with enhanced stemness through cell-autonomous and cell-non-autonomous mechanisms (which can include senescence propagation, tissue remodelling and regulation of immune responses via SASP factors). This association could reflect a key built-in feature of senescent cells in supporting the recovery phase of wound healing3,4,5, when tissue-insulting stresses are resolved and parenchymal cellularity needs to be replenished6. Senescent cardiac fibroblasts contribute to cardiac regeneration in neonatal mice7, and are crucial for heart repair in both neonatal and adult zebrafish8. The presence of senescent human-derived cultured cells limits fibrosis during skin wound healing9, and similar antifibrotic effects of senescent cells have been observed in the kidneys, liver and lungs in mouse models10,11,12.
A gradual but highly variable accumulation of senescent cells can be observed during the natural ageing process. In aged organisms, senescent cells can contribute to a variety of age-related pathologies, including pulmonary fibrosis, biliary liver damage, arteriosclerosis, retinopathy, osteoarthritis, dementia and diabetes mellitus13,14,15,16,17,18,19. Clearance of senescent cells during ageing extends the lifespan and healthspan of aged transgenic mice20, whereas in other mouse studies, transplantation of a small proportion of senescent cells or injection of chimeric antigen receptor (CAR) T cells with a senescent phenotype accelerated physical dysfunction and reduced lifespan21,22. Conversely, some senescent cell types have indispensable structural functions in organ integrity and their forced clearance in mice leads to tissue damage and organ impairment23. Importantly, premature accumulation of senescent cells can happen as a consequence of endogenous and exogenous stresses, such as microbial attack (Box 1). More central to this Review, exposure to constitutive mitogenic signals (such as OIS) or to DNA-damaging and other non-genotoxic therapeutic agents induces senescence in susceptible neoplastic cells and other tissue components. Such stable proliferation arrest of (pre)neoplastic cells seems to operate as a potent tumour suppressive mechanism while simultaneously promoting the survival of the SASP-producing senescent cells and their crosstalk with the tumour microenvironment (TME). Moreover, eventual re-entry of these damaged and mutagenized cells into the cell cycle would result in expansion of biologically altered post-senescent cells.
In summary, senescent cells have rather heterogeneous phenotypes and can exhibit both antitumour and tumour-promoting features. Whether these different subsets of senescent cells are the consequence of distinct intrinsic programmes or are instead instructed by their varying environmental contexts remains to be explored. In this Review, we present the available evidence supporting the contrasting roles of senescent cells in cancer, and discuss how these features can be exploited with therapeutic intent.
Hallmarks of senescent cells
Proliferation arrest and unresolved DDR
In response to internal or external stresses linked to DNA alterations, proteotoxicity (a consequence of premature protein oxidation and low-fidelity synthesis of high amounts of SASP factors) and aberrant mitogenic signals, cells activate complex mechanisms to prevent propagation of damage. A common outcome is the induction of cell senescence, a viability-protective, lastingly stable cell cycle arrest phenomenon (Box 2). The major mediators of senescence-associated proliferation arrest (SAPA) are cyclin-dependent kinase (CDK) inhibitors 1 and 2A (commonly known as p21 and p16INK4a), which block the formation of CDK–cyclin complexes involved in the cell cycle checkpoints at the G1–S phase transition24. CDKs can phosphorylate different Rb family members, leading to the release and subsequent activation of the transcription factor E2F. Upon p53 activation, Rb cooperates with retinoblastoma-like proteins 1 and 2 (commonly known as p107 and p130, respectively) to suppress the transcription of genes involved in this phase transition25, which collectively promote a senescent state26. p21 suppresses CDK2–cyclin E activity, thereby retaining Rb in its hypophosphorylated G1 form that inhibits transcription of E2F target genes, which are typically involved in DNA replication and thus promote S phase entry. Further stabilized by Rb-mediated repressive chromatin marks, the expression of E2F target genes is firmly silenced, locking the cell into a lasting G1 phase arrest. p16INK4a directly interacts with and inhibits CDK4/6. p16INK4a expression is considered a common and robust, albeit not necessarily specific, marker of senescence, and its promoter activity and transcriptional activation are extensively exploited as reporters of senescent cells in vivo20,23,27,28. Thus, when p21 and p16INK4a are chronically activated to block CDKs, Rb proteins remain hypophosphorylated. and neutralize E2F transcription factors, thus locking the cell into an indefinite proliferative halt24.
Chronic activation of the DNA damage response (DDR) is a common inducer of the senescence response and SAPA. DNA double-strand breaks are robust activators of the DDR via initial recruitment of the kinase ATM to the site of DNA damage29. The recruitment of ATM to DNA lesions drives phosphorylation of histone H2AX, which facilitates the assembly of specific DNA repair complexes. In addition to H2AX phosphorylation, histone methylation also contributes to the assembly of DDR components. A complex encompassing transcription intermediary factor 1β, Rb-bound heterochromatin protein 1 (HP1) isoforms and the H3K9 methyltransferase SUV39H1 is loaded directly onto the chromatin at DNA double-strand breaks, leading to Rb-dependent local trimethylation of histone H3K9. The resulting product, H3K9me3, functions as a seed mark for senescence-associated heterochromatin foci in the vicinity of E2F target gene promoters. Moreover, H3K9me3 also activates histone acetyltransferase KAT5, which subsequently acetylates ATM29. Acetylated ATM kinase phospho-activates CHEK1 and CHEK2, which, in turn, further spread the DNA damage signal by phosphorylating numerous proteins (such as BRCA1, PML, p53, CDC25A and TLK1) involved in DNA repair, damage-induced transcription, cell cycle arrest, apoptosis and chromatin remodelling30,31. Ultimately, persistent DDR signalling leads to p53 phosphorylation at multiple serine residues, which eventually enables its role as a positive regulator of the transcription of many genes, including p21 (ref.32).
Structural and metabolic changes
Chronic activation of DDR signalling and persistent SAPA are associated with structurally and/or functionally defective organelles. A study in cultured human fibroblasts revealed that OIS is associated with multinucleation or enlarged nuclear size33 that are consequences of incomplete mitosis and failed cytokinesis. These processes are characterized by structural nuclear alterations in senescent cells, among which the most consistent is loss of lamin-B1 (ref.34). A characteristic feature of senescent cells in vitro is their enlarged cell body, which either acts as a cellular insult that triggers senescence or results as a consequence of ongoing cell growth in the absence of cell division35. Upregulation of senescence-associated β-galactosidase (SA-β-gal) activity36 and accumulation of lipid-containing granules (known as lipofuscin) are important markers of senescent cells, reflecting lysosomal and autophagic abnormalities and dysregulated mTOR signalling. Indeed, activation of mTOR complex 1 (mTORC1), which integrates various stress signals and modulates cell growth accordingly37, occurs in response to senescence-inducing stimuli38. In PTEN-loss in vitro models of induced senescence, the activated mTOR kinase in mTORC1 or mTORC2 stabilizes p53 and triggers the senescent state39. In a different in vitro model, however, only mTORC1, and not mTORC2, was essential to establish RAS-induced senescence and replicative senescence40. Moreover, mTOR signalling is a major regulator of autophagy41. The expression of senescence markers and susceptibility to induction of liver cancer were higher in mice in which the essential autophagy gene Atg5 was silenced than in wild-type mice42. Although autophagy and senescence are undoubtedly closely linked, this relationship is not yet fully understood43. In preclinical models, proteotoxic stress not only drives entry into a senescent state, but also activates autophagy, which consequently buffers toxicity from misfolded SASP factors44,45. In mice harbouring a BrafV600E mutation and Pten loss, deletion of Atg7, another autophagy-related gene, facilitates melanoma formation by disabling senescence46. In cultured human fibroblasts, inhibition of RAS effector pathways, including autophagy-promoting PI3K–AKT signalling, enables senescence through its effect on FOXO transcription factors47. These findings suggest that mTOR inhibitors, commonly referred to as rapalogs, might induce or prevent senescence in a context-dependent manner45,48,49.
The mitochondria of senescent cells typically have defects in the respiratory chain and excessive production of reactive oxygen species (ROS). Of note, cellular senescence is associated with an increased number of mitochondria, but the membrane potential of these mitochondria is decreased, leading to enhanced ROS production50. Interestingly, mitochondrial dysfunction can be a direct inducer of senescence51,52. Multiple factors involved in senescence induction, such as oxidative stress and proteotoxic stress, lead to protein misfolding, which in turn evokes endoplasmic reticulum stress via the unfolded protein response45,53. Accordingly, senescent cells have an increased unfolded protein response45,54. Changes in lysosomes, mitochondria and the endoplasmic reticulum can be cause and consequence of senescence-associated metabolic alterations. Senescent cells are hypermetabolic; that is, they have enhanced oxygen consumption, and use glycolysis, fatty acid oxidation and oxidative phosphorylation in an aberrant manner to maximize energy production, which is needed to maintain ATP-consuming homeostatic processes45,55,56. Cellular AMP to ATP and ADP to ATP ratios are known to increase during senescence57. Enhanced glucose uptake can be visualized by [18F]-fluorodeoxyglucose PET in senescent tumours in vivo, which display a lack of proliferative activity when imaged for the DNA synthesis-indicating tracer [18F]-fluorothymidine45. The relative and potentially dynamically changing contributions of glycolysis, the tricarboxylic acid (TCA) cycle, oxidative phosphorylation and fatty acid oxidation during long-term maintenance of senescence, as well as their cooperation and complementation in energy production and related metabolic pathways, remain to be elucidated in greater detail58.
Reconversion of lactate to pyruvate by l-lactate dehydrogenase B fuels the TCA cycle out of the senescence-enforced enhancement of non-oxidative glucose metabolism (commonly referred to as the Warburg effect)45. Moreover, Rb can promote the conversion of pyruvate into acetyl-CoA by activating pyruvate dehydrogenase via transcriptional upregulation of pyruvate dehydrogenase (acetyl-transferring) phosphatase 2 (ref.56). In this way, Rb increases the flux of pyruvate through the TCA cycle and, subsequently, oxidative phosphorylation59.
Senescent cells also have an altered plasma membrane composition, with the most consistent change being upregulation of caveolin-1, an important component of cholesterol-enriched microdomains referred to as caveolae60. Other plasma membrane proteins whose expression has also been reported to change during senescence are receptor-type tyrosine protein phosphatase DEP-1, β2-microglobulin, membrane-bound oxidized vimentin, dipeptidyl peptidase 4 and urokinase-type plasminogen activator receptor (uPAR)22,61,62. The changes in plasma membrane composition not only have cell-autonomous signalling implications but also participate in the communication between senescent cells and their microenvironment, in concert with mediators of the SASP.
SASP
The NF-κB, p38, mTOR and C/EBPβ signalling pathways are major components of the SASP63,64,65,66,67. These pathways are mainly involved in chronic DDR signalling, and include pro-inflammatory cytokines (for example, IL-1α, IL-1β, IL-6 and IL-8), chemokines (for example, CCL2, CCL5 and CXCL1), growth factors (for example, HGF, EGF and TGFα), matrix-remodelling enzymes (for example, matrix metalloproteinases (MMP) 1 and 3)68 and various oxylipins69. SASP factors are the major paracrine messengers between senescent cells and their surrounding cells, including stromal bystanders, immune cells, premalignant and cancer cells. Like many other senescence-associated features, however, the SASP is not stable, instead being highly dynamic6,70,71. Indeed, senescence-associated phenotypes are temporal and early-stage senescent cells are phenotypically different from late-stage senescent cells72,73,74. Induction of p21 and a Notch-driven SASP are considered early events70,75,76, whereas upregulation of p16INK4a and an NF-κB-driven SASP are evident at later stages of senescence71,77,78. This temporal regulation of senescence-associated phenotypes seems to be, at least in part, controlled by the sequential and dynamic activation of diverse transcriptional programmes and hierarchies72,73,74.
Stability of senescence
Continuous transcriptional or, more generally, functional changes in senescent cells, and the permanent need to actively maintain senescence-supporting transcription puts the stability of cell cycle arrest at risk, occasionally leading to the restoration of proliferative properties with a potentially detrimental effect on tumour suppression5,79. Specifically, cancer cells that undergo latent stem-like reprogramming during senescence might exert this transcriptional feature upon cell cycle re-entry, driving particularly aggressive relapses, as demonstrated in studies in a mouse model of lymphoma and human-derived lymphoma cells5. Of note, most of the evidence supporting the dynamic and temporal dependence of senescence-associated phenotypes comes from studies in culture systems and, therefore, efforts are needed to prove that these events occur in physiological and pathological contexts in vivo.
Senescence in patients with neoplasia
The accumulation of SA-β-gal-positive senescent cells in the skin of older individuals (aged ≥65 years) was first described almost three decades ago80. Hallmarks of senescent cells were subsequently identified in dermal premalignant lesions, specifically human melanocytic naevi81,82. These discoveries sparked unprecedented interest in the measurement of senescence in neoplastic and malignant human tissues (Table 1). Given that no single marker exists that robustly and unequivocally recognizes and discriminates this unique state, accurate labelling of senescent cells in preclinical studies has been difficult83. Only a consistent profile of numerous senescence indicators — the most prominent of which are damaged DNA, activated DDR and/or MAPK signalling, halted cell cycle, expanded lysosomal compartment and histone modifications — faithfully define senescence. Therefore, studies claiming to have detected senescence in tissue samples on the basis of a single marker must be interpreted with caution.
Senescent cells in premalignant lesions
Cellular senescence is a common feature in human neoplastic tissues. For example, the senescence marker CXCR2 was detected in prostate intraepithelial neoplasia-derived samples84, and PML and ERK were found in benign human prostatic hyperplasia-derived samples85,86. Senescent cells expressing both SA-β-gal and p16INK4a were detected in neurofibroma-derived samples47. Colon adenomas, which are stable precursor lesions of invasive colon cancer, were found to be negative for the proliferation marker Ki67 and positive for SA-β-gal87,88. Moreover, colon adenoma-derived samples also have elevated expression of p16INK4a, HP1γ (an H3K9me3-enriched senescence-associated heterochromatin focus-promoting scaffold that connects H3K9 methyltransferases to Rb as another binding partner), and focal γH2AX expression, indicating enhanced DNA damage87,89. p16INK4a, p21 and a variety of SASP factors were detected in patient-derived non-malignant tissue surrounding the tumour boundaries of hepatocellular carcinoma lesions90. Nevertheless, the mere detectability of senescent cells is not an indication of whether these cells will have a tumour-suppressive or pro-tumorigenic functional role.
Therapy-induced senescence
Senescence has key implications not only for tumour development but also for responses to anticancer therapy. Indeed, in various preclinical models, exposure to chemotherapy drugs or radiation increased the presence of senescence marker-positive cells91,92. Confirmation of these results in samples derived from patients with cancer undergoing treatment indicated that anticancer therapy might lead to the induction and accumulation of senescent cells in malignant and non-malignant tissues in clinical settings93,94. These findings are not surprising, given that most common anticancer therapies can cause DNA damage, the major inducer of senescence in both non-malignant and cancer cells — albeit with the caveat that the thresholds to enter senescence seem to vary between malignant and non-malignant cells, as described for apoptosis. Additionally, the vast majority of anticancer drugs are administered systemically, thus potentially inducing senescence in multiple tissues or compartments. Alkylating agents27,95,96 (for example, cisplatin, cyclophosphamide and temozolomide), topoisomerase inhibitors27,65,91,95 (such as doxorubicin, etoposide and camptothecin) and γ-irradiation78, but also microtubule inhibitors27,97,98 (for example, paclitaxel) and, to a lesser extent, vinca alkaloids78 (for example, vincristine), have all been identified as senescence inducers in preclinical models. An analysis of biopsy-derived samples from patients with prostate cancer treated with mitoxantrone revealed upregulation of the senescence markers p16INK4a and p21 as well as the SASP factors IL-6 and IL-8 (ref.68). Staining of samples derived from patients with breast cancer who received chemotherapy (various regimens) revealed the presence of various senescence markers (p16INK4a, p21, p53 and SA-β-gal) within malignant lesions93. Another study showed that cytotoxic chemotherapy increases cellular senescence (higher p16INK4a expression) in the haematopoietic compartment in patients with breast cancer and induces long-lasting elevation of the SASP factors VEGFA and CCL2 (ref.99).
Interestingly, chemotherapy promotes senescence not only in tumours but also, to a lesser extent and in a rather temporary way, in non-malignant tissues, with potential long-term implications for the recovery of non-malignant tissue after successful elimination of the malignant cell population. In this regard, the detection of an increased number of T cells overexpressing p16INK4a in patients with breast cancer treated with different chemotherapeutic agents is a finding worth highlighting because it indicates that immunosenescence can occur as a bystander effect of treatment99,100. Neoadjuvant chemotherapy is also associated with elevated expression of p16INK4a in the mammary duct, lobular and adipose tissues of patients with breast cancer101. In tumour-free mice, exposure to the topoisomerase II inhibitor doxorubicin leads to systemically elevated SASP and functional impairment reminiscent of accelerated ageing, which is only explained in part by direct organ toxicity of this chemotherapeutic agent27.
Similar to chemotherapy, radiation is used for cancer treatment owing to its ability to generate an acute burst of DNA damage. The advantage of radiotherapy is its more local delivery, although not in a cancer-specific manner. Radiation has been shown to induce senescence in human head-and-neck squamous cell carcinoma-derived cells102. Analysis of non-malignant submandibular gland samples derived from irradiated patients revealed upregulation of p16INK4a (ref.103). SA-β-gal-positive cells have been found in non-malignant lung tissues from patients with non-small-cell lung cancer who had received chemotherapy, but not in tumour and non-malignant lung tissues obtained prior to chemotherapy94. Consistent elevated expression of p21 was identified by microarray hybridization analysis in the white blood cells of patients who had received radiotherapy104.
Treatment-induced premature accumulation of senescent cells can also be observed in patients exposed to targeted therapies. The CDK4/6 inhibitors palbociclib, ribociclib and abemaciclib have a function similar to that of p16INK4a, thereby eliciting a stable proliferation arrest indicative of cellular senescence105,106,107,108. Whether this type of senescence is independent of DNA damage is under debate109,110. CDK4/6 inhibitors are able to induce a p53-dependent proliferation arrest with features of senescence in non-malignant cells in culture and in vivo111. The pan-inhibitor of histone deacetylases vorinostat, induces cellular senescence in cultured urothelial carcinoma and leukaemia cells112,113. Decitabine is a mild DNA-damaging agent and potent DNA methyltransferase inhibitor that is able to activate, among other loci, p16INK4a by modulating methylation at its CpG-enriched regions; this agent induced SA-β-gal activity in cultured malignant pleural mesothelioma cells114. Drugs targeting angiogenesis, such as VEGF inhibitors, can induce senescence in preclinical models of colorectal cancer115 and renal carcinoma116. Furthermore, antiangiogenic therapies can also lead to the elevation of serum levels of cytokines in patients117, possibly reflecting a SASP-like response to such agents. Finally, therapeutic antibodies, especially those not conjugated with a directly DNA-damaging and thus cytotoxic payload (such as the anti-CD20 antibody rituximab), promote senescence in cultured lymphoma cells118. In a study with results published in 2021, the induced senescence responses of 13 cancer cell lines to the cytotoxic agent etoposide and the Aurora kinase A inhibitor alisertib was profiled at the transcriptomic level, resulting in a classifier termed SENCAN that can be used as a tool to detect cancer cell senescence in vitro119.
Taken together, broad evidence indicates that senescence is an integral effector mechanism induced by most anticancer treatment modalities, especially but not solely those resulting in DNA damage. Therefore, the complex senescence process is a key long-term treatment outcome in clinical oncology. Additional studies are needed to quantify the extent of senescence induction in non-malignant cell compartments as a side effect of various therapeutic modalities, to document the temporary nature and/or persistence of such cells, and to gain deeper insights into their potential beneficial or detrimental role in long-term tumour control and organ regeneration in patients with cancer.
Senescence and tumour suppression
Growth arrest
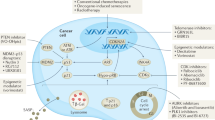
Senescent cells are in a stable state of cell cycle arrest, which presents a natural barrier to tumorigenesis (Fig. 1). OIS is a senescence programme driven by activated oncogenes, especially members of the RAS and BRAF families. Such mitogenic moieties overwhelm non-malignant cellular growth control mechanisms by fuelling various aberrant signalling pathways and unscheduled DNA replication in particular. The cellular counter-response upon sensing unleashed pro-mitotic activity is to trigger a firm proliferation arrest, highlighting the directly protective role of senescence as a switch against tumour initiation and development120. Studies in various mouse models, supported by correlative evidence from human-derived tissue samples, have underscored the tumour-suppressive role of OIS81,120,121,122,123.

Oncogene-induced senescence (OIS) is a senescence programme driven by activated oncogenes (for example, NRASG12V and BRAFV600E). OIS forms a natural barrier to tumorigenesis by inducing stable growth arrest of premalignant cells, reinforced by cyclin-dependent kinase inhibitors (such as p16INK4a and p21). Thus, OIS operates as a cell-intrinsic tumour-suppressive mechanism. Cells undergoing senescence (including OIS) acquire metabolic changes and the senescence-associated secretory phenotype (SASP), both of which mediate tumour suppression in a cell-extrinsic manner. SASP factors, including chemokines, cytokines, growth factors and enzymes, can induce paracrine senescence or a stable proliferation arrest in neighbouring cancer cells. Some SASP factors also enhance immune surveillance, which, in turn, accounts for the clearance of senescent cells. Moreover, SASP factors can induce secondary cell death in the cancer cell population. NK, natural killer; SA-β-gal, senescence-associated β-galactosidase.
In mice harbouring oncogenic Ras but lacking intact alleles of Suv39h1 or carrying a p16INK4a-insensitive constitutively active CDK4 mutant, tumour development occurred that was otherwise controlled by Ras-induced senescence120,124. Hence, disruption of senescence-essential genes, particularly those encoding key mediators of cell cycle arrest, predisposes organisms to develop cancer. Whether senescence induction and/or maintenance is fully dependent on individual gene activities, such that their inactivation would prevent or terminate the process, is not entirely clear; for example, particularly strong pro-senescence triggers can still evoke senescence in the absence of functional TP53 alleles78. Conversely, inactivation of Tp53, the genes encoding either one of the three Rb family members or Suv39h1 in already senescent cells enables cell cycle re-entry out of the terminal arrest condition5,125,126. Tp53-knockout mice are born at normal Mendelian ratios and lack apparent phenotypic alterations but are prone to develop spontaneous tumours at around 6 months of age127,128. Similarly, about two-thirds of mice with knockout of Cdkn2a (the locus encoding p16INK4a and the p53 upstream regulator p19ARF) develop fibrosarcomas, sarcomas or lymphomas at 5–9 months of age129. Both models, however, recapitulate a compound defect of senescence and apoptosis: mice with Tp53 knockout and, to a much lesser extent, those with Cdkn2a knockout also have impaired DNA repair and thus, accumulate DNA damage. This scenario complicates assessment of the selective contribution of senescence to tumour suppression in these models. Of note, mouse models solely lacking senescence-mediating gene activities as a lost tumour-suppressor principle still need the activation of an oncogenic driver in such a ‘reverse tumorigenesis’ sequence; for example, spontaneous selection for activation of Ras or another oncogene in senescence-incapable cells. By contrast, genetic models driven by constitutively activated Ras and in which senescence is genetically ablated typically develop macroscopic tumour lesions with much shorter latencies. A variety of spontaneous tumours were observed at an average age of 16 months in mice with knockout of Cip1 (the gene encoding p21)130. Double knockout of Cdkn2a (with the locus encoding p19ARF left intact) and Cip1 rendered mice extremely susceptible to carcinogen-induced skin cancer, typically driven by Hras mutations131. Of note, 38–50% of human cancers carry inactivating alterations in TP53 (ref.132), 48–80% in CDKN2A133 and 14% in CIP1 (ref.134), while 15–20% and up to 8% harbour oncogenic mutations in RAS and BRAF, respectively135,136.
Importantly, the tumour-suppressive function of senescence is further enhanced by cell-extrinsic mechanisms. Indeed, senescent cells are able to promote senescence of adjacent cells through both the SASP and direct cell–cell interactions, thus also limiting the propagation of not yet senescent premalignant or fully malignant cells in their vicinity63,84,137,138,139. In some cases, SASP factors enforce apoptosis or necrosis of surrounding cells. For example, the SASP factor TNFα can induce ROS-dependent apoptosis in human-derived cancer cell lines140, while IL-6 triggers apoptosis in cultured neoplastic T lymphocytes141. Hence, senescence operates as an antitumour barrier in vivo, and SASP-mediated paracrine control of potential precursor lesions in the environment further enhances its robustness.
Immune surveillance
An increasing body of evidence supports the important role of oncogene-driven senescent cells in promoting cancer immunosurveillance. Premalignant senescent cells seem to be primed for clearance by the immune system; for example, macrophages recruited via secretion of CCL2 and further activated through CD4+ T cell assistance eliminate NrasG12V-senescent premalignant hepatocytes in mice142. Global remodelling of the super-enhancer landscape in HRASG12V-induced senescent human cultured fibroblasts, specifically via recruitment of the chromatin reader bromodomain-containing protein 4 to SASP-gene-adjacent super-enhancer sites in the genome, was found to have a crucial role in immunosurveillance when these fibroblasts were injected into mice143. Indeed, inhibition of bromodomain-containing protein 4 suppressed the SASP programme and disrupted the immune clearance of these premalignant OIS cells143. The tumour suppressor p53–p21 axis cell-autonomously controls the OIS response, but additionally mediates communication between senescent cells and immune cells, presumably via the SASP144,145,146. Specifically, restoration of Tp53 in mouse models of lymphoma and sarcoma led to tumour regression with features of cellular senescence, thereby implying that p53-mediated apoptosis and senescence-evoked immune clearance are the underlying mechanisms144. Interestingly, similar findings were observed in a mouse model of liver cancer upon restoration of p53. Notably, this process was mediated by the innate immune clearance triggered by the SASP factors CSF1, CCL2, IL-15 and CXCL1 (ref.145). p53 cooperates with the NF-κB pathway to regulate SASP factors that can activate macrophages to form a tumour-suppressive microenvironment147,148. In a different mouse model of liver cancer, sustained expression of p21 was found to induce a senescence programme with a peculiar secretory phenotype including CXCL14 and IGFBP3, which contributed to macrophage and lymphocyte recruitment to the tumour site, thus activating immunosurveillance mechanisms146. Notably, oncogenic Ras overexpression also activated immune surveillance functions that protected from oncogenic growth via p21-dependent senescence146.
As opposed to apoptosis-mediated tumour suppression, senescence-mediated tumour suppression is difficult to study in p53-dependent settings, and has been genetically dissected in a Myc-driven mouse model of lymphoma, in which apoptotic remnants of lymphoma cells triggered the activation of macrophages, which subsequently secreted senescence-promoting TGFβ that induced tumour-suppressive senescence in the lymphoma cell compartment121. These steps could be attributed to lymphoma cells (through Bcl2-mediated apoptotic block or Suv39h1 knockout-based senescence incapability) and macrophages (through interference with TGFβ production), respectively, and all led to accelerated lymphoma growth in vivo121. As seen in response to p53 and/or p21 reactivation, restoration of melanoma senescence by pharmacological inhibition or genetic inactivation of H3K9me3 demethylases led to the recruitment of macrophages to tumour sites in vivo149. In mouse models of aggressive B cell lymphomas harbouring NF-κB-deregulating mutations, activating Myd88 or Card11 mutations accelerated lymphomagenesis despite enforcing OIS in a substantial proportion of Eµ-myc lymphoma cultured cells. Conversely, these cells constituted the senescence-associated immunogenic tumour population that underwent selective and direct elimination by primed CD8+ T cells upon inhibition of PD-L1. This finding provided the first demonstration of an immunogenic switch of senescent cells recognized by the adaptive immune system and leading to delayed tumour progression in mice150. Thus, therapeutic interference with key factors modulating innate or adaptive immune responses is a promising strategy to enhance the clearance of premalignant OIS cells and prevent tumour progression.
Immunogenic effects can also be observed in the context of anticancer therapy, in particular, in tumours that enter therapy-induced senescence (TIS) in response to a variety of different antineoplastic agents. Combinations of inhibitors of MEK and CDK4/6 potently induced cellular senescence accompanied by NF-κB-driven SASP in a Kras-mutant-driven mouse model of lung cancer151. In these mice, the SASP components TNFα and ICAM1 subsequently elicited natural killer (NK) cell-mediated immunosurveillance that contributed to tumour regression151. Dual inhibition of MEK and CDK4/6 also led to senescence phenotypes in a mouse model of Kras-mutant pancreatic ductal adenocarcinoma (PDAC)152. Importantly, SASP factors secreted by the senescent PDAC cells contributed to vascular remodelling, which facilitated drug delivery and promoted the accumulation of CD8+ T cells whose cytotoxicity could be enhanced through antibody-mediated inhibition of PD-1 (ref.152). Moreover, in a mouse model of breast cancer, CDK4/6 inhibition-driven cellular senescence also triggered antitumour immunity, mediated by suppression of regulatory T cells and re-expression of endogenous retroviral elements, which elicited an interferon response106,153. Complementary findings unveiled enhanced CD8+ T cell cytotoxicity following CDK4/6 inhibition in other breast cancer models154. Finally, chemotherapy-mediated NF-κB induction in lymphoma TIS mouse models controlled tumour growth at least in part through the recruitment of innate immune cells, namely macrophages64,155.
Collectively, these findings suggest that TIS functions as a potent tumour suppressor not only by cell-intrinsic growth control but also through immune-mediated cell-extrinsic mechanisms. Nevertheless, the characteristics of the SASP-modulated immune cell activity, and in particular the actual senescence-associated neoepitopes recognized by the adaptive immune system, remain to be characterized.
Senescence and tumour promotion
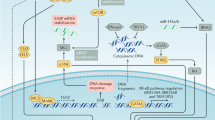
Cell-extrinsic mechanisms of senescence, mainly the SASP, might have paradoxical tumour-promoting properties (Fig. 2). Several SASP factors are associated with pro-tumorigenic processes, including chronic inflammation, mitogenic signalling, stemness, angiogenesis, migration and invasion, genotoxicity and immunosuppression84,156 (Supplementary Table 1 and Fig. 2). Pioneering studies showed that senescent cells promote the malignant conversion of otherwise non-malignant cells both in vitro and in vivo157, and that they stimulate the proliferation of fully transformed breast cancer cells in immunocompromised mice157,158. Orthotopic co-transplantation of cells with senescence induced by BRAFV600E and thyrocytes with thyroid cancer cells in mice increased the tumour invasion ability of the latter159. Doxorubicin-induced systemic senescence contributed to breast cancer metastasis in an orthotopic mouse model. Moreover, these detrimental effects were neutralized via genetic or pharmacological clearance of senescent cells27. Doxorubicin treatment also induced senescence in the MMTV-Wnt1 breast carcinoma mouse model, in which the senescence response was linked to impaired tumour growth and recurrence by competing with and thus protecting against apoptosis as an ultimate ‘cytolytic end point’, further aggravated by mitogenic SASP effects160. Non-malignant brain cells with radiation-induced senescence contributed to the growth of glioma cells in mice, which was blunted with the senotherapeutic agent navitoclax161. Correlative evidence for pro-tumorigenic functions of senescence has also been found in studies of patient-derived samples. For example, the SASP of cultured human melanoma cells was shown to exert pro-tumorigenic and pro-metastatic properties in a xenograft mouse model162. Moreover, a senescence-associated gene signature was identified in the peritumoural area of hepatocellular carcinomas both in mice and humans, with the presence of this signature in the latter correlating with poor overall survival90.

Following induction of senescence by intrinsic or therapeutic stresses, non-malignant or cancer cells can secrete senescence-associated factors that mediate secondary effects on tumour progression (Supplementary Table 1). Different SASP factors contribute to cancer stemness, proliferation, migration, invasion and metastasis, thus enhancing the malignant potential of the cancer cell population. Moreover, senescence-associated factors also modulate the tumour microenvironment by promoting tumour angiogenesis and preventing the antitumour roles of immune cells. MMP, matrix metalloproteinase; SASP, senescence-associated secretory phenotype.
SASP-mediated tumorigenesis
Many studies have highlighted the pro-tumorigenic activity of individual SASP factors. IL-6 and IL-8, two well-characterized and abundant SASP factors, are known drivers of cancer proliferation163,164. CCL5 can promote cancer cell proliferation through the activation of c-MYC and cyclin D1 (refs.165,166). HGF stimulates mitogenic signalling cascades in cancer cells and cooperates with MMPs to further accelerate cancer progression158. The ability of senescent cells to support metastatic growth is associated with tissue remodelling properties that can be attributed to various SASP factors. For example, IL-6 promotes angiogenesis and, conversely, the fraction of CD31-expressing endothelial cells in tumours was reduced in Il6 knockdown mice163. Other SASP factors, such as CXCL5 and VEGF, increase blood vessel density in tumour xenograft models167,168. In addition to angiogenesis, SASP factors can promote migration and invasion. MMPs operate as master regulators of cancer invasiveness through degradation of extracellular matrix components, thereby facilitating tumour dissemination to secondary sites169. Accordingly, in prostate tumours with Pten-loss-induced senescence, genetic ablation of Timp1 (encoding a metalloproteinase inhibitor factor) operates as a switch favouring the development of metastases via effects on the function and relative abundance of certain SASP factors, such as MMPs, GDF-15, FGF1 and IGFBP5 (ref.170). Of note, IL-6 and IL-8 can contribute to MMP induction via activation of the transcription factor STAT3 (refs.171,172). In a mouse model of Cdkn1b overexpression-induced senescence, IL-6 promoted osteoclast formation and, thus, a more penetrable TME for breast cancer bone metastasis173. Furthermore, CXCL12 release from senescent thyrocytes stimulated thyroid cancer cells to invade lymph nodes in a mouse xenograft model159. Moreover, soluble E-cadherin secreted by p19ARF-expressing senescent cells enhanced melanoma cell metastasis and invasiveness in vitro and in vivo174. Importantly, many of the aforementioned effects ascribed to the senescent state reflect well-established biological properties of these growth factors. Aspects that remain to be determined are whether the local or systemic elevation of these factors is truly the result of an accumulation of senescent cells, and whether the burden of senescent ‘persister’ cells (either residual malignant or stressed non-malignant cells) is sufficiently high to account for the attributed effects in patients with cancer.
SASP-mediated immune evasion
Another important mechanism by which senescent cells and their SASP indirectly contribute to cancer progression and relapse is the negative modulation of the immune system. In contrast to the already discussed immune-activating properties of senescent cells in specific TMEs, studies in several mouse models have shown that SASP factors can suppress host immunity under certain circumstances. Via secretion of CCL2, hepatocytes with NrasG12V-induced senescence attracted a subset of CCR2+ myeloid cells that engaged cytotoxic NK cells, eventually blocking tumour immune surveillance90. IL-6-secreting senescent cells residing in the tumour stroma were shown to recruit myeloid suppressor cells (expressing CD11b and Gr-1) and reduce antitumour T cell immune surveillance175. Moreover, IL-6 and IL-8 also enhanced the surface expression of HLA-E that interacts with the inhibitory receptor NKG2A, thereby blunting the activity of cytotoxic NK cells and mature CD8+ T-cells176. In Pten-null senescent prostate tumours, activation of the Jak2–Stat3 pathway led to an immunosuppressive TME mediated by various SASP factors. In turn, SASP reduction and remodelling by JAK2 inhibitors restored antitumour immunity177. Thus, fine-tuning the release of SASP factors is a strategy that could help re-establish or improve cancer immunosurveillance.
Reversibility of senescence in cancer
SAPA might not always mark an irreversible end point for all cells within a population of presumably senescent cancer cells, and those that escape cell cycle arrest are likely to contribute to a clinical cancer relapse5,178. This phenomenon is distinct from partial, defective or irregular states of senescence that collectively account for states lacking certain aspects of the full-featured senescence response (that can be referred to as ‘light senescence’, senescence-like or pseudo-senescent states) (Box 2). Not surprisingly, reversible senescence is often, albeit not exclusively, observed in the context of TIS. Unlike the continuous pro-senescent DNA replication stress enforced by constitutively active mitogenic oncogenes, cells entering senescence after just a single dose of a genotoxic agent undergo DNA damage stress to a lesser extent, especially if previous DNA damage events have been largely resolved by DNA repair attempts. Upon a single exposure to chemotherapy, maintenance-essential DDR signalling can only emanate from remaining damage sites, particularly the irreparable telomeres179. Besides, senescent cells are metabolically highly active and turn around crucial maintenance factors of the senescence response. For example, they need to renew transcriptionally repressive H3K9me3 marks in the vicinity of E2F target gene promoters caused by nucleosome turnover149,180. Of note, genetic interference with senescence-essential factors, especially using inducible systems, underscored that, similar to other biological states, senescence is not necessarily irreversible125,126,149. In a lymphoma mouse model, spontaneous cell cycle re-entry was tracked through staining of several senescence and proliferation markers, and observed in a rare subpopulation of faithfully senescent cells that regained DNA replication capacity during TIS5. Even cancer cells with major genetic defects in senescence programmes, such as TP53 mutations, could potentially enter senescence if exposed to a sufficiently high dose of DNA-damaging agent91, although this response might not be as deep and lasting as that elicited in cells lacking these alterations. Indeed, a small proportion of H1299 TP53-null lung cancer-derived cells with senescence induced by the topoisomerase I inhibitor camptothecin were able to resume proliferation 18–24 days after treatment in a process mediated by CDK1 (refs.94,181). Doxorubicin-induced senescent MCF7 breast cancer cells retaining wild-type TP53 but harbouring a PIK3CAE545K mutation were able to form colonies upon drug removal182. Chemotherapy-induced senescent H460 lung cancer cells and HCT116 colon cancer cells also regained proliferative capacity as early as 6 days after removal of etoposide or doxorubicin. At this time point, however, cultured cells are typically expected to exhibit full-featured TIS, thereby leading to questioning whether a full senescence state had been established183. The PARP inhibitor olaparib induced a senescence-like proliferation arrest in ovarian cancer-derived cultured cells that relied on the continuous provision of the drug. Indeed, this state was reversible upon removal of the PARP inhibitor, but nevertheless led to the proposition that the temporary senescent state switch may be a novel therapeutic opportunity (discussed below)184.
Escape from proliferation arrest is not exclusively found in TIS and has also been reported in the context of OIS. For example, reactivation of telomerase activity or deletion of CDKN2A contributed to escape from HRASG12V-induced or BRAFV600E-induced senescence programmes in various human cell types in culture185. Unlike loss of p16INK4a and p19ARF expression, however, overexpression of hTERT prior to the activation of these oncogenes did not prevent the onset of senescence63,186. Fibroblasts with HRASG12D-induced senescence and melanocytes with BRAFV600E-induced senescence resumed proliferation in culture upon induction of KDM4C, an H3K9me3-active demethylase, out of senescence149. Similarly, inactivation of a regulatable variant of Suv39h1 in mice permitted lymphoma cells with doxorubicin-induced senescence to re-enter the cell cycle5,178. Moreover, a proportion of oncogenic cultured cells with CDC6-induced senescence had spontaneously escaped from proliferation arrest 4 weeks after senescence induction. These ‘escapers’ harboured genomic alterations caused by chromosome inversion that favoured senescence evasion and an aggressive cancer phenotype187.
Of note, culture adaptation is a strong selector against an intact senescence response, especially if multi-passage cell lines and not primary cells are used188. Even with formally intact TP53 or CDKN2A alleles, cancer cell lines often harbour other mutations that impair maximal senescence capacity, making it virtually impossible to judge whether their regrowth potential out of a senescence-like arrest condition truly reflects senescence escape, or re-progression from a never fully entered senescence state (a process referred to as ‘senescence bypass’). Biological heterogeneity as well as unequal distribution of therapeutic agents in tissues might account for a similar phenomenon in vivo that, despite being likely, remains to be investigated in greater detail. Nevertheless, robust evidence from studies in primary cell cultures supports the view that fully senescent cells might indeed re-acquire proliferative capacity while remaining, to some extent, locked into other features of cellular senescence5,149. Such evidence not only sheds light on a potential mechanism underlying clinical relapses but also emphasizes that the state after senescence is a dissociated one that combines partial preservation and partial reversal of senescence-associated phenotypes. Thus, escape from senescence could occur if loss of maintenance-essential genes permits cell cycle re-entry despite an epigenetically firmly secured senescent arrest. Such a transition, however, should be considered as a biological progression rather than as actual reversibility of senescence. The unequivocal demonstration of the existence of senescence reversibility mechanisms requires in vivo tracking systems that are currently not available.
Senescence-associated stemness
Adding to the stemness-instructive paracrine effects that SASP factors can exert3,4, other reprogramming mechanisms can underlie cell-intrinsic senescence-associated stemness; that is, the de novo formation of cancer stem cells that drive aggressive relapse via their tumour re-initiating self-renewal potential. One such process was described in a TIS mouse model in which WNT pathway mediators and activation of MEK–MAPK and PI3K–AKT signalling, which subsequently inhibited glycogen synthase kinase-3β and thus β-catenin degradation, activated stemness properties5. In a mouse model of oncogene-driven breast cancer, expression of the NF-κB cascade-activating receptor RANK in mammary epithelial cells elicited both senescence and stemness. Although tumour onset was delayed, tumour growth and aggressiveness were favoured in the long term189. The gain of stemness and aggressive growth properties has also been observed in other settings in which previously fully or partially senescent cancer cells resumed proliferation178. Similar characteristics were seen in CIP1-overexpressing TP53-null cultured cancer cells, which entered a transient senescent state but quickly escaped with enhanced cancer stem cell properties and an aggressive phenotype190. Further supporting these findings, senescence-like resilient phenotypes induced by cytarabine were observed in cultured acute myeloid leukaemia cells that might hypothetically act as leukaemia-reinitiating stem cells and promote relapse in patients191. Importantly, retained senescence-associated gain of stemness in senescence escapers has been demonstrated in samples derived from patients with relapsed diffuse large B cell lymphoma that exhibited strong enrichment for WNT signalling activity reminiscent of senescence-associated stemness5.
Implication for treatment-related AEs
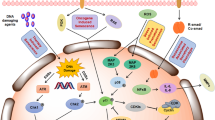
In addition to their potential pro-tumorigenic role, SASP factors might explain or contribute to some of the adverse events (AEs) associated with anticancer therapies (Fig. 3). In patients with cancer, older age (>65 years) is associated with reduced tolerance to anticancer treatment27 and a greater risk of developing serious AEs192, as well as genetic alterations that might lead to reduced sensitivity to treatment; together these features constitute major hurdles for the successful administration of antineoplastic therapies at efficient doses. Interestingly, high expression of p16INK4a in circulating T cells before treatment, presumably a surrogate biomarker of system-wide senescent cell burden, correlates with severe chemotherapy-induced fatigue in patients with breast cancer, suggesting that senescent cells might contribute to the reduced tolerance of older individuals to anticancer therapies27. Ageing exacerbates the toxicity of these treatments and, in addition, some of them can also accelerate the onset of age-related dysfunctions, rendering chemotherapy a potential inducer of premature ageing via induction of senescence and SASP193,194.

Endogenous senescent cells in different tissues of older individuals (>65 years) reduce tolerance to cancer therapy and contribute to cancer progression. By contrast, cancer therapies can also induce senescence in different tissues and organs, thereby mimicking accelerated ageing. These senescent cells and associated detrimental SASP factors contribute to treatment-related adverse events, such as fatigue, frailty, cardiac dysfunction, hepatic failure, renal failure, general inflammation, bone loss and muscle weakness. SASP, senescence-associated secretory phenotype.
Cancer survivors are at an increased risk of developing secondary primary malignancies and non-cancer chronic diseases, such as chronic heart failure, coronary heart disease and pulmonary fibrosis, earlier and faster than the general population as a result of impaired organ function195,196. Examples of such accelerated ageing phenotypes have been observed in survivors of childhood cancers who are cured from their malignant diseases but tend to develop chronic conditions — such as cardiovascular dysfunction, ischaemic cardiac failure, renal and hepatic disorders, diabetes, poor fitness, muscular weakness and cognitive decline — two decades earlier than individuals with no history of paediatric cancer treatment197,198. Indeed, experimental evidence suggests that these manifestations of accelerated ageing in cancer survivors are associated with systemic senescence and SASP factors, probably as a result of anticancer therapy199. Childhood cancer survivors have premature accumulation of T lymphocytes expressing high levels of p16INK4a and have higher circulating concentrations of the pro-inflammatory SASP factors IL-10 and IL-17 (refs.193,197). Acceleration of age-associated dysfunctions and frailty can also be observed in survivors of cancers in adulthood, who tend to have a higher incidence of hospitalization, chronic diseases and non-cancer-related mortality200, although a causal link between anticancer therapy and senescence dependency are technically difficult to demonstrate. As in children and adolescents, TIS is also believed to be a major driving force of frailty and other AEs in adults owing to its detrimental effect on tissue function via chronic inflammation, similar to ‘inflammaging’, which is a chronic mild inflammatory process that develops with age198,200,201.
Studies in mouse models have demonstrated that TIS promotes cardiac dysfunction, inflammation, bone loss, muscle weakness, renal and hepatic failure, haematopoietic insufficiency, and overall frailty27,111,202. Despite the presumably temporary presence of TIS cells in non-malignant tissues, a fact that should not be overlooked is that certain cancer types and stages require continuous dosing or extensive treatment cycles, followed by next-line protocols if no longer effective, often resulting in many months or even years of chronic drug exposure.
Senotherapies in cancer
The preclinical evidence of increased secondary tumour incidence, aggressive relapses and chronic treatment-related toxicity as a putative result of persisting TIS cells, further aggravated by the ageing-related accumulation of endogenously senescent cells, underscores the rationale for senolytic therapeutic approaches (Fig. 4). Senotherapy can refer to the selective elimination of senescent cells using agents termed senolytics (Supplementary Table 2) or to the reduced production and secretion of SASP factors using drugs called senomorphics (Supplementary Table 3).

Three strategies are summarized. a | Senolytic therapies selectively kill senescent cells that have detrimental pro-tumorigenic effects (see Supplementary Table 2). b | Senomorphic therapy inhibits the signalling pathways that regulate the senescence-associated secretory phenotype (SASP), such as NF-κB, mTOR and p38, or individual SASP factors that promote tumour progression (see Supplementary Table 3). c | Alternatively, pro-senescent cancer therapies can promote secretion of less-detrimental SASP factors and, thus, a non-deleterious senescence phenotype.
Senolytic therapy
In the first demonstration of effective senolysis in a cancer context, specifically delayed tumour growth in a mouse model, the autophagy inhibitor bafilomycin promoted caspase-dependent lymphoma cell death via the endoplasmic reticulum-associated proteotoxic pathway, following TIS using cyclophosphamide45. Importantly, the same therapeutic sequence failed to delay tumour growth beyond the effect of cyclophosphamide alone in mice harbouring lymphomas that were genetically senescence-incapable. Senolytic drugs mainly target anti-apoptotic pathways that are upregulated in senescent cells to ensure survival despite enhanced stress signalling. Navitoclax, a BCL-2 family inhibitor targeting the proteins BCL-2, BCL-XL and BCL-W, selectively kills senescent cells in culture (including replicative lifespan-exhausted, (pre)malignant and virus-induced senescent cells), and in sublethally irradiated or aged mice. Navitoclax-related depletion of senescent cells also ‘rejuvenated’ the regenerative capacity of aged haematopoietic stem cells and muscle stem cells in mice without cancer203. In the context of cancer, exposure to navitoclax following doxorubicin or etoposide effectively induced tumour regression in a mouse xenograft model204. Navitoclax was also able to efficiently kill ovarian and breast cancer cells that underwent PARP inhibitor-related senescence in vitro and in mouse xenografts184. Galacto-conjugated nanoparticles with navitoclax (nav–gal) can be specifically activated by a high SA-β-gal activity and can selectively kill senescent cells. Co-exposure to cisplatin and nav–gal resulted in enhanced elimination of lung cancer cells both in vitro and in vivo205. In mice with doxorubicin-induced senescence, the BET family protein degrader ARV-825 had effective senolytic activity against senescent hepatic stellate cells and delayed liver cancer development206. In a mouse model of liver cancer, the mTOR inhibitor AZD8055 also showed senolytic potential against cancer cells with senescence induced by small-molecule inhibitors of CDC7 (ref.207). Cardiac glycosides, including the widely prescribed digoxin, were able to selectively kill TIS cancer cells in culture and in vivo208,209.
The immune system can also elicit endogenous senolytic effects following TIS. For example, dual inhibition of MEK and CDK4/6 induced senescence in a mouse model of Kras-mutant PDAC, favoured tumour vascularization and endothelial cell activation, and ultimately enhanced the efficacy of anti-PD-1 antibodies in this model152. Moreover, CAR T cells targeting uPAR on the surface of senescent cells have been developed as promising cell-based senolytics22. Mice with lung adenocarcinomas were first exposed to a combination of MEK and CDK4/6 inhibitors to induce senescence, and then received uPAR-targeting CAR T cells to selectively kill those senescent cells, leading to a marked delay in tumour growth22.
Other senolytic drugs have been widely used in the settings of ageing and other disease models. ABT-737 is another BCL-2, BCL-XL and BCL-W inhibitor that induced apoptosis in senescent cells within the lung upon radiation-induced DNA damage and in lung epidermal cells with p53 hyperactivation caused by transgenic overexpression of p14ARF (the human homologue of p19ARF)210. Removal of senescent cells by ABT-737 in the liver also enhanced liver regeneration in adult mice without cancer211. The BCL-XL-selective inhibitors A1155463 and A1331852 have been developed as promising senolytic drugs212. The broad-spectrum tyrosine kinase receptor inhibitor dasatinib combined with quercetin, a flavonoid with extended activity against various kinases, including SRC and PI3K, has been extensively investigated as a senolytic combination regimen213. In mice, this combination selectively killed transplanted senescent cells, delayed the decline in physical activities and extended lifespan21. Notably, dasatinib–quercetin has already been administered to patients with diabetic kidney disease as a putative senolytic regimen in clinical trials214. Exposure to this combination reduced the abundance of senescent cells and the pro-inflammatory SASP factors in human adipose tissues21, and improved organ function of patients diagnosed with idiopathic pulmonary fibrosis215. Similar to quercetin, fisetin is a flavonoid that was identified as a senolytic drug that can remove senescent cells, thereby improving the healthspan and lifespan of progeroid mice upon long-term exposure216. Moreover, a study using an SA-β-gal screening system identified the HSP90 inhibitor 17-demethoxy-geldanamycin as a potent senolytic drug217. FOXO4-DRI is a peptide designed to disrupt the p53–FOXO4 interaction, thereby selectively inducing apoptosis in senescent cells. Removal of senescent cells using FOXO4-DRI neutralized doxorubicin-induced toxicity in vivo and restored general fitness as well as renal function in aged animals218. Senolytic drugs have also shown activity in the context of SARS-CoV-2 infection (Box 1). Given all these exciting findings, more preclinical but, ultimately, clinical studies (some of which are ongoing) are needed to demonstrate whether the long-term benefits of senolytics outweigh their potential toxicities, and to identify the most effective dose schedules, especially regarding one-time versus repeated administration of senolytic agents.
Senomorphic therapy
Given that the majority of tumour-promoting and chemotoxicity-promoting functions of senescent cells seem to be causally related to the SASP, senomorphic SASP inhibitors could be used as effective alternatives to senolytics, thereby potentially preserving the less SASP-dependent pro-immunogenic functions of senescent cells, especially tumour immunosurveillance in the context of cancer cell senescence. SASP inhibitors, however, might require long-term administration because most of their effects are likely to vanish upon drug discontinuation. NF-κB-mediated signalling is the master regulator of the pro-inflammatory role of the SASP. Metformin, to some extent, prevents nuclear translocation of NF-κB pathway components and their subsequent transactivation at target gene promoters, thereby reducing expression of various SASP factors and potentially explaining, at least in part, the anti-ageing and antitumour effects of metformin in both mouse models and patients with diabetes219. In various preclinical studies, the mTOR inhibitor rapamycin might have the context-dependent potential to induce senescence45, diminish NF-κB activity65,66, suppress the pro-inflammatory SASP at the translational level, and limit the growth-promoting effect of senescent bystander fibroblasts on prostate tumours66. Hypoxia mimetics (such as roxadustat) interfere with the expression of various SASP factors by attenuating mTOR activation, and have been shown to reduce senescence-mediated AEs in a mouse model of doxorubicin-induced senescence220. Inhibitors of the p38 pathway were also found to suppress the SASP, leading to reduced bone loss and metastasis in a mouse model of TIS breast cancer202,221,222. Inhibition of the JAK signalling pathway in aged animals reduced inflammaging and alleviated age-related frailty; a number of JAK inhibitors are being tested in clinical trials involving patients with myelofibrosis, acute myeloid leukaemia or lymphomas223,224.
Antibodies against SASP factors also hold the potential to limit detrimental senescence-associated functions. Siltuximab, a neutralizing anti-IL-6 monoclonal antibody approved for the treatment of multicentric Castleman disease, is being tested in various oncological settings225. Canakinumab, an antibody inhibiting IL-1β that is approved for the treatment of a variety of pyrexia-featured inflammatory syndromes, has shown some activity in various trials involving patients with non-small-cell lung cancer226. These and other agents require in-depth follow-up investigations in appropriate model systems and clinical trials to pinpoint their specific anticancer efficacy as senescence-dependent, SASP-suppressing agents beyond unspecific anti-inflammatory potential.
The phenotype of senescent cells is highly heterogeneous, with subsets of senescent cells exhibiting only a partial SASP, and thus a fundamentally different strategy to capitalize on the beneficial side of senescence might be the identification of compounds that promote activation of less-toxic senescence programmes. CDK4/6 inhibitors operate as potent senescence inducers in cancer cells and, to some extent, in non-malignant cells, including lymphocytes. CDK4/6 inhibitor-induced senescent non-malignant and malignant cells largely lack pro-inflammatory SASP factors111,153. The proportion of senescent cancer cells in mice exposed to the CDK4/6 inhibitor abemaciclib was comparable to that in mice exposed to doxorubicin, but mice exposed abemaciclib suffered less from detrimental effects owing to a reduction in pro-inflammatory SASP factors111. Importantly, despite the marked reduction in the inflammatory SASP response, CDK4/6 inhibition triggers antitumour immunity and favours senescent cell removal111,153. Whether other fundamental and potentially deleterious facets of the senescent phenotype, in particular epigenetic remodelling into a latent stemness programme5, occur in response to these agents requires further investigation.
Conclusions
Cellular senescence is an inherent and virtually unavoidable consequence of treatment in patients with cancer. Cancer cell senescence mainly refers to surviving cancer cells that enter stable and durable cell cycle arrest, but can also be triggered in non-malignant cells in various organ systems across the body. Given the complex cell-extrinsic effects that senescent cells can exert in their surroundings, and the fundamental cell-intrinsic rewiring that profoundly alters cellular functionality and can account for stem-like reprogramming, the consequences of senescence are far more complex than those of apoptosis. Thus, managing residual senescent cancer cells as well as the consequences of senescence of non-malignant cells in patients receiving pro-senescent antitumour therapies is a clinical challenge. Weighing the balance between the ‘bright’ and ‘dark’ sides of senescence is difficult, given that tumour-suppressive and tumour-promoting effects linked to senescent cancer cells can coexist in the same patient. Specifically, neither a dependable quantitative assessment of the different contributions that such effects could have on long-term outcome nor marker-based detection and selective targeting of less-desirable senescent cell populations is currently feasible in the clinic. Pharmacological suppression or modulation of the SASP might work to a certain extent, but is unlikely to robustly change tumour fate. By and large, premature cancer cell senescence has acutely beneficial but chronically detrimental ramifications. Most cytotoxic and cytostatic cancer treatments currently available induce senescence, whether intended or not, as a collateral effect in a certain proportion of the surviving cancer cell population. Thus far, senolysis (that is, senescence-related opportunities to eliminate drug-exposed malignant cells that failed to undergo apoptotic cell death in the first place but contributed to the initial treatment response via proliferative arrest) seems to be the preferred strategy because it seems the only definitive option towards tumour eradication. Although numerous promising candidate senolytics are being identified, some of which have entered clinical trials227, prospective results of large-cohort oncology trials remain to be reported. Such studies should provide insights as to whether protection from post-senescent cancer relapse and concurrent elimination of organ function-disabling senescent cells in non-malignant tissues can be established as key objectives of therapeutic senolytic approaches in patients with cancer. When these goals are achieved, hopefully in the near future, another major challenge not discussed here remains the drug-exposed death-resistant and never-senescent cancer cell.
References
Muñoz-Espín, D. et al. Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118 (2013).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014).
Ritschka, B. et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 31, 172–183 (2017).
Mosteiro, L. et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 354, aaf4445 (2016).
Milanovic, M. et al. Senescence-associated reprogramming promotes cancer stemness. Nature 553, 96–100 (2018).
Lee, S. & Schmitt, C. A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 21, 94–101 (2019).
Feng, T. et al. CCN1-induced cellular senescence promotes heart regeneration. Circulation 139, 2495–2498 (2019).
Sarig, R. et al. Transient p53-mediated regenerative senescence in the injured heart. Circulation 139, 2491–2494 (2019).
Jun, J.-I. & Lau, L. F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685 (2010).
Krizhanovsky, V. et al. Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667 (2008).
Wolstein, J. M. et al. INK4a knockout mice exhibit increased fibrosis under normal conditions and in response to unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 299, F1486–F1495 (2010).
Li, Y. et al. Hyaluronan synthase 2 regulates fibroblast senescence in pulmonary fibrosis. Matrix Biol. 55, 35–48 (2016).
Minamino, T. et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 15, 1082–1087 (2009).
Childs, B. G. et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477 (2016).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017).
Schafer, M. J. et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 8, 14532 (2017).
Bussian, T. J. et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582 (2018).
Ferreira-Gonzalez, S. et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat. Commun. 9, 1020 (2018).
Binet, F. et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 369, eaay5356 (2020).
Baker, D. J. et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020).
Grosse, L. et al. Defined p16High senescent cell types are indispensable for mouse healthspan. Cell Metab. 32, 87–99.e6 (2020).
Hernandez-Segura, A., Nehme, J. & Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 (2018).
Schade, A. E., Fischer, M. & DeCaprio, J. A. RB, p130 and p107 differentially repress G1/S and G2/M genes after p53 activation. Nucleic Acids Res. 47, 11197–11208 (2019).
Sage, J., Miller, A. L., Pérez-Mancera, P. A., Wysocki, J. M. & Jacks, T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 424, 223–228 (2003).
Demaria, M. et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 (2017).
Omori, S. et al. Generation of a p16 reporter mouse and its use to characterize and target p16high cells in vivo. Cell Metab. 32, 814–828.e6 (2020).
Ayrapetov, M. K., Gursoy-Yuzugullu, O., Xu, C., Xu, Y. & Price, B. D. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl Acad. Sci. USA 111, 9169–9174 (2014).
Bekker-Jensen, S. et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 173, 195–206 (2006).
Lukas, C., Falck, J., Bartkova, J., Bartek, J. & Lukas, J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol. 5, 255–260 (2003).
Turenne, G. A., Paul, P., Laflair, L. & Price, B. D. Activation of p53 transcriptional activity requires ATM’s kinase domain and multiple N-terminal serine residues of p53. Oncogene 20, 5100–5110 (2001).
Dikovskaya, D. et al. Mitotic stress is an integral part of the oncogene-induced senescence program that promotes multinucleation and cell cycle arrest. Cell Rep. 12, 1483–1496 (2015).
Freund, A., Laberge, R.-M., Demaria, M. & Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 23, 2066–2075 (2012).
Neurohr, G. E. et al. Excessive cell growth causes cytoplasm dilution and contributes to senescence. Cell 176, 1083–1097 (2019).
Sugrue, M. M., Shin, D. Y., Lee, S. W. & Aaronson, S. A. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc. Natl Acad. Sci. USA 94, 9648–9653 (1997).
Lloyd, A. C. The regulation of cell size. Cell 154, 1194–1205 (2013).
Blagosklonny, M. V. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging 4, 159–165 (2012).
Jung, S. H. et al. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 38, 1639–1650 (2019).
Kolesnichenko, M., Hong, L., Liao, R., Vogt, P. K. & Sun, P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle 11, 2391–2401 (2014).
Jung, C. H., Ro, S.-H., Cao, J., Otto, N. M. & Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295 (2010).
Cassidy, L. D. et al. Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk. Nat. Commun. 11, 307 (2020).
Bernard, M. et al. Autophagy drives fibroblast senescence through MTORC2 regulation. Autophagy 16, 2004–2016 (2020).
Young, A. R. J. et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 23, 798–803 (2009).
Dörr, J. R. et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501, 421–425 (2013).
Xie, X., Koh, J. Y., Price, S., White, E. & Mehnert, J. M. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 5, 410–423 (2015).
Courtois-Cox, S. et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 10, 459–472 (2006).
Wall, M. et al. The mTORC1 inhibitor everolimus prevents and treats Eμ-Myc lymphoma by restoring oncogene-induced senescence. Cancer Discov. 3, 82–95 (2013).
Walters, H. E., Deneka-Hannemann, S. & Cox, L. S. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging 8, 231–243 (2016).
Passos, J. F. et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 5, e110 (2007).
Wiley, C. D. et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314 (2016).
Igelmann, S. et al. A hydride transfer complex reprograms NAD metabolism and bypasses senescence. Mol. Cell 81, 3848–3865.e19 (2021).
Pluquet, O., Pourtier, A. & Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 308, C415–C425 (2015).
Cormenier, J. et al. The ATF6α arm of the unfolded protein response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E2 intracrine pathway. Mech. Ageing Dev. 170, 82–91 (2018).
Quijano, C. et al. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 11, 1383–1392 (2012).
Kaplon, J. et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112 (2013).
James, E. L. et al. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 14, 1854–1871 (2015).
Wiley, C. D. & Campisi, J. From ancient pathways to aging cells — connecting metabolism and cellular senescence. Cell Metab. 23, 1013–1021 (2016).
Takebayashi, S. et al. Retinoblastoma protein promotes oxidative phosphorylation through upregulation of glycolytic genes in oncogene-induced senescent cells. Aging Cell 14, 689–697 (2015).
Zou, H., Stoppani, E., Volonte, D. & Galbiati, F. Caveolin-1, cellular senescence and age-related diseases. Mech. Ageing Dev. 132, 533–542 (2011).
Althubiti, M. et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 5, e1528 (2014).
Kim, K. M. et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 31, 1529–1534 (2017).
Kuilman, T. et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008).
Chien, Y. et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 25, 2125–2136 (2011).
Herranz, N. et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 17, 1205–1217 (2015).
Laberge, R.-M. et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 17, 1049–1061 (2015).
Freund, A., Patil, C. K. & Campisi, J. p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J. 30, 1536–1548 (2011).
Coppé, J.-P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, e301 (2008).
Wiley, C. D. et al. Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis. Cell Metab. 33, 1124–1136.e5 (2021).
Hoare, M. et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 18, 979–992 (2016).
Kolesnichenko, M. et al. Transcriptional repression of NFKBIA triggers constitutive IKK‐ and proteasomeindependent p65/RelA activation in senescence. EMBO J. 40, e104296 (2021).
Hernandez-Segura, A. et al. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27, 2652–2660.e4 (2017).
Casella, G. et al. Transcriptome signature of cellular senescence. Nucleic Acids Res. 47, 11476 (2019).
Martínez-Zamudio, R. I. et al. AP-1 imprints a reversible transcriptional programme of senescent cells. Nat. Cell Biol. 22, 842–855 (2020).
Parry, A. J. et al. NOTCH-mediated non-cell autonomous regulation of chromatin structure during senescence. Nat. Commun. 9, 1840 (2018).
Hsu, C.-H., Altschuler, S. J. & Wu, L. F. Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell 178, 361–373.e12 (2019).
Takahashi, A. et al. Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 8, 1291–1297 (2006).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009).
Mastri, M. et al. A transient pseudosenescent secretome promotes tumor growth after antiangiogenic therapy withdrawal. Cell Rep. 25, 3706–3720.e8 (2018).
Dimri, G. P. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA 92, 9363–9367 (1995).
Michaloglou, C. et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720–724 (2005).
Gray-Schopfer, V. C. et al. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br. J. Cancer 95, 496–505 (2006).
Sharpless, N. E. & Sherr, C. J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 15, 397–408 (2015).
Acosta, J. C. et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 (2008).
Vernier, M. et al. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 25, 41–50 (2011).
Deschênes-Simard, X. et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 27, 900–915 (2013).
Bartkova, J. et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 (2006).
Haugstetter, A. M. et al. Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br. J. Cancer 103, 505–509 (2010).
Fujita, K. et al. p53 isoforms Δ133p53 and p53β are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 11, 1135–1142 (2009).
Eggert, T. et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30, 533–547 (2016).
Chang, B. D. et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 59, 3761–3767 (1999).
Schmitt, C. A. et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109, 335–346 (2002).
te Poele, R. H., Okorokov, A. L., Jardine, L., Cummings, J. & Joel, S. P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 62, 1876–1883 (2002).
Roberson, R. S., Kussick, S. J., Vallieres, E., Chen, S.-Y. J. & Wu, D. Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 65, 2795–2803 (2005).
Han, Z. et al. Role of p21 in apoptosis and senescence of human colon cancer cells treated with camptothecin. J. Biol. Chem. 277, 17154–17160 (2002).
Peiris-Pagès, M., Sotgia, F. & Lisanti, M. P. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog-GLI signalling in breast cancer cells. Oncotarget 6, 10728–10745 (2015).
Hu, W. et al. Mechanistic investigation of bone marrow suppression associated with palbociclib and its differentiation from cytotoxic chemotherapies. Clin. Cancer Res. 22, 2000–2008 (2016).
Ota, H. et al. Sirolimus and everolimus induce endothelial cellular senescence via sirtuin 1 down-regulation therapeutic implication of cilostazol after drug-eluting stent implantation. J. Am. Coll. Cardiol. 53, 2298–2305 (2009).
Sanoff, H. K. et al. Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J. Natl Cancer Inst. 106, dju057 (2014).
Mitin, N. et al. A biomarker of aging, p16, predicts peripheral neuropathy in women receiving adjuvant taxanes for breast cancer. Preprint at MedRxiv https://doi.org/10.1101/2022.02.10.22270086 (2022).
Febres-Aldana, C. A., Kuritzky, N., Krishnamurthy, K., Poppiti, R. & Howard, L. Evaluation of the expression of P16INK4A by immunohistochemistry in post-neoadjuvant chemotherapy hormone receptor negative breast cancer specimens. Breast Dis. 39, 51–59 (2020).
Schoetz, U. et al. Early senescence and production of senescence-associated cytokines are major determinants of radioresistance in head-and-neck squamous cell carcinoma. Cell Death Dis. 12, 1162 (2021).
Peng, X. et al. Cellular senescence contributes to radiation-induced hyposalivation by affecting the stem/progenitor cell niche. Cell Death Dis. 11, 854 (2020).
Amundson, S. A. et al. Human in vivo radiation-induced biomarkers: gene expression changes in radiotherapy patients. Cancer Res. 64, 6368–6371 (2004).
Perez, M. et al. Efficacy of bortezomib in sarcomas with high levels of MAP17 (PDZK1IP1). Oncotarget 7, 67033–67046 (2016).
Goel, S. et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell 29, 255–269 (2016).
Llanos, S. et al. Lysosomal trapping of palbociclib and its functional implications. Oncogene 38, 3886–3902 (2019).
Hafner, M. et al. Multiomics profiling establishes the polypharmacology of FDA-approved CDK4/6 inhibitors and the potential for differential clinical activity. Cell Chem. Biol. 26, 1067–1080.e8 (2019).
Guan, X. et al. Stromal senescence by prolonged CDK4/6 inhibition potentiates tumor growth. Mol. Cancer Res. 15, 237–249 (2017).
Crozier, L. et al. CDK4/6 inhibitors induce replication stress to cause long‐term cell cycle withdrawal. EMBO J. https://doi.org/10.15252/embj.2021108599 (2022).
Wang, B. et al. Pharmacological CDK4/6 inhibition reveals a p53‐dependent senescent state with restricted toxicity. EMBO J. https://doi.org/10.15252/embj.2021108946 (2022).
Elknerova, K. et al. Epigenetic modulation of gene expression of human leukemia cell lines–induction of cell death and senescence. Neoplasma 58, 35–44 (2011).
Kaletsch, A. et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin. Epigenetics 10, 100 (2018).
Amatori, S., Bagaloni, I., Viti, D. & Fanelli, M. Premature senescence induced by DNA demethylating agent (Decitabine) as therapeutic option for malignant pleural mesothelioma. Lung Cancer 71, 113–115 (2011).
Hasan, M. R., Ho, S. H. Y., Owen, D. A. & Tai, I. T. Inhibition of VEGF induces cellular senescence in colorectal cancer cells. Int. J. Cancer 129, 2115–2123 (2011).
Zhu, Y. et al. Sunitinib induces cellular senescence via p53/Dec1 activation in renal cell carcinoma cells. Cancer Sci. 104, 1052–1061 (2013).
Jain, R. K. et al. Biomarkers of response and resistance to antiangiogenic therapy. Nat. Rev. Clin. Oncol. 6, 327–338 (2009).
Däbritz, J. H. M. et al. CD20-targeting immunotherapy promotes cellular senescence in B-cell lymphoma. Mol. Cancer Ther. 15, 1074–1081 (2016).
Jochems, F. et al. The cancer SENESCopedia: a delineation of cancer cell senescence. Cell Rep. 36, 109441 (2021).
Braig, M. et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436, 660–665 (2005).
Rane, S. G., Cosenza, S. C., Mettus, R. V. & Reddy, E. P. Germ line transmission of the Cdk4(R24C) mutation facilitates tumorigenesis and escape from cellular senescence. Mol. Cell Biol. 22, 644–656 (2002).
Collado, M. et al. Senescence in premalignant tumours. Nature 436, 642 (2005).
Denchi, E. L., Attwooll, C., Pasini, D. & Helin, K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol. Cell Biol. 25, 2660–2672 (2005).
Reddy, H. K. D. L. et al. Requirement of Cdk4 for v-Ha-ras–induced breast tumorigenesis and activation of the v-ras–induced senescence program by the R24C mutation. Genes Cancer 1, 69–80 (2010).
Beauséjour, C. M. et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 22, 4212–4222 (2003).
Sage, J. et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 14, 3037–3050 (2000).
Donehower, L. A. et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221 (1992).
Jacks, T. et al. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4, 1–7 (1994).
Serrano, M. et al. Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27–37 (1996).
Martín-Caballero, J., Flores, J. M., García-Palencia, P. & Serrano, M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 61, 6234–6238 (2001).
Takeuchi, S. et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res. 70, 9381–9390 (2010).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008 (2010).
Foulkes, W. D., Flanders, T. Y., Pollock, P. M. & Hayward, N. K. The CDKN2A (p16) gene and human cancer. Mol. Med. 3, 5–20 (1997).
Shamloo, B. & Usluer, S. p21 in cancer research. Cancers 11, 1178 (2019).
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002).
Chen, K., Zhang, Y., Qian, L. & Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 14, 116 (2021).
Acosta, J. C. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990 (2013).
Biran, A. et al. Senescent cells communicate via intercellular protein transfer. Genes Dev. 29, 791–802 (2015).
Di, X. et al. A chemotherapy-associated senescence bystander effect in breast cancer cells. Cancer Biol. Ther. 7, 864–872 (2008).
Kim, J. J., Lee, S. B., Park, J. K. & Yoo, Y. D. TNF-α-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-XL. Cell Death Differ. 17, 1420–1434 (2010).
Regis, G. et al. IL-6, but not IFN-γ, triggers apoptosis and inhibits in vivo growth of human malignant T cells on STAT3 silencing. Leukemia 23, 2102–2108 (2009).
Kang, T.-W. et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551 (2011).
Tasdemir, N. et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 6, 612–629 (2016).
Ventura, A. et al. Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665 (2007).
Xue, W. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007).
Sturmlechner, I. et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 374, eabb3420 (2021).
Reimann, M. et al. Tumor stroma-derived TGF-β limits myc-driven lymphomagenesis via Suv39h1-dependent senescence. Cancer Cell 17, 262–272 (2010).
Lujambio, A. et al. Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460 (2013).
Yu, Y. et al. Targeting the senescence-overriding cooperative activity of structurally unrelated H3K9 demethylases in melanoma. Cancer Cell 33, 785 (2018).
Reimann, M. et al. Adaptive T-cell immunity controls senescence-prone MyD88- or CARD11-mutant B-cell lymphomas. Blood 137, 2785–2799 (2021).
Ruscetti, M. et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362, 1416–1422 (2018).
Ruscetti, M. et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 181, 424–441.e21 (2020).
Goel, S. et al. CDK4/6 inhibition triggers anti-tumor immunity. Nature 548, 471–475 (2017).
Heckler, M. et al. Inhibition of CDK4/6 promotes CD8 T-cell memory formation. Cancer Discov. 11, 2564–2581 (2021).
Jing, H. et al. Opposing roles of NF-κB in anti-cancer treatment outcome unveiled by cross-species investigations. Genes Dev. 25, 2137–2146 (2011).
Wang, B., Kohli, J. & Demaria, M. Senescent cells in cancer therapy: friends or foes? Trends Cancer 6, 838–857 (2020).
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P.-Y. & Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl Acad. Sci. USA 98, 12072–12077 (2001).
Liu, D. & Hornsby, P. J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67, 3117–3126 (2007).
Kim, Y. H. et al. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 8, 15208 (2017).
Jackson, J. G. et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 21, 793–806 (2012).
Fletcher-Sananikone, E. et al. Elimination of radiation-induced senescence in the brain tumor microenvironment attenuates glioblastoma recurrence. Cancer Res. 81, 5935–5947 (2021).
Ohanna, M. et al. Senescent cells develop a PARP-1 and nuclear factor-κB-associated secretome (PNAS). Genes Dev. 25, 1245–1261 (2011).
Ancrile, B., Lim, K.-H. & Counter, C. M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 21, 1714–1719 (2007).
Ortiz-Montero, P., Londoño-Vallejo, A. & Vernot, J.-P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 15, 17 (2017).
Eyman, D., Damodarasamy, M., Plymate, S. R. & Reed, M. J. CCL5 secreted by senescent aged fibroblasts induces proliferation of prostate epithelial cells and expression of genes that modulate angiogenesis. J. Cell Physiol. 220, 376–381 (2009).
Aldinucci, D. & Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm. 2014, 292376 (2014).
Coppé, J.-P., Kauser, K., Campisi, J. & Beauséjour, C. M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 281, 29568–29574 (2006).
Chen, C. et al. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 10, 178 (2019).
Kessenbrock, K., Plaks, V. & Werb, Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 (2010).
Guccini, I. et al. Senescence reprogramming by TIMP1 deficiency promotes prostate cancer metastasis. Cancer Cell 39, 68–82.e9 (2021).
Fisher, D. T., Appenheimer, M. M. & Evans, S. S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 26, 38–47 (2014).
Waugh, D. J. J. & Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 14, 6735–6741 (2008).
Luo, X. et al. Stromal-initiated changes in the bone promote metastatic niche development. Cell Rep. 14, 82–92 (2016).
Kawaguchi, K., Komoda, K., Mikawa, R., Asai, A. & Sugimoto, M. Cellular senescence promotes cancer metastasis by enhancing soluble E-cadherin production. iScience 24, 103022 (2021).
Ruhland, M. K. et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 7, 11762 (2016).
Pereira, B. I. et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 10, 2387 (2019).
Toso, A. et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 9, 75–89 (2014).
Saleh, T., Tyutyunyk-Massey, L. & Gewirtz, D. A. Tumor cell escape from therapy-induced senescence as a model of disease recurrence after dormancy. Cancer Res. 79, 1044–1046 (2019).
Fumagalli, M. et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 (2012).
Narita, M. et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716 (2003).
Wang, Q. et al. Polyploidy road to therapy‐induced cellular senescence and escape. Int. J. Cancer 132, 1505–1515 (2013).
Elmore, L. W., Di, X., Dumur, C., Holt, S. E. & Gewirtz, D. A. Evasion of a single-step, chemotherapy-induced senescence in breast cancer cells: implications for treatment response. Clin. Cancer Res. 11, 2637–2643 (2005).
Saleh, T. et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharmacol. 162, 202–212 (2019).
Fleury, H. et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 10, 2556 (2019).
Patel, P. L., Suram, A., Mirani, N., Bischof, O. & Herbig, U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc. Natl Acad. Sci. 113, E5024–E5033 (2016).
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997).
Zampetidis, C. P. et al. A recurrent chromosomal inversion suffices for driving escape from oncogene-induced senescence via subTAD reorganization. Mol. Cell 81, 4907–4923.e8 (2021).
Nobori, T. et al. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 368, 753–756 (1994).
Benítez, S. et al. RANK links senescence to stemness in the mammary epithelia, delaying tumor onset but increasing tumor aggressiveness. Dev. Cell 56, 1727–1741.e7 (2021).
Galanos, P. et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 18, 777–789 (2016).
Duy, C. et al. Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer Discov. 11, 1542–1561 (2021).
Tralongo, A. C. et al. Anti-cancer treatment strategies in the older population: time to test more? Geriatrics 6, 42 (2021).
Smitherman, A. B. et al. Accelerated aging among childhood, adolescent, and young adult cancer survivors is evidenced by increased expression of p16INK4a and frailty. Cancer 126, 4975–4983 (2020).
Jabbour, W. & Wion, D. Biomarkers of aging associated with past treatments in breast cancer survivors: when therapy-induced pathways turn out to be potential therapeutic targets. NPJ Breast Cancer 4, 4 (2018).
Zhu, J. et al. Accelerated aging in breast cancer survivors and its association with mortality and cancer recurrence. Breast Cancer Res. Treat. 180, 449–459 (2020).
Bhakta, N. et al. The cumulative burden of surviving childhood cancer: an initial report from the St. Jude Lifetime Cohort Study. Lancet 390, 2569–2582 (2017).
Ariffin, H. et al. Young adult survivors of childhood acute lymphoblastic leukemia show evidence of chronic inflammation and cellular aging. Cancer 123, 4207–4214 (2017).
Ness, K. K. et al. Frailty in childhood cancer survivors. Cancer 121, 1540–1547 (2015).
Krawczuk-Rybak, M. & Latoch, E. Risk factors for premature aging in childhood cancer survivors. Dev. Period. Med. 23, 97–103 (2019).
Ness, K. K. & Wogksch, M. D. Frailty and aging in cancer survivors. Transl. Res. 221, 65–82 (2020).
Franceschi, C. et al. The network and the remodeling theories of aging: historical background and new perspectives. Exp. Gerontol. 35, 879–896 (2000).
Yao, Z. et al. Therapy-induced senescence drives bone loss. Cancer Res. 80, 1171–1182 (2020).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Saleh, T. et al. Clearance of therapy‐induced senescent tumor cells by the senolytic ABT‐263 via interference with BCL‐XL–BAX interaction. Mol. Oncol. 14, 2504–2519 (2020).
González‐Gualda, E. et al. Galacto‐conjugation of navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 19, e13142 (2020).
Wakita, M. et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 11, 1935 (2020).
Wang, C. et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 574, 268–272 (2019).
Triana-Martínez, F. et al. Identification and characterization of cardiac glycosides as senolytic compounds. Nat. Commun. 10, 4731 (2019).
Guerrero, A. et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 1, 1074–1088 (2019).
Yosef, R. et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 7, 11190 (2016).
Ritschka, B. et al. The senotherapeutic drug ABT-737 disrupts aberrant p21 expression to restore liver regeneration in adult mice. Genes Dev. 34, 489–494 (2020).
Zhu, Y. et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 9, 955–963 (2017).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Hickson, L. J. et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. Ebiomedicine 47, 446–456 (2019).
Justice, J. N. et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. Ebiomedicine 40, 5544–5563 (2019).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. Ebiomedicine 36, 18–28 (2018).
Fuhrmann-Stroissnigg, H. et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 8, 422 (2017).
Baar, M. P. et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147.e16 (2017).
Moiseeva, O. et al. Metformin inhibits the senescence‐associated secretory phenotype by interfering with IKK/NF‐κB activation. Aging Cell 12, 489–498 (2013).
van Vliet, T. et al. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 81, 2041–2052.e6 (2021).
Alimbetov, D. et al. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology 17, 305–315 (2016).
Murali, B. et al. Inhibition of the stromal p38MAPK/MK2 pathway limits breast cancer metastases and chemotherapy-induced bone loss. Cancer Res. 78, 5618–5630 (2018).
Xu, M. et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl Acad. Sci. USA 112, E6301–E6310 (2015).
Harrison, C. N. et al. Health‐related quality of life and symptoms in patients with myelofibrosis treated with ruxolitinib versus best available therapy. Br. J. Haematol. 162, 229–239 (2013).
Chen, R. & Chen, B. Siltuximab (CNTO 328): a promising option for human malignancies. Drug Des. Dev. Ther. 9, 3455–3458 (2015).
Schenk, K. M., Reuss, J. E., Choquette, K. & Spira, A. I. A review of canakinumab and its therapeutic potential for non-small cell lung cancer. Anticancer Drugs 30, 879–885 (2019).
Wang, L., Lankhorst, L. & Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer https://doi.org/10.1038/s41568-022-00450-9 (2022).
Camell, C. D. et al. Senolytics reduce coronavirus-related mortality in old mice. Science 373, eabe4832 (2021).
Lee, S. et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 599, 283–289 (2021).
Pierro, F. D. et al. Possible therapeutic effects of adjuvant quercetin supplementation against early-stage COVID-19 infection: a prospective, randomized, controlled, and open-label study. Int. J. Gen. Med. 14, 2359–2366 (2021).
Pierro, F. D. et al. Potential clinical benefits of quercetin in the early stage of COVID-19: results of a second, pilot, randomized, controlled and open-label clinical trial. Int. J. Gen. Med. 14, 2807–2816 (2021).
Marescal, O. & Cheeseman, I. M. Cellular mechanisms and regulation of quiescence. Dev. Cell 55, 259–271 (2020).
Mathiassen, S. G., Zio, D. D. & Cecconi, F. Autophagy and the cell cycle: a complex landscape. Front. Oncol. 7, 51 (2017).
Ishimwe, N. et al. Autophagy regulation as a promising approach for improving cancer immunotherapy. Cancer Lett. 475, 34–42 (2020).
Jiang, G.-M. et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 18, 17 (2019).
Phan, T. G. & Croucher, P. I. The dormant cancer cell life cycle. Nat. Rev. Cancer 20, 398–411 (2020).
Hangauer, M. J. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017).
Cabanos, H. F. & Hata, A. N. Emerging insights into targeted therapy-tolerant persister cells in cancer. Cancers 13, 2666 (2021).
Oren, Y. et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 596, 576–582 (2021).
Rehman, S. K. et al. Colorectal cancer cells enter a diapause-like DTP state to survive chemotherapy. Cell 184, 226–242.e21 (2021).
Acknowledgements
The authors thank the Dutch Cancer Foundation (KWF) for support, and all members of the Demaria and Schmitt laboratories for insightful discussions.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, contributed substantially to discussions of content and wrote the article. C.A.S. and M.D. reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
M.D. is a founder and shareholder of Cleara Biotech. C.A.S. and B.W. declare no competing interests.
Peer review
Peer review information
Nature Reviews Clinical Oncology thanks G. Ferbeyre, T. Tchkonia and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Schmitt, C.A., Wang, B. & Demaria, M. Senescence and cancer — role and therapeutic opportunities. Nat Rev Clin Oncol 19, 619–636 (2022). https://doi.org/10.1038/s41571-022-00668-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41571-022-00668-4
This article is cited by
-
The senescence journey in cancer immunoediting
Molecular Cancer (2024)
-
Implications of ZNF334 gene in lymph node metastasis of lung SCC: potential bypassing of cellular senescence
Journal of Translational Medicine (2024)
-
Blocking methionine catabolism induces senescence and confers vulnerability to GSK3 inhibition in liver cancer
Nature Cancer (2024)
-
Mitofusin 1 silencing decreases the senescent associated secretory phenotype, promotes immune cell recruitment and delays melanoma tumor growth after chemotherapy
Scientific Reports (2024)
-
RAB27B-regulated exosomes mediate LSC maintenance via resistance to senescence and crosstalk with the microenvironment
Leukemia (2024)
