
Abstract
Over the past decade, various liquid biopsy techniques have emerged as viable alternatives to the analysis of traditional tissue biopsy samples. Such surrogate ‘biopsies’ offer numerous advantages, including the relative ease of obtaining serial samples and overcoming the issues of interpreting one or more small tissue samples that might not reflect the entire tumour burden. To date, the majority of research in the area of liquid biopsies has focused on blood-based biomarkers, predominantly using plasma-derived circulating tumour DNA (ctDNA). However, ctDNA can also be obtained from various non-blood sources and these might offer unique advantages over plasma ctDNA. In this Review, we discuss advances in the analysis of ctDNA from non-blood sources, focusing on urine, cerebrospinal fluid, and pleural or peritoneal fluid, but also consider other sources of ctDNA. We discuss how these alternative sources can have a distinct yet complementary role to that of blood ctDNA analysis and consider various technical aspects of non-blood ctDNA assay development. We also reflect on the settings in which non-blood ctDNA can offer distinct advantages over plasma ctDNA and explore some of the challenges associated with translating these alternative assays from academia into clinical use.
Key points
-
Plasma-based circulating tumour DNA (ctDNA) assays can provide invaluable information on the status of a patient’s cancer; however, numerous alternative sources of ctDNA are available that might offer unique advantages in certain settings.
-
Non-blood sources of ctDNA include urine, cerebrospinal fluid, pleural or peritoneal fluid, saliva, stool, and seminal fluid, among others.
-
Analysing ctDNA from non-blood sources might provide a more sensitive method than plasma-based ctDNA assays for particular tumour types or anatomical locations.
-
Non-blood ctDNA assays might be complementary to plasma assays when used for the detection of driver alterations or mechanisms of resistance.
-
Challenges to the clinical implementation of non-blood-based ctDNA assays include difficulties relating to standardization of pre-analytical factors, lack of commercially available assays and the invasive procedures required to obtain certain sample types.
Similar content being viewed by others

Introduction
The past decades have witnessed an explosion in therapeutic options for both early stage and advanced-stage malignancies. This increase in available therapies has been accompanied by growing interest in developing more personalized treatment approaches, thus increasing the demand for molecular profiling strategies that enable the successful delivery of precision medicine.
The term ‘liquid biopsies’ refers to biomarkers found within biological fluids, traditionally blood, that can be sampled to provide clinically valuable information on both the patient and their underlying malignancy1. This approach offers an alternative to traditional needle-based biopsy sampling methods that enables minimally invasive repeat biopsy sampling, even for patients with tumours that are anatomically difficult to sample directly. Liquid biopsy approaches can also overcome issues such as a lack of representativeness, which is associated with the analysis of small tissue biopsy samples2. To date, the majority of liquid biopsy research has focused on blood-based biomarkers, including circulating tumour DNA (ctDNA), circulating tumour cells (CTCs) and circulating microRNAs3. However, data published over the past decades indicate that these cancer-derived cells and molecules can also be found within non-blood biological fluids, including, among others, cerebrospinal fluid (CSF), urine, pleural fluid and peritoneal fluid4,5,6,7,8,9,10. This expanding interest in liquid biopsies from a wider range of sources is supported by the improved availability of ctDNA detection and analysis technologies. In this Review, we discuss the role of non-blood biological fluids as sources of ctDNA and their potentially distinct and/or complementary roles in oncology (Box 1) as well as exploring some of the challenges associated with translating these alternative biomarker assays from the laboratory to the clinic.
Definitions and assay considerations
The term cell-free DNA (cfDNA) refers to fragments of DNA that are present outside of cells that can be detected within bodily fluids. In blood plasma, cfDNA typically consists of double-stranded DNA fragments of around 140–170 base pairs (bp) in length that mostly originate from leukocytes11,12. ctDNA refers to the portion of cfDNA derived from cancer cells, which typically comprises strands of <145 bp in length and is responsible for the substantially higher plasma cfDNA concentrations often seen in patients with cancer11,13,14,15 (Box 2). Various studies have confirmed a high level of concordance between genomic alterations detected in plasma ctDNA and those found in tumour tissues16,17. Nonetheless, the mechanisms of ctDNA release are still poorly defined and various hypotheses include tumour cell apoptosis, direct release from tumour cells, secretion from macrophages following phagocytosis of tumour cells and, to a lesser degree, release from CTCs13.
ctDNA can also arise from direct contact with tumour tissues in non-blood biological fluids, and these fluids might therefore contain a higher proportion of ctDNA than plasma. Furthermore, the comparative absence of cfDNA originating from haematopoietic cells in non-blood sources means that ctDNA might be present at higher relative concentrations and at higher variant allele frequencies (VAFs) compared with ctDNA isolated from blood18. This relative lack of cfDNA can be advantageous when quantifying ctDNA and when attempting to detect alterations present at low relative frequencies owing to a lack of the background noise created by clonal haematopoiesis, which typically increases with advancing age and/or previous radiotherapy19. Furthermore, ctDNA from non-blood sources might be more reflective of the primary tumour (as seen in pleural fluid samples obtained from patients with advanced-stage thoracic malignancies) or, alternatively, might enable the characterization of a genetically distinct population of tumour cells, such as those located in the CSF in patients with intracranial disease18,20.
A variety of assay types can be used to measure ctDNA and these have been extensively reviewed elsewhere1,21. Assay choice is influenced by the sample type1,21. Another key consideration is the relative amount of ctDNA present within a sample, which dictates the required assay sensitivity. ctDNA assays include those designed to detect specific preselected mutations using digital PCR (dPCR)-based techniques, targeted next-generation sequencing (NGS) panels (comprising up to 1,000 genes), or even broader approaches involving whole-exome sequencing or the analysis of global methylation status22. Considerations regarding the choice of assay apply equally to ctDNA derived from plasma and from non-blood sources. However, the high ctDNA to cfDNA ratio within most non-blood samples relative to plasma suggests that these samples might be less susceptible to dilution by non-tumour-derived DNA. Obtaining larger sample volumes, enabling both a greater quantity of ctDNA to be extracted and analysed and an improved level of sensitivity, is also easier when sampling certain fluids such as urine23.
A plethora of alternative sources of ctDNA are available, and several roles for these samples are now emerging (Fig. 1 and Box 1). Each has advantages and disadvantages when compared to ctDNA from blood plasma (Box 3). In the following sections, we discuss existing evidence supporting the utility of ctDNA obtained from non-blood sources and highlight how the information provided from these samples might augment blood-based analysis. Of note, we use the term ctDNA to refer more broadly to these non-blood sources of tumour-derived cfDNA.

Owing to the advantages of direct contact with one or more tumour types, non-blood sources of circulating tumour DNA (ctDNA) have certain advantages that could either supplant or complement plasma ctDNA. CNS, central nervous system; NSCLC, non-small-cell lung cancer.
Urinary ctDNA
Transrenal tumour DNA
The presence of DNA within urine and its potential clinical use have been recognized since the early 2000s. Urine provides a promising source of ctDNA that can be sampled entirely non-invasively9,24. ctDNA within the urine is comprised of two distinct fractions9. The first is transrenal tumour DNA (trtDNA), which originates from plasma and enters the urine through glomerular filtration and is therefore limited in size (typically <250 bp)25. The low concentration of trtDNA has previously restricted the analysis of this type of ctDNA, although these issues have been partially overcome using modern analysis approaches such as NGS23. The second fraction originates from tumour cells shedding directly from the urinary tract and, by virtue of not undergoing renal filtration, can be of larger molecular weight26.
The most notable advantage of analysing urinary ctDNA is the ability to obtain samples entirely non-invasively, without the need for venesection or for a health-care professional to be present during sampling27. This advantage makes serial sampling much simpler and enables samples to be obtained within the patient’s home, which is of particular interest given the focus on ‘virtual’ care following the COVID-19 pandemic28. Urine samples are potentially also less susceptible to dilution of ctDNA with cfDNA from leukocyte lysis29 although cfDNA shed from the urinary tract epithelia might have a similar effect30. Nonetheless, urine has the unique benefit of enabling the direct assessment of DNA released from urological cancers.
Despite these theoretical advantages, urinary ctDNA assays are substantially less developed than their plasma-based alternatives and certain disadvantages exist and should be highlighted. Firstly, trtDNA must pass through the glomerulus, which limits the molecular weight of the fragments to <250 bp. Secondly, the glomerular filtration rate controls the rate of urinary trtDNA accumulation and can be highly variable, particularly in patients receiving systemic anticancer therapy. Indeed, an analysis of urine samples obtained at various times of day suggests substantially lower trtDNA yields from samples obtained <1.5 h after a previous void27. The use of preservatives and optimized storage and transport temperatures is also crucial given the short half-life of unpreserved DNA27,31. A further potential drawback is the often large volume of voided urine and the dilutional effects this will have on ctDNA. Processing of large-volume samples using cfDNA isolation protocols is more technically challenging and, while some studies describe the detection of ctDNA in small volumes of urine (entry volumes of 1–2 ml), others used larger volumes and added a sample concentration step to the workflow30,32,33.
Most research into clinical uses of trtDNA is from patients with non-small-cell lung cancer (NSCLC), testing for alterations in EGFR, including the T790M mutation, and KRAS23,34,35. EGFR mutation status was examined through dPCR and NGS analysis of urine samples from patients receiving third-generation EGFR inhibitors in clinical trials23. In a cohort of 63 patients from the Tiger-X trial23, comparisons with tissue analysis demonstrated that trtDNA had a detection sensitivity of 67–75% depending on the specific mutation subtype, which increased to 80–93% when larger volumes of urine were available (compared with 87–100% for plasma samples)23. Combined analysis of urinary and plasma ctDNA identified 89% of all T790M mutations compared with 75% following tissue-only analyses23. These data highlight a potential synergistic effect of combining different liquid biopsy methods and provide early evidence of concordance between trtDNA and tissue EGFR status. This approach also enables the identification of EGFR alterations that were not detected in tumour samples and could be used to monitor both treatment response and the development of acquired resistance. Similarly, analysis of the presence of KRAS mutations in trtDNA has shown high levels of concordance of around 70–77% relative to tissue sampling, which correlates favourably with plasma ctDNA36, as well as providing prognostic information for patients with elevated KRAS-mutant trtDNA at baseline35,37.
trtDNA analysis has also shown utility across several other cancer types, including gastric, colorectal, pancreatic and ovarian cancers, as well as central nervous system (CNS) lymphomas38,39,40. dPCR-based analysis of KRAS and BRAF mutations in ctDNA from plasma and urine samples from patients with colorectal cancer (CRC) revealed sensitivity and specificity values of 85.5% and 100%, respectively, for urinary ctDNA, with overall agreement with tissue analysis of 91%, which was similar to plasma ctDNA (sensitivity 87.3%, specificity 100%, overall agreement 92%)41. Whole-exome sequencing has also been shown to be a feasible method of analysing trtDNA in urine samples obtained from patients with CRC; however, in a study involving 24 patients, only 4 had levels of trtDNA sufficient for analysis, thus highlighting the current challenges associated with this approach30. Nonetheless, liquid biopsy of trtDNA is garnering increasing interest as an early detection method42,43. For example, oncogenic KRAS mutations can be detected in urine samples from a subset of patients with stage I CRC despite the lower levels of ctDNA inherent to early stage disease reducing the sensitivity of the method, suggesting a potential role in CRC diagnosis43.
Owing to the entirely non-invasive sampling process, the utility of trtDNA has been investigated as a method of monitoring disease status following curative-intent treatment. trtDNA levels have been found to decline following surgery in women with early stage breast cancer (2.1-fold reduction within 6 months of surgery), with both a higher absolute concentration at 6 months (HR 1.71, 95% CI 1.28–2.28) and greater change from baseline (HR 2.42, 95% CI 1.88–3.33) found to be predictive of disease relapse within 3 years44. These results were similar to those obtained with plasma ctDNA analysis, although the authors observed that urinary DNA might be a slightly better risk-stratification method. Similarly, trtDNA has been detected as early as 9 months prior to radiological evidence of disease recurrence in patients with hepatocellular carcinoma following tumour ablation with a curative intent45. Therefore, regular urine sampling (which can be performed at home, with the samples returned by post) following curative treatment could enable early detection of disease recurrence with a greater level of compliance compared with strategies that involve regular clinic visits.
Urinary cell-free DNA
Bladder, prostate and renal cancers, which occur within the urinary tract, can release DNA fragments directly into urine. Research by various groups has demonstrated higher concentrations of urinary cfDNA (ucfDNA) in patients with bladder cancer, especially in those with advanced-stage disease compared to those without cancer or with benign urological conditions46,47,48. Indeed, an analysis of ucfDNA using NGS identified a diagnostic model based on the detection of mutations in five genes (TERT, FGFR3, TP53, PIK3CA and KRAS) that had excellent diagnostic accuracy for urothelial carcinoma in individuals with haematuria (AUC 0.94), although this method might be best deployed in combination with other methods owing to a number of false-negative findings49.
The use of ucfDNA for disease monitoring and prediction of relapse in patients with urothelial carcinoma has also been investigated50,51. In one of these studies, higher concentrations of ucfDNA were found in patients with disease progression even when plasma ctDNA was undetectable51. High concentrations of ucfDNA were also detected several months before clinical progression51. Urine can also be used for genomic analysis of bladder cancer52,53. In one prospective study, 59 patients with suspected urothelial carcinoma were identified using cystoscopy, and comparisons between tumour DNA from the cystoscopy specimen, ctDNA from plasma and ucfDNA revealed robust concordance (specificity 99.3%, sensitivity 86.7%) between tumour DNA and ucfDNA but not ctDNA (sensitivity 10.3%)29. These data highlight two key benefits of ucfDNA testing. Firstly, the authors identified a much lower frequency of urothelial carcinoma-associated genomic alterations (such as TERT promoter mutations and those in TP53, KDM6A, FGFR3, PIK3CA and ARID1A) and a disproportionately larger number of clonal haematopoiesis-related mutations (such as those involving CHEK2, DNMT3A, TET2 and ASXL1) in plasma samples compared with urine. Secondly, the urine samples could consistently be successfully analysed after being collected at home and stored in preservative containers.
The non-invasive nature of ucfDNA analysis, in addition to the early evidence that detection is feasible, has increased the level of interest in its use for the early detection of urological cancers. UroSEEK, a specific multiplex PCR-based assay (ten-gene multiplex, TERT singleplex and aneuploidy) has been developed specifically for the early detection of urothelial carcinoma and tested in a cohort of 570 patients with symptoms suggestive of this disease, such as haematuria or lower urinary tract symptoms54. UroSEEK alone identified 83% of patients who went on to be diagnosed with bladder cancer, this sensitivity increasing to 95% when combined with urinary cytology, with a specificity of 93%. UroSEEK positivity preceded a clinical diagnosis of urothelial carcinoma by an average of 2.3 months and by >1 year in eight patients. In addition to UroSEEK, urinary CAPPseq-based techniques have also demonstrated potential for disease monitoring in patients at risk of disease recurrence54,55. In one study, surveillance following local therapy revealed detectable tumour DNA in urine samples from 91% of patients who would go on to have disease recurrence55. The detection of ctDNA in urine preceded clinical disease recurrence in 92% of patients (median 2.7 months), which has the potential to provide considerable clinical benefit. In summary, ucfDNA provides a promising method of early cancer detection and/or monitoring for disease progression or recurrence in patients with urothelial carcinomas.
Renal cell carcinomas (RCCs) are known to shed comparatively less ctDNA than most other tumour types56. Studies comparing plasma and urine samples from patients with RCC have found only limited overlap between the ctDNA content of both fluids (owing to few patients having detectable ctDNA in both plasma and urine); however, similar to plasma ctDNA, ucfDNA can also overcome the issues of tumour heterogeneity associated with analysis of a single tumour biopsy sample57. Furthermore, analysis of the DNA methylome of ucfDNA derived from patients with RCC has been shown to enable the detection of RCC with a high level of sensitivity (AUC 0.86) although lower than that for the equivalent analysis of blood plasma (AUC 0.99)58. In another study, levels of STOX1 detected in ucfDNA from patients with RCC decreased substantially following nephrectomy, highlighting possible prognostic implications59.
Serum prostate-specific antigen (PSA) is routinely used for both screening and disease monitoring in men with suspected or known prostate cancer; however, this biomarker comes with a high risk of overdiagnosis, especially when used in isolation, and ucfDNA might provide a more accurate alternative60. A real-time PCR-based assay designed to detect >250 bp fragments of the genes encoding MYC, HER2 and AR showed promising levels of sensitivity (0.79) and specificity (0.84) in a pilot study comparing ucfDNA from 29 patients with prostate cancer with that obtained from 25 volunteers without cancer; however, the diagnostic performance of this assay was lower than that of serum PSA when tested in a larger cohort that included patients with benign urogenital conditions33,61. These results might be explained by a number of patients in the control group having conditions, such as calculi or renal tract inflammation, that might have led to increased cfDNA shedding into urine, leading to false-positive results. Emerging interest exists in the analysis of epigenetic alterations, including DNA methylation, in ucfDNA obtained from men with prostate cancer62.
In summary, ucfDNA analysis has been demonstrated to be feasible and to provide results that are highly concordant with those obtained from tissue and plasma ctDNA sequencing. These observations suggest clinical validity in cancer detection, personalized treatment-related decision-making, serial monitoring and/or following curative therapy in a variety of cancer subtypes63. Analyses focusing on ucfDNA have several advantages: similar to plasma DNA, the effects of tumour heterogeneity on the accuracy of a single tissue biopsy sample can be avoided; furthermore, the ease of serial monitoring without the need for hospital visits provides a unique benefit for patients with urological cancers. The main challenges to the implementation of ucfDNA-based assays are analytical, with differences in processing methodologies and collection timings limiting the widespread adoption of these methods. Utilizing these techniques within the clinic would require extensive prospective testing, which is currently lacking.
CSF ctDNA
For patients with extracranial malignancies, the aim of a liquid biopsy is generally not to supplant the gold standard of diagnosis based on tumour histology but rather to provide information on treatment response or disease relapse or to identify targetable alterations. However, obtaining tumour tissue samples from patients with CNS malignancies is both considerably more challenging and potentially dangerous64. Both the extent of surgical intervention and the overall treatment strategy are dependent on prognosis (and therefore tumour type), which potentially necessitates an intraoperative histological diagnosis, thereby both delaying and potentially complicating any surgical procedures65. In these scenarios, liquid biopsy sampling could have a valuable role in both the diagnosis of primary CNS disease and in confirming possible secondary leptomeningeal spread.
Plasma ctDNA has been shown to be present in only a minority (<10–45% depending on primary histology) of patients with both primary CNS malignancies and isolated CNS metastases and is typically detected at lower concentrations than those in patients with non-CNS solid tumours, likely owing to the blood–brain barrier limiting the translocation of both ctDNA and CTCs66,67,68,69. By contrast, CSF circulates around the brain parenchyma within the intracerebral ventricles, sub-arachnoid cisterns, sulci, and spinal canal and is, therefore, an excellent source of ctDNA from CNS tumours. Furthermore, the CSF generally contains fewer circulating immune cells relative to blood plasma, which might reduce the overall cfDNA content of the CSF, thus permitting the more sensitive detection of cancer-specific mutations present at low VAFs70.
The comparative abundance of ctDNA in the CSF compared with plasma (given the relative ease in obtaining plasma samples) is a key consideration for the use of liquid biopsy sampling in patients with CNS-restricted disease. Alterations present in ctDNA can consistently be detected at lower VAFs in the CSF compared with plasma and, when assessed longitudinally, changes in the abundance of ctDNA in the CSF are also more representative of tumour burden8,71,72. For example, in one study involving 19 patients with gliomas with detectable ctDNA present in the CSF (as determined by NGS), tumour-associated mutations were only detected in plasma samples from three patients (16%). In these three patients, the VAF of the alteration was substantially lower in plasma relative to CSF (mean VAF 0.58% versus 23.96%)73.
CSF ctDNA was also more abundant in patients with meningeal involvement or in those with parenchymal tumours that were in close contact with the CSF reservoir66,72. Fragmentation patterns in CSF ctDNA obtained from patients with glioma also demonstrated a relative enrichment of shorter DNA fragments (~145 bp) similar to that seen in plasma ctDNA from patients with a high systemic tumour burden14,74.
Several established molecular markers have been integrated into the classification of gliomas, including 1p/19q codeletions and alterations in IDH1/2, H3F3A, HIST1H3B/C, ATRX, RELA, TP53, TERT and BRAF75. Alterations in a number of these genes have been studied in CSF ctDNA in cohorts of patients with diffuse gliomas using dPCR-based approaches76,77. These studies demonstrated that analysis of CSF ctDNA enables tumours to be successfully classified into molecular subtypes and provides prognostic information. Indeed, enrolment in an ongoing clinical trial testing the tyrosine-kinase inhibitor anlotinib in patients with glioblastoma (NCT04004975) is restricted to patients with mutations in one of four target genes (VEGFR, KIT, PDGFR or FGFR) detected in both CSF ctDNA and a pathological tumour specimen.
Diffuse midline gliomas are associated with a particularly dismal prognosis. These tumours are frequently associated with the histone mutation H3K27M, which is often used as a criterion for clinical trial enrolment and has shown promise as a potential CSF biomarker76,77,78,79. This approach to stratification has potential utility both in offering diagnostic confirmation when biopsy sampling is not feasible and in enriching the cohort with patients who are more likely to respond to a specific targeted therapy. Substantial reductions in H3K27M in CSF ctDNA were concordant with radiological responses in 10 of 12 patients according to longitudinal analysis78.
Research interest in the role of CSF cfDNA in medulloblastoma has also increased over the past few years71,80. Liu et al.80 collected serial CSF samples from a total of 123 patients from surgical resection and throughout the course of adjuvant therapy in a prospective study. Copy number variants in CSF matching those detected in the primary tumour were used as a marker of minimal residual disease (MRD). Baseline MRD status correlated strongly with metastatic status (85% of patients with metastatic disease had detectable MRD versus 54% with localized disease) and serial ctDNA measurements revealed detectable MRD ≥3 months prior to radiological progression in 16 of 32 patients who had a complete response and subsequent disease relapse. A robust association with inferior post-radiotherapy progression-free survival in patients with MRD was also observed (HR 6.53, 95% CI 3.22–13.26; P <0.0001). A particular feature of medulloblastomas that might make these tumours amenable to CSF cfDNA assays is that patients often present with hydrocephalus requiring therapeutic drainage prior to surgery and might also require the insertion of further drainage devices for therapeutic purposes, thereby allaying some of the concerns around the invasiveness of repeat diagnostic lumbar punctures71.
Interest also exists in the potential of a ctDNA-based CSF biomarker for patients with primary CNS lymphomas (PCNSLs), both for diagnostic purposes in patients with equivocal disease histology or in whom tumour material is unobtainable and for monitoring of response and/or detection of disease relapse. CSF sampling is performed routinely in all patients with suspected CNS lymphoma, both for prognostic and staging purposes. However, CSF cytology is normally only positive in patients with leptomeningeal involvement or in whom the disease is connecting with the ventricular system.
The majority of studies involving patients with PCNSLs have used dPCR-based approaches for analysis, with driver mutations selected based on tissue genome or exome analyses or using other preselected target genes. Of these, MYD88 is the most commonly studied genomic feature of PCNSLs. Nonetheless, the sensitivity of CSF-detected MYD88 mutations varies considerably between studies, ranging from 25% to 81% in unselected patients with PCNSL40,81,82,83.
A subset of patients with PCNSLs have a good initial radiological response to induction chemotherapy but subsequently have very early disease relapse after consolidative autologous stem cell transplantation. If CSF ctDNA can be used to identify this group before the emergence of radiological evidence of disease relapse, an alternative consolidation therapy, such as whole-brain radiotherapy (for presumed chemoresistant disease), could be offered earlier in the course of disease84. Biomarker analyses performed in the context of a phase Ib trial using an NGS panel designed specifically for patients with haematological malignancies were able to demonstrate a positive correlation between a radiological complete response and disappearance of CSF ctDNA, with CSF ctDNA recurrence preceding radiological relapse in one patient83. In another study using variant-specific digital-droplet PCR, investigators were able to detect CSF ctDNA as early as 8 months prior to CNS relapse being confirmed radiologically85.
Contrasting results have been observed when PCNSL ctDNA has been examined in paired plasma and CSF samples. In one study, ctDNA was detectable in the CSF but not in plasma from patients with CNS-restricted PCNSLs85. By contrast, MYD88L265P was detected in samples obtained from the CSF, plasma and urine of patients with PCNSL in a separate study40. These investigators were able to detect this alteration at 100% sensitivity (based on tissue positivity for MYD88L265P) in both plasma and CSF samples and, intriguingly, at 88% sensitivity (in 8 of 9 patients) in urine, although mutant copies were present at higher fractional abundances in CSF samples than in either plasma or urine40. These contrasting results might be related to the timing of sampling in relation to primary surgery (either biopsy sampling or resection).
ctDNA could have a valuable role in the detection, monitoring and characterization of specific resistance mechanisms unique to the intracranial lesions in patients with CNS metastases from extracranial primary tumours. Discrepancies in response to therapy between intracranial and extracranial lesions are not uncommon (even in patients receiving drugs with good CNS penetrance) and this observation might reflect the existence of one or more subclonal populations harbouring different mechanisms of resistance8,86. In this setting, biopsy sampling of CNS disease is unlikely to be justifiable and many of these alterations are likely to be difficult to detect in ctDNA obtained from plasma samples87.
Leptomeningeal metastases are often particularly difficult to diagnose and are associated with a poor prognosis. Studies using NGS-based methods to assess the utility of CSF ctDNA for this purpose have attempted to characterize the mutational landscape of leptomeningeal metastases in patients with primary NSCLC88,89. The ctDNA detection rate and VAFs of detected alleles were both higher in CSF samples than in plasma (sensitivity 81.5% and 62.5% for CSF and plasma, respectively). The maximum allelic fraction was 43.6% for CSF versus 4.6% for plasma and a unique genomic profile was observed in the CSF, primarily owing to the presence of CSF-specific copy number variants20,88,90. In patients with NSCLC, analysis of CSF ctDNA might enable more sensitive detection of alterations than analysis of plasma given that EGFR alterations are typically detected at a sensitivity of 70–90% in plasma samples from patients with advanced-stage EGFR-mutant disease91,92,93,94,95. Analysis of CSF ctDNA has also been demonstrated to provide insights into mechanisms of acquired resistance that might be confined to the CNS and might better represent the genomic landscape of leptomeningeal disease20,96,97. A number of case reports describing patients with melanoma suggest that CSF ctDNA might provide a useful method of monitoring intracranial disease response or identifying CNS-restricted targetable alterations, although these findings have not been validated in larger cohorts thus far89,98,99.
Certain practical elements require further consideration in order for CSF ctDNA-based assays to be used more widely. Lumbar punctures are routinely performed in certain settings, such as in patients with leptomeningeal disease and in those with haematological cancers with CNS involvement, although they are not usually mandated for those with secondary brain parenchymal metastases. Owing to the invasive nature of a lumbar puncture, obtaining samples for translational research (in the absence of a therapeutic indication such as hydrocephalus) might be ethically challenging. Other limitations to the development of ctDNA-based assays for the analysis of CSF include the small sample sizes, biases created by the exclusion of patients for whom a lumbar puncture is contraindicated and technical issues such as contamination with blood during the procedure and variability in sample processing times.
Pleural and peritoneal fluid ctDNA
Pleural and peritoneal fluids provide a thin lubricating layer that reduces friction between pleural linings, abdominal organs and the peritoneum. They are ultrafiltrates of the blood that are relatively enriched in non-haematopoietic cfDNA owing to an absence of peripheral blood cells. Malignant pleural effusions (MPEs) and ascites involve tumour cells that infiltrate the pleura and/or peritoneum via direct, haematogenous or lymphatic spread. The fluid is therefore in close proximity to tumour tissue and might contain ctDNA from tumour cells100.
Even when cytology is negative, MPEs can have a high cfDNA content and can be more amenable to genomic analysis than both tissue and plasma samples18,101. The most sensitive platforms for plasma ctDNA profiling have only 70–80% sensitivity for samples obtained from patients with advanced-stage NSCLC and <50% for those with early stage disease102. Negative results therefore necessitate tissue biopsy confirmation, which can be challenging in those with early stage disease who have a lower tumour burden and possibly poorly accessible primary lesions. An advantage of analysing pleural ctDNA is that samples (obtained via pleural effusion) are immediately available for genetic testing, whereas tissue samples usually require conventional histopathological processing as per standard-of-care diagnostic workflows. Parallel histological analysis of tissue samples and molecular profiling of targetable mutations in ctDNA from pleural effusions might enable earlier detection of targetable alterations and thus permit earlier selection of the most appropriate therapy103. One study used an NGS-based panel of 416 genes to analyse pleural fluid, plasma, and tissue samples and reported higher VAFs and mutation detection rates in pleural fluid cfDNA (98.4% versus 87% in plasma ctDNA)18. Furthermore, 93% of tissue-determined driver alterations were detectable in pleural fluid ctDNA compared with 62% in plasma18. In another study, actionable variants were identified in 87% of MPE samples versus only 48% of paired plasma samples103.
A number of studies have investigated the utility of ctDNA in peritoneal fluid, either obtained from ascites that have developed as a consequence of malignancy or from peritoneal lavage during surgical resection104,105. This approach might be particularly useful for the detection and monitoring of peritoneal disease106,107,108. A study involving 20 patients with CRC and isolated peritoneal disease demonstrated that peritoneal ctDNA was detectable in all patients, whereas only 20% of patients had detectable plasma ctDNA, with significantly higher VAFs in peritoneal cfDNA (16.4% versus 0.28%; P = 0.0019)108. Smaller-cohort studies have investigated ctDNA obtained from presurgical or postsurgical peritoneal lavage as a predictor of disease recurrence, with promising initial results reported107,109.
Similar considerations regarding the feasibility of sample collection apply to both pleural or peritoneal aspiration and CSF sampling: pleural and/or peritoneal aspiration is a moderately invasive procedure requiring a trained operator. Aspiration for diagnostic or therapeutic purposes at initial presentation is the standard-of-care approach in patients with unexplained effusions/ascites and might also be appropriate if effusion occurs on disease relapse or progression; however pre-planned repeat sampling to monitor treatment response might be less practical.
Salivary ctDNA
Saliva is another body fluid that can be non-invasively obtained without requiring the presence of a health-care professional110. Saliva contains cells, proteins and nucleic acids, but is considerably less complex than blood111. The ability to detect germline DNA in saliva has been used in forensic science for decades as well as for certain medical indications: for example, germline BRCA1/2 mutations can be identified in saliva112. Nonetheless, the lower concentrations of ctDNA relative to germline DNA require more advanced detection technologies113. Similar to other non-blood forms of ctDNA, salivary ctDNA (sctDNA) is ideally placed (owing to direct tumour contact via cellular exfoliation or apoptosis) to inform on the status of local tumours such as head and neck cancers. Furthermore, sctDNA can also originate from more distal malignancies either through passive diffusion, active transport or ultrafiltration of ctDNA from the blood across the mucosal membranes114.
Analysis of sctDNA has the ability to differentiate patients with head and neck squamous cell carcinomas (HNSCCs) from those without this disease5. HNSCC is commonly associated with human papillomavirus (HPV) infection. In a PCR-based analysis of plasma and saliva samples from patients with oropharyngeal squamous cell carcinomas, HPV16 DNA was detected prior to treatment with a sensitivity of 52.8% in saliva (67.2% in plasma) and a specificity of 100%115. Furthermore, detectable HPV DNA and the concentration of HPV DNA in saliva following treatment were both associated with a significantly higher risk of disease recurrence (HR 10.7, 95% CI 2.36–48.50; P = 0.002) and inferior OS (HR for death 25.9, 95% CI 3.23–208; P = 0.002)115 and, in a separate study, were demonstrated to correlate with tumour burden and to be predictive of treatment response116. Results from pretreatment and post-treatment samples suggest saliva alone is the ideal sample for the assessment of malignancies of the oral cavity, although this should be used in combination with plasma ctDNA for assessments of tumours located in the oropharynx, hypopharynx and larynx, all of which have less direct exposure to saliva117.
The analysis of sctDNA is limited by several technical challenges118. In patients with NSCLC, dPCR-based analysis of EGFR mutations in plasma and saliva samples revealed high levels of concordance, although EGFR alterations were not detected in saliva DNA samples from several patients with detectable EGFR alterations in plasma. This observation might be explained by the low concentrations of ctDNA entering the saliva from plasma118. Other research suggests that sctDNA fragments are ultrashort (40–60 bp) and might not be amplifiable using conventional PCR techniques; this observation led to the development of an electric field-induced release and measurement (EFIRM) assay to detect EGFR alterations in saliva119,120,121. Direct comparisons suggest that EFIRM is a more sensitive method of detection than dPCR, and might therefore be the optimal method of assessing saliva samples from patients with malignancies other than HNSCC122.
Stool ctDNA
Human DNA is hypothesized to enter the stool via a combination of cellular shedding and colonocyte apoptosis123. Nonetheless, owing to the large and often diverse gastrointestinal microbiome, human DNA accounts for only around 0.01% of the total DNA content of stool, with the remainder derived from bacteria present in the gastrointestinal tract and/or diet123,124.
Screening for CRC is an important current focus of research into the role of stool DNA. This aspect is one of the most advanced areas where non-blood ctDNA testing is being translated into clinical use. The rationale is that early stage colorectal lesions develop predominantly within the mucosa with epithelial shedding of DNA into the lumen of the colon. Analyses of stool DNA samples using sequencing panels comprising 3 mutations in KRAS, 8 in TP53, 10 in APC, BAT-26 and a marker of elongated DNA strands have demonstrated increased sensitivity for detection of CRC compared to faecal immunochemistry and occult blood testing, albeit with an increased risk of false-positive results7,125. This assay has subsequently been developed into a standalone FDA-approved screening test (Cologuard), with evidence of increased uptake among individuals who were previously not compliant with stool screening tests126,127. However, this technique is less cost-effective than the alternatives and therefore might not be appropriate for use in large-scale screening programmes128. The Cologuard assay is also being studied as an alternative to colonoscopic surveillance post removal of polyps/adenomas, which could reduce both costs and burden on patients129.
The utility of stool DNA analysis has also been investigated in patients with other tumour types. Pancreatic cancer is notorious for its usually late diagnosis and poor prognosis; therefore, substantial efforts are currently focused on early diagnosis. Pancreatic secretions directly enter the gastrointestinal tract and the feasibility of detecting KRAS mutations in stool samples obtained from patients with pancreatic adenocarcinomas or cholangiocarcinomas has been studied; five of the six mutations found in stool were identical to those present in the resected carcinomas130. Mutations in KRAS and DNA methylation markers were detected in stool samples from patients with pancreatic cancer using dPCR, although only 19% of patients enrolled in this study agreed to provide a stool sample, highlighting compliance as a limitation131. Indeed, several studies have identified participant aversion to stool-based sampling, which might hinder the adoption of such methods132,133.
Outside of screening, NGS-based analysis of stool DNA samples has been successful in the detection of a range of hotspot mutations in established cancer-associated genes in samples from patients with gastrointestinal cancers, thus highlighting a role for stool DNA analysis in precision medicine and prognostication134. Analysis of stool DNA might also have utility beyond the detection of tumour DNA: analysis of 16S ribosomal DNA in stool samples from patients with NSCLC suggests that the composition of the microbiome can be associated with both responsiveness and toxicities in patients with immune-checkpoint inhibitors and could act as a predictive biomarker135.
In summary, stool DNA has the potential to improve outcomes via analysis as part of cancer screening programmes and to provide information on the genomic profiles of individual tumours, thus informing precision medicine approaches. Current challenges include the rarity of detectable tumour DNA within stool, an insufficient understanding of the stool microbiome and patient aversion to providing faecal samples.
Seminal fluid ctDNA
Seminal fluid, originating from the seminal vesicles and contributing to the liquid component of the male ejaculate, is another bodily fluid that can be used to obtain ctDNA for analysis. Cell-free seminal fluid DNA (cfsDNA) generally exists at higher concentrations relative to cfDNA in other bodily fluids; this relative abundance of cfsDNA might be explained by a combination of sexual abstinence (3–5 days) before providing a sample (permitting secreted DNA to accumulate) and reduced DNase activity owing to the effects of other semen content136. DNA fragments detected in the seminal fluid range from 180 bp to 15 kbp in length136. Owing to the abundance of prostate secretions in the seminal fluid, growing research interest exists in the utility of cfsDNA in men with prostate cancer. Comparisons of cfsDNA from men with prostate cancer to that from men without indicate significantly higher median concentrations in the former (428.45 ng/ml versus 25.4 ng/ml; P < 0.001), with those with benign prostatic hypertrophy having an intermediate median concentration (77.4 ng/ml)6,137. Notably, men with prostate cancer can have median cfsDNA concentrations of up to 2,243 ng/ml, which is approximately 100-fold higher than blood cfDNA concentrations (180 ng/ml) reported for patients with cancer11. More comprehensive analyses of csfDNA from men with prostate cancer (such as those involving NGS) are warranted given the high concentration of DNA available within this fluid, especially as this fluid can also be obtained non-invasively.
Limitations of the clinical utility of cfsDNA-based assays currently include the lack of defined protocols for the collection and analysis of samples; these issues would need to be addressed before widespread adoption. Furthermore, difficulties might emerge in obtaining seminal fluid samples from patients who have developed erectile dysfunction following androgen deprivation or in whom semen volume is reduced following brachytherapy and/or radiotherapy138. The utility of this approach is also potentially abrogated in those who have undergone radical prostatectomy with seminal vesicle removal.
Emerging sources of cfDNA
Beyond the fluids already discussed, several other possible sources of cfDNA are available. cfDNA has been identified in bile from patients with biliary tract cancers and was shown to be superior to plasma ctDNA for the detection of cancer-related somatic mutations4,139,140. Uterine lavage fluid has been studied as a source of cfDNA in a cohort of 107 women undergoing hysteroscopy and curettage following post-menopausal bleeding. Mutations in cancer-associated genes were identified in all seven women subsequently diagnosed with endometrial malignancies (six of whom were diagnosed with stage IA disease), suggesting potential as a relatively non-invasive screening method141. However, almost half of patients (n = 51) had cancer-associated mutations at detectable allele frequencies despite no histological evidence of cancer, suggesting a high risk of false-positive findings. Similarly, patterns of hypermethylation typically associated with endometrial cancer can be detected in vaginal fluid samples collected from tampons from women with endometrial carcinoma and also, to a lesser extent, from those with benign conditions10. These examples highlight the potential scope of novel approaches involving the application of ctDNA technologies that are already used to analyse plasma ctDNA to other bodily fluids.
Challenges and opportunities
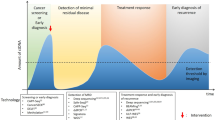
Several challenges must be overcome in attempts to translate the analysis of non-blood ctDNA into the clinic. Some of these challenges are specific to certain sample types and others apply more broadly, including to plasma (Box 1). The half-life of ctDNA is relatively short (approximately 2 h)142 indicating a need for rapid processing, which presents a challenge for samples obtained using more-invasive procedures, and there remains a need to streamline the sampling process from collection to processing. Preservative-containing stabilization tubes are available and can extend the processing time for ctDNA present in plasma samples to up to 48 h without compromising the sensitivity of detection143,144. However, the effects of these preservatives on ctDNA from non-blood sources have not been established. A number of other pre-analytical variables relating to transport and storage conditions that might be harder to standardize for samples from non-blood sources also remain unaddressed. The incorporation of invasive sampling procedures, such as pleural aspiration and lumbar puncture, into clinical trial protocols might also be difficult to justify, especially before the assays are fully validated. Conversely, samples of urine and saliva can be obtained entirely non-invasively and repeated over many timepoints with minimal risks to patients.
The majority of studies discussed in this Review were conducted in academic settings and used ‘in-house’ sequencing platforms. The lack of standardized analytical approaches currently limits the use of non-blood ctDNA to specific research purposes and/or within the context of a clinical trial. If such assays are to be implemented clinically, the chosen assay will first require robust analytical and clinical validation. Access to accredited laboratories (ISO 17025/FDA accredited) is usually required for analytical validation; these are rarely present in universities and contracting to commercial research laboratories is costly. Furthermore, a separate process would be needed for validation of each sample type, although consideration should also be given as to whether these processes could be streamlined. For example, the same NGS assay could potentially be validated for the analysis of cfDNA from multiple sample types in parallel. The past few years have seen an explosion in the number of biotech companies offering a wide range of plasma liquid biopsy tests (such as FoundationOne and Guardant360)145,146. Despite this, commercial tests enabling the analysis of ctDNA from non-blood sources are limited to Cologuard, which is approved for stool-based CRC screening129. Further clinical validation in clinical trials in which non-blood ctDNA testing is used to determine either treatment eligibility, timing or choice of therapy will be required.
Analyses of ctDNA obtained from a number of the non-blood sources discussed in this manuscript have demonstrated levels of sensitivity comparable to those achieved with plasma ctDNA (Supplemental Table 1). However, the collection of ctDNA from multiple biological fluids might be complementary and could be used to increase overall sensitivity compared with the analysis of plasma ctDNA alone as well as offering greater insight into tumour characteristics across different anatomical sites and times. A challenge for the development of plasma ctDNA-based assays has been defining the gold standard against which to assess sensitivity and specificity given the underlying rationale that liquid biopsies might reveal mutations that are not detected in a single tumour biopsy sample143. Adding further sample types might achieve increased sensitivity but care must also be taken to avoid a concomitant increase in false-positive findings. Recognized standards for assessing the performance of newly developed assays in the wide range of available non-blood sample types will need to be developed.
Clonal haematopoiesis is common in patients with cancer and especially in those of advancing age or who previously received radiotherapy. This feature can confound genomic analysis, especially the specificity of plasma ctDNA19. The reduced influence of clonal haematopoiesis on non-blood sample types might therefore offer advantages in the context of screening programmes and early detection (in which the relevant allele might be present at very low frequencies and be more restricted to the primary site and its immediate milieu). Indeed, the sensitivity of plasma ctDNA for the detection of early stage malignancies (the target of screening programmes) can be lower than for advanced-stage disease, which highlights where assays involving biofluids that are in closer proximity to the primary tumour might be advantageous7,29,137,147,148. Such an approach would clearly be limited to particular tumour types (prostate, bladder, endometrial and/or colorectal cancers) compared with plasma, which might offer the ability to screen multiple cancer types using a single blood test149,150. This lower specificity would likely be less disadvantageous in the context of later-stage disease monitoring or in the detection of specific resistance mutations23.
All liquid biopsies, including those involving ctDNA from blood and non-blood sources, are vulnerable to the effects of varying anatomical disease distribution on ctDNA concentrations. For example, in patients with extrathoracic metastases from NSCLC, the concordance of results obtained from analysis of plasma ctDNA is substantially higher than in those without such metastases151. Similarly, in patients with CRC, plasma ctDNA can be detected at substantially lower levels in those with isolated peritoneal disease than in those with liver metastases (in contrast to peritoneal cfDNA)108. Thus, identifying sources of genetic material beyond plasma or tissue might help circumvent these effects of anatomical variations, although crucially, the disease distribution in a particular patient will need to be considered carefully when deciding which assays are likely to yield the most useful results and how best to interpret those results.
The majority of published data available at this point have used mutation-based approaches to study ctDNA and this is reflected in the scope of our Review. The most obvious direct clinical application of these methods currently seems to be the detection of targetable driver or resistance mutations. However, growing research interest exists in the detection of DNA methylation and fragmentation in ctDNA; evidence of the ability to apply such methods to the various biofluids discussed in this Review is still emerging58,62,148,152.
Conclusions
In summary, the analysis of ctDNA obtained from non-blood bodily fluids has several potential benefits, from enabling exploratory research through to routine clinical use, and provides unique benefits such as enabling more detailed analysis of specific tumour types in certain anatomical locations and the potential for entirely non-invasive serial sampling. However, well-designed clinical trials in which the results are used to inform treatment decisions and that demonstrate meaningful benefits to patients will be necessary before non-blood ctDNA assays are to be broadly implemented clinically.
Change history
16 December 2022
In the version of this article initially published, the peer reviewer name Y. Nakamura was misspelled and is now amended in the HTML and PDF versions of the article.
References
Siravegna, G. et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 30, 1580–1590 (2019).
De Mattos-Arruda, L. et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: A proof-of-principle. Ann. Oncol. 25, 1729–1735 (2014).
Heitzer, E., Haque, I. S., Roberts, C. E. S. & Speicher, M. R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 20, 71–88 (2019).
Gou, Q. et al. Cell-free DNA from bile outperformed plasma as a potential alternative to tissue biopsy in biliary tract cancer. ESMO Open 6, 100275 (2021).
Sethi, S., Benninger, M. S., Lu, M., Havard, S. & Worsham, M. J. Noninvasive molecular detection of head and neck squamous cell carcinoma: an exploratory analysis. Diagn. Mol. Pathol. 18, 81–87 (2009).
Ponti, G. et al. Seminal cell-free DNA assessment as a novel prostate cancer biomarker. Pathol. Oncol. Res. 24, 941–945 (2018).
Imperiale, T. F. et al. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 370, 1287–1297 (2014).
De Mattos-Arruda, L. et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 6, 8839 (2015).
Su, Y.-H. et al. Human urine contains small, 150 to 250 nucleotide-sized, soluble DNA derived from the circulation and may be useful in the detection of colorectal cancer. J. Mol. Diagn. 6, 101–107 (2004).
Bakkum-Gamez, J. N. et al. Detection of endometrial cancer via molecular analysis of DNA collected with vaginal tampons. Gynecol. Oncol. 137, 14–22 (2015).
Schwarzenbach, H., Hoon, D. S. B. & Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 11, 426–437 (2011).
Lo, Y. M. D. et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2, 61ra91 (2010).
Thierry, A. R., El Messaoudi, S., Gahan, P. B., Anker, P. & Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 35, 347–376 (2016).
Underhill, H. R. et al. Fragment length of circulating tumor DNA. PLoS Genet 12, e1006162 (2016).
Mouliere, F. et al. High fragmentation characterizes tumour-derived circulating DNA. PLoS One 6, e23418 (2011).
Higgins, M. J. et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin. Cancer Res. 18, 3462–3469 (2012).
Wyatt, A. W. et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J. Natl Cancer Inst. 109, djx118 (2017).
Tong, L. et al. Tumor-derived DNA from pleural effusion supernatant as a promising alternative to tumor tissue in genomic profiling of advanced lung cancer. Theranostics 9, 5532–5541 (2019).
Ptashkin, R. N. et al. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncol. 4, 1589–1593 (2018).
Li, Y. S. et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: a new medium of liquid biopsy. Ann. Oncol. 29, 945–952 (2018).
Pessoa, L. S., Heringer, M. & Ferrer, V. P. ctDNA as a cancer biomarker: a broad overview. Crit. Rev. Oncol. Hematol. 155, 103109 (2020).
Adashek, J. J., Janku, F. & Kurzrock, R. Signed in blood: circulating tumor DNA in cancer diagnosis, treatment and screening. Cancers 13, 3600 (2021).
Reckamp, K. L. et al. A highly sensitive and quantitative test platform for detection of NSCLC EGFR mutations in urine and plasma. J. Thorac. Oncol. 11, 1690–1700 (2016).
Botezatu, I. et al. Genetic analysis of DNA excreted in urine: a new approach for detecting specific genomic DNA sequences from cells dying in an organism. Clin. Chem. 46, 1078–1084 (2000).
Melkonyan, H. S. et al. Transrenal nucleic acids: from proof of principle to clinical tests. Ann. N. Y. Acad. Sci. 1137, 73–81 (2008).
Green, E. A. et al. Clinical utility of cell-free and circulating tumor DNA in kidney and bladder cancer: a critical review of current literature. Eur. Urol. Oncol. https://doi.org/10.1016/j.euo.2021.04.005 (2021).
Augustus, E. et al. The art of obtaining a high yield of cell-free DNA from urine. PLoS One 15, e0231058 (2020).
Leung, M. S. T., Lin, S. G., Chow, J. & Harky, A. COVID-19 and oncology: service transformation during pandemic. Cancer Med. 9, 7161–7171 (2020).
Zhang, R. et al. Urinary molecular pathology for patients with newly diagnosed urothelial bladder cancer. J. Urol. 206, 873–884 (2021).
Crisafulli, G. et al. Whole exome sequencing analysis of urine trans-renal tumour DNA in metastatic colorectal cancer patients. ESMO Open. 4, e000572 (2019).
Yao, W., Mei, C. & Nan, X. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: a qualitative study. Gene 590, 142–148 (2016).
Kim, W. T. et al. Urinary cell-free nucleic acid IQGAP3: a new non-invasive diagnostic marker for bladder cancer. Oncotarget 9, 14354–14365 (2018).
Salvi, S. et al. Urine cell-free DNA integrity analysis for early detection of prostate cancer patients. Dis. Markers 2015, 574120 (2015).
Berz, D., Raymond, V. M., Garst, J. H. & Erlander, M. G. Non-invasive urine testing of EGFR activating mutation and T790M resistance mutation in non-small cell lung cancer. Exp. Hematol. Oncol. 5, 24 (2016).
Wang, X. et al. Investigation of transrenal KRAS mutation in late stage NSCLC patients correlates to disease progression. Biomarkers 22, 654–660 (2017).
Guibert, N. et al. Monitoring KRAS mutations in circulating DNA and tumor cells using digital droplet PCR during treatment of KRAS-mutated lung adenocarcinoma. Lung Cancer 100, 1–4 (2016).
Xie, F., Li, P., Gong, J., Tan, H. & Ma, J. Urinary cell-free DNA as a prognostic marker for KRAS-positive advanced-stage NSCLC. Clin. Transl. Oncol. 20, 591–598 (2018).
Shi, L. et al. EGFR mutation status analysis in cerebrospinal fluid and plasma of advanced lung adenocarcinoma with brain metastases. J. Thorac. Oncol. 12, S938–S939 (2017).
Du, Z.-H., Bi, F.-F., Wang, L. & Yang, Q. Next-generation sequencing unravels extensive genetic alteration in recurrent ovarian cancer and unique genetic changes in drug-resistant recurrent ovarian cancer. Mol. Genet. Genom. Med. 6, 638–647 (2018).
Watanabe, J. et al. Comparison of circulating tumor DNA between body fluids in patients with primary central nervous system lymphoma. Leuk. Lymphoma 60, 3587–3589 (2019).
Yu, H., Han, L., Yuan, J. & Sun, Y. Circulating tumor cell free DNA from plasma and urine in the clinical management of colorectal cancer. Cancer Biomark. 27, 29–37 (2019).
Chen, W. et al. Potential use of transrenal DNA for non-invasive monitoring and prognosis of colorectal cancer. Biomarkers 24, 524–529 (2019).
Tian, F., Liao, Y. & Zhang, Y. Variations in transrenal DNA and comparison with plasma DNA as a diagnostic marker for colorectal cancer. Int. J. Biol. Markers 32, 434–440 (2017).
Zuo, Z., Tang, J., Cai, X., Ke, F. & Shi, Z. Probing of breast cancer using a combination of plasma and urinary circulating cell-free DNA. Biosci. Rep. 40, BSR20194306 (2020).
Hann, H.-W. et al. Detection of urine DNA markers for monitoring recurrent hepatocellular carcinoma. Hepatoma Res. 3, 105–111 (2017).
Chang, H. W. et al. Urinary cell-free DNA as a potential tumor marker for bladder cancer. Int. J. Biol. Markers 22, 287–294 (2007).
Brisuda, A. et al. Total amount of cell free DNA in urine of patients harbouring urothelial carcinoma and in urine of controls. Eur. Urol. Suppl. 12, e1221 (2013).
Zancan, M. et al. Free DNA in urine: a new market for bladder cancer? Preliminary data. Int. J. Biol. Markers 20, 134–136 (2005).
Ou, Z. et al. Detection of bladder cancer using urinary cell‐free DNA and cellular DNA. Clin. Transl. Med. 9, 4 (2020).
Christensen, E. et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J. Clin. Oncol. 37, 1547–1557 (2019).
Birkenkamp-Demtröder, K. et al. Genomic alterations in liquid biopsies from patients with bladder cancer. Eur. Urol. 70, 75–82 (2016).
Togneri, F. S. et al. Genomic complexity of urothelial bladder cancer revealed in urinary cfDNA. Eur. J. Hum. Genet. 24, 1167–1174 (2016).
Christensen, E. et al. Liquid biopsy analysis of FGFR3 and PIK3CA hotspot mutations for disease surveillance in bladder cancer. Eur. Urol. 71, 961–969 (2017).
Springer, S. U. et al. Non-invasive detection of urothelial cancer through the analysis of driver gene mutations and aneuploidy. Elife 7, e32143 (2018).
Dudley, J. C. et al. Detection and surveillance of bladder cancer using urine tumor DNA. Cancer Discov. 9, 500–509 (2019).
Zill, O. A. et al. The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin. Cancer Res. 24, 3528 (2018).
Smith, C. G. et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome Med. 12, 23 (2020).
Nuzzo, P. V. et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 26, 1041–1043 (2020).
Julia, O. et al. MP21-11 diagnostic and prognostic value of urine circulating cell-free DNA in renal cell carcinoma. J. Urol. 201, e296 (2019).
Jahn, J. L., Giovannucci, E. L. & Stampfer, M. J. The high prevalence of undiagnosed prostate cancer at autopsy: implications for epidemiology and treatment of prostate cancer in the prostate-specific antigen-era. Int. J. Cancer 137, 2795–2802 (2015).
Casadio, V. et al. Urine cell-free DNA integrity as a marker for early prostate cancer diagnosis: a pilot study. Biomed. Res. Int. 2013, 270457 (2013).
Silva, R. et al. Evaluating liquid biopsies for methylomic profiling of prostate cancer. Epigenetics 15, 715–727 (2020).
Lu, T. & Li, J. Clinical applications of urinary cell-free DNA in cancer: current insights and promising future. Am. J. Cancer Res. 7, 2318–2332 (2017).
Khatab, S., Spliet, W. & Woerdeman, P. A. Frameless image-guided stereotactic brain biopsies: emphasis on diagnostic yield. Acta Neurochir. 156, 1441–1450 (2014).
Seoane, J., De Mattos-Arruda, L., Le Rhun, E., Bardelli, A. & Weller, M. Cerebrospinal fluid cell-free tumour DNA as a liquid biopsy for primary brain tumours and central nervous system metastases. Ann. Oncol. 30, 211–218 (2019).
De Mattos-Arruda, L. et al. Abstract 930: analysis of cell-free tumor DNA in cerebrospinal fluid to characterize and monitor the genetic alterations of brain tumors. Cancer Res. 75, 930 (2015).
Chen, W. W. et al. Beaming and droplet digital PCR analysis of mutant idh1 mRNA in glioma patient serum and cerebrospinal fluid extracellular vesicles. Mol. Ther. Nucleic Acids 2, e109 (2013).
Moya, I. et al. P05.09 Detection of clinically relevant mutations in the cerebrospinal fluid of patients with central nervous system metastases. Neuro-Oncology 20, iii304 (2018).
Bettegowda, C. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra24 (2014).
Pan, W., Gu, W., Nagpal, S., Gephart, M. H. & Quake, S. R. Brain tumor mutations detected in cerebral spinal fluid. Clin. Chem. 61, 514–522 (2015).
Escudero, L. et al. Circulating tumour DNA from the cerebrospinal fluid allows the characterisation and monitoring of medulloblastoma. Nat. Commun. 11, 5376 (2020).
Pentsova, E. I. et al. Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J. Clin. Oncol. 34, 2404–2415 (2016).
Miller, A. M. et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 565, 654–658 (2019).
Mouliere, F. et al. Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol. Med. 10, e9323 (2018).
Wesseling, P. & Capper, D. WHO 2016 classification of gliomas. Neuropathol. Appl. Neurobiol. 44, 139–150 (2018).
Fujioka, Y. et al. Molecular diagnosis of diffuse glioma using a chip-based digital PCR system to analyze IDH, TERT, and H3 mutations in the cerebrospinal fluid. J. Neurooncol. 152, 47–54 (2021).
Martínez-Ricarte, F. et al. Molecular diagnosis of diffuse gliomas through sequencing of cell-free circulating tumor DNA from cerebrospinal fluid. Clin. Cancer Res. 24, 2812–2819 (2018).
Panditharatna, E. et al. Clinically relevant and minimally invasive tumor surveillance of pediatric diffuse midline gliomas using patient-derived liquid biopsy. Clin. Cancer Res. 24, 5850–5859 (2018).
Li, D. et al. Standardization of the liquid biopsy for pediatric diffuse midline glioma using ddPCR. Sci. Rep. 11, 5098 (2021).
Liu, A. P. Y. et al. Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell 39, 1519–1530.e4 (2021).
Rimelen, V. et al. Tumor cell-free DNA detection in CSF for primary CNS lymphoma diagnosis. Acta Neuropathol. Commun. 7, 43 (2019).
Watanabe, J. et al. High detection rate of MYD88 mutations in cerebrospinal fluid from patients with CNS lymphomas. JCO Precis. Oncol. https://doi.org/10.1200/PO.18.00308 (2019).
Grommes, C. et al. Phase 1b trial of an ibrutinib-based combination therapy in recurrent/refractory CNS lymphoma. Blood 133, 436–445 (2019).
Mutter, J. A. et al. Profiling of circulating tumor DNA for noninvasive disease detection, risk stratification, and MRD monitoring in patients with CNS lymphoma. Blood 138, 6 (2021).
Bobillo, S. et al. Cell free circulating tumor DNA in cerebrospinal fluid detects and monitors central nervous system involvement of B-cell lymphomas. Haematologica 106, 513–521 (2021).
Woodcock, D. J. et al. Prostate cancer evolution from multilineage primary to single lineage metastases with implications for liquid biopsy. Nat. Commun. 11, 5070 (2020).
Wong, S. Q. et al. Circulating tumor DNA analysis and functional imaging provide complementary approaches for comprehensive disease monitoring in metastatic melanoma. JCO Precis. Oncol. https://doi.org/10.1200/po.16.00009 (2017).
Ying, S. et al. Unique genomic profiles obtained from cerebrospinal fluid cell-free DNA of non-small cell lung cancer patients with leptomeningeal metastases. Cancer Biol. Ther. 20, 562–570 (2019).
Azad, T. et al. Next generation sequencing of cerebrospinal fluid to improve diagnostic sensitivity, detect spatial heterogeneity, and predict outcomes for advanced lung cancer patients with leptomeningeal carcinomatosis. J. Neurosurg. 132, 110 (2020).
Zhao, Y. et al. Evaluating the cerebrospinal fluid ctDNA detection by next-generation sequencing in the diagnosis of meningeal Carcinomatosis. BMC Neurol. 19, 331 (2019).
Liu, Y. et al. Cell-free DNA from cerebrospinal fluid can be used to detect the EGFR mutation status of lung adenocarcinoma patients with central nervous system metastasis. Transl. Lung Cancer Res. 10, 914–925 (2021).
Zheng, M.-M. et al. Clinical utility of cerebrospinal fluid cell-free DNA as liquid biopsy for leptomeningeal metastases in ALK-rearranged NSCLC. J. Thorac. Oncol. 14, 924–932 (2019).
Yang, H. et al. Sensitive detection of EGFR mutations in cerebrospinal fluid from lung adenocarcinoma patients with brain metastases. J. Mol. Diagn. 16, 558–563 (2014).
Oxnard, G. R. et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J. Clin. Oncol. 34, 3375–3382 (2016).
Oxnard, G. R. et al. Noninvasive detection of response and resistance in egfrmutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 20, 1698–1705 (2014).
Wu, X. et al. Genetic profiling of cerebrospinal fluid cfDNA from NSCLC patients with leptomeningealmetastases reveals EGFR-TKIs resistant mutations independent of extracranial lesions (cases series). Cancer Res. 80 (Suppl. 16), 723 (2020).
Jiang, B. et al. Unique genetic profiles from circulating Cell-Free DNA of cerebrospinal fluid in leptomeningeal metastases of EGFR Mutant NSCLC. J. Thorac. Oncol. 12, S1771 (2017).
Melms, J. et al. Implementation of cell‑free tumor DNA sequencing from the cerebrospinal fluid to guide treatment in a patient with primary leptomeningeal melanoma: a case report. Mol. Clin. Oncol. 9, 58–61 (2018).
Li, Y. et al. Tumor DNA in cerebral spinal fluid reflects clinical course in a patient with melanoma leptomeningeal brain metastases. J. Neurooncol. 128, 93–100 (2016).
Skok, K., Hladnik, G., Grm, A. & Crnjac, A. Malignant pleural effusion and its current management: a review. Medicina 55, 490 (2019).
Sriram, K. B. et al. Pleural fluid cell-free DNA integrity index to identify cytologically negative malignant pleural effusions including mesotheliomas. BMC Cancer 12, 428 (2012).
Durin, L. et al. Liquid biopsy of non-plasma body fluids in non-small cell lung cancer: look closer to the tumor! Cells 9, 2486 (2020).
Zhou, Z. et al. Lung cancer genomic alterations in cell free DNA in pleural effusion compared to plasma. J. Clin. Oncol. 35 (Suppl. 15), e23206 (2017).
Han, M.-R. et al. Clinical implications of circulating tumor DNA from ascites and serial plasma in ovarian cancer. Cancer Res. Treat. 52, 779–788 (2020).
Shi, C. et al. Analysis of mutation detection in cell-free DNA in ascites using comprehensive NGS panel. J. Clin. Oncol. 37 (Suppl. 15), e13029 (2019).
Leick, K. M. et al. Peritoneal cell-free tumor DNA as biomarker for peritoneal surface malignancies. Ann. Surg. Oncol. 27, 5065–5071 (2020).
López-Rojo, I. et al. Liquid biopsy in peritoneal fluid and plasma as a prognostic factor in advanced colorectal and appendiceal tumors after complete cytoreduction and hyperthermic intraperitoneal chemotherapy. Ther. Adv. Med. Oncol. 12, 1758835920981351 (2020).
Van’t Erve, I. et al. Detection of tumor-derived cell-free DNA from colorectal cancer peritoneal metastases in plasma and peritoneal fluid. J. Pathol. Clin. Res. https://doi.org/10.1002/cjp2.207 (2021).
Leick, K. M. et al. Peritoneal cell-free tumor DNA as a biomarker of peritoneal recurrence in resected pancreatic cancers. Ann. Surg. Oncol. 27, S125–S126 (2020).
Kaczor-Urbanowicz, K. E. et al. Saliva diagnostics-current views and directions. Exp. Biol. Med. 242, 459–472 (2017).
Zimmermann, B. G., Park, N. J. & Wong, D. T. Genomic targets in saliva. Ann. N. Y. Acad. Sci. 1098, 184–191 (2007).
Dean, M. et al. Addressing health disparities in Hispanic breast cancer: accurate and inexpensive sequencing of BRCA1 and BRCA2. Gigascience 4, 50 (2015).
Wang, X., Kaczor-Urbanowicz, K. E. & Wong, D. T. W. Salivary biomarkers in cancer detection. Med. Oncol. 34, 7 (2017).
Kaczor-Urbanowicz, K. E. et al. Clinical validity of saliva and novel technology for cancer detection. Biochim. Biophys. Acta Rev. Cancer 1872, 49–59 (2019).
Ahn, S. M. et al. Saliva and plasma quantitative polymerase chain reaction-based detection and surveillance of human papillomavirus-related head and neck cancer. JAMA Otolaryngol. Head Neck Surg. 140, 846–854 (2014).
Hanna, G. J. et al. Salivary HPV DNA informs locoregional disease status in advanced HPV-associated oropharyngeal cancer. Oral. Oncol. 95, 120–126 (2019).
Wang, Y. et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci. Transl. Med. 7, 293ra104 (2015).
Ding, S. et al. Saliva-derived cfDNA is applicable for EGFR mutation detection but not for quantitation analysis in non-small cell lung cancer. Thorac. Cancer 10, 1973–1983 (2019).
Wei, F. et al. Noninvasive saliva-based EGFR gene mutation detection in patients with lung cancer. Am. J. Respir. Crit. Care Med. 190, 1117–1126 (2014).
Pu, D. et al. Evaluation of a novel saliva-based epidermal growth factor receptor mutation detection for lung cancer: a pilot study. Thorac. Cancer 7, 428–436 (2016).
Li, F. et al. EFIRM liquid biopsy (eLB): detection of ultrashort circulating tumor DNA (usctDNA) in plasma and saliva of non-small cell lung cancer (NSCLC) patients. J. Clin. Oncol. 36 (Suppl. 15), e24062 (2018).
Li, F. et al. Ultra-short circulating tumor DNA (usctDNA) in plasma and saliva of non-small cell lung cancer (NSCLC) patients. Cancers 12, 2041 (2020).
Aghagolzadeh, P. & Radpour, R. New trends in molecular and cellular biomarker discovery for colorectal cancer. World J. Gastroenterol. 22, 5678–5693 (2016).
Klaassen, C. H. W. et al. Quantification of human DNA in feces as a diagnostic test for the presence of colorectal cancer. Clin. Chem. 49, 1185–1187 (2003).
Imperiale, T. F., Ransohoff, D. F., Itzkowitz, S. H., Turnbull, B. A. & Ross, M. E. Fecal DNA versus fecal occult blood for colorectal-cancer screening in an average-risk population. N. Engl. J. Med. 351, 2704–2714 (2009).
Prince, M., Lester, L., Chiniwala, R. & Berger, B. Multitarget stool DNA tests increases colorectal cancer screening among previously noncompliant medicare patients. World J. Gastroenterol. 23, 464–471 (2017).
Redwood, D. G. et al. Stool DNA testing for screening detection of colorectal neoplasia in Alaska native people. Mayo Clin. Proc. 91, 61–70 (2016).
Naber, S. K. et al. Cost-effectiveness of a multitarget stool DNA test for colorectal cancer screening of medicare beneficiaries. PLoS One 14, e0220234–e0220234 (2019).
van Lanschot, M. C. J. et al. Molecular stool testing as an alternative for surveillance colonoscopy: a cross-sectional cohort study. BMC Cancer 17, 116 (2017).
Caldas, C. et al. Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res. 54, 3568 LP–3563573 (1994).
Kisiel, J. B. et al. Stool DNA testing for the detection of pancreatic cancer: assessment of methylation marker candidates. Cancer 118, 2623–2631 (2012).
van Dam, L. et al. What influences the decision to participate in colorectal cancer screening with faecal occult blood testing and sigmoidoscopy? Eur. J. Cancer 49, 2321–2330 (2013).
Osborne, J. M. et al. The impact of sample type and procedural attributes on relative acceptability of different colorectal cancer screening regimens. Patient Prefer. Adherence 12, 1825 (2018).
Youssef, O. et al. Gene mutations in stool from gastric and colorectal neoplasia patients by next-generation sequencing. World J. Gastroenterol. 23, 8291–8299 (2017).
Zhang, F. et al. Analysis of the gut microbiota: an emerging source of biomarkers for immune checkpoint blockade therapy in non-small cell lung cancer. Cancers 13, 2514 (2021).
Li, H.-G., Huang, S.-Y., Zhou, H., Liao, A.-H. & Xiong, C.-L. Quick recovery and characterization of cell-free DNA in seminal plasma of normozoospermia and azoospermia: implications for non-invasive genetic utilities. Asian J. Androl. 11, 703–709 (2009).
Ponti, G. et al. Quick assessment of cell-free DNA in seminal fluid and fragment size for early non-invasive prostate cancer diagnosis. Clin. Chim. Acta 497, 76–80 (2019).
Tran, S., Boissier, R., Perrin, J., Karsenty, G. & Lechevallier, E. Review of the different treatments and management for prostate cancer and fertility. Urology 86, 936–941 (2015).
Shen, N. et al. Bile cell‑free DNA as a novel and powerful liquid biopsy for detecting somatic variants in biliary tract cancer. Oncol. Rep. 42, 549–560 (2019).
Han, J.-Y. et al. Liquid biopsy from bile-circulating tumor DNA in patients with biliary tract cancer. Cancers 13, 4581 (2021).
Nair, N. et al. Genomic analysis of uterine lavage fluid detects early endometrial cancers and reveals a prevalent landscape of driver mutations in women without histopathologic evidence of cancer: a prospective cross-sectional study. PLoS Med. 13, e1002206 (2016).
Diehl, F. et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 14, 985–990 (2008).
Merker, J. D. et al. Circulating tumor DNA analysis in patients with cancer: American society of clinical oncology and college of American pathologists joint review. J. Clin. Oncol. 36, 1631–1641 (2018).
Kang, Q. et al. Comparative analysis of circulating tumor DNA stability in K3EDTA, Streck, and CellSave blood collection tubes. Clin. Biochem. 49, 1354–1360 (2016).
Woodhouse, R. et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS One 15, e0237802 (2020).
Bauml, J. M. et al. Clinical validation of Guardant360 CDx as a blood-based companion diagnostic for sotorasib. Lung Cancer 166, 270–278 (2021).
Abbosh, C., Birkbak, N. J. & Swanton, C. Early stage NSCLC — challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 15, 577–586 (2018).
Liu, M. C. et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 31, 745–759 (2020).
Cohen, J. D. et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926 (2018).
Cristiano, S. et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 570, 385 (2019).
Aggarwal, C. et al. Clinical implications of plasma-based genotyping with the delivery of personalized therapy in metastatic non-small cell lung cancer. JAMA Oncol. 5, 173–180 (2019).
Li, J. et al. Reliable tumor detection by whole-genome methylation sequencing of cell-free DNA in cerebrospinal fluid of pediatric medulloblastoma. Sci. Adv. 6, eabb5427 (2020).
Acknowledgements
The work of A.T. is supported by an NIHR Academic Clinical Fellowship. The work of D.R. and C.D. is funded by Cancer Research UK (CRUK) via core funding to the CRUK Manchester Institute (grant A27412) and the CRUK Manchester Centre (grant A25254). Support was received by the NIHR Manchester Biomedical Research Centre and the Manchester Experimental Cancer Medicine Centre.
Author information
Authors and Affiliations
Contributions
A.T., M.C. and N.C. researched data for article and wrote the manuscript. All authors contributed to discussions of content and reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
C.D. has acted as a consultant and/or adviser of AstraZeneca, Boehringer Ingelheim, Biocartis, GRAIL and Merck, and has received research funding from Angle, Amgen, Astex Pharmaceuticals, AstraZeneca, Bayer, Bioven, Bristol Myers Squibb, Boehringer Ingelheim, Carrick Therapeutics, Celgene, Clearbridge Biomedics, Epigene Therapeutics, GlaxoSmithKline, Menarini, Merck AG, Neomed Therapeutics, Novartis, Roche, Taiho Oncology and Thermo Fisher Scientific. N.C. has received research funding from AstraZeneca, Avacta, Bayer, Boehringer, Eisai, Merck, Orion, Pfizer, RedX, Roche, Starpharma, Stemline Tarveda, Taiho Oncology and UCB. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Clinical Oncology thanks J. Seoane, Y. Nakamura and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tivey, A., Church, M., Rothwell, D. et al. Circulating tumour DNA — looking beyond the blood. Nat Rev Clin Oncol 19, 600–612 (2022). https://doi.org/10.1038/s41571-022-00660-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41571-022-00660-y
This article is cited by
-
The role of piRNAs in predicting and prognosing in cancer: a focus on piRNA-823 (a systematic review and meta-analysis)
BMC Cancer (2024)
-
Multicancer screening test based on the detection of circulating non haematological proliferating atypical cells
Molecular Cancer (2024)
-
Circulating tumor DNA validity and potential uses in metastatic breast cancer
npj Breast Cancer (2024)
-
Single-cell low-pass whole genome sequencing accurately detects circulating tumor cells for liquid biopsy-based multi-cancer diagnosis
npj Precision Oncology (2024)
-
Liquid-based biomarkers in breast cancer: looking beyond the blood
Journal of Translational Medicine (2023)
