
Abstract
Endothelial cells are at the interface between circulating blood and tissues. This position confers on them a crucial role in controlling oxygen and nutrient exchange and cellular trafficking between blood and the perfused organs. The endothelium adopts a structure that is specific to the needs and function of each tissue and organ and is subject to tissue-specific signalling input. In adults, endothelial cells are quiescent, meaning that they are not proliferating. Quiescence was considered to be a state in which endothelial cells are not stimulated but are instead slumbering and awaiting activating signals. However, new evidence shows that quiescent endothelium is fully awake, that it constantly receives and initiates functionally important signalling inputs and that this state is actively regulated. Signalling pathways involved in the maintenance of functionally quiescent endothelia are starting to be identified and are a combination of endocrine, autocrine, paracrine and mechanical inputs. The paracrine pathways confer a microenvironment on the endothelial cells that is specific to the perfused organs and tissues. In this Review, we present the current knowledge of organ-specific signalling pathways involved in the maintenance of endothelial quiescence and the pathologies associated with their disruption. Linking organ-specific pathways and human vascular pathologies will pave the way towards the development of innovative preventive strategies and the identification of new therapeutic targets.
Key points
-
Quiescent endothelial cells require active maintenance to preserve normalcy in a tissue-specific manner.
-
Dysregulation of signalling pathways involved in endothelial normalcy maintenance leads to endothelial dysfunction and vascular pathologies.
-
Endothelial quiescence and normalcy are important for disease resilience.
-
Identification of organ-specific signalling pathways that maintain endothelial normalcy and quiescence will lead to new therapeutic targets supporting disease resilience and treatment of associated vascular pathologies.
Similar content being viewed by others

Introduction
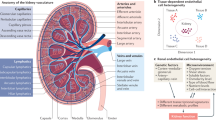
The endothelium forms the innermost layer of blood vessels and lymphatic vessels and is best viewed as a multifunctional organ with both systemic and tissue-specific roles. At the whole-organism level, the endothelium regulates oxygen and nutrient supply, immune cell trafficking and inflammation1, haemostasis and coagulation2, vasomotor tone3, blood vessel permeability4 and angiogenesis5. In addition, the endothelium has a number of organ-specific functions including regulation of organ size and function (myocardial hypertrophy6, liver size and function7, pulmonary alveolar repair8 and kidney function9,10).
Given this heterogeneity of endothelial cell function, it is not surprising that studies show a remarkable heterogeneity of gene expression profiles in endothelial cells from different organs11. Interestingly, these expression profiles are functionally matched to local tissue needs. Microenvironment stimuli (shear stress, hypoxia and the presence of specific growth factors, cytokines and hormones) and epigenetics define and continuously optimize local characteristics of endothelial cells. Epigenetic signatures that regulate the basal expression of endothelial-specific genes in different organs are specified during embryonic development and conserved during mitotic cycles12. Transcriptome analysis of endothelial cells from different tissues revealed heterogeneous gene expression signatures even after several passages in cell culture, indicating that tissue-specific epigenetic modifications participate in the regulation of organotypic transcriptomic profiles13,14. However, after long-term cell culture, which removes endothelial cells from their in vivo microenvironment, approximately 50% of gene expression patterns are lost15, and major architectural characteristics, such as fenestrae, also disappear16.
To characterize organotypic endothelial specificity as close to in vivo conditions as possible, many groups have utilized microarray or RNA sequencing (RNA-seq) of endothelial cells isolated by flow cytometry without the cell culture step11,17. Single-cell RNA-seq of endothelial cells isolated from adult male mice identified transcriptomic signatures of quiescent arterial, venous, capillary and lymphatic endothelial cells in 11 different tissues11. Interestingly, lymphatic endothelial cells from all the tissues cluster together, suggesting that the molecular signature of lymphatic endothelial cells is not tissue-specific. By contrast, arterial and venous endothelial cells from a specific tissue clustered together, showing that vascular endothelial cell heterogeneity comes mainly from tissue specificity rather than arterial, capillary or venous identity. Moreover, capillary endothelial cells that are involved in gas, ion, metabolite and hormone exchange between the blood and tissues have the highest heterogeneity among tissues11.
Structural differences in the capillary endothelium were first described in the 1960s with the use of electron microscopy18. Three major types of capillaries exist (continuous, fenestrated and discontinuous). The capillary type of an organ is related to its functions19. Most organs have barrier-forming, continuous capillaries (lungs, brain, skin and heart) with tightly connected endothelial cells surrounded by a continuous basement membrane. This architecture permits diffusion of water, small solutes and lipid-soluble materials, while precluding the passage of cells or pathogens. By contrast, fenestrated capillaries have intracellular pores (windows) with a diaphragm and are found in renal glomeruli, exocrine glands, endocrine glands and intestinal mucosa. These fenestrae increase permeability to fluids and solutes, but not macromolecules20. Sinusoids are fenestrated capillaries with gaps instead of pores between endothelial cells and a thinner basement membrane than in continuous or fenestrated endothelia and are present in the liver, spleen and bone marrow. The gaps found in sinusoids facilitate selective exchange of materials.
Structural differences notwithstanding, normal endothelial cells everywhere are quiescent. This quiescent state is defined by minimal or absent endothelial proliferation and migration, minimal or no vascular leakage across the endothelial barrier and minimal (or fully absent) expression of leukocyte adhesion molecules. Indeed, the half-life of a normal endothelial cell is an estimated 6 years in the heart as measured by 14C incorporation21, and proliferative activity is absent (except in the liver and spleen, where about 1% of endothelial cells proliferate in a quiescent state11). However, this ‘quiescent’ endothelium performs lots of active work, from secretion of paracrine and endocrine factors to active support of barrier maintenance for cell survival. Remarkably, little attention has been paid to what controls this ‘active quiescence’ and what maintains vascular normalcy under physiological conditions (Fig. 1). Although much effort has been expended on exploration of signalling pathways underlying endothelial cell activation and proliferation, almost no attention has been given to the signalling events that maintain endothelial normalcy and quiescence. The latest advances in this area are the subject of this Review.

Quiescent endothelial cells receive autocrine, endocrine and paracrine signalling inputs. Paracrine stimulation confers a tissue-specific microenvironment to the endothelium. For example, vascular endothelial growth factor (VEGF) signalling is crucial to the formation and maintenance of fenestrated endothelia, whereas paracrine WNT signalling induces formation and maintenance of continuous endothelial lining, which is crucial for vascular integrity in general and that of the blood–brain barrier in particular. Autocrine transforming growth factor-β (TGFβ) signalling is a signature of endothelial cell dysfunction. ANG1, angiopoietin 1; ACVR2A, activin A receptor type 2A; ALK, activin receptor-like kinase; BMP9, bone morphogenetic protein 9; BMPR2, bone morphogenetic protein receptor type 2; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; LRP, lipoprotein receptor-related protein; TGFβR, transforming growth factor-β receptor; TIE2, angiopoietin 1 receptor; VEGFR, vascular endothelial growth factor receptor.
FGF signalling
The fibroblast growth factor (FGF) signalling cascade includes a family of 18 ligands, four receptor tyrosine kinases (RTK) (FGFR1–FGFR4) and several accessory molecules such as Klotho proteins and syndecans22. After FGFs bind to their high-affinity RTKs, several intracellular pathways are activated, including the phosphoinositide 3-kinase (PI3K)–AKT pathway and mitogen-activated protein kinase (MAPK) pathways mediated by extracellular signal-regulated kinase 1 (ERK1) and ERK2 (ref.22).
The extensive structural overlap and cross-reactivity among FGF ligands and receptors represent a real challenge to identifying the function of FGF signalling in the endothelium. Results from studies in mice with knockout of individual Fgf genes or individual Fgfr genes are hard to interpret because of functional redundancy, whereas attempts to use FGFR chemical inhibitors are hampered by the low specificity and cross-reactivity of these compounds. Successful strategies to circumvent the redundancy in the FGF family and investigate FGF signalling include mice with knockout of multiple Fgfr genes (Fgfr1−/−Fgfr2−/− (ref.22) and Fgfr1−/−Fgfr3−/− (ref.23)), the use of soluble FGFR traps that target various FGF family members24, and endothelial cell-specific expression of a dominant-negative FGFR1 construct that can inactivate all four FGF receptors25. Mice with conditional endothelial cell-specific deletion of Fgfr1 and Fgfr2 are viable, with no vascular developmental defects and no alterations in vascular homeostasis26. However, postnatal endothelial cell-specific knockout of Fgfr1 in mice with global knockout of Fgfr3 results in impaired development of blood and lymphatic vessels23. A soluble receptor trap strategy was tested with the use of a soluble FGFR1 trap (sFGFR1) that binds to a large number of FGFs24. In this study, transient FGF inhibition was achieved in vivo in mice via adenovirus-mediated systemic expression of sFGFR1. This FGF inhibition led to an increase in vascular permeability and, eventually, pulmonary and myocardial haemorrhages, demonstrating the necessity for FGF signalling in the maintenance of vascular integrity24 (Table 1). A particularly interesting finding was the disrupted endothelial cell–cell junctions in large vessels, such as the femoral artery, carotid artery and jugular vein (Table 1). One possible explanation for the disrupted endothelial cell–cell junctions is that the loss of FGF signalling decreases the expression of the phosphatase SHP2 (also known as PTPN11), thereby increasing phosphorylation of the junctional protein VE-cadherin on tyrosine 658, which, in turn, results in loss of the VE-cadherin–β-catenin interaction27. The intracellular kinase SRC can also phosphorylate VE-cadherin, especially in venous endothelial cells28. Phosphorylated VE-cadherin is internalized and ubiquitinated in response to inflammatory mediators28. However, phosphorylation of VE-cadherin in the absence of inflammatory mediators is not sufficient for induction of vascular permeability28.
The FGF signalling cascade also maintains endothelial cell identity. FGF2 stimulation decreases expression of the transforming growth factor-β (TGFβ) receptors (TGFβRs), TGFβ ligands (especially TGFβ2) and the major intracellular TGFβ signal transducer SMAD2 (refs29,30). FGF-driven suppression of TGFβ signalling reduces endothelial-to-mesenchymal transition (EndMT)31,32 (Table 1), a state in which endothelial cells lose the expression of important endothelial genes and acquire mesenchymal cell-like characteristics, including increased migration and proliferation32. Mechanistically, FGF signalling controls the cellular levels of the let-7 microRNA family30, which targets a number of TGFβ family members. A reduction in let-7 expression results in substantial increases in the level and activity of TGFβ ligands and TGFβRs in vitro and in vivo30. Also, addition of FGF2 to human umbilical vein endothelial cells in vitro increases the expression of miR-20a, which targets the TGFβRs33, another mechanism involved in FGF–TGFβ crosstalk. In addition, FGF signalling is crucial for the expression of the endothelial receptor vascular endothelial growth factor receptor 2 (VEGFR2), the primary signalling receptor of vascular endothelial growth factor A (VEGFA)25.
Endothelial cells are located at the interface with the circulating blood and, therefore, are subject to shear stress resulting from the flow of blood in the vasculature. Shear stress can be linear, pulsatile or disturbed depending on location (for example, near arterial bifurcations and along the aortic arch inner curvature) and vary in strength. Endothelial cell sensing of shear stress is a complex process that is reviewed elsewhere34. The proteoglycan syndecan 4 is one element of the shear stress-sensing complex. Indeed, loss of endothelial syndecan 4 in mice results in impaired alignment of endothelial cells under flow35. Given that syndecan 4 is an important modulator of FGF2 signalling36,37,38, this study suggests that flow modulates FGF signalling. Furthermore, shear stress can directly affect the expression of endothelial FGF receptors. In particular, decreased FGFR1 expression has been observed in endothelial cells in areas of disturbed shear stress39. In vitro studies have confirmed the relationship between the level of endothelial FGFR1 expression and the magnitude and type of shear stress39.
Loss of FGF signalling in endothelial cells in areas of high shear stress has been linked to increased atherosclerotic plaque growth39 (Table 2). Atherosclerosis is a progressive disease characterized by gradual intracellular lipid deposition in the vasculature leading to the formation of atherosclerotic plaques40,41. At these sites, endothelial cells acquire a pro-inflammatory phenotype, which predisposes to atherosclerotic plaque development41. Disturbed flow-induced expression of pro-inflammatory genes in endothelial cells affects a number of signalling pathways. Human cultured endothelial cells under oscillating shear stress, but not under laminar shear stress, lose FGFR1 expression and activate the TGFβ pathway, leading to extensive EndMT39. TGFβ signalling further promotes an inflammatory phenotype in endothelial cells, thereby establishing a feed-forward loop between inflammation and TGFβ pathway activation39,42. In patients with coronary artery disease, a strong correlation exists between disease progression and loss of FGFR1 expression and activation of TGFβ signalling in endothelial cells of the left main coronary artery, with up to 70% of endothelial cells overlying atherosclerotic plaques expressing mesenchymal markers39 (Table 2). Syndecan 4, a proteoglycan that increases FGF2 signalling, protects against atherosclerotic plaque formation in mice35, highlighting the protective role of endothelial FGF signalling against atherosclerosis.
VEGF signalling
VEGF is the most-studied angiogenic growth factor. All members of the VEGF family have important functions in the endothelium. VEGFA and VEGFC are key drivers of angiogenesis and lymphangiogenesis, respectively43. VEGFB is involved in the regulation of endothelial cell metabolism44, whereas the function of VEGFD is less clear45. A closely related growth factor, placental growth factor, is crucial for placental angiogenesis46. All the VEGF ligands function via three related high-affinity tyrosine kinase receptors (VEGFR1–VEGFR3) and a host of auxiliary signalling molecules including neuropilins and syndecans43. VEGFR2 is the principal signalling VEGFR in blood endothelial cells, whereas both VEGFR2 and VEGFR3 are involved in lymphatic endothelial cell VEGF signalling45. Similar to FGF ligand binding to FGFRs, VEGF ligand binding to VEGFR2 activates several intracellular pathways, including PI3K–AKT and MAPK (including ERK1, ERK2 and p38 MAPK) pathways, among a number of others43. Of these VEGF-induced pathways, VEGF-mediated activation of ERK1 and ERK2 is thought to be crucial during embryonic vascular development and in angiogenic settings in adult tissues43,47.
The role of VEGF–VEGFR2 signalling in angiogenesis is well established. However, the role of this signalling cascade in the quiescent endothelium remains unclear. Interestingly, VEGF ligands are expressed by a number of specialized cell types, such as podocytes, choroid plexus epithelium and hepatocytes in adult mice48. Studies using mice expressing the VEGF–lacZ reporter construct showed that VEGFA is expressed in cells overlying fenestrated and sinusoidal blood vessels, such as podocytes in the kidney and hepatocytes in the liver, as well as in tissues with secretory functions48. Furthermore, VEGFR2 in the adjacent endothelial cells was phosphorylated, demonstrating paracrine activity of the non-endothelial VEGF in these specialized environments.
Local effects
The importance of paracrine VEGFA signalling in quiescent endothelium is well documented in glomerulus endothelium49 (Fig. 1; Table 1). Renal glomeruli are composed of fenestrated capillary endothelial cells and highly specialized epithelial cells (podocytes) separated by a glomerular basement membrane. Glomerular podocytes continuously express high levels of VEGFA. Constitutive heterozygous deletion of Vegfa in podocytes in mice leads to endotheliosis (swelling of endothelial cells with a partial loss of fenestrations) by 2.5 weeks of age49. By 6.5 weeks of age, mice with heterozygous deletion of Vegfa have an expanded glomerular basement membrane and a near-complete loss of endothelial fenestration. The outcome is terminal renal failure with nephrotic syndrome at 9 weeks of age (Table 1). Interestingly, constitutive overexpression of Vegfa in podocytes in mice leads to a different kidney disease49. In this case, by 5 days of age, mice have a collapsing glomerulopathy characterized by kidney haemorrhages, proteinuria and complete collapse of the capillary loops, with no or few multinucleated endothelial cells (Table 1). Induction of Vegfa overexpression in podocytes of adult mice induces a different phenotype from that of mice with non-inducible Vegfa overexpression49,50. Inducible Vegfa overexpression in podocytes in adult mice also leads to kidney failure, proteinuria, glomerular basement membrane thickening, slit diaphragm loss and podocyte effacement, although endothelial cells are mostly unaffected (no endotheliosis or loss of fenestration)50 (Table 1). Together, these data show the crucial requirement for finely balanced VEGF signalling in the filtration barrier of the glomeruli at different developmental stages.
Similar to the kidney, ablation of VEGF in endocrine glands leads to the loss of endothelial fenestrations51. Deletion of Vegfa52 or overexpression of a soluble form of Vegfr1 (ref.53) in pancreatic β-cells in mice results in a loss of endothelial fenestrations in pancreatic islets (Table 1). A similar loss of endothelial fenestrations is observed in thyroid capillaries following pan-endothelial Vegfr2 deletion in mice54 (Table 1).
In addition to paracrine activation of VEGF signalling, an autocrine VEGF loop is also crucial to the integrity of the quiescent endothelium55 (Table 1). VEGFA expression in pulmonary, aortic and intestinal blood vessels in adult mice is patchy55. Constitutive homozygous deletion of Vegfa in endothelial cells results in lethality at any age, including around 25% lethality in adult mice, with a peak in death at 20–25 weeks after birth, as a result of multiorgan haemorrhage (in the spleen, kidney, brain, intestines, heart and lungs), intestinal perforations and myocardial infarction55. These haemorrhages and thrombotic events are the consequence of endothelial cell apoptosis55. Interestingly, endothelial-specific knockout of Vegfa in mice does not induce a loss of endothelial fenestrations in renal glomeruli55, unlike in mice with loss of paracrine VEGFA signalling. These differences in phenotype imply differences in VEGF signalling circuits when activated in an autocrine versus paracrine manner. However, the identity of these circuits remains unknown. Finally, small amounts of circulating VEGF ligands are found in blood. Whether the presence of VEGFs in blood implies the existence of endocrine signalling and what that might entail has not been established.
The VEGF pathway is also activated by shear stress. VEGFR2 is part of a complex with VE-cadherin and platelet endothelial cell adhesion molecule56, which allows endothelial cells to sense shear stress. Increased shear stress leads to ligand-independent activation of VEGFR2 by SRC, activation of AKT and then activation of endothelial nitric oxide synthase (eNOS; also known as NOS3) to induce vasodilatation57.
Systemic hypertension
In addition to specific effects in various organs, VEGFs have a number of systemic effects that became evident with the widespread clinical use of VEGF-neutralizing agents and tyrosine kinase inhibitors that target VEGFR2. Use of anti-VEGF therapies can often trigger the development of systemic hypertension owing to decreased synthesis of the vasodilator nitric oxide (NO) and increased expression of the vasoconstrictor endothelin 1 (ET1)58,59,60,61,62 (Table 2). The decrease in blood NO levels is due to a reduction in the expression of eNOS, which catalyses the synthesis of NO from l-arginine. However, specifically how decreased VEGF signalling leads to increased ET1 expression and whether a specific vascular bed is predominantly responsible for NO or ET1 synthesis is unclear. Other adverse effects of anti-VEGF therapies include proteinuria and membranous glomerulonephropathy, which can potentially advance to renal failure63 and are attributable to the requirement for VEGF in the maintenance of the renal vasculature and filtration units (Table 2). Finally, haemorrhages have been described in patients with cancer who were treated with sunitinib or sorafenib, two tyrosine kinase inhibitors targeting VEGFR2 signalling64 (Table 2).
Lack of VEGF signalling is responsible for the hypertension in women with pre-eclampsia, a common maternal complication of pregnancy associated with oedema, renal failure and systemic hypertension (Table 2). Syncytiotrophoblasts, a placental cell type, secrete a soluble form of VEGFR1 (sVEGFR1) and if this secretion is abnormally elevated, increased amounts of circulating sVEGFR1 sequestrate VEGFA, initiating the disease process65,66,67. Adenovirus-mediated overexpression of sVEGFR1 in pregnant rats induced systemic hypertension, proteinuria and glomerular endotheliosis, similar to what is observed in patients with pre-eclampsia65, and validating the requirement for sVEGFR1 in the pathogenesis of pre-eclampsia. Similar to the hypertension induced by the use of VEGF inhibitors, systemic hypertension in patients with pre-eclampsia is also driven by decreased NO production and increased ET1 levels in plasma68,69. Interestingly, elevated plasma TGFβ2 levels in patients with systemic hypertension70 and the changes in renal histology in patients with pre-eclampsia71 are similar to findings in mice with a systemic deletion of Erk1 and an inducible, endothelial-specific deletion of Erk2 (Erk1−/−Erk2iEC−/−)10 (Table 1).
Both FGF and VEGF signalling cascades activate endothelial ERK1 and ERK2 signalling43. A study from 2019 elucidated the function of ERK1 and ERK2 in the quiescent endothelium10. Inducible, endothelial-specific deletion of Erk2 in adult Erk1-null mice led to universal lethality within 4 weeks10. Interestingly, the phenotypes of these mice are a combination of the phenotypes found after inhibition of the FGF pathway and the VEGF pathway (Table 1). Erk1−/−Erk2iEC−/− mice have increased TGFβ signalling as a result of a decrease in let-7 microRNA expression, leading to EndMT. This TGFβ-induced EndMT has previously been observed after endothelial FGF pathway inhibition30. These Erk1−/−Erk2iEC−/− mice also develop systemic hypertension due to a loss of eNOS expression and increased ET1 expression, lose fenestrations in endocrine gland and kidney endothelium, and develop kidney failure with proteinuria and endotheliosis of the glomerulus endothelium10. These phenotypes are very similar to those found in mice with VEGF pathway inhibition. Taken together, these results demonstrate the crucial function of the ERK1–ERK2 pathway in the maintenance of quiescent vasculature integrity and highlight the differences between VEGF-mediated and FGF-mediated activation of ERK1–ERK2.
Pulmonary vasculature
The function of VEGF in the pulmonary vasculature is less well-defined. Pulmonary capillaries are continuous, which means they do not have fenestrations. Nevertheless, VEGF is important for the maintenance of the pulmonary vasculature. Inhibition of VEGF signalling in rats by treatment with the VEGFR blocker SU5416 leads to pruning of the pulmonary arterial vasculature, which, in turn, induces alveolar cell apoptosis and emphysema at high doses72 (Table 1). Interestingly, Notch signalling can attenuate the angiogenic sprouting effect of VEGF signalling73,74. Consistent with this notion, mice with a heterozygous deletion of Dll4 (which encodes the Delta-like protein 4, a ligand of the Notch pathway), which is highly expressed in arterial endothelial cells and is a target gene of VEGF signalling, have pulmonary haemorrhages, suggesting a crucial role of VEGF signalling and the downstream Notch pathway in pulmonary endothelial cells75 (Table 1). Furthermore, some patients treated with neutralizing antibodies against DLL4 (demcizumab or enoticumab) or a bispecific antibody against DLL4 and VEGF (navicixizumab) develop pulmonary hypertension76,77,78,79 (Table 2). The cancer therapy drug dasatinib inhibits ephrin receptor signalling, which is directly connected to the VEGF signalling pathway by modulating VEGFR2 endocytosis80 and activation of downstream pathways81. The interaction of these signalling pathways and the fact that multiple intracellular and cell-surface kinases are simultaneously inhibited by dasatinib might explain why this therapy is associated with the development of pulmonary hypertension in some patients82,83 and aggravates pulmonary hypertension in rats84 (Table 2).
VEGF signalling also has an important role in vascular protection in patients with acute respiratory distress syndrome (ARDS)85,86 (Table 2). ARDS is characterized by diffuse alveolar damage leading to impaired gas exchange and is common in several pulmonary diseases, including viral pneumonitis (such as those caused by H1N1 influenza virus infection), severe acute respiratory syndrome and coronavirus disease 2019 (COVID-19), in which ARDS is associated with intense bronchial and lung parenchyma inflammation87,88. The early, exudative phase of ARDS is characterized by diffuse alveolar damage, disruption and loss of epithelial and endothelial cell–cell junctions, and alveolar oedema89. The exudative phase is followed by a proliferative phase that involves the formation of hyaline membranes on the epithelial side of the basement membrane and extensive cellular infiltrates in the alveolar spaces90. VEGFs are present in the normal alveolar space and probably serve to maintain alveolar function85. The loss of this protection, such as occurs with increased levels of VEGFA trap sVEGFR1 in the lungs, is predictive of the development of ARDS and of an increased risk of adverse outcomes among patients with ARDS86. At the same time, these protective effects of VEGF signalling are counterbalanced by the capacity of the same VEGFA to induce oedema and promote inflammation85,90,91,92. Indeed, studies of patients with ARDS associated with viral vasculitis, such as those caused by hantavirus infection, have demonstrated a strong association between VEGF levels in the lungs and pulmonary oedema93,94,95.
This association renders VEGF an unappealing agent for ARDS therapy and emphasizes the need for a drug that selectively activates VEGF protective pathways while inhibiting its pro-inflammatory signalling. However, the absence of a drug that differentiates between the beneficial (cell survival) and detrimental (oedema) effects of VEGF signalling prevents the development of a promising new therapeutic modality.
WNT and Hedgehog signalling
Whereas the VEGF–ERK signalling axis is central to the maintenance of endothelial fenestrae of the kidney and endocrine glands, WNT signalling has an equally important role in the maintenance of tight junctions and the continuous endothelium of the central nervous system96. The WNT family is composed of ten WNT receptors (Frizzled 1–10), four WNT co-receptors (LDL receptor-related protein 5 (LRP5), LRP6, RTK-like orphan receptor 2 and receptor-like tyrosine kinase), and 16 WNT ligands (WNT1–WNT16). Canonical WNT signalling involves the binding of WNT ligands to Frizzled receptors and the co-receptors LRP5 and LRP6. Phosphorylation of LRP5 and LRP6 recruits the β-catenin destruction complex (Axin, casein kinase 1, glycogen synthase kinase 3 and adenomatous polyposis coli protein) from the cytoplasm to the plasma membrane, where the complex cannot degrade β-catenin. This stabilized form of β-catenin accumulates and translocates to the nucleus to activate the transcription of WNT target genes. Glia and neurons produce WNT7a and WNT7b ligands (Fig. 1), and binding of these WNT ligands to the receptor Frizzled 4 activates canonical WNT signalling in endothelial cells of the central nervous system97,98,99. Upregulation of β-catenin in endothelial cells leads to increased expression of genes encoding tight junction components (claudin 1, claudin 3 and claudin 5) and the glucose transporter 1 (GLUT1)97. Simultaneously, expression of Plvap, the gene encoding plasmalemma vesicle protein 1 (PV1), which is the principal component of endothelial fenestrae, is repressed in endothelial cells97,100 (Fig. 2). This combined effect of WNT signalling is crucial to the integrity of the blood–brain barrier (BBB). Indeed, endothelial cell-specific deletion of Ctnnb1, the gene encoding β-catenin, in adult mice leads to severe seizures, brain haemorrhages and death96 (Table 1). Endothelial deletion of Fzd4, which encodes the receptor Frizzled 4, in adult mice leads to increased PV1 levels and decreased claudin 5 levels in retinal, cerebellar, spinal cord and olfactory bulb endothelial cells98 (Fig. 2; Table 1). Therefore, the WNT pathway is a perfect example of how the cellular microenvironment can induce the final differentiation step of endothelial cells, leading to a highly specialized organotypic endothelium.

a | Regulation of endothelial fenestration is achieved by a combination of signalling pathways regulating Plvap expression. Inhibition of the transforming growth factor-β (TGFβ) signalling is controlled by several signalling pathways including bone morphogenetic protein (BMP), fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF) signalling circuits. b | Pathological endothelium. Decreased FGF or VEGF signalling input leads to activation of an autocrine TGFβ signalling loop that, in turn, induces inflammation, hypertension and endothelial-to-mesenchymal transition (EndMT). Excessive VEGF signalling and autocrine angiopoietin 2 (ANG2) signalling induce pathological angiogenesis. ACVR2A, activin A receptor type 2A; ALK, activin receptor-like kinase; BMPR2, bone morphogenetic protein receptor type 2; eNOS, endothelial nitric oxide synthase; ERK, extracellular signal-regulated kinase; FGFR, fibroblast growth factor receptor; GLUT1, glucose transporter 1; LRP, lipoprotein receptor-related protein; PLVAP, plasmalemma vesicle-associated protein; SMAD, mothers against decapentaplegic homologue; TIE2, angiopoietin 1 receptor; VEGFR, vascular endothelial growth factor receptor.
Similar to the paracrine WNT signalling pathway, a paracrine sonic hedgehog (SHH) signalling pathway is also activated in the BBB101. Astrocytes in the BBB express the ligand SHH, which binds to and inactivates the receptor protein patched homologue 1 (PTCH1), which is expressed in brain endothelial cells101 (Table 1). Inactivation of PTCH1, in turn, results in the inactivation of the G protein-coupled receptor Smoothened (SMO), which leads to the activation of the glioma-associated oncogene (GLI1). Genetic endothelial-specific deletion of Smo specifically led to an increase in BBB permeability in adult mice, manifested by plasma protein leakage and decreased expression of junctional proteins (occludin, claudin 3, claudin 5 and tight junction protein ZO1)101. This increased BBB permeability induces a pro-inflammatory phenotype in BBB endothelial cells with upregulation of intercellular adhesion molecule 1 (ICAM1) and recruitment of circulating inflammatory cells101. Furthermore, SHH signalling also induces the expression of netrin 1 in the BBB endothelial cells, a laminin-related protein that is critical for cell–cell junction and cell–substrate adhesion102.
Dysregulation of the WNT–β-catenin pathway has been implicated in various central nervous system disorders that involve BBB breakdown, including multiple sclerosis103, Alzheimer disease104 and Huntington disease105. The blood–retina barrier shares high similarity with the BBB. Mutations in NDP, a gene encoding the WNT ligand Norrin and expressed in the blood–retina barrier, are linked to Norrie disease, a condition in which the blood–retina barrier integrity is compromised, leading to blindness106,107 (Table 2). Dysregulation of the SHH pathway is found in HIV-associated neurocognitive disorders with disruption of BBB integrity108.
Angiopoietin signalling
Angiopoietins (ANGs) are a family of secreted factors comprising ANG1, ANG2 and ANG3 (ANG4 in humans). Unlike the FGF and VEGF signalling pathways, which are involved in both pro-angiogenic processes and maintenance of endothelial cell quiescence signalling, ANG1 signalling is purely a quiescence signalling pathway in endothelial cells. ANG1 is expressed in mural cells and binds to the angiopoietin 1 receptor (TIE2) expressed on the surface of endothelial cells109 (figs 1,2). After ANG1 binding, TIE2 clusters and autophosphorylates to activate downstream pathways. Phosphorylated TIE2 is found in all adult vasculatures110. The main pathway activated downstream of TIE2 is the AKT pathway111. During inflammation or hypoxia, endothelial cells can deactivate the quiescence signal of TIE2 by expressing ANG2 that competes with ANG1 for binding to TIE2 (ref.112) and predominantly functions as an antagonist of TIE2 (ref.113) (Fig. 2; Table 1). The loss of TIE2 signalling reactivates the endothelium by weakening endothelial cell–cell junctions114,115, inducing the expression of pro-inflammatory adhesion molecules, including ICAM1 and vascular cell adhesion molecule 1 (VCAM1)112, and increasing the levels of procoagulant proteins on the luminal surface of endothelial cells116,117 (Table 1). Intriguingly, ANG2–TIE2 interactions are context-dependent — ANG2 acts as a TIE2 agonist under pathogen-free conditions but as an antagonist under inflammatory conditions (such as infection and tumour necrosis factor (TNF) or lipopolysaccharide stimulation)118. TIE1 is an orphan receptor unable to bind any angiopoietin or other known ligands. TIE1 and TIE2 interact in the absence of a ligand119. Endothelial TIE1 is required for the agonist effects of paracrine ANG1 and autocrine ANG2 on TIE2 activation. During inflammation, the ectodomain of TIE1 in endothelial cells is cleaved, resulting in the loss of ANG2 agonist activity, thereby promoting vascular remodelling and leakiness120. TIE1 cleavage reduces, but does not abolish, ANG1 agonist activity.
Despite AKT signalling functioning in different aspects of cellular regulation in multiple cell types, in the quiescent endothelium, AKT is considered to function as a survival pathway121. In vitro transduction of endothelial cells with a dominant-negative Akt variant decreases endothelial viability by opposing the pro-survival effects of VEGF122. However, a 2016 study in mice showed that the main function of the endothelial AKT pathway is not endothelial cell survival but maintenance of adequate interactions between endothelial cells, pericytes and vascular smooth muscle cells123 (Table 1). Indeed, endothelial cell-specific deletion of Akt1 in mice with global Akt2 deletion alters Jagged 1–Notch signalling between endothelial and mural cells, leading to apoptosis of vascular smooth muscle cells and pericytes and subsequent vessel regression, particularly in coronary arteries123. However, endothelial cell apoptosis was not detected in these Akt2−/− mice with endothelial cell-specific Akt1 deletion. The regulation of Notch signalling by AKT following ANG1 stimulation in endothelial cells is mediated by the endothelial transcriptional regulator ERG124. Inducible, endothelial cell-specific deletion of Erg in mice leads to a phenotype similar to that produced by the deletion of Akt1 and Akt2, with a loss of vascular smooth muscle cell coverage and vascular regression125.
Clinical data have added a layer of complexity to our understanding of ANG–TIE signalling. Multiple cutaneous and mucosal venous malformations have been reported in patients carrying gain-of-function genetic variants in TEK (encoding TIE2)126,127. This vascular malformation is characterized by the development of soft, blue, compressive and localized lesions128. Histological features of these venous lesions include uneven endothelial cell lining, disorganized extracellular matrix structure, enlarged lumen and sparse mural cell coverage128. These lesions can be present at birth or present around puberty128. These gain-of-function TEK genetic variants result in autophosphorylation of TIE2 and excessive activation of the downstream AKT pathway in endothelial cells129. At the same time, secretion by endothelial cells of platelet-derived growth factor B, which is responsible for mural cell recruitment, is downregulated129. These observations suggest that, although TIE2 signalling is important for the switch from an activated to a quiescent endothelium, overactivation of this pathway is deleterious. The mechanism responsible for ensuring the proper extent of TIE2 activation is currently unclear. Vascular endothelial protein tyrosine phosphatase (VEPTP), a vascular phosphatase, seems to be crucial in limiting TIE2 activation, because neutralization of VEPTP in vivo in mice results in vascular lesions similar to those seen in mice with gain-of-function TEK genetic variants130,131.
BMP signalling
The TGFβ superfamily includes a large pool of ligands, such as TGFβ1–TGFβ3, bone morphogenetic proteins (BMPs), growth differentiation factors (GDFs), activins and inhibins and nodal. These ligands bind to a complex of two dimers of a combination of type I receptors named activin receptor-like kinase 1 (ALK1)–ALK7 and two dimers of type II receptors (BMP receptor type 2 (BMPR2), TGFβR, activin receptor type 2A and activin receptor type 2B), leading to the activation of a number of different canonical (SMAD-dependent) and non-canonical signalling cascades132. The major distinction between canonical BMP and TGF pathways is the phosphorylation of SMAD2–SMAD3 by TGFβ, activins, inhibins and nodal, whereas BMPs and certain GDFs phosphorylate SMAD1, SMAD5 and SMAD8.
BMP9 and BMP10 are circulating BMPs produced by the liver and the heart, respectively133. Both ligands bind the high-affinity receptor ALK1, which is specifically expressed by endothelial cells134. ALK1 signalling is crucial for developmental angiogenesis135,136,137,138. In addition, ALK1 also has a crucial role in the quiescent endothelium. Endothelial deletion of Alk1 in adult mice (aged 2 months) is lethal within 9–21 days of deletion, although the exact cause of death is still unknown139 (Table 1). Major autopsy findings included cardiac enlargement and haemorrhages in the lungs and the gastrointestinal tract139. Deletion of Alk1 in endothelial cells in adult mice also led to the spontaneous formation of arteriovenous malformations (AVMs) — direct shunts between arteries and veins — in the gastrointestinal tract and uterus139. AVMs were also suspected to be present in the lungs, but were difficult to assess because of the multiple haemorrhages in the lungs139. Furthermore, wounding can also induce de novo AVM formation in the skin of adult mice with Alk1 deletion139. Endoglin, the co-receptor for ALK1, is also important for vascular quiescence. Endothelial-specific deletion of Eng (which encodes endoglin) in adult mice resulted in wound-induced AVMs in the skin, but no visceral AVMs were found140. However, spontaneous pelvic AVMs were shown in another study of mice with endothelial-specific Eng deletion141 (Table 1). Interestingly, the pelvic area where the AVMs form after Eng deletion has high VEGFA levels141. Anti-VEGF treatment blocked the formation and maturation of AVMs in Alk1-knockout mice and Eng-knockout mice141,142. Together, these data support the concept that ALK1 and endoglin are required for the maintenance of endothelial quiescence in adult life to counteract an over-exuberant endothelial proliferative response to VEGF signalling.
The molecular mechanism by which ALK1–endoglin signalling maintains the integrity of the quiescent endothelium is still unknown. BMP9 and BMP10 induce endothelial quiescence by inhibiting endothelial cell migration and proliferation in microvascular endothelial cells134,143. Moreover, the BMP9–ALK1 signalling pathway inhibits the pro-angiogenic VEGF–AKT1 pathway144. BMP9, which is produced by hepatic stellate cells, induces the fenestration of the sinusoidal endothelial cells145 (Figs 1,2). Deletion of Bmp9 in 129/Ola mice triggers hepatic fibrosis following sinusoidal capillarization (transformation of the fenestrated hepatic sinusoids into continuous capillaries, with synthesis of a basement membrane of collagen between the endothelial cells and the hepatocytes)146 (Table 1).
The BMP9–ALK1 pathway is also modulated by shear stress. Endoglin increases BMP9 signalling through ALK1 in endothelial cells during shear stress147. This increased BMP9 activity is accomplished by an increase in the association between endoglin and ALK1 before their binding to the ligand147. Loss of eng in zebrafish leads to defective blood flow-induced cell shape changes, resulting in enlarged vessels148. The primary cilia, which can function as a sensor of blood flow-induced mechanical forces on endothelial cells, can also regulate BMP signalling. In vitro, the loss of BMP9 signalling through the cilium was shown to increase endothelial cell migration149. Together, these studies show that shear stress increases BMP9 signalling in quiescent endothelial cells.
Interestingly, stimulation of human pulmonary endothelial cells with BMP9 in vitro inhibits the TGFβ pathway by inducing the expression of inhibitory SMADs (SMAD6 and SMAD7) and by decreasing TGFβR2 expression150. These results identify a second pathway that inhibits TGFβ signalling in endothelial cells10,30 (Fig. 2). Further evidence supporting the importance of TGFβ pathway inhibition in quiescent endothelium is the increased expression of SMAD6 and SMAD7 observed in pulmonary endothelial cells of adult mice compared with pulmonary endothelial cells from infant mice, which are proliferative cells151. These studies firmly support the notion that BMP9–ALK1 signalling inhibits the TGFβ pathway in quiescent pulmonary endothelial cells. The endothelial-specific transcription factor ERG can also activate the BMP pathway by upregulating SMAD1 as well as inhibiting the TGFβ pathway by downregulating SMAD3 expression in the quiescent endothelium of the hepatic vasculature152.
Disruption of normal BMP signalling in quiescent endothelial cells is the molecular basis for hereditary haemorrhagic telangiectasia (HHT), a rare genetic vascular disease (Table 2). HHT is characterized by recurrent nosebleeds, mucous telangiectasia and formation of AVMs153,154. In most patients, HHT is caused by loss-of-function genetic variants in ENG or ACVRL1 (encoding ALK1)155,156, and these variants decrease BMP9 signalling157,158. Interestingly, a second variant on the somatically non-mutated allele of ACVRL1 or ENG can be found in some lesions in patients with HHT159. These results establish HHT as a disease of decreased BMP9 and BMP10 signalling. AVMs in patients with HHT are predominantly found in the liver and lungs and, to a lesser extent, in the brain160. Interestingly, AVMs in the liver are more frequent in patients with HHT type 2 (patients carrying an ACVRL1 variant) than in patients with HHT type 1 (carrying an ENG variant), whereas the opposite is true for AVMs in the lung160. Whether AVMs are congenital or acquired during adult life is unclear. Given that most AVMs are asymptomatic, sparse data exist on AVM frequency in children and younger adults. One study found that, in patients with HHT type 2, hepatic AVMs were present in 67% of patients aged <45 years and in 93% of patients aged >45 years161. A similar difference was found in patients with HHT type 1 (hepatic AVMs were present in 46% of patients aged <45 years and in 78% of patients aged >45 years)161. With regard to pulmonary AVMs, a Canadian study compared the frequency of pulmonary AVMs in children (aged <18 years) and their parents162. In patients with HHT type 1, the frequency of pulmonary AVMs was similar in both groups, whereas among patients with HHT type 2, 8.3% of children had pulmonary AVMs compared with 25.9% of the parents162. Moreover, the incidence of HHT symptoms increases with age, and symptomatic AVMs in the liver are found in adult patients (aged >30 years)163,164. Taken together, these data suggest that at least some AVMs can develop in adulthood because of alterations in endothelial quiescence and that AVM size increases with age leading to symptomatic AVMs in older patients. The hypothesis of an increase in VEGFA signalling as a result of the loss of BMP signalling owing to ACVRL1 or ENG variants was validated by data from a clinical trial showing that inhibition of VEGF in patients with HHT decreases the symptoms of HHT165.
The BMP pathway is also involved in the development and progression of pulmonary arterial hypertension (PAH) (Table 2). Heterozygous germline variants in BMPR2 underlie the main genetic susceptibility for PAH, found in 53–86% of patients with a family history of PAH and 14–35% of patients with idiopathic PAH166. Although the presence of a BMPR2 variation is neither necessary nor sufficient to cause PAH, a reduction in BMPR2 activity is currently viewed as the major molecular defect conferring a predisposition to develop PAH as well as an increased risk of progression of the disease166,167. Constitutive deletion of Bmpr2 in endothelial cells in mice predisposed the animals to develop spontaneous PAH168, supporting the notion that disrupting BMP signalling in the endothelium is a risk factor for PAH. However, given the potential inhibitory role of BMPR2 in BMP signalling169, the exact effect of BMPR2 variants on BMP signalling in the pulmonary endothelium is unclear. The discovery in some patients with PAH of variants in GDF2 (encoding BMP9), leading to decreased circulating BMP9 level, and variants in BMP10 revealed another layer of complexity of the regulation of the pulmonary endothelium and PAH pathogenesis170,171,172,173. Therapy with BMP9 has been proposed as a strategy to compensate for the loss of one BMPR2 allele in patients with PAH150. However, Bmp9-null mice do not develop spontaneous pulmonary hypertension, and these mice were even protected in experimental models of pulmonary hypertension174. Given these contradictory findings, further research is needed to clarify the role of BMP signalling in PAH. In particular, understanding how the reduction in the BMPR2 activity predisposes to PAH and how the BMP9–BMP10–BMPR2 axis contributes to the pathophysiology of PAH is essential. An early event that seems to be facilitated by dysfunction in the BMP9–BMP10–BMPR2 axis is the pro-inflammatory phenotype of endothelial cells in PAH175. In PAH, during the process of vascular remodelling, quiescent pulmonary endothelial cells become activated and express high levels of adhesion molecules, such as VCAM1 and ICAM1, and secrete high levels of chemokines, such as IL-6 and CC-chemokine ligand 2 (CCL2; also known as MCP1)176.
TGFβ signalling
TGFβ ligands bind to TGFβR1 (also known as ALK5) and TGFβR2. Type III receptors (TGFβR3) increase ligand binding to their cognate receptors. Although endothelial ALK5 and TGFβR2 are crucial for cerebral vascular development177 and endothelial TGFβR3 for coronary vessel development178 in mouse embryos, Alk5 or Tgfbr2 deletion in endothelial cells in adult mice does not affect vascular morphogenesis179. Activation of the TGFβ pathway in endothelial cells in adult mice and humans is associated with a change in endothelial cell identity referred to as EndMT, a cell fate change event underlying a number of pathological processes32 (Figs1,2). When endothelial cells undergo EndMT, they acquire mesenchymal characteristics including fibroblast-like morphology, cell junction rearrangement, increased mobility and proliferation, a thrombogenic and inflammatory phenotype, and increased secretion of the extracellular matrix proteins fibronectin and collagen180. To date, at least three pathways that inhibit TGFβ signalling in quiescent endothelia have been identified: the VEGF/FGF–ERK–let-7 pathway and the BMP9–ALK1 pathway discussed above, and the cerebral cavernous malformation (CCM)–MEKK3 pathway10,30,150,181. Postnatal deletion of any of the three known CCM genes in mice leads to overactivation of the MEKK3 pathway182,183, which induces the expression of Klf4 (which encodes the transcription factor Krüppel-like factor 4 (KLF4))181 and Klf2 (which encodes KLF2)184. The exact mechanism of how KLF2 and KLF4 induce CCM lesion formation is unclear. One study showed that KLF4 induces an autocrine loop that involves BMP6, which activates the TGFβ pathway leading to EndMT181. However, another study found that CCM gene deletion induces MEKK3-mediated overactivation of KLF2 and KLF4, but that this overactivation does not induce EndMT183. Interestingly, an ensemble computational intelligence strategy, comprising deep learning and probabilistic programming of RNA-seq data, causally linked the loss of ERK1 and ERK2 in human endothelial cells in vitro to the activation of an autocrine loop driven by TGFβ2 (ref.10). Verified in mice, this autocrine loop resulted not only in the induction of EndMT (seen in vitro and in vivo in Erk1−/−Erk2iEC−/− mice), but also in systemic hypertension. The latter was induced by suppression of eNOS expression (and therefore NO production) and induction of vasoconstrictor ET1 expression. A decrease in endothelial fenestration was also observed (caused by decreased PV1 expression as seen in vitro and in vivo in Erk1−/−Erk2iEC−/− mice)10 (Fig. 2). Systemic hypertension and the loss of endothelial fenestrations are features of VEGF–ERK pathway inhibition. This in silico analysis suggested that the phenotypes seen after the loss of VEGF signalling are, at least partially, due to increased TGFβ signalling.
Fibrosis is a devastating process characterized by myofibroblast cell proliferation and abnormal extracellular matrix accumulation, leading to organ failure. Endothelial cell injury often precedes the development of fibrosis and is suspected to be an initiating event185 (Table 2). Endothelial cells produce profibrotic mediators, such as TGFβ, plasminogen activator inhibitor 1 and connective tissue growth factor, which induce fibroblast growth and differentiation and collagen synthesis by fibroblasts186. In addition, endothelial cells can also differentiate into fibroblast-like cells and secrete collagen as a result of EndMT32. However, the exact contribution of EndMT as the source of myofibroblasts is controversial, and lineage tracing experiments in animal models of cardiac and renal fibrosis show that EndMT is not a major source of myofibroblasts187,188,189,190,191. Liver sinusoidal endothelial cells (LSECs) have a major role in liver fibrosis192,193. After liver injury, LSECs rapidly switch from a fenestrated to a capillarized phenotype and acquire a pro-vasoconstrictive, pro-inflammatory, pro-angiogenic and pro-fibrotic phenotype, which induces hepatic stellate cell activation that leads to liver fibrosis. VEGF and BMP9 can both function to maintain the fenestrated quiescent state of LSECs145,194. Inflammatory cells also contribute to the development of fibrosis. Activated endothelial cells provide important signals, such as the expression of adhesion molecules (for example, ICAM1 and VCAM1) and secretion of various cytokines and chemokines (such as IL-6, CCL2 and CXC-chemokine ligand 12), to recruit leukocytes and perpetuate inflammation. This pro-inflammatory phenotype of endothelial cells and the recruitment of inflammatory cells contribute to the pro-fibrotic environment by inducing the secretion of collagen195. Activation of the TGFβ pathway in endothelial cells triggers an endothelial inflammatory phenotype39. In addition, TGFβ secreted by endothelial cells can induce resident fibroblasts to become myofibroblasts196. Finally, activation of the TGFβ pathway is also a major trigger of plaque formation in atherosclerosis, as a consequence of decreased FGF signalling (see the section on FGF signalling)39,42.
Conclusions
Endothelial quiescence has emerged as an important area of investigation in the field of vascular biology research. The vascular endothelium is central to the regulation of tissue and organ homeostasis and is crucial for disease resilience. Understanding the signalling pathways that regulate the numerous functions of the quiescent endothelium in different organs is central to the understanding of normal physiology as well as the pathophysiology of numerous diseases, and addressing important knowledge gaps is the current challenge in the field of vascular biology (Box 1).
Although many functions of the quiescent endothelium are organ-specific (Fig. 1), other functions are general to all quiescent endothelial cells. Thus, the TGFβ signalling pathway is inhibited in the healthy quiescent endothelium regardless of organ or location, and activation of TGFβ is linked to the loss of normal endothelial cell fate (via EndMT) and to the development and progression of disease states (Figs 1,2). Indeed, the capacity of an endothelial bed to avoid the activation of TGFβ signalling is closely linked to its capacity to resist disease development and might account for different disease susceptibilities in different patient populations. It is interesting to speculate that an increased susceptibility of endothelial cells to TGFβ activation might be a risk factor for some of the most common vascular diseases, such as atherosclerosis. The ability to understand and assess this endothelial cell susceptibility, both genetically and functionally, would allow better risk assessment and the development of therapies aimed at disease prevention. The importance of keeping TGFβ signalling in check is further illustrated by the variety of signalling cascades that inhibit TGFβ signalling in endothelial cells, including VEGF–ERK, FGF–ERK, BMP9–ALK1 and CCM–MEKK3 (Fig. 2). The existence of these signalling cascades suggests that control of TGFβ signalling is crucial for maintaining cell homeostasis and that abnormalities in this pathway can trigger specific diseases.
Endothelial cell senescence and ageing are crucially linked to endothelial cell quiescence, and endothelial normalcy is probably one of the most crucial factors contributing to a healthy lifespan. Examples of such a link include arterial stiffness and hypertension, two hallmarks of the ageing process. Although this subject is outside the scope of this Review, the mechanisms of ageing-related endothelial cell senescence have been well described previously197.
A thorough knowledge of the dynamic control of endothelial quiescence is required. To this end, we need to understand how a signalling pathway that is involved in angiogenic stimulation, such as VEGF signalling, can also maintain endothelial quiescence. This dichotomy could be a result of different VEGF dosages, differential VEGF signalling through different co-receptors, such as neuropilin 1 (refs198,199) and syndecan 2 (ref.200), alterations in the duration of VEGF stimulation, paracrine versus endocrine versus autocrine activation of VEGF signalling, or crosstalk with other signalling pathways. All these factors might differentially affect VEGF stimulation and point towards the existence of regulators that modulate the effects of VEGF signalling to achieve the desired physiological objective201,202.
We also need to understand the molecular basis of the organotypic effects of endothelial cell signalling. For example, although CCM proteins are expressed in all endothelial cells, variants in CCM genes seem to affect only the central nervous system vasculature. Another related problem is that organ-specific mutation of genes in a given signalling pathway does not have a consistent phenotype across organs. Genetic variants in ACVRL1 and ENG lead to the development of HHT (with AVM mainly in the lungs and liver), whereas variants in BMPR2 predispose to the development of pulmonary hypertension, with no effect on the vasculature of other organs.
Advances in research into endothelial cell metabolism show a difference in the metabolic signature between quiescent and activated endothelial cells203. Of note, alterations in endothelial cell metabolism could be very important to regulate cell quiescence and warrant further investigation.
Another important unknown is the link between endothelial cell quiescence and disease resilience. Emerging data from single-cell RNA-seq studies highlight the heterogeneity of endothelial cells between tissues but also within each tissue11 (Fig. 3a,b). These single-cell RNA-seq data suggest that in many cases, disease progression is due to the expansion of a single population of normal cells (for example, endothelial or smooth muscle cells) that are susceptible to a particular disease stimulus42,204,205 (Fig. 3c). These findings also suggest that other normal populations of these cell types are disease-resistant. An in-depth understanding of this phenomenon is crucially important. Further studies to characterize the genetic, molecular and metabolic signatures of disease-resistant versus disease-prone cell populations are also needed (Box 1; Fig. 3c). To summarize, the understanding of the active regulation of the organotypic endothelial quiescence is currently one of the biggest challenges in vascular biology research.

a | Quiescent endothelial cell (EC) heterogeneity in structure, function, immune regulation (interferon response and leukocyte adhesion molecule expression) and metabolism between tissues and within tissues. The information shown in this panel a is from ref.11. b | Heterogeneity in healthy capillary ECs between organs. c | Development of endothelial dysfunction. This is a stepwise process, progressing from activation of ECs to the development of endothelial-to-mesenchymal transition to the full-blown pathological end state. This sequence of events leads to ECs losing their normal fate and acquiring features of mesenchymal cell types, including fibroblasts, smooth muscle cells and macrophages, in a process known as endothelial-to-mesenchymal transition. These events result in the initiation and propagation of inflammation, loss of normal endothelial structures and function, increased vascular permeability and formation of pathological lesions, such as atherosclerotic plaques.
References
Al-Soudi, A., Kaaij, M. H. & Tas, S. W. Endothelial cells: from innocent bystanders to active participants in immune responses. Autoimmun. Rev. 16, 951–962 (2017).
Yau, J. W., Teoh, H. & Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 15, 130 (2015).
Konukoglu, D. & Uzun, H. Endothelial dysfunction and hypertension. Adv. Exp. Med. Biol. 956, 511–540 (2017).
Komarova, Y. A., Kruse, K., Mehta, D. & Malik, A. B. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ. Res. 120, 179–206 (2017).
Chen, W. et al. The endothelial tip-stalk cell selection and shuffling during angiogenesis. J. Cell Commun. Signal. 13, 291–301 (2019).
Tirziu, D. et al. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J. Clin. Invest. 117, 3188–3197 (2007).
Greene, A. K. et al. Endothelial-directed hepatic regeneration after partial hepatectomy. Ann. Surg. 237, 530–535 (2003).
Cao, Z. et al. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat. Med. 22, 154–162 (2016).
Eremina, V., Baelde, H. J. & Quaggin, S. E. Role of the VEGF – a signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol. 106, p32–p37 (2007).
Ricard, N. et al. Endothelial ERK1/2 signaling maintains integrity of the quiescent endothelium. J. Exp. Med. 216, 1874–1890 (2019).
Kalucka, J. et al. Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779.e20 (2020).
Minami, T. & Aird, W. C. Endothelial cell gene regulation. Trends Cardiovasc. Med. 15, 174–184 (2005).
Chi, J. T. et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl Acad. Sci. USA 100, 10623–10628 (2003).
Marcu, R. et al. Human organ-specific endothelial cell heterogeneity. iScience 4, 20–35 (2018).
Aird, W. C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2, a006429 (2012).
DeLeve, L. D., Wang, X., Hu, L., McCuskey, M. K. & McCuskey, R. S. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G757–G763 (2004).
Nolan, D. J. et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 26, 204–219 (2013).
Florey. The endothelial cell. Br. Med. J. 2, 487–490 (1966).
Augustin, H. G. & Koh, G. Y. Organotypic vasculature: from descriptive heterogeneity to functional pathophysiology. Science 357, eaal2379 (2017).
Levick, J. R. & Smaje, L. H. An analysis of the permeability of a fenestra. Microvasc. Res. 33, 233–256 (1987).
Bergmann, O. et al. Dynamics of cell generation and turnover in the human heart. Cell 161, 1566–1575 (2015).
Ornitz, D. M. & Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266 (2015).
Yu, P. et al. FGF-dependent metabolic control of vascular development. Nature 545, 224–228 (2017).
Murakami, M. et al. The FGF system has a key role in regulating vascular integrity. J. Clin. Invest. 118, 3355–3366 (2008).
Murakami, M. et al. FGF-dependent regulation of VEGF receptor 2 expression in mice. J. Clin. Invest. 121, 2668–2678 (2011).
Oladipupo, S. S. et al. Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc. Natl Acad. Sci. USA 111, 13379–13384 (2014).
Hatanaka, K., Lanahan, A. A., Murakami, M. & Simons, M. Fibroblast growth factor signaling potentiates VE-cadherin stability at adherens junctions by regulating SHP2. PLoS ONE 7, e37600 (2012).
Orsenigo, F. et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 3, 1208 (2012).
Fafeur, V., Terman, B. I., Blum, J. & Bohlen, P. Basic FGF treatment of endothelial cells down-regulates the 85-kDa TGFβ receptor subtype and decreases the growth inhibitory response to TGF-β1. Growth Factors 3, 237–245 (1990).
Chen, P. Y. et al. FGF regulates TGF-β signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2, 1684–1696 (2012).
Ramos, C. et al. FGF-1 reverts epithelial-mesenchymal transition induced by TGF-β1 through MAPK/ERK kinase pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 299, L222–L231 (2010).
Dejana, E., Hirschi, K. K. & Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 8, 14361 (2017).
Correia, A. C., Moonen, J. R., Brinker, M. G. & Krenning, G. FGF2 inhibits endothelial-mesenchymal transition through microRNA-20a-mediated repression of canonical TGF-β signaling. J. Cell Sci. 129, 569–579 (2016).
Baeyens, N. Fluid shear stress sensing in vascular homeostasis and remodeling: towards the development of innovative pharmacological approaches to treat vascular dysfunction. Biochem. Pharmacol. 158, 185–191 (2018).
Baeyens, N. et al. Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc. Natl Acad. Sci. USA 111, 17308–17313 (2014).
Horowitz, A., Tkachenko, E. & Simons, M. Fibroblast growth factor-specific modulation of cellular response by syndecan-4. J. Cell Biol. 157, 715–725 (2002).
Tkachenko, E., Lutgens, E., Stan, R. V. & Simons, M. Fibroblast growth factor 2 endocytosis in endothelial cells proceed via syndecan-4-dependent activation of Rac1 and a Cdc42-dependent macropinocytic pathway. J. Cell Sci. 117, 3189–3199 (2004).
Zhang, Y., Li, J., Partovian, C., Sellke, F. W. & Simons, M. Syndecan-4 modulates basic fibroblast growth factor 2 signaling in vivo. Am. J. Physiol. Heart Circ. Physiol. 284, H2078–H2082 (2003).
Chen, P. Y. et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Invest. 125, 4514–4528 (2015).
Hong, Y. M. Atherosclerotic cardiovascular disease beginning in childhood. Korean Circ. J. 40, 1–9 (2010).
Heo, K. S., Fujiwara, K. & Abe, J. Shear stress and atherosclerosis. Mol. Cell 37, 435–440 (2014).
Chen, P. Y. et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 1, 912–926 (2019).
Simons, M., Gordon, E. & Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 17, 611–625 (2016).
Zafar, M. I. et al. The role of vascular endothelial growth factor-B in metabolic homoeostasis: current evidence. Biosci. Rep. 37, BSR20171089 (2017).
Apte, R. S., Chen, D. S. & Ferrara, N. VEGF in signaling and disease: beyond discovery and development. Cell 176, 1248–1264 (2019).
Chau, K., Hennessy, A. & Makris, A. Placental growth factor and pre-eclampsia. J. Hum. Hypertens. 31, 782–786 (2017).
Srinivasan, R. et al. Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS ONE 4, e8283 (2009).
Maharaj, A. S., Saint-Geniez, M., Maldonado, A. E. & D’Amore, P. A. Vascular endothelial growth factor localization in the adult. Am. J. Pathol. 168, 639–648 (2006).
Eremina, V. et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Invest. 111, 707–716 (2003).
Veron, D. et al. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 77, 989–999 (2010).
Zhang, Y. et al. Endocrine vasculatures are preferable targets of an antitumor ineffective low dose of anti-VEGF therapy. Proc. Natl Acad. Sci. USA 113, 4158–4163 (2016).
Iwashita, N. et al. Impaired insulin secretion in vivo but enhanced insulin secretion from isolated islets in pancreatic beta cell-specific vascular endothelial growth factor-A knock-out mice. Diabetologia 50, 380–389 (2007).
D’Hoker, J. et al. Conditional hypovascularization and hypoxia in islets do not overtly influence adult β-cell mass or function. Diabetes 62, 4165–4173 (2013).
Jang, J. Y. et al. VEGFR2 but not VEGFR3 governs integrity and remodeling of thyroid angiofollicular unit in normal state and during goitrogenesis. EMBO Mol. Med. 9, 750–769 (2017).
Lee, S. et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell 130, 691–703 (2007).
Coon, B. G. et al. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J. Cell Biol. 208, 975–986 (2015).
Jin, Z. G. et al. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ. Res. 93, 354–363 (2003).
Touyz, R. M. & Herrmann, J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis. Oncol. 2, 13 (2018).
Touyz, R. M., Herrmann, S. M. S. & Herrmann, J. Vascular toxicities with VEGF inhibitor therapies–focus on hypertension and arterial thrombotic events. J. Am. Soc. Hypertens. 12, 409–425 (2018).
Facemire, C. S., Nixon, A. B., Griffiths, R., Hurwitz, H. & Coffman, T. M. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension 54, 652–658 (2009).
Kappers, M. H. et al. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension 56, 675–681 (2010).
de Jesus-Gonzalez, N. et al. Regorafenib induces rapid and reversible changes in plasma nitric oxide and endothelin-1. Am. J. Hypertens. 25, 1118–1123 (2012).
Eremina, V. et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 358, 1129–1136 (2008).
Je, Y., Schutz, F. A. & Choueiri, T. K. Risk of bleeding with vascular endothelial growth factor receptor tyrosine-kinase inhibitors sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. Lancet Oncol. 10, 967–974 (2009).
Maynard, S. E. et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Invest. 111, 649–658 (2003).
Shibuya, M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 153, 13–19 (2013).
Armaly, Z., Jadaon, J. E., Jabbour, A. & Abassi, Z. A. Preeclampsia: novel mechanisms and potential therapeutic approaches. Front. Physiol. 9, 973 (2018).
Nova, A., Sibai, B. M., Barton, J. R., Mercer, B. M. & Mitchell, M. D. Maternal plasma level of endothelin is increased in preeclampsia. Am. J. Obstet. Gynecol. 165, 724–727 (1991).
Bernardi, F., Constantino, L., Machado, R., Petronilho, F. & Dal-Pizzol, F. Plasma nitric oxide, endothelin-1, arginase and superoxide dismutase in pre-eclamptic women. J. Obstet. Gynaecol. Res. 34, 957–963 (2008).
Shaarawy, M., El Meleigy, M. & Rasheed, K. Maternal serum transforming growth factor beta-2 in preeclampsia and eclampsia, a potential biomarker for the assessment of disease severity and fetal outcome. J. Soc. Gynecol. Investig. 8, 27–31 (2001).
Phipps, E., Prasanna, D., Brima, W. & Jim, B. Preeclampsia: updates in pathogenesis, definitions, and guidelines. Clin. J. Am. Soc. Nephrol. 11, 1102–1113 (2016).
Kasahara, Y. et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Invest. 106, 1311–1319 (2000).
Siekmann, A. F. & Lawson, N. D. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445, 781–784 (2007).
Benedito, R. et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137, 1124–1135 (2009).
Rostama, B. et al. DLL4/Notch1 and BMP9 interdependent signaling induces human endothelial cell quiescence via P27KIP1 and thrombospondin-1. Arterioscler. Thromb. Vasc. Biol. 35, 2626–2637 (2015).
Smith, D. C. et al. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin. Cancer Res. 20, 6295–6303 (2014).
Coleman, R. L. et al. Demcizumab combined with paclitaxel for platinum-resistant ovarian, primary peritoneal, and fallopian tube cancer: The SIERRA open-label phase Ib trial. Gynecol. Oncol. 157, 386–391 (2020).
Jimeno, A. et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Invest. New Drugs 37, 461–472 (2019).
Chiorean, E. G. et al. A phase I first-in-human study of enoticumab (REGN421), a fully human delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin. Cancer Res. 21, 2695–2703 (2015).
Wang, Y. et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486 (2010).
Groppa, E. et al. EphrinB2/EphB4 signaling regulates non-sprouting angiogenesis by VEGF. EMBO Rep. 19, e45054 (2018).
Montani, D. et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 125, 2128–2137 (2012).
Weatherald, J. et al. Pulmonary complications of Bcr-Abl tyrosine kinase inhibitors. Eur. Respir. J. 56, e2000279 (2020).
Guignabert, C. et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J. Clin. Invest. 126, 3207–3218 (2016).
Barratt, S., Medford, A. R. & Millar, A. B. Vascular endothelial growth factor in acute lung injury and acute respiratory distress syndrome. Respiration 87, 329–342 (2014).
Wada, T. et al. The role of angiogenic factors and their soluble receptors in acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) associated with critical illness. J. Inflamm. 10, 6 (2013).
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020).
Yang, F. et al. Analysis of 92 deceased patients with COVID-19. J. Med. Virol. 92, 2511–2515 (2020).
Herrero, R., Sanchez, G. & Lorente, J. A. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann. Transl. Med. 6, 32 (2018).
Sharp, C., Millar, A. B. & Medford, A. R. Advances in understanding of the pathogenesis of acute respiratory distress syndrome. Respiration 89, 420–434 (2015).
Azamfirei, L. et al. Vascular endothelial growth factor: a possible mediator of endothelial activation in acute respiratory distress syndrome. Minerva Anestesiol. 76, 609–616 (2010).
Kosmidou, I., Karmpaliotis, D., Kirtane, A. J., Barron, H. V. & Gibson, C. M. Vascular endothelial growth factors in pulmonary edema: an update. J. Thromb. Thrombolysis 25, 259–264 (2008).
Alkharsah, K. R. VEGF upregulation in viral infections and its possible therapeutic implications. Int. J. Mol. Sci. 19, 1642 (2018).
Gavrilovskaya, I., Gorbunova, E., Koster, F. & Mackow, E. Elevated VEGF levels in pulmonary edema fluid and PBMCs from patients with acute hantavirus pulmonary syndrome. Adv. Virol. 2012, 674360 (2012).
Li, Y. et al. Elevated vascular endothelial growth factor levels induce hyperpermeability of endothelial cells in hantavirus infection. J. Int. Med. Res. 40, 1812–1821 (2012).
Tran, K. A. et al. Endothelial β-catenin signaling is required for maintaining adult blood-brain barrier integrity and central nervous system homeostasis. Circulation 133, 177–186 (2016).
Liebner, S. et al. Wnt/β-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 183, 409–417 (2008).
Wang, Y. et al. Norrin/Frizzled4 signaling in retinal vascular development and blood brain barrier plasticity. Cell 151, 1332–1344 (2012).
Wang, Y. et al. Interplay of the Norrin and Wnt7a/Wnt7b signaling systems in blood-brain barrier and blood-retina barrier development and maintenance. Proc. Natl Acad. Sci. USA 115, E11827–E11836 (2018).
Zhou, Y. et al. Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Invest. 124, 3825–3846 (2014).
Alvarez, J. I. et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 334, 1727–1731 (2011).
Podjaski, C. et al. Netrin 1 regulates blood-brain barrier function and neuroinflammation. Brain 138, 1598–1612 (2015).
Lengfeld, J. E. et al. Endothelial Wnt/β-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc. Natl Acad. Sci. USA 114, E1168–E1177 (2017).
Liu, L., Wan, W., Xia, S., Kalionis, B. & Li, Y. Dysfunctional Wnt/β-catenin signaling contributes to blood-brain barrier breakdown in Alzheimer’s disease. Neurochem. Int. 75, 19–25 (2014).
Lim, R. G. et al. Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood-brain barrier deficits. Cell Rep. 19, 1365–1377 (2017).
Berger, W. et al. Isolation of a candidate gene for Norrie disease by positional cloning. Nat. Genet. 2, 84 (1992).
Xu, Q. et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 116, 883–895 (2004).
Bohannon, D. G., Ko, A., Filipowicz, A. R., Kuroda, M. J. & Kim, W. K. Dysregulation of sonic hedgehog pathway and pericytes in the brain after lentiviral infection. J. Neuroinflammation 16, 86 (2019).
Suri, C. et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87, 1171–1180 (1996).
Wong, A. L. et al. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ. Res. 81, 567–574 (1997).
Papapetropoulos, A. et al. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J. Biol. Chem. 275, 9102–9105 (2000).
Fiedler, U. et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat. Med. 12, 235–239 (2006).
Maisonpierre, P. C. et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277, 55–60 (1997).
Gamble, J. R. et al. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ. Res. 87, 603–607 (2000).
Frye, M. et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J. Exp. Med. 212, 2267–2287 (2015).
Daly, C. et al. Angiopoietins bind thrombomodulin and inhibit its function as a thrombin cofactor. Sci. Rep. 8, 505 (2018).
Higgins, S. J. et al. Tie2 protects the vasculature against thrombus formation in systemic inflammation. J. Clin. Invest. 128, 1471–1484 (2018).
Kim, M. et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J. Clin. Invest. 126, 3511–3525 (2016).
Seegar, T. C. et al. Tie1-Tie2 interactions mediate functional differences between angiopoietin ligands. Mol. Cell 37, 643–655 (2010).
Korhonen, E. A. et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J. Clin. Invest. 126, 3495–3510 (2016).
Claesson-Welsh, L. VEGF receptor signal transduction – a brief update. Vasc. Pharmacol. 86, 14–17 (2016).
Fujio, Y. & Walsh, K. Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J. Biol. Chem. 274, 16349–16354 (1999).
Kerr, B. A. et al. Stability and function of adult vasculature is sustained by Akt/Jagged1 signalling axis in endothelium. Nat. Commun. 7, 10960 (2016).
Shah, A. V. et al. The endothelial transcription factor ERG mediates angiopoietin-1-dependent control of Notch signalling and vascular stability. Nat. Commun. 8, 16002 (2017).
Birdsey, G. M. et al. The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/β-catenin signaling. Dev. Cell 32, 82–96 (2015).
Vikkula, M. et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell 87, 1181–1190 (1996).
Wouters, V. et al. Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. Eur. J. Hum. Genet. 18, 414–420 (2010).
Boon, L. M., Mulliken, J. B., Enjolras, O. & Vikkula, M. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch. Dermatol. 140, 971–976 (2004).
Uebelhoer, M. et al. Venous malformation-causative TIE2 mutations mediate an AKT-dependent decrease in PDGFB. Hum. Mol. Genet. 22, 3438–3448 (2013).
Winderlich, M. et al. VE-PTP controls blood vessel development by balancing Tie-2 activity. J. Cell Biol. 185, 657–671 (2009).
Souma, T. et al. Context-dependent functions of angiopoietin 2 are determined by the endothelial phosphatase VEPTP. Proc. Natl Acad. Sci. USA 115, 1298–1303 (2018).
Dituri, F., Cossu, C., Mancarella, S. & Giannelli, G. The interactivity between TGFβ and BMP signaling in organogenesis, fibrosis, and cancer. Cells 8, 1130 (2019).
Tillet, E. et al. A heterodimer formed by bone morphogenetic protein 9 (BMP9) and BMP10 provides most BMP biological activity in plasma. J. Biol. Chem. 293, 10963–10974 (2018).
David, L., Mallet, C., Mazerbourg, S., Feige, J. J. & Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109, 1953–1961 (2007).
Oh, S. P. et al. Activin receptor-like kinase 1 modulates transforming growth factor-β1 signaling in the regulation of angiogenesis. Proc. Natl Acad. Sci. USA 97, 2626–2631 (2000).
Urness, L. D., Sorensen, L. K. & Li, D. Y. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 26, 328–331 (2000).
Larrivee, B. et al. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 22, 489–500 (2012).
Ricard, N. et al. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 119, 6162–6171 (2012).
Park, S. O. et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Invest. 119, 3487–3496 (2009).
Garrido-Martin, E. M. et al. Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models–brief report. Arterioscler. Thromb. Vasc. Biol. 34, 2232–2236 (2014).
Tual-Chalot, S. et al. Loss of endothelial endoglin promotes high-output heart failure through peripheral arteriovenous shunting driven by VEGF signaling. Circ. Res. 126, 243–257 (2020).
Han, C. et al. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis 17, 823–830 (2014).
David, L. et al. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 102, 914–922 (2008).
Ola, R. et al. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat. Commun. 7, 13650 (2016).
Desroches-Castan, A. et al. Bone morphogenetic protein 9 is a paracrine factor controlling liver sinusoidal endothelial cell fenestration and protecting against hepatic fibrosis. Hepatology 70, 1392–1408 (2019).
Desroches-Castan, A. et al. Differential consequences of Bmp9 deletion on sinusoidal endothelial cell differentiation and liver fibrosis in 129/Ola and C57BL/6 mice. Cells 8, 1079 (2019).
Baeyens, N. et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 214, 807–816 (2016).
Sugden, W. W. et al. Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat. Cell Biol. 19, 653–665 (2017).
Vion, A. C. et al. Primary cilia sensitize endothelial cells to BMP and prevent excessive vascular regression. J. Cell Biol. 217, 1651–1665 (2018).
Long, L. et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 21, 777–785 (2015).
Schlereth, K. et al. The transcriptomic and epigenetic map of vascular quiescence in the continuous lung endothelium. eLife 7, e34423 (2018).
Dufton, N. P. et al. Dynamic regulation of canonical TGFβ signalling by endothelial transcription factor ERG protects from liver fibrogenesis. Nat. Commun. 8, 895 (2017).
Dupuis-Girod, S., Bailly, S. & Plauchu, H. Hereditary hemorrhagic telangiectasia: from molecular biology to patient care. J. Thromb. Haemost. 8, 1447–1456 (2010).
Shovlin, C. L. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 24, 203–219 (2010).
McAllister, K. A. et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 8, 345–351 (1994).
Johnson, D. W. et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 13, 189–195 (1996).
Ricard, N. et al. Functional analysis of the BMP9 response of ALK1 mutants from HHT2 patients: a diagnostic tool for novel ACVRL1 mutations. Blood 116, 1604–1612 (2010).
Mallet, C. et al. Functional analysis of endoglin mutations from hereditary hemorrhagic telangiectasia type 1 patients reveals different mechanisms for endoglin loss of function. Hum. Mol. Genet. 24, 1142–1154 (2015).
Snellings, D. A. et al. Somatic mutations in vascular malformations of hereditary hemorrhagic telangiectasia result in Bi-allelic loss of ENG or ACVRL1. Am. J. Hum. Genet. 105, 894–906 (2019).
Lesca, G. et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: data from the French-Italian HHT network. Genet. Med. 9, 14–22 (2007).
Sabba, C. et al. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J. Thromb. Haemost. 5, 1149–1157 (2007).
Latino, G. A. et al. Prevalence of pulmonary arteriovenous malformations in children versus adults with hereditary hemorrhagic telangiectasia. J. Pediatr. 163, 282–284 (2013).
Plauchu, H., de Chadarevian, J. P., Bideau, A. & Robert, J. M. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am. J. Med. Genet. 32, 291–297 (1989).
Garcia-Tsao, G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J. Hepatol. 46, 499–507 (2007).
Dupuis-Girod, S. et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA 307, 948–955 (2012).
Guignabert, C., Bailly, S. & Humbert, M. Restoring BMPRII functions in pulmonary arterial hypertension: opportunities, challenges and limitations. Expert. Opin. Ther. Targets 21, 181–190 (2017).
Evans, J. D. et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir. Med. 4, 129–137 (2016).
Hong, K. H. et al. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 118, 722–730 (2008).
Olsen, O. E. et al. BMPR2 inhibits activin and BMP signaling via wild-type ALK2. J. Cell Sci. 131, jcs213512 (2018).
Wang, X. J. et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 53, 1801609 (2019).
Graf, S. et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 9, 1416 (2018).
Hodgson, J. et al. Characterization of GDF2 mutations and levels of BMP9 and BMP10 in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 201, 575–585 (2020).
Eyries, M. et al. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur. Respir. J. 53, 1801371 (2019).
Tu, L. et al. Selective BMP-9 inhibition partially protects against experimental pulmonary hypertension. Circ. Res. 124, 846–855 (2019).
Majka, S. et al. Physiologic and molecular consequences of endothelial Bmpr2 mutation. Respir. Res. 12, 84 (2011).
Le Hiress, M. et al. Proinflammatory signature of the dysfunctional endothelium in pulmonary hypertension. Role of the macrophage migration inhibitory factor/CD74 complex. Am. J. Respir. Crit. Care Med. 192, 983–997 (2015).
Nguyen, H. L. et al. TGF-β signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab. Invest. 91, 1554–1563 (2011).
Compton, L. A., Potash, D. A., Brown, C. B. & Barnett, J. V. Coronary vessel development is dependent on the type III transforming growth factor beta receptor. Circ. Res. 101, 784–791 (2007).
Park, S. O. et al. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood 111, 633–642 (2008).
Kalluri, R. & Weinberg, R. A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428 (2009).
Cuttano, R. et al. KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol. Med. 8, 6–24 (2016).
Zhou, Z. et al. The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev. Cell 32, 168–180 (2015).
Zhou, Z. et al. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 532, 122–126 (2016).
Renz, M. et al. Regulation of β1 integrin-Klf2-mediated angiogenesis by CCM proteins. Dev. Cell 32, 181–190 (2015).
Sun, X., Nkennor, B., Mastikhina, O., Soon, K. & Nunes, S. S. Endothelium-mediated contributions to fibrosis. Semin. Cell Dev. Biol. 101, 78–86 (2020).
Yin, Q. et al. Pulmonary microvascular endothelial cells from bleomycin-induced rats promote the transformation and collagen synthesis of fibroblasts. J. Cell Physiol. 226, 2091–2102 (2011).
LeBleu, V. S. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053 (2013).
Djudjaj, S. & Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 65, 16–36 (2019).
Ali, S. R. et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 115, 625–635 (2014).
Kanisicak, O. et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260 (2016).
Frangogiannis, N. G. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 65, 70–99 (2019).
Lafoz, E., Ruart, M., Anton, A., Oncins, A. & Hernandez-Gea, V. The endothelium as a driver of liver fibrosis and regeneration. Cells 9, 929 (2020).
Hammoutene, A. & Rautou, P. E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 70, 1278–1291 (2019).
Deleve, L. D., Wang, X. & Guo, Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 48, 920–930 (2008).
Williamson, J. D., Sadofsky, L. R., Crooks, M. G., Greenman, J. & Hart, S. P. Bleomycin increases neutrophil adhesion to human vascular endothelial cells independently of upregulation of ICAM-1 and E-selectin. Exp. Lung Res. 42, 397–407 (2016).
Phan, S. H., Gharaee-Kermani, M., Wolber, F. & Ryan, U. S. Stimulation of rat endothelial cell transforming growth factor-beta production by bleomycin. J. Clin. Invest. 87, 148–154 (1991).
Jia, G., Aroor, A. R., Jia, C. & Sowers, J. R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1802–1809 (2019).
Lanahan, A. A. et al. PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 130, 902–909 (2014).
Lampropoulou, A. & Ruhrberg, C. Neuropilin regulation of angiogenesis. Biochem. Soc. Trans. 42, 1623–1628 (2014).
Corti, F. et al. N-terminal syndecan-2 domain selectively enhances 6-O heparan sulfate chains sulfation and promotes VEGFA165-dependent neovascularization. Nat. Commun. 10, 1562 (2019).
Kholodenko, B., Yaffe, M. B. & Kolch, W. Computational approaches for analyzing information flow in biological networks. Sci. Signal. 5, re1 (2012).
Kolch, W., Halasz, M., Granovskaya, M. & Kholodenko, B. N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 15, 515–527 (2015).
Li, X., Sun, X. & Carmeliet, P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. 30, 414–433 (2019).
Chen, P. Y. et al. Smooth muscle cell reprogramming in aortic aneurysms. Cell Stem Cell 26, 542–557.e11 (2020).
Misra, A. et al. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat. Commun. 9, 2073 (2018).
Acknowledgements
M.S. is supported, in part, by NIH grants HL135582, HL149343 and HL107205.
Author information
Authors and Affiliations
Contributions
All the authors researched data for the article, provided substantial contributions to discussions of its content, wrote the article, and reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Cardiology thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ricard, N., Bailly, S., Guignabert, C. et al. The quiescent endothelium: signalling pathways regulating organ-specific endothelial normalcy. Nat Rev Cardiol 18, 565–580 (2021). https://doi.org/10.1038/s41569-021-00517-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-021-00517-4
This article is cited by
-
Single-cell RNA sequencing reveals hub genes of myocardial infarction-associated endothelial cells
BMC Cardiovascular Disorders (2024)
-
Vascular organoids: unveiling advantages, applications, challenges, and disease modelling strategies
Stem Cell Research & Therapy (2023)
-
Dynamic nomogram prediction model for diabetic retinopathy in patients with type 2 diabetes mellitus
BMC Ophthalmology (2023)
-
Extracellular vesicles derived from endothelial cells modulate macrophage phenotype in vitro
European Journal of Medical Research (2023)
-
A peripheral signature of Alzheimer’s disease featuring microbiota-gut-brain axis markers
Alzheimer's Research & Therapy (2023)
