
Abstract
Senescence is a cellular response to a variety of stress signals that is characterized by a stable withdrawal from the cell cycle and major changes in cell morphology and physiology. While most research on senescence has been performed on non-cancer cells, it is evident that cancer cells can also mount a senescence response. In this Review, we discuss how senescence can be induced in cancer cells. We describe the distinctive features of senescent cancer cells and how these changes in cellular physiology might be exploited for the selective eradication of these cells (senolysis). We discuss activation of the host immune system as a particularly attractive way to clear senescent cancer cells. Finally, we consider the challenges and opportunities provided by a ‘one-two punch’ sequential treatment of cancer with pro-senescence therapy followed by senolytic therapy.
Similar content being viewed by others


Introduction
Cells progressively accumulate damage during their lifetime due to a multitude of endogenous and exogenous stressors. Although an array of repair mechanisms exists, the damage can sometimes be persistent. Replicative cells also require defence mechanisms to reduce the impact of these persistent stresses on tissue degeneration and to restrict cancer development. One of these defence mechanisms involves switching to a stable non-proliferative, metabolically active survival state named cellular senescence. Cellular senescence can be induced by a variety of stress and damage signals. The phenomenon was first described in 1961 by Hayflick and Moorhead1, who observed that replicative cells have a limited number of divisions — the ‘Hayflick limit’ — that we now understand to be caused by telomere shortening; this is sensed by the cells as a persistent DNA damage response2. As a consequence, these cells undergo senescence to avoid further genomic instability and accumulation of DNA damage3. In addition to telomere shortening, various other stress stimuli can trigger senescence, including oncogenic stress4, oxidative stress5, lysosomal or endoplasmic reticulum stress6,7, nutrient depletion8, and genotoxic stress induced by cancer therapies9 or even from pathogens, including coronavirus10,11. Induction of senescence in response to stress stimuli other than telomere attrition is sometimes referred to as stress-induced premature senescence12,13.
Induction of senescence in response to stress stimuli results in stable cell cycle arrest14. Therefore, senescence represents a defence mechanism against cancer15,16. However, cell cycle withdrawal is also a feature of several other cell states such as quiescence, terminal differentiation, dormancy or drug-tolerant persistence17. It is difficult to unambiguously differentiate between these related cellular states, mainly because we lack ‘gold standard’ markers of the senescent state. Heterochromatinization of proliferative genes is often seen in senescent cells, and this is thought to enforce a durable cell cycle arrest that cannot be readily reversed by pro-mitogenic stimuli18. However, as epigenetic modifications are reversible in principle, it is probably incorrect to consider senescence as a strictly irreversible arrest. Indeed, several studies have highlighted the reversible nature of senescence18,19,20,21. In addition to resistance to proliferative stimuli, another hallmark of senescent cells is resistance to cell death via resistance to apoptotic stimuli or upregulation of pro-survival pathways. In this way, senescent cells remain in a viable state. Apoptotic resistance is regulated by persistent activation of anti-apoptotic proteins, such as the BCL-2 protein family, as well as epigenetic repression of pro-apoptotic proteins such as BAX22,23. Senescent cells are often also characterized by increased lysosomal capacity14. This is linked to increased activity of the lysosomal enzyme senescence-associated β-galactosidase (SA-β-gal), which is one of the most widely used markers for senescence although it is neither required nor specific for the senescence phenotype24,25. Other hallmarks of senescence include altered cell metabolism by mitochondrial dysfunction, macromolecular damage, endoplasmic reticulum stress, loss of lamin B1 (a protein of the nuclear lamina), and the overexpression or activation of cell cycle inhibitors INK4A and ARF (both encoded by CDKN2A), p21 (encoded by CDKN1A) and p53 (encoded by TP53)12.
Arguably, one of the more distinctive, yet not exclusive, features of senescence is the secretion of pro-inflammatory cytokines, growth factors and matrix metalloproteinases (MMPs), collectively termed the senescence-associated secretory phenotype (SASP)26,27. The first evidence of a SASP produced by senescent cells was a gene expression study performed in 1999 demonstrating the expression of inflammation-associated genes in senescent fibroblasts28, although the term ‘SASP’ was not used until 2008 (ref.26). The SASP is generated as a result of activation of the nuclear factor κB (NF-κB)29, cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING)30, GATA4 (ref.31), CCAAT/enhancer-inding protein-βa (CEBPβ)32, NOTCH33,34, IL-6 (ref.35), Janus kinase (JAK)–signal transducer and activator of transcription (STAT)36, p38 MAPK37 and mTOR pathways38. The SASP factors can reinforce senescence in an autocrine fashion and influence the tissue microenvironment by paracrine signalling to adjacent tumour, non-tumour and immune cells39,40,41. Once the levels of senescent cells achieve a certain threshold, they can even produce systemic effects, causing various ageing-related diseases14,27,42,43,44.
A substantial number of anticancer interventions induce senescence in cancer cells by triggering genotoxic stress, hyperactivation of mitogenic signalling or oxidative stress, leading to stable cell cycle arrest and SASP induction42,45. Therefore, therapy-induced senescence serves as an initial antitumour mechanism to halt proliferation and prevent further genomic instability46,47 (Fig. 1). Through their SASP, senescent cancer cells can also arrest neighbouring cancer cells, improve the vasculature for drug delivery and recruit immune cells that can contribute further to tumour suppression29,35,48,49,50,51,52,53. However, persistence of therapy-induced senescent cells may be detrimental in the long term by creating a pro-inflammatory, immunosuppressive microenvironment54. Moreover, the SASP can promote angiogenesis to advance tumour growth and an epithelial to mesenchymal transition in neighbouring cancer cells, which can enhance migration and promote metastasis55,56,57,58. Selective killing of senescent cells — senolytic therapy — has initially been developed to reduce age-related symptoms and improve healthy longevity59,60,61,62 but senolytic therapy may also have utility for the eradication of senescent cancer cells. Recently, nine Cancer Grand Challenges were identified, one of which was to understand and exploit senescence to improve cancer treatment63. Here, we discuss the opportunities and challenges provided by this ‘one-two punch’ approach for the treatment of cancer consisting of pro-senescence and senolytic therapy (Box 1).

Persistent DNA damage caused by hyperactive oncogenic signals, conventional chemotherapies, radiotherapy, and CDC7 inhibitor (XL413 or TAK931) or telomerase inhibitor (GRN163L or BIBR15) treatment often results in induction of senescence. Massive DNA damage response triggers ATM or ATR signalling and results in p53 and p21 activation. Consequently, cells undergo senescence via hypophosphorylation of RB (hypo-pRB). p53–p21 signalling-mediated senescence can also be triggered by other drugs, such as the PTEN inhibitor VO-OHpi, MDM2–p53 interaction disruptors nutlin 3, RG7112 or UBX0101, or the histone deacetylase inhibitor vorinostat. Vorinostat or the DNA methyltransferase inhibitor decitabine can induce CDKN2A expression, which activates both p53-mediated and cyclin-dependent kinase (CDK) inhibition-mediated senescence through its transcripts encoding ARF and INK4A. CDK inhibition-mediated senescence can be induced by inhibitors of CDK4 and CDK6 (CDK4/6) such as palbociclib, abemaciclib or ribociclib. Co-inhibiting CDK2 with CDK4/6 using CDK2/4/6 inhibitor PF-06873600 can achieve more potent senescence induction. Aurora kinases (AURK) and PLK1 inhibitors block the G2/M progression of the cell cycle and this can also induce senescence. These senescence-related pathways are interdependent and reinforce each other. Together, they contribute to several senescence phenotypes, including p21 and p16 upregulation, stable cycle arrest, β-galactosidase (β-gal) activation, and production of the senescence-associated secretory phenotype (SASP).
Induction of senescence in tumours
Oncogene-induced senescence
Cellular senescence serves as a powerful protective mechanism against tumorigenesis16. The activation of oncogenes, such as HRASV12, triggers a growth arrest, referred to as oncogene-induced senescence (OIS), which was first demonstrated in vivo in 1997 (refs4,64,65,66) (Fig. 1). After this, in 2005, the concept of OIS was extended in multiple carcinogenesis models, including lymphomas, prostate cancer, lung adenomas, hyperplastic pituitary gland and melanocytic naevi66,67,68,69,70. Melanocytic naevi induced by oncogenic BRAF mutations generally remain senescent for decades to evade progression into melanoma68. Likewise, the loss of tumour suppressor genes, such as Pten, can also induce senescence in primary prostate epithelium, referred to as PTEN-loss-induced cellular senescence (PICS)67. The p53 pathway plays a central role in OIS and PICS71. In OIS, activation of oncogenes leads to DNA damage71; this in turn activates p53, which induces senescence. This is different from PICS, in which p53 is activated via the mTOR pathway in the absence of clear DNA damage72. In addition, OIS can also be mediated by activation of the INK4A–RB pathway, independent of p53 activation and DNA damage signalling4,64,73. Moreover, senescence can also be triggered by other oncogenic pathways such as activated MYC, which increases levels of the ARF-encoding transcript from the CDKN2A locus, resulting in stabilized p53 (ref.74), and hyperactivated WNT–β-catenin signalling, which triggers a DNA damage response via the p53–p21 pathway75. Dysregulation of these pathways by acquired genetic alterations is a frequent event during tumorigenesis as it enables cells to evade a senescence response76,77,78,79,80.
Chemotherapies and radiotherapies
Despite the ability of malignant tumours to evade senescence, they can still be forced to enter a senescent state using therapeutics leading to therapy-induced senescence16,81. Conventional anticancer therapeutics, such as chemotherapy or radiotherapy, are known to induce senescence in cancer cells81,82,83,84.
Low doses of chemotherapy particularly trigger a senescent cell state in human cancer cells, while apoptosis is induced at higher doses85,86,87. This finding might explain why often only a subset of cancer cells become senescent in response to conventional chemotherapies or radiotherapies as the senescence response is only triggered in a specific window of DNA damage. Mechanistically, many chemotherapies cause DNA damage in cancer cells, which triggers senescence through ATM–CHK2 and ATR–CHK1 kinase-mediated activation of the interconnected p53–RB pathways88,89 (Fig. 1). Topoisomerase I and II inhibitors, such as doxorubicin, etoposide and camptothecin, are widely used for the treatment of a variety of cancer types and have been shown to dysregulate re-ligation of DNA strands after supercoil unwinding. This leads to massive DNA damage and increased expression of p53 and its downstream targets CDKN1A and PAI1 (also known as SERPINE1), subsequently inducing senescence90,91,92. Platinum-based compounds, such as cisplatin, carboplatin and oxaliplatin, also induce extensive DNA damage through DNA cross-linking, resulting in senescence induction93,94. Similarly, alkylating agents, such as temozolomide, dacarbazine and busulfan, form DNA crosslinks by reacting with atoms in DNA, triggering a DNA damage-mediated senescence response95. Microtubule inhibitors, such as paclitaxel, docetaxel and vinca alkaloids, dysregulate the normal microtubule spindle dynamics to impair metaphase–anaphase transition and arrest the cells at mitosis. This cell cycle dysfunction may also cause extensive DNA damage and trigger a p53–p21-facilitated senescence response96,97. Methotrexate and gemcitabine both induce genotoxic stress by blocking DNA synthesis, thereby inducing cellular senescence98,99. It is important to keep in mind that, while quite a few existing chemotherapeutics have some ability to induce senescence, the apoptotic response is dominant in most cancers100. As such, most chemotherapies are unable to induce senescence in a significant fraction of cancer cells in vivo81.
Radiotherapy is applied broadly for the treatment of multiple cancer types. This anticancer treatment can induce an irreparable DNA damage response that activates ATM or ATR and p53–p21 pathway-mediated apoptosis and cellular senescence89,101,102 (Fig. 1). As, unlike chemotherapy, the treatment can be applied locally, there is less collateral damage to normal tissues and, consequently, potentially also less secondary cancer103. Nevertheless, the tissue surrounding the cancer can show an increase in senescent cell burden, resulting in an array of local side effects, including immunosuppressive effects27,55,104.
Cell cycle inhibition
A hallmark of senescent cells is the upregulation of cyclin-dependent kinase (CDK) inhibitor proteins, such as INK4A and p21, to induce cell cycle arrest. By contrast, cancer cells often have upregulated levels of CDKs for progression through the cell cycle105. Therefore, drugs that inhibit CDKs or enhance levels of CDK inhibitor proteins are currently being investigated for use in senescence-inducing cancer therapy106. In particular, CDK4 and CDK6 (hereafter referred to as CDK4/6) are important for progression from G1 to S phase of the cell cycle and are overexpressed in a number of human cancers106,107. CDK4/6 inhibitors mimic the function of INK4A and are able to induce senescence in various cancer cells108,109,110,111,112,113 (Fig. 1). Three CDK4/6 inhibitors — palbociclib, abemaciclib and ribociclib — are approved by the FDA for the treatment of advanced breast cancer. It is important to point out, however, that approval was based on tumour control and not specifically on an ability to induce senescence42.
CDK2 is another potential therapeutic target for pro-senescence therapy. CDK2 can promote the bypass of OIS and result in tumour progression114. The majority of clinically relevant resistance to CDK4/6 inhibitors, such as loss of RB1 (which encodes RB) and amplification of CCNE1 (which encodes cyclin E1) or CDK6, could be overcome by CDK2 inhibition to induce senescence115,116,117. As such, CDK2 depletion was shown to re-sensitize CDK4/6 inhibitor-resistant cells117,118,119,120. Unfortunately, the lack of a specific CDK2 inhibitor has, until now, prevented exploiting the synergy between CDK2 inhibition and CDK4/6 inhibition. A triple CDK2/4/6 inhibitor (PF-06873600) for which clinical development is ongoing in breast cancer in combination with endocrine therapy, is a potent senescence inducer in various cancer models119,121 (Fig. 1). Unfortunately, the compound also has an affinity for CDK1, which might lead to toxicity as seen with other multi-CDK inhibitors119.
Aurora kinases (AURKs) and polo-like kinases (PLKs) are serine/threonine kinases that regulate several mitotic processes and are potential targets for senescence-inducing therapy. In particular, PLK1 and AURKA and AURKB (also referred to as the AURK–PLK1 axis) are frequently overexpressed in cancer and tightly regulated122,123,124. Multiple PLK1 inhibitors, such as BI-2535 and BI-6727, and AURK inhibitors, such as alisertib or barasertib, are being tested as potent inducers of senescence in various tumour models in vitro and in mice, and are also currently being investigated in clinical trials125,126,127,128,129,130,131,132,133 (Fig. 1).
Finally, it was shown that inhibition of DNA replication through small molecule inhibition of the kinase CDC7 using XL413 or TAK-931 leads to senescence induction in liver cancer. The senescence response was seen only in TP53-mutant tumours, presumably because TP53-wild-type tumours retain the ability to mount cell cycle arrest upon CDC7 inhibition, whereas TP53-mutant cells will instead undergo senescence as a result of replication stress due to inhibited DNA replication origin firing134.
Telomerase inhibition
As mentioned above, replicative senescence occurs in response to telomere attrition. Cancer cells circumvent this most commonly by reactivating telomerase activity135. To date, numerous compounds that inhibit the telomerase complex have been identified as candidates for anticancer therapy136. Among these, BIBR15 and GRN163L are potent telomerase inhibitors that effectively induce senescence and suppress cancer cell proliferation137,138 (Fig. 1). However, the use of GRN163L for pro-senescence therapy should be examined further as this compound also induced apoptosis in human pancreatic cancer cells138.
Epigenetic modulators
Another approach to induce senescence is by modulating the epigenome of cancer cells. Decitabine inhibits DNA methyltransferases causing demethylation of the CpG-enriched regions of the CDKN2A promoter, inducing INK4A and ARF expression and senescence139. Vorinostat, a histone deacetylase inhibitor, can also upregulate the expression of multiple tumour suppressor genes, such as CDKN2A and TP53, and can induce senescence via these two major pathways in various cancer cell lines140,141 (Fig. 1).
Targeting tumour suppressors or oncogenes
Initiation and maintenance of cellular senescence are dependent on p53, which is frequently impaired in cancer cells. In mouse models, genetic restoration of Trp53 results in regression of sarcomas and liver carcinomas by inducing a senescence response, while an apoptotic response was observed for lymphoma regression50,142,143. In addition, senescence induction was accompanied by upregulation of the SASP and recruitment of immune cells to the tumour, suggesting efficient clearance of senescent cancer cells50. Currently, several compounds activating p53 are being developed. For example, the MDM2 inhibitors nutlin 3 and RG7112 can interfere with the p53–MDM2 interaction and show promising results in inducing senescence in tumours that retain wild-type TP53 in human cancer cell line models144,145,146. Notably, disruption of the p53–MDM2 complex could also induce apoptosis instead of senescence as demonstrated by the senolytic drug UBX0101 or the forkhead box protein O4 (FOXO4)-DRI peptide in non-cancer models, which is discussed in more detail below147,148. Whether this approach will also work for cancer has yet to be investigated (Fig. 1).
Inactivation of PTEN, for example, with the PTEN inhibitor VO-OHpic, has potential for pro-senescence cancer therapy in vitro and in mice72,149 (Fig. 1). It was demonstrated in vitro that PTEN status is a significant determinant of cell fate in glioma cells after ionizing radiation; PTEN-mutant cells underwent premature senescence while cancer cells expressing PTEN underwent apoptosis150. In addition, the inactivation of PTEN resulted in p53-mediated senescence and suppression of tumorigenesis in mice67.
Given the frequent mutation of the TP53 and RB1 pathways in human cancer and the notion that these pathways are central to the senescence response, one would think that cancer cells are mostly resistant to induction of senescence. However, this is not the case: nearly all cancer cell lines can be rendered senescent in vitro using DNA damaging agents or AURK inhibitors, and this was independent of RB1 and TP53 mutation status151. Apparently, while these genes are mediators in the senescence response of cancer cells, they are not essential for senescence induction in cancer.
Effects of senescent cells in tumours
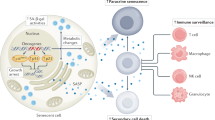
The SASP factors suppress cancer in part by reinforcing the senescent growth arrest and/or by promoting immune surveillance29. Oncogene-induced and therapy-induced senescent cells secrete the inflammatory cytokine IL-1α, which is a crucial SASP initiator and regulator152. IL-1α triggers an autocrine inflammatory response through activation of NF-κB, which leads to the transcription of IL-6 and IL-8 (ref.152) (Fig. 2a). Subsequently, these inflammatory cytokines reinforce senescence proliferation arrest through increased production of reactive oxygen species and a sustained DNA damage response, particularly in oncogene-induced senescent cells152,153. Furthermore, IL-1α also mediates paracrine senescence in neighbouring cells to suppress tumour progression40. Moreover, IL-1α, IL-6 and IL-8 mediate the recruitment of M1-like macrophages, T helper 1 cells and natural killer (NK) cells to the tumour microenvironment (Fig. 2a). These infiltrating immune cells drive the elimination of senescent cancer cells and might also eliminate non-senescent cancer cells through a bystander effect, although this is not yet proven51,154. In addition, immune cells, such as T helper 1 cells, can also trigger senescence in cancer cells through the secretion of inflammatory cytokines155.

Nuclear factor-κB (NF-κB) signalling is activated in therapy-induced senescent cancer cells and this drives production of IL-1α, IL-6, IL-8, CCL2, matrix metalloproteinases (MMPs) and other senescence-associated secretory phenotype (SASP) factors. a | The positive effects are that some of these cytokines, such as IL-6, IL-8 and CCL2, can recruit natural killer (NK) cells and T cells contributing to immune surveillance. In addition, IL-6 and IL-8 can reinforce cellular senescence in an autocrine fashion through increased reactive oxygen species (ROS) production and a sustained DNA damage response. These interleukins (ILs) can spread senescence to surrounding cancer cells in a paracrine fashion, which further controls tumour growth. b | As the negative effects, IL-6 either secreted by senescent cancer cells or released from the extracellular matrix (ECM) by MMPs recruits myeloid-derived suppressor cells (MDSCs), resulting in an immunosuppressive microenvironment. Moreover, the cleaved ECM components release growth factors that can promote further tumour growth and epithelial-to-mesenchymal transition (EMT), leading to metastasis. Vascular endothelial growth factor (VEGF) from the SASP stimulates blood vessel formation that also contributes to metastasis. IL-Rs, interleukin receptors.
Although the SASP of senescent cancer cells is initially tumour suppressive by reinforcing growth arrest and promoting immune clearance, it is suggested to be mostly detrimental in the long term27,156. Early evidence for this notion was found two decades ago by an in vivo study demonstrating increased proliferation and tumorigenesis of both premalignant and malignant epithelial cells when co-injected with human senescent fibroblasts in mice157; the SASP was an important contributor to this effect157. Another study observed that MMPs secreted by senescent human fibroblasts were of primary importance in promoting tumorigenesis158. These prominent SASP factors are involved in the processing and degradation of the extracellular matrix, which can promote cancer cell growth and invasion159. In addition, MMPs also promote the release of many other cytokines and growth factors supporting tumorigenesis such as vascular endothelial growth factor (VEGF), which promotes tumour-driven angiogenesis160 (Fig. 2b). Furthermore, senescent cells also secrete the chemokine CXCL1, which promotes tumour growth161.
The SASP factors IL-6 and IL-8 are also important mediators of the pro-tumorigenic effects of senescent cells because they create a chronic inflammatory microenvironment that supports cancer growth26,162. In addition, they drive the transcription of genes encoding MMPs and drive epithelial to mesenchymal transition, thereby promoting tumour invasiveness163,164,165,166 (Fig. 2b). Another pro-tumorigenic effect of senescent cells mediated by IL-6 is the recruitment of myeloid-derived suppressor cells (MDSCs) to the tumour microenvironment55. These MDSCs block IL-1α signalling and therefore antagonize the establishment of senescence in cancer cells167. Additionally, MDSCs block immune surveillance by inhibiting CD8+ T cells (mediated by IL-6 (ref.55)) and NK cells (mediated by the chemokine CCL2 (ref.154)). In this way, the SASP creates an immunosuppressive environment, facilitating tumour growth55,168 (Fig. 2b). However, the pro-tumorigenic and anti-tumorigenic effects of senescent cancer cells are likely mediated by a complex interplay between multiple SASP factors and the immune microenvironment. Moreover, the effects of SASP factors will be influenced by tissue type, resident immune cells, inflammatory networks or by the nature of the senescence inducer. As a result, it is presently very difficult to predict whether the effects of senescent cancer cells are pro-tumorigenic or anti-tumorigenic.
Perhaps even more surprisingly, several studies have demonstrated that senescence-inducing therapies are associated with complex reprogramming that could eventually drive stemness in both tumour and normal cells21,169. Moreover, remaining senescent cancer cells that are not cleared by the immune system can spontaneously escape proliferation arrest under certain circumstances and re-enter the cell cycle20,21,170. Another study observed that oncogene-induced senescent cells could also re-enter the cell cycle, particularly by restoring telomerase activity through de-repression of the telomerase reverse transcriptase (TERT) gene171. Importantly, senescent cells that resume growth have a WNT-dependent enhanced growth and tumour initiating potential21. This senescence-associated stemness results in a highly aggressive tumour, driven by WNT pathway activation independent of the WNT ligand via the SASP and is found to be enriched in relapsed tumours21. Moreover, expression of β-catenin in pituitary stem cells provokes a signature of senescence and SASP and can induce craniopharyngioma tumours in a paracrine fashion. Importantly, mice with reduced senescence burden and SASP responses showed decreased tumorigenic potential, indicating that the SASP may drive tumour induction172. In the past few years, the finding of extracellular vesicles and exosomes as part of the SASP is generating wide interest in the field of senescence. Small extracellular vesicles from senescent cells can also be tumour promoting and reinforce cancer cell proliferation173,174,175.
Taken together, the role of cellular senescence in tumours and the outcome of senescence-inducing therapies are complex and often unpredictable, mainly because of the dual role of the SASP. The effect of the SASP is highly dependent on context and cell type and variable during the different stages of cancer progression27,29,151. For example, SASP-mediated immunosuppression promotes tumour growth, especially in later stages of tumour progression, while the SASP acts a tumour suppressor in the early stages of tumorigenesis154. In addition, TP53 status may be a factor in determining whether the senescence-induced inflammatory response will be tumour suppressive or tumour promoting, respectively176. In the long term, the SASP of senescent cancer cells is suggested to be primarily detrimental in promoting neoplastic growth, therapy resistance, immunosuppression, metastasis and angiogenesis27,55,157,177. Moreover, senescent cancer cells can potentially remain dormant for a long time, thereby evading therapy and posing a risk for tumour relapse16,20,178. It has also been reported that many genotoxic chemotherapies have debilitating side effects caused by inducing senescence in normal tissues. The senescent normal cells remain chronically present and promote local and systemic inflammation caused by the SASP, which results in or exacerbates chemotherapy side effects177. Hence, it could be helpful to combine pro-senescence therapy with senolytic therapy in the context of cancer. Besides direct targeting of the cancer cells by delivering a one-two punch and decreasing the side effects of chemo-radiotherapies on normal tissues, senolytics should eliminate incipient preneoplastic senescent cells, or other senescent cells in the tumour microenvironment, to suppress the detrimental effects of the SASP156,162.
Senolytics for anticancer therapy
As discussed above, persistence of therapy-induced senescent cells after treatment may be detrimental. This argues for a strategy in which therapy-induced senescent cells are eliminated for the long-term good, minimizing risk of tumour progression and avoiding adverse effects156.
Targeting senescent cancer cells with senolytic agents
One of the hallmarks of senescent cells is a change in chromatin structure that results in changes in gene expression. These alterations can affect fundamental processes like regulation of apoptosis, resulting in acquisition of new vulnerabilities specific to the senescent cells that can be selectively targeted by drugs that could act as senolytic agents156,179,180,181. Such compounds may have clinical utility in combination with or when used sequentially with senescence-inducing therapies (Fig. 3; Table 1).

Changes in senescent cells can induce vulnerabilities that can be targeted by senolytic agents to eliminate these cells. a | Anti-apoptotic family proteins are upregulated in senescent cells to confer resistance to apoptosis. Navitoclax, as an anti-apoptotic BCL-2 family inhibitor, can induce intrinsic apoptosis through cytochrome c release that in turn activates apoptosis executioner caspases 9, 3 and 7. Galacto-conjugated navitoclax (Nav-Gal) is processed by senescence-associated β-galactosidase (SA-β-gal) to deploy active navitoclax in senescent cells. PZ15227 hijacks the cereblon E3 ligase to degrade BCL-XL protein. b | Doxorubicin can be released by SA-β-gal from encapsulated nanocarriers to induce cytotoxicity in senescent cancer cells. c | Dasatinib targets, among others, the SRC kinase and PI3K–AKT signalling nodes. Quercetin or fisetin interfere with the anti-apoptotic protein BCL-XL through inhibition of upstream pathways including PI3K. mTOR inhibition using AZD8055 or temsirolimus can act downstream of receptor tyrosine kinases (RTKs)–SRC–PI3K and induce senolysis through activation of apoptosis. d | Cardiac glycosides, such as digoxin and ouabain, can activate the pro-apoptotic BCL-2 family protein NOXA to induce intrinsic apoptosis. Moreover, cardiac glycosides trigger cell depolarization and acidification through inhibition of Na+/K+ pumps. Both mechanisms can promote senolysis. e | UBX0101 or nutlin 3 can lead to p53 accumulation by disrupting the ubiquitin degradation mechanism mediated by MDM2. The forkhead box protein O4 (FOXO4)-DRI peptide interferes with the binding of FOXO4 to p53, which occurs in senescent cells, and this activates senolysis. The proteolysis-targeting chimaera drug ARV-825 degrades bromodomain proteins (BRD2, BRD3 and BRD4), which disrupts 53BP1 recruitment and interferes with non-homologous end joining (NHEJ), resulting in massive DNA damage-mediated senolysis.
Senescent cells often have increased levels of the anti-apoptotic BCL-2 family proteins182. Compounds targeting this family of proteins have been studied intensively for senolytic therapy. Several studies have shown that navitoclax (also known as ABT263) and the related ABT737, selective inhibitors of BCL-2, BCL-XL and BCL-W, effectively eliminated senescent cells of multiple types, including senescent cancer cells, by re-activating the apoptotic pathway128,151,183,184 (Fig. 3a). In non-cancer-bearing mice, this was associated with beneficial effects such as rejuvenation and attenuation of age-related diseases182,184. Navitoclax has also shown activity in cancer cell lines and mouse models in combination with various senescence-inducing therapies (Table 1), including a class of AURK inhibitors128,151, etoposide128,185,186, doxorubicin185, olaparib187 and ionizing radiation185,188, with evidence of reduced side effects177. Consistent with this, a phase II study showed that rituximab, as a senescence inducer, combined with navitoclax demonstrated higher effectiveness than rituximab monotherapy for patients with chronic lymphocytic leukaemia189. Furthermore, this combination was well tolerated and resulted in prolonged progression-free survival. As expected, patients with higher levels of BCL-2 responded better to navitoclax treatment189.
Despite this positive result, several previous clinical studies revealed major side-effects of navitoclax related to on-target effects in haematological cells such as thrombocytopenia and neutropenia190. Navitoclax has been tested in clinical trials in combination with several conventional chemotherapies. The chemotherapy doses used in these studies were high enough to induce cell death rather than senescence; possibly as a consequence of this, the outcome of these studies was rather disappointing. Most trials were stopped after phase I due to the lack of efficacy or the high toxicities of the combinations (Table 1).
Thrombocytopenia following navitoclax has been shown to result from BCL-XL inhibition; therefore, another BCL-2-selective inhibitor, venetoclax (also known as ABT199), was re-engineered to spare platelets191. However, the senolytic effect of navitoclax relies on BCL-XL inhibition in some cancer types. Consequently, venetoclax is mostly inactive as a senolytic in cancer cells128,182. Other attempts have been made to lower the toxicity of navitoclax. For example, a galacto-conjugated navitoclax prodrug, Nav-Gal, has been recently proposed to enhance the delivery of navitoclax192. Navitoclax remains inactive in prodrug form but becomes active when the galacto moiety is cleaved off by SA-β-gal. As senescent cells display high levels of SA-β-gal, navitoclax delivered via Nav-Gal will only be activated by SA-β-gal in senescent cells, thus limiting off-target effects in other cells192 (Fig. 3b). Moreover, it was also shown in mice that nanocarrier-encapsulated doxorubicin can be activated by SA-β-gal to specifically eliminate palbociclib-induced senescent cancer cells while preventing systemic toxicities193. This exciting strategy using nanocarriers targeting senescent cells has been also further validated in other studies194,195. Another approach limiting the platelet toxicity of navitoclax is the use of the proteolysis-targeting chimaera (PROTAC) drug PZ15227, a BCL-XL specific PROTAC that hijacks the cereblon E3 ubiquitin ligase to degrade BCL-XL proteins196. Cereblon is expressed across human cancer cells but expressed minimally in platelets. As a result, the senolytic specificity of targeting BCL-2 anti-apoptotic proteins in senescent cancer cells is increased and the undesired toxicities, such as thrombocytopenia, are reduced (Fig. 3a).
In addition, the senolytic activity of navitoclax is cell type-dependent as upregulation of BCL-2 or other anti-apoptotic BCL-2 family proteins is not a universal senescence vulnerability151,179. The senolytic response to navitoclax is highly divergent among cancer cell lines in vitro, and biomarkers of response remain elusive other than the insensitivity of cells with mutations in genes encoding pro-apoptotic BCL2-family members, such as BAX and BAK, to navitoclax treatment151. Thus, loss-of-function mutations in the genes encoding intrinsic pro-apoptotic BCL-2-family members might be a common resistance mechanism for the navitoclax-class of senolytic agents.
Another example of a PROTAC senolytic drug is the bromodomain and extraterminal domain (BET) family degrader ARV825, which is composed of a potent inhibitor of BET proteins BRD2, BRD3, and BRD4 and the E3 ligase binder pomalidomide197,198. ARV825 provokes senolysis through two independent pathways. ARV825 downregulates XRCC4 gene expression and blocks the recruitment of p53-binding protein 1 (53BP1), which interferes with non-homologous end-joining DNA double-strand break repair and activates apoptosis198,199 (Fig. 3e). Moreover, ARV825 also induces autophagy, which may contribute to senolysis198.
Increased expression of pro-survival and apoptosis resistance networks, including ephrins, SRC, PI3K, p21, BCL-XL and PAI2, was seen in senescent cells183. This study also showed that dasatinib, a multi-tyrosine kinase inhibitor that targets, amongst others, the SRC kinase and PI3K–AKT signalling nodes, can kill senescent cells. Quercetin, a natural flavonoid, also has senolytic activity183,200,201. It is not clear how exactly quercetin induces senolysis, but it has been reported that it interferes with the anti-apoptotic protein BCL-XL through inhibition of upstream pathways including PI3K. Administration of a combination of dasatinib and quercetin to aged mice eliminated senescent cells and improved cardiovascular function and survival202. The combination of dasatinib and quercetin as the first-in-human senolytic therapy showed efficacy in treating idiopathic pulmonary fibrosis, a fatal disease associated with the appearance of senescent cells in the lungs203 (Fig. 3c). Fisetin, another natural flavonol with potent senolytic activity, is twice as potent as quercetin47,204. The efficacy of fisetin alone or in combination with other anticancer agents has been studied in a wide range of cancer types. Fisetin treatment has anti-proliferative and pro-apoptotic effects in cancer cells in vitro and in mice and suppresses inflammation, migration and metastases in mice204. Although dasatinib, quercetin and fisetin are potent senolytic compounds, they act on a wide range of pathways implicated in various biological processes47. The lack of mechanistic insight into how these drugs induce senolysis may be an impediment for their clinical use. Moreover, these senolytic therapies have not shown effectiveness in combination with pro-senescence therapies in animal models of liver cancer205. Nevertheless, the combination of dasatinib plus quercetin has shown initial promise in the treatment of patients with idiopathic pulmonary fibrosis203. These drugs are currently also being tested in age-related diseases and in infectious diseases that cause senescence-like phenotypes (Table 1).
Compounds that modulate p53 activity can also act as senolytics. For example, the peptide FOXO4-DRI interferes with the interaction between p53 and FOXO4, resulting in p53 nuclear exclusion followed by apoptosis selectively in senescent non-cancer cells148 (Fig. 3e). In mice, the FOXO4-DRI peptide was well tolerated, improved health span and neutralized doxorubicin-induced chemotoxicity caused by therapy-induced senescent cell burden148. Although this peptide demonstrates promising senolytic activity in mice, its therapeutic use still needs to be clinically evaluated. The senolytic compound UBX0101 also modulates p53 activity. UBX0101 selectively kills senescent cells by disrupting the interaction between p53 and MDM2147 (Fig. 3e). A limitation of drugs targeting the p53–MDM2 interaction is that they are not specific for cancer cells or senescent cells, and may therefore cause side effects in normal cells206. However, this could potentially be limited by local drug administration. For example, intra-articular injection of UBX0101 into arthritic joints effectively cleared senescent cells, reduced the SASP and improved joint function in mice147. Nevertheless, UBX0101 was discontinued after evaluation of the phase I clinical trial data for lack of efficacy207. Whether drugs like UBX0101 have utility in cancer remains to be determined. However, such local administration is mostly impossible in cancer therapy. It should also be kept in mind that p53-targeted senolytic therapy can only be effective in TP53-wild-type tumours, which excludes approximately 50% of all cancers208,209.
Two independent studies identified the cardiac glycosides digoxin and ouabain as senolytic agents with activity in multiple cancer models210,211. Cardiac glycosides can inhibit Na+/K+ ATPase pump activity resulting in an imbalanced electrochemical gradient within the cell, causing depolarization and acidification. Senescent cells have a depolarized plasma membrane and higher concentrations of hydrogen ions, making them more vulnerable to cardiac glycosides211 (Fig. 3d). Cardiac glycosides can also alter the pro-apoptotic BCL-2 family protein NOXA through the inhibition of Na+/K+ transport, resulting in apoptosis210. Digoxin behaves as a senolytic at concentrations close to those observed in the plasma of patients treated for heart failure with this drug (20–33 nM)210. However, the narrow dosage window of cardiac glycosides might require further optimization for their clinical use as senolytics.
mTOR is an essential regulator of the SASP and mTOR inhibitors have shown senolytic effects in senescent cancer cells38,134,212,213. Treatment with docetaxel and the mTOR inhibitor temsirolimus suppressed prostate and breast cancer growth in mice214. Moreover, the mTOR inhibitor AZD8055 acted as a potent senolytic agent for liver cancer cells rendered senescent through CDC7 inhibition (Figs 1,3c). This drug combination has been validated in mice by showing its synergistic activity in controlling liver cancer134. However, these studies were limited to specific tumour types and the senolytic application of mTOR inhibition remains to be tested in other cancers.
Immune response-mediated senolysis
SASP cytokines, chemokines and other factors that modulate immune cells can either promote or inhibit senescent cell clearance29,215 (Fig. 4; Table 1). Early studies of the effect of SASP factors on the immune system acknowledged that several factors promote clearance of senescent cells, indicating that the SASP facilitates the removal of senescent pre-neoplastic cells, thus preserving tissue homeostasis. Evidence to support this comes from studies of liver fibrosis, which is associated with an increased risk of cancer. In a carbon tetrachloride-induced liver fibrosis model, carbon tetrachloride treatment can induce DNA damage and lead to upregulation of a ligand for an NK cell activating receptor, NKG2D; this promotes NK cell-mediated cytotoxicity to eradicate senescent cells50,52,216,217. In the NrasG12V oncogene-driven hepatocellular carcinoma mouse model, the restoration of Trp53 expression promoted senescence in cancer cells associated with CCL2 secretion. This chemokine attracts NK cells and, together with NKG2D upregulation on senescent cells, results in NK cell-mediated senolysis154 (Fig. 4a). As a therapeutic strategy, the MEK inhibitor trametinib and the CDK4/6 inhibitor palbociclib have been used to induce senescence in the KRASG12D;Trp53-/- lung cancer mouse model. Tumour necrosis factor (TNF) and intercellular adhesion molecule 1 (ICAM1), as part of the SASP, were regulated by NF-κB and produced by the therapy-induced senescent cells, and both served as NK cell-activating molecules, promoting cancer cell eradication48. This study suggests that, although chronic inflammation caused by SASP induction can have detrimental effects in some scenarios26,27,59, senescence and the SASP can also favour immune system recognition and cancer cell killing. The timely clearance of senescent cancer cells by NK cells in this model underscores that senescence induction can be beneficial48.

a | Senescent cells secrete senescence-associated secretory phenotype (SASP) factors, such as CCL2, and upregulate surface proteins, such as NKG2D and intercellular adhesion molecule 1 (ICAM1), which drive the recruitment of natural killer (NK) cells to promote immune surveillance. However, matrix metalloproteinases (MMPs) may cleave NKG2D ligand expressed on NK cells to dampen the cytotoxic effect of NK cells. b | Other SASP factors, such as CCL5, CXCL1 and IL-6, recruit CD8+ T cells for senescent cell clearance. Opposite to these immune-stimulatory scenarios, IL-6 can also attract myeloid-derived suppressor cells (MDSCs) to suppress the senolytic effect of NK cells and T cells. Senescent cells may present specific antigens on major histocompatibility complex (MHC) class I to activate CD8+ cells. However, as an immune suppressive mechanism, senescent cells can also upregulate PDL1 to attenuate T cell responses. c | Chimeric antigen receptor (CAR) T cell therapies armed with chimeric receptors can interact with the surface protein antigens present on senescent cells. Therefore, CAR T cells can specifically recognize and eliminate these senescent cells. d | Another SASP factor, vascular endothelial growth factor (VEGF), promotes blood vessel remodelling and can improve drug delivery and immune cell recruitment.
To study the possibility that increased immune surveillance through therapy targeting the immune checkpoint PD1 can be senolytic, trametinib and palbociclib were combined to induce senescence in a KRAS-mutant pancreatic ductal adenocarcinoma mouse model49. Besides direct senolytic effects of anti-PD1 antibody therapy in combination with palbociclib and trametinib, the study also demonstrated an impact of therapy-induced senescence on improving tumour vasculature function through SASP-facilitated vascular remodelling. This response increases blood vessel density and permeability, leading to enhanced uptake and activity of the chemotherapeutic agent gemcitabine, which is the standard-of-care therapy for pancreatic cancer patients (Fig. 4d). Moreover, in the tumour microenvironment, SASP induction facilitates increased endothelial cell expression of VCAM1, a cell surface protein that stimulates lymphocyte adhesion and extravasation into tissues49. Other SASP components, such as the pro-angiogenic factor VEGF and the pro-inflammatory cytokines and chemokines CCL5, CXCL1 and IL-6, also facilitate an increase of CD8+ T cell infiltration into tumours and improve the efficacy of checkpoint blockade anti-PD1 therapy49 (Fig. 4b). These data are intriguing and establish a link between therapy-induced SASP, vascular remodelling and improvement of T cell-based immunotherapies. The previous work of the same group using a mouse Kras mutant lung cancer model identified an NK cell surveillance programme without involvement of T cells48. Both studies used mouse models driven by the same KRAS and Trp53 mutations and senescence inducers. However, immune surveillance programmes can be somewhat different between tissues. The lungs are generally rich in NK cells but these cells are scarcer in the pancreas218, which may explain the different involvement of immune cells218. Differences in the SASP produced by different cancer types may further contribute to tissue-specific differences in immune surveillance. Moreover, the effects of pro-senescence therapy on immune responses and anti-PD1 efficacy has been demonstrated in multiple preclinical studies in various cancer types219,220,221,222,223,224 (Table 1).
The presence of neo-antigens is a requirement for recognition of any cancer cell by cytotoxic T cells. This begs the question of which neo-antigens are recognized on senescent cancer cells in cases where anti-PD1 therapy shows synergy with pro-senescence therapy. One possibility is that the epigenetic reprogramming that takes place during senescence leads to re-expression of cancer/testis antigens and upregulation of antigen-presentation genes, including those encoding major histocompatibility complex (MHC) class I molecules, which can further enhance T cell recognition of neo-antigens on senescent cancer cells49,225. An interesting other possibility stems from the finding that mRNA transcripts in senescent cells often retain introns226. Pharmacological perturbation of RNA splicing leads to the generation of neo-antigens that can be recognized by T cells227. Therefore, senescent cells may create neo-antigens by mis-splicing events. The recruitment of immune cells by the SASP may be synergistic with the expression of such neo-antigens, which could be further stimulated by therapies that enhance T cell function such as anti-PD1 antibodies.
In contrast to the activation of immunity, some studies have reported suppression of immune surveillance by senescent cancer cells. IL-6, as one of the SASP factors, can increase the number of tumour-infiltrating MDSCs, which can suppress the function of lymphocytes and result in a tumour-permissive microenvironment29,55 (Fig. 4b). This is similar to the results of other studies, which demonstrated that IL-8 and IL-6 are associated with reduced efficacy of immune checkpoint blockade in patients228,229,230. In a PTEN-null senescent prostate tumour model, the SASP facilitates the activation of the JAK2–STAT3 pathway through the protein tyrosine phosphatase SHP2 to inhibit immune surveillance by establishing an immunosuppressive SASP-enriched tumour microenvironment that contributes to tumour growth231. Moreover, MMPs in the SASP can cleave the NK activation ligand NKG2D on NK cells to avoid immune surveillance232 (Fig. 4a).
Together, these studies indicate that combining senescence induction therapies with immunotherapies can potentially improve therapeutic outcomes. Through their SASP, therapy-induced senescence can impact cancer cells and their surrounding tissues by inducing inflammation, recruiting immune cells, spreading senescence to other cells, and altering immune cell function in both positive and negative ways. Additionally, the SASP may vary among senescent cancer cells depending on the tissue type, how senescence was induced, over time, and in response to hormones and, in turn, this may influence the efficacy of immunotherapies29,43,215. To partially overcome these context-dependency issues of immunotherapies, chimeric antigen receptor (CAR) T cell therapies can be developed with chimeric receptors that recognize cell surface proteins upregulated in senescence. This strategy was shown to be feasible with urokinase-type plasminogen activator receptor (uPAR) as a cell surface membrane protein broadly upregulated during senescence; uPAR-specific CAR T cells effectively eliminated senescent cells in vitro and in a lung cancer mouse model233 (Fig. 4c). There are other cell surface proteins that are upregulated in senescence that might also be targeted by similar immunotherapies, including NOTCH1 (ref.34), NOTCH3 (ref.234), DEP1 (ref.235), B2M235, DPP4 (ref.236), NKG2D237 and ICAM1 (ref.238). CAR T cell-based senolytic therapies hold promise although cost issues may limit their clinical application239. Perhaps, antibody–drug conjugates that combine monoclonal antibodies specific to surface antigens with toxins could potentially be a viable application to target senescent cancer cells that show upregulation of certain membrane proteins. Proof of concept for this approach was recently delivered with a B2M–duocarmycin antibody–drug conjugate, which used a B2M-targeting antibody linked to duocarmycin as a toxin240.
Future perspectives and challenges
It is generally accepted that combination therapies can forestall resistance development. The concept of combination therapies for cancer has been used clinically since 1958 (ref.241). However, toxicity is a major factor limiting the efficacy of drug combinations in oncology. Sequential drug treatment regimens would, at least in principle, allow for a larger array of drugs to be combined as direct combination toxicity is avoided. A one-two punch sequential therapy of pro-senescence therapy followed by senolytic therapy appears feasible in principle as senescence is a stable cellular state that persists after the senescence inducer is withdrawn128,242. As such, pro-senescence therapy is well suited for use in a sequential treatment regimen. Thus, a first unmet need is a lack of drugs that are highly effective in inducing senescence in a high proportion of cancer cells. In addition, such drugs should show selectivity for cancer cells over normal cells as inducing senescence in normal tissues (for example, as occurs with many genotoxic chemotherapies) can cause detrimental side effects177 (Fig. 5).

To achieve better antitumour responses and to inhibit tumour progression mediated by senescence-associated secretory phenotype (SASP) factors, senescence-inducing therapies can be combined with senolytic treatments. Senolysis can also be mediated by recruitment of immune cells by the SASP factors produced by senescent cells. Detection of senescent cells within the patient’s tumour is important to assess the efficacy of pro-senescence therapies. One way this can potentially be achieved is by a PET-CT scan using an 18F-labelled β-galactosidase tracer (18F-β-gal). As potential non-invasive approaches, senescence-associated proteins or metabolites can be detected to measure senescence burden in blood. These technologies provide possibilities to evaluate the efficacy of pro-senescence–senolytic treatment responses and could help guide future clinical trials. NK, natural killer.
Another challenge is the lack of gold standard biomarkers of the senescent state. It is evident that no single marker can unambiguously discriminate between senescence and other growth-arrested states. Several laboratories have developed multi-gene signatures to describe senescence in primary cells. For example, we have recently used a panel of senescent cancer cells to identify the SENCAN classifier for cancer senescence151. In vivo, detection of senescent cells could be achieved by using galacto-conjugated fluorescent nanoparticles, which have been tested in models of chemotherapy-induced senescence193,243. To measure efficiency of senescence induction in tumours of patients on therapy, non-invasive imaging modalities would be ideal. One option might be to use a radioactive β-gal PET tracer for this purpose, although this is still in early development207,244 (Fig. 5). However, β-gal associated detection strategies might be insufficient to define all senescent cells. Cells from some tissue types do not induce SA-β-gal activity when they become senescent245. Macrophages also tend to exhibit increased SA-β-gal activity246,247; therefore, false positives may result from macrophages inside inflamed tumour tissues. Other non-invasive strategies may also detect clearance of senescent cells. For example, oxylipin biosynthesis is significantly increased during senescence248. In particular, the intracellular prostaglandin dihomo-15D-PGJ2, one type of oxylipin, specifically accumulates in senescent cells and is released during senolysis. Importantly, dihomo-15D-PGJ2 could be measured from urine and blood samples from humans248 (Fig. 5). In addition, other markers, such as cleaved uPAR, could potentially be used in non-invasive approaches to detect senescent cancer cells233,249. However, it should be further investigated whether these markers can be used to detect senescent cancer cells in different contexts, such as cells that have diverse genetic backgrounds, arise in different tissue types or have undergone senescence by different agents. In addition, non-invasive detection of SASP factors in plasma could potentially reveal senescent cell burden250. Non-invasive detection of senescent cells should be invaluable in future clinical trials of pro-senescence and senolytic combinations.
A further issue that needs to be solved is the lack of broadly acting senolytic drugs. Most senolytic drugs were developed with the aim to reverse the effects of ageing and were consequently tested mostly on primary cells. Our recent study of the most commonly used senolytic (navitoclax) on a panel of senescent cancer cells showed that it has widely variable activity as a senolytic151. Thus, apart from the lack of an absolute marker of the senescent state, the field is also in need of a druggable and broadly present vulnerability of senescent cancer cells. We have developed an inducible CRISPR–Cas9-based genetic screening platform in which gene-editing is activated only after senescence is induced in cells. This allows for performance of drop-out screens to identify novel senolytic targets on the genome scale. If universal vulnerabilities of senescent cancer cells exist, unbiased genetic screens should allow their identification.
A further challenge that is certainly not unique to senescence-based therapies is the issue of tumour heterogeneity. Tumour heterogeneity leads to variable drug responses that may limit the effectiveness of senescence induction within tumours. Will it be necessary to provoke a senescence response in most inherently heterogeneous cancer cells, or will senescent cells aid in the killing of non-senescent cancer cells through a bystander effect? Senescent cells can spread the senescent phenotype through the SASP to the surrounding non-senescent cells within tumours251, which may sensitize non-senescent cancer cells to senolytic treatments; however, this has not been proven so far. Moreover, such bystander effects could be fortified by the local tumour microenvironment shaped by the SASP of the senescent cells. For instance, it is thought that the clinical efficacy of immunotherapy is, at least in part, caused by the killing of subpopulations of resistant cancer cells through a bystander effect. While the contribution of bystander effects in the context of senescence-inducing therapies are understood only poorly to date, it may represent an opportunity to overcome tumour heterogeneity.
It will be crucial to better understand how the SASP produced by senescent cancer cells impacts the interaction between senescent cancer cells and the immune system. Some early data point towards a synergy between pro-senescence therapy and checkpoint immunotherapy48,49. It is important to keep in mind that there is a publication bias for experiments that yield positive results. It is therefore possible that not all pro-senescence therapies and not all cancer types will benefit from combination with checkpoint immunotherapy. Such notion is supported by the substantial heterogeneity in SASP factors produced by different senescent cells151. Thus, it will be important to understand when a SASP provokes an immune response that can be enhanced by checkpoint immunotherapy and when it does not. It may become possible to use so called senomorphic drugs such as the NF-κB-inhibiting drugs apigenin and kaempferol or the mTOR inhibitor rapamycin212,252, which can modify the SASP of senescent cells to become more responsive to checkpoint therapy clearance253.
Finally, ablation of senescent normal cells by senolytic therapy in aged individuals needs to be carefully evaluated. In the elderly, senescent cells can be a high percentage of the net number of cells in some tissues and this might conceptually jeopardize tissue structural integrity or affect vascular endothelial cells, leading to blood–tissue barrier disorder that may, in turn, lead to liver and perivascular tissue fibrosis and health collapse254,255. This potential issue highlights the need for cancer-selective senolytics to be developed.
Thus, while many questions currently remain unanswered, key advantages of senescence-based therapies, such as sequential treatment opportunities and attraction of immune cells by senescent cells, warrant further investigation of this approach.
References
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961). This was the first study to define cellular senescence.
Fumagalli, M. et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 (2012).
Deng, Y. & Chang, S. Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab. Invest. 87, 1071–1076 (2007).
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a). Cell 88, 593–602 (1997).
Moiseeva, O., Bourdeau, V., Roux, A., Deschênes-Simard, X. & Ferbeyre, G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 29, 4495–4507 (2009).
Pluquet, O., Pourtier, A. & Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 308, 415–425 (2015).
Dörr, J. R. et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501, 421–425 (2013).
Carroll, B. et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J. Cell Biol. 216, 1949–1957 (2017).
Chang, B. D. et al. Role of p53 and p21(waf1/cip1) in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 18, 4808–4818 (1999).
Nehme, J., Borghesan, M., Mackedenski, S., Bird, T. G. & Demaria, M. Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell 19, e13237 (2020).
Lee, S. et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 599, 283–289 (2021).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019). This review defines key features of senescence and recommends their use as biomarkers.
Itahana, K., Campisi, J. & Dimri, G. P. Mechanisms of cellular senescence in human and mouse cells. Biogerontology 5, 1–10 (2004).
Hernandez-Segura, A., Nehme, J. & Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 (2018).
Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705 (2013).
Collado, M. & Serrano, M. Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer 10, 51–57 (2010).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011 (2017).
Beauséjour, C. M. et al. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 22, 4212–4222 (2003).
Dirac, A. M. G. & Bernards, R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J. Biol. Chem. 278, 11731–11734 (2003).
Lee, S. & Schmitt, C. A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 21, 94–101 (2019).
Milanovic, M. et al. Senescence-associated reprogramming promotes cancer stemness. Nature 553, 96–100 (2018).
Sanders, Y. Y. et al. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 1, 8–16 (2013).
Ryu, S. J., Oh, Y. S. & Park, S. C. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 14, 1020–1028 (2007).
Lee, B. Y. et al. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 5, 187–195 (2006).
Kopp, H. G., Hooper, A. T., Shmelkov, S. V. & Rafii, S. β-galactosidase staining on bone marrow. The osteoclast pitfall. Histol. Histopathol. 22, 971–976 (2007).
Coppé, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 (2008).
Coppé, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 5, 99–118 (2010). This review summarizes the beneficial and detrimental effects of the SASP.
Shelton, D. N., Chang, E., Whittier, P. S., Choi, D. & Funk, W. D. Microarray analysis of replicative senescence. Curr. Biol. 9, 939–945 (1999).
Faget, D. V., Ren, Q. & Stewart, S. A. Unmasking senescence: context-dependent effects of SASP in cancer. Nat. Rev. Cancer 19, 439–453 (2019). This review discusses how SASP production and functions can differ between cancer types.
Yang, H., Wang, H., Ren, U., Chen, Q. & Chena, Z. J. CGAS is essential for cellular senescence. Proc. Natl Acad. Sci. USA 114, E4612–E4620 (2017).
Kang, C. et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, 6255 (2015).
Huggins, C. J. et al. C/EBPγ suppresses senescence and inflammatory gene expression by heterodimerizing with C/EBPβ. Mol. Cell. Biol. 33, 3242–3258 (2013).
Teo, Y. V. et al. Notch signaling mediates secondary senescence. Cell Rep. 27, 997–1007.e5 (2019).
Hoare, M. et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 18, 979–992 (2016).
Kuilman, T. et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008).
Madani, A. Y. et al. Signal transducer and activator of transcription 3 (Stat3) suppresses stat1/interferon signaling pathway and inflammation in senescent preadipocytes. Antioxidants 10, 334 (2021).
Iwasa, H., Han, J. & Ishikawa, F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes. Cell 8, 131–144 (2003).
Laberge, R. M. et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 17, 1049–1061 (2015).
Ritschka, B. et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 31, 172–183 (2017).
Acosta, J. C. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990 (2013).
Di Mitri, D. & Alimonti, A. Non-cell-autonomous regulation of cellular senescence in cancer. Trends Cell Biol. 26, 215–226 (2016).
Wang, B., Kohli, J. & Demaria, M. Senescent cells in cancer therapy: friends or foes? Trends Cancer 6, 838–857 (2020).
Hernandez-Segura, A. et al. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27, 2652–2660.e4 (2017). This review summarizes the expression of different SASP proteins as a function of tissue context.
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Saleh, T. et al. Therapy-induced senescence: an “old” friend becomes the enemy. Cancers 12, 822 (2020).
Nardella, C., Clohessy, J. G., Alimonti, A. & Pandolfi, P. P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 11, 503–511 (2011).
Wyld, L. et al. Senescence and cancer: a review of clinical implications of senescence and senotherapies. Cancers 12, 2134 (2020).
Ruscetti, M. et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362, 1416–1422 (2018). This study highlights NK cell-mediated senolysis.
Ruscetti, M. et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 181, 424–441.e21 (2020). This study highlights T cell-mediated senolysis.
Xue, W. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007).
Kang, T. W. et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551 (2011).
Krizhanovsky, V. et al. Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667 (2008).
Lujambio, A. et al. Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460 (2013).
Ruhland, M. K. & Alspach, E. Senescence and immunoregulation in the tumor microenvironment. Front. Cell Dev. Biol. 9, 754069 (2021).
Ruhland, M. K. et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 7, 11762 (2016).
Watanabe, S., Kawamoto, S., Ohtani, N. & Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 108, 563–569 (2017).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009).
Yang, L., Fang, J. & Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 3, 17049 (2017).
Childs, B. G., Durik, M., Baker, D. J. & Van Deursen, J. M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 21, 1424–1435 (2015).
Baker, D. J. et al. Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011). This study provides evidence that senolysis has anti-ageing effects.
Baker, D. J. et al. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Van Deursen, J. M. The role of senescent cells in ageing. Nature 509, 439–446 (2014).
Cancer Research UK. Cancer Grand Challenges: Senescence https://cancergrandchallenges.org/challenges/senescence (2021).
Muñoz-Espín, D. & Serrano, M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 (2014).
Sharpless, N. E. & Sherr, C. J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 15, 397–408 (2015).
Braig, M. et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436, 660–665 (2005).
Chen, Z. et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730 (2005).
Michaloglou, C. et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720–724 (2005). This study provided evidence of senescence as a barrier to cancer development.
Collado, M. et al. Tumour biology: senescence in premalignant tumours. Nature 436, 642 (2005).
Denchi, E. L., Attwooll, C., Pasini, D. & Helin, K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol. Cell. Biol. 25, 2660–2672 (2005).
Halazonetis, T. D., Gorgoulis, V. G. & Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 (2008).
Alimonti, A. et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Invest. 120, 681–693 (2010).
Coppé, J. P. et al. Tumor suppressor and aging biomarker p16 INK4a induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 286, 36396–36403 (2011).
Wu, C. H. et al. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Natl Acad. Sci. USA 104, 13028–13033 (2007).
Zhang, D. Y., Wang, H. J. & Tan, Y. Z. Wnt/β-catenin signaling induces the aging of Mesenchymal stem cells through the DNA damage response and the P53/P21 pathway. PLoS One 6, e21397 (2011).
Sarkisian, C. J. et al. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol. 9, 493–505 (2007).
Sharpless, N. E., Ramsey, M. R., Balasubramanian, P., Castrillon, D. H. & DePinho, R. A. The differential impact of p16INK4a or p19ARF deficiency on cell growth and tumorigenesis. Oncogene 23, 379–385 (2004).
Carrire, C. et al. Deletion of Rb accelerates pancreatic carcinogenesis by oncogenic kras and impairs senescence in premalignant lesions. Gastroenterology 141, 1091–1101 (2011).
Qiu, W. et al. Disruption of p16 and activation of kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget 2, 862–873 (2011).
Morton, J. P. et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl Acad. Sci. USA 107, 246–251 (2010).
Ewald, J. A., Desotelle, J. A., Wilding, G. & Jarrard, D. F. Therapy-induced senescence in cancer. J. Natl Cancer Inst. 102, 1536–1546 (2010).
Chang, B. D. et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 59, 3761–3767 (1999).
Gewirtz, D. A., Holt, S. E. & Elmore, L. W. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochem. Pharmacol. 76, 947–957 (2008).
Schmitt, C. A. et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109, 335–346 (2002).
Roninson, I. B. Tumor cell senescence in cancer treatment. Cancer Res. 63, 2705–2715 (2003).
Lee, M. & Lee, J. S. Exploiting tumor cell senescence in anticancer therapy. BMB Rep. 47, 51–59 (2014).
Petrova, N. V., Velichko, A. K., Razin, S. V. & Kantidze, O. L. Small molecule compounds that induce cellular senescence. Aging Cell 15, 999–1017 (2016).
Maréchal, A. & Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 5, a012716 (2013).
Lakin, N. D. & Jackson, S. P. Regulation of p53 in response to DNA damage. Oncogene 18, 7644–7655 (1999).
Nitiss, J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350 (2009).
Pommier, Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6, 789–802 (2006).
Kortlever, R. M., Higgins, P. J. & Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 8, 878–884 (2006).
Rottenberg, S., Disler, C. & Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 21, 37–50 (2021).
Dasari, S. & Tchounwou, P. B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 740, 364–378 (2014).
Hall, A. G. & Tilby, M. J. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 6, 163–173 (1992).
Perez, E. A. Microtubule inhibitors: differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Therapeutics 8, 2086–2095 (2009).
Klein, L. E., Freeze, B. S., Smith, A. B. & Horwitz, S. B. The microtubule stabilizing agent discodermolide is a potent inducer of accelerated cell senescence. Cell Cycle 4, 501–507 (2005).
Dabrowska, M., Skoneczny, M., Uram, L. & Rode, W. Methotrexate-induced senescence of human colon cancer cells depends on p53 acetylation, but not genomic aberrations. Anticancer. Drugs 30, 374–382 (2019).
Song, Y., Baba, T. & Mukaida, N. Gemcitabine induces cell senescence in human pancreatic cancer cell lines. Biochem. Biophys. Res. Commun. 477, 515–519 (2016).
Kaufmann, S. H. & Earnshaw, W. C. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 256, 42–49 (2000).
Sabin, R. J. & Anderson, R. M. Cellular senescence — its role in cancer and the response to ionizing radiation. Genome Integr. 2, 7 (2011).
Fei, P. & El-Deiry, W. S. P53 and radiation responses. Oncogene 22, 5774–5783 (2003).
Carli, P. M. et al. Increase therapy-related leukemia secondary to breast cancer. Leukemia 14, 1014–1017 (2000).
Li, M., You, L., Xue, J. & Lu, Y. Ionizing radiation-induced cellular senescence in normal, non-transformed cells and the involved DNA damage response: a mini review. Front. Pharmacol. 9, 522 (2018).
Canavese, M., Santo, L. & Raje, N. Cyclin dependent kinases in cancer: potential for therapeutic intervention. Cancer Biol. Ther. 13, 451–457 (2012).
Malumbres, M. & Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 (2009).
Hamilton, E. & Infante, J. R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 45, 129–138 (2016).
Yoshida, A., Lee, E. K. & Diehl, J. A. Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 76, 2990–3002 (2016).
Klein, M. E. et al. PDLIM7 and CDH18 regulate the turnover of MDM2 during CDK4/6 inhibitor therapy-induced senescence. Oncogene 37, 5066–5078 (2018).
Rader, J. et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 19, 6173–6182 (2013).
Leonard, J. P. et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 119, 4597–4607 (2012).
Guha, M. Blockbuster dreams for Pfizer’s CDK inhibitor. Nat. Biotechnol. 31, 187 (2013).
Dickson, M. A. et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J. Clin. Oncol. 31, 2024–2028 (2013).
Campaner, S. et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 12, 54–59 (2010).
Pandey, K. et al. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: a review. Int. J. Cancer 145, 1179–1188 (2019).
McCartney, A. et al. Mechanisms of resistance to CDK4/6 inhibitors: potential implications and biomarkers for clinical practice. Front. Oncol. 9, 666 (2019).
Pandey, K. et al. Combined cdk2 and cdk4/6 inhibition overcomes palbociclib resistance in breast cancer by enhancing senescence. Cancers 12, 3566 (2020).
Herrera-Abreu, M. T. et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 76, 2301–2313 (2016).
Kumarasamy, V., Vail, P., Nambiar, R., Witkiewicz, A. K. & Knudsen, E. S. Functional determinants of cell cycle plasticity and sensitivity to CDK4/6 inhibition. Cancer Res. 81, 1347–1360 (2021).
Gong, X. et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell 32, 761–776.e6 (2017).
Freeman-Cook, K. D. et al. Discovery of PF-06873600, a CDK2/4/6 inhibitor for the treatment of cancer. J. Med. Chem. 64, 9056–9077 (2021).
Seki, A., Coppinger, J. A., Jang, C. Y., Yates, J. R. & Fang, G. Bora and the kinase Aurora A cooperatively activate the kinase Plk1 and control mitotic entry. Science 320, 1655–1658 (2008).
Das, K. et al. Aurora-A expression, hormone receptor status and clinical outcome in hormone related cancers. Pathology 42, 540–546 (2010).
Murga-Zamalloa, C., Inamdar, K. V. & Wilcox, R. A. The role of aurora A and polo-like kinases in high-risk lymphomas. Blood Adv. 3, 1778–1787 (2019).
Driscoll, D. L. et al. Plk1 inhibition causes post-mitotic DNA damage and senescence in a range of human tumor cell lines. PLoS One 9, e111060 (2014).
Wang, L. X. et al. Aurora A kinase inhibitor AKI603 induces cellular senescence in chronic myeloid leukemia cells harboring T315I mutation. Sci. Rep. 6, 35533 (2016).
Huck, J. J. et al. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol. Cancer Res. 8, 373–384 (2010).
Wang, L. et al. High-throughput functional genetic and compound screens identify targets for senescence induction in cancer. Cell Rep. 21, 773–783 (2017). This study demonstrates that sequential use of one-two punch pro-senescence and senolytic therapy can be used to kill cancer cells.
Liu, Y. et al. Combining an aurora kinase inhibitor and a death receptor ligand/agonist antibody triggers apoptosis in melanoma cells and prevents tumor growth in preclinical mouse models. Clin. Cancer Res. 21, 5338–5348 (2015).
Kim, H. J., Cho, J. H. & Kim, J. R. Downregulation of polo-like kinase 1 induces cellular senescence in human primary cells through a p53-dependent pathway. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 68, 1145–1156 (2013).
Capparelli, C. et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, ‘fueling’ tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 11, 3599–3610 (2012).
Qin, S., Schulte, B. A. & Wang, G. Y. Role of senescence induction in cancer treatment. World J. Clin. Oncol. 9, 180–187 (2018).
Falchook, G. S., Bastida, C. C. & Kurzrock, R. Aurora kinase inhibitors in oncology clinical trials: current state of the progress. Semin. Oncol. 42, 832–848 (2015).
Wang, C. et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 574, 268–272 (2019).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Mengual Gomez, D. L., Armando, R. G., Cerrudo, C. S., Ghiringhelli, P. D. & Gomez, D. E. Telomerase as a cancer target. Development of new molecules. Curr. Top. Med. Chem. 16, 2432–2440 (2016).
Pascolo, E. et al. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J. Biol. Chem. 277, 15566–15572 (2002).
Burchett, K. M., Yan, Y. & Ouellette, M. M. Telomerase inhibitor imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PLoS One 9, e85155 (2014).
Amatori, S., Bagaloni, I., Viti, D. & Fanelli, M. Premature senescence induced by DNA demethylating agent (Decitabine) as therapeutic option for malignant pleural mesothelioma. Lung Cancer 71, 113–115 (2011).
Elknerova, K. et al. Epigenetic modulation of gene expression of human leukemia cell lines - induction of cell death and senescence. Neoplasma 58, 35–44 (2011).
Kaletsch, A. et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin. Epigenetics 10, 100 (2018).
Martins, C. P., Brown-Swigart, L. & Evan, G. I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 127, 1323–1334 (2006).
Ventura, A. et al. Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665 (2007).
Vu, B. et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4, 466–469 (2013).
Efeyan, A. et al. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. 67, 7350–7357 (2007).
Vassilev, L. T. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017).
Baar, M. P. et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147.e16 (2017).
Augello, G. et al. A PTEN inhibitor displays preclinical activity against hepatocarcinoma cells. Cell Cycle 15, 573–583 (2016).
Lee, J. J. et al. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 18, 666–677 (2011).
Jochems, F. et al. The Cancer SENESCopedia: a delineation of cancer cell senescence. Cell Rep. 3, 109441 (2021).
Orjalo, A. V., Bhaumik, D., Gengler, B. K., Scott, G. K. & Campisi, J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl Acad. Sci. USA 106, 17031–17036 (2009).
Acosta, J. C. et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 (2008).
Eggert, T. et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30, 533–547 (2016).
Braumüller, H. et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 494, 361–365 (2013).
Sieben, C. J., Sturmlechner, I., van de Sluis, B. & van Deursen, J. M. Two-step senescence-focused cancer therapies. Trends Cell Biol. 28, 723–737 (2018).
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P. Y. & Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl Acad. Sci. USA 98, 12072–12077 (2001).
Liu, D. & Hornsby, P. J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67, 3117–3126 (2007).
Khalilgharibi, N. & Mao, Y. To form and function: on the role of basement membrane mechanics in tissue development, homeostasis and disease. Open Biol. 11, 200360 (2021).
Donnini, S. et al. Pyrazolo-pyrimidine-derived c-Src inhibitor reduces angiogenesis and survival of squamous carcinoma cells by suppressing vascular endothelial growth factor production and signaling. Int. J. Cancer 120, 995–1004 (2007).
Yang, G. et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl Acad. Sci. USA 103, 16472–16477 (2006).
Lasry, A. & Ben-Neriah, Y. Senescence-associated inflammatory responses: aging and cancer perspectives. Trends Immunol. 36, 217–228 (2015).
Wang, L. et al. Activation of IL-8 via PI3K/AKT-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol. Ther. 16, 1220–1230 (2015).
Goulet, C. R. et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 19, 137 (2019).
Fisher, D. T., Appenheimer, M. M. & Evans, S. S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 26, 38–47 (2014).
Waugh, D. J. J. & Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 14, 6735–6741 (2008).
Di Mitri, D. et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 515, 134–137 (2014).
Lian, J., Yue, Y., Yu, W. & Zhang, Y. Immunosenescence: a key player in cancer development. J. Hematol. Oncol. 13, 155 (2020).
Triana-Martínez, F., Loza, M. I. & Domínguez, E. Beyond tumor suppression: senescence in cancer stemness and tumor dormancy. Cells 9, 346 (2020).
Roberson, R. S., Kussick, S. J., Vallieres, E., Chen, S. Y. J. & Wu, D. Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 65, 2795–2803 (2005).
Patel, P. L., Suram, A., Mirani, N., Bischof, O. & Herbig, U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc. Natl Acad. Sci. USA 113, E5024–E5033 (2016).
Gonzalez-Meljem, J. M. et al. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat. Commun. 8, 1819 (2017).
Takasugi, M. et al. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 8, 15729 (2017).
Lehmann, B. D. et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 68, 7864–7871 (2008).
Jakhar, R. & Crasta, K. Exosomes as emerging pro-tumorigenic mediators of the senescence-associated secretory phenotype. Int. J. Mol. Sci. 20, 2547 (2019).
Pribluda, A. et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 24, 242–256 (2013).
Demaria, M. et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 (2017). This study provides evidence that senescent normal cells contribute to the side-effects of chemotherapies.
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105 (2021).
Carpenter, V. J., Saleh, T. & Gewirtz, D. A. Senolytics for cancer therapy: is all that glitters really gold? Cancers 13, 723 (2021).
Zhu, Y. et al. The achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Childs, B. G. et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov. 16, 718–735 (2017). This review summarises senescence-associated diseases and the therapeutic options.
Yosef, R. et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 7, 11190 (2016).
Zhu, Y. et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 (2016).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016). This study identifies navitoclax as a senolytic agent.
Saleh, T. et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-XL–BAX interaction. Mol. Oncol. 14, 2504–2519 (2020).
Hann, C. L. et al. Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer. Cancer Res. 68, 2321–2328 (2008).
Fleury, H. et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 10, 2556 (2019).
Wu, H. et al. Ionizing radiation sensitizes breast cancer cells to Bcl-2 inhibitor, ABT-737, through regulating Mcl-1. Radiat. Res. 182, 618–625 (2014).
Kipps, T. J. et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk. Lymphoma 56, 2826–2833 (2015).
Kaefer, A. et al. Mechanism-based pharmacokinetic/pharmacodynamic meta-analysis of navitoclax (ABT-263) induced thrombocytopenia. Cancer Chemother. Pharmacol. 74, 593–602 (2014).
Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 (2013).
González-Gualda, E. et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 19, e13142 (2020).
Muñoz-Espín, D. et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 10, e9355 (2018).
Estepa-Fernández, A. et al. Senolysis reduces senescence in veins and cancer cell migration. Adv. Ther. 4, 2100149 (2021).
Galiana, I. et al. Preclinical antitumor efficacy of senescence-inducing chemotherapy combined with a nanosenolytic. J. Control. Rel. 323, 624–634 (2020).
He, Y. et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun. 11, 1996 (2020).
Raina, K. et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl Acad. Sci. USA 113, 7124–7129 (2016).
Wakita, M. et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 11, 1935 (2020).
Lu, J. et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22, 755–763 (2015).
Kirkland, J. L. & Tchkonia, T. Senolytic drugs: from discovery to translation. J. Intern. Med. 288, 518–536 (2020).
Piscitani, L., Sirolli, V., Di Liberato, L., Morroni, M. & Bonomini, M. Nephrotoxicity associated with novel anticancer agents (Aflibercept, dasatinib, nivolumab): case series and nephrological considerations. Int. J. Mol. Sci. 21, 4878 (2020).
Paez-Ribes, M., González-Gualda, E., Doherty, G. J. & Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 11, e10234 (2019).
Justice, J. N. et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 (2019).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018).
Kovacovicova, K. et al. Senolytic cocktail dasatinib+quercetin (D+Q) does not enhance the efficacy of senescence-inducing chemotherapy in liver cancer. Front. Oncol. 8, 459 (2018).
van Deursen, J. M. Senolytic therapies for healthy longevity. Science 364, 636–637 (2019).
Dolgin, E. Send in the senolytics. Nat. Biotechnol. 38, 1371–1377 (2020).
Levine, A. J. & Oren, M. The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer 9, 749–758 (2009).
Efeyan, A. & Serrano, M. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle 6, 1006–1010 (2007).
Guerrero, A. et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 1, 1074–1088 (2019).
Triana-Martínez, F. et al. Identification and characterization of cardiac glycosides as senolytic compounds. Nat. Commun. 10, 4731 (2019).
Herranz, N. et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 17, 1205–1217 (2015).
Kucheryavenko, O., Nelson, G., von Zglinicki, T., Korolchuk, V. I. & Carroll, B. The mTORC1-autophagy pathway is a target for senescent cell elimination. Biogerontology 20, 331–335 (2019).
Fung, A. S., Wu, L. & Tannock, I. F. Concurrent and sequential administration of chemotherapy and the mammalian target of rapamycin inhibitor temsirolimus in human cancer cells and xenografts. Clin. Cancer Res. 15, 5389–5395 (2009).
Prata, L. G. P. L., Ovsyannikova, I. G., Tchkonia, T. & Kirkland, J. L. Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin. Immunol. 40, 101275 (2018).
Iannello, A., Thompson, T. W., Ardolino, M., Lowe, S. W. & Raulet, D. H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 210, 2057–2069 (2013).
Soriani, A. et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 113, 3503–3511 (2009).
Salmon, H., Remark, R., Gnjatic, S. & Merad, M. Host tissue determinants of tumour immunity. Nat. Rev. Cancer 19, 215–227 (2019).
Goel, S. et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475 (2017).
Zhang, J. et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95 (2018).
Deng, J. et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8, 216–233 (2018).
Jerby-Arnon, L. et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e24 (2018).
Knudsen, E. S. et al. Targeting dual signalling pathways in concert with immune checkpoints for the treatment of pancreatic cancer. Gut 70, 127–138 (2021).
Brenner, E. et al. Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat. Commun. 11, 1335 (2020).
Whitehurst, A. W. Cause and consequence of cancer/testis antigen activation in cancer. Annu. Rev. Pharmacol. Toxicol. 54, 251–272 (2014).
Yao, J. et al. Prevalent intron retention fine-tunes gene expression and contributes to cellular senescence. Aging Cell 19, e13276 (2020).
Lu, S. X. et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell 184, 4032–4047.e31 (2021).
Schalper, K. A. et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 26, 688–692 (2020).
Yuen, K. C. et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 26, 693–698 (2020).
Laino, A. S. et al. Serum interleukin-6 and C-reactive protein are associated with survival in melanoma patients receiving immune checkpoint inhibition. J. Immunother. Cancer 8, e000842 (2020).
Toso, A. et al. Enhancing chemotherapy efficacy in pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 9, 75–89 (2014).
Muñoz, D. P. et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight 4, e124716 (2019).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020). This study highlights the feasibility of targeting senescent cells with CAR T cells.
Cui, H., Kong, Y., Xu, M. & Zhang, H. Notch3 functions as a tumor suppressor by controlling cellular senescence. Cancer Res. 73, 3451–3459 (2013).
Althubiti, M. et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 5, e124716 (2014).
Kim, K. M. et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 31, 1529–1534 (2017).
Sagiv, A. et al. NKG2D ligands mediate immunosurveillance of senescent cells. Aging 8, 328–344 (2016).
Gorgoulis, V. G. et al. p53-Dependent ICAM-1 overexpression in senescent human cells identified in atherosclerotic lesions. Lab. Investig. 85, 502–511 (2005).
Lyman, G. H., Nguyen, A., Snyder, S., Gitlin, M. & Chung, K. C. Economic evaluation of chimeric antigen receptor T-cell therapy by site of care among patients with relapsed or refractory large B-cell lymphoma. JAMA Netw. Open 3, e202072 (2020).
Ekpenyong-Akiba, A. E., Poblocka, M. & Macip, S. In Senolytics in Disease, Ageing and Longevity (Springer, 2020).
Frei, E. et al. A comparative study of two regimens of combination chemotherapy in acute leukemia. Blood 13, 1126–1148 (1958).
Wang, L. & Bernards, R. Taking advantage of drug resistance, a new approach in the war on cancer. Front. Med. 12, 490–495 (2018).
Lozano-Torres, B. et al. An OFF-ON two-photon fluorescent probe for tracking cell senescence in vivo. J. Am. Chem. Soc. 139, 8808–8811 (2017).
Krueger, M. A. et al. Abstract 1146: [18F]FPyGal: a novel ß-galactosidase specific PET tracer for in vivo imaging of tumor senescence. 1146 (AACR Annual Meeting, 2019).
Kuilman, T., Michaloglou, C., Mooi, W. J. & Peeper, D. S. The essence of senescence. Genes Dev. 24, 2463–2479 (2010).
Hall, B. M. et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 9, 1867–1884 (2017).
de Mera-Rodríguez, J. A. et al. Is senescence-associated β-galactosidase a reliable in vivo marker of cellular senescence during embryonic development? Front Cell Dev. Biol. 9, 623175 (2021).
Wiley, C. D. et al. Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis. Cell Metab. 33, 1124–1136.e5 (2021).
Schafer, M. J. et al. The senescence-associated secretome as an indicator of age and medical risk. JCI Insight 5, e133668 (2020).
Basisty, N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020).
Nelson, G. et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349 (2012).
Tilstra, J. S. et al. NF-κB inhibition delays DNA damage — induced senescence and aging in mice. J. Clin. Invest. 122, 2601–2612 (2012).
Niedernhofer, L. J. & Robbins, P. D. Senotherapeutics for healthy ageing. Nat. Rev. Drug Discov. 17, 377 (2018).
Grosse, L. et al. Defined p16High senescent cell types are indispensable for mouse healthspan. Cell Metab. 32, 87–99.e6 (2020).
Omori, S. et al. Generation of a p16 reporter mouse and its use to characterize and target p16high cells in vivo. Cell Metab. 32, 814–828.e6 (2020).
Puglisi, M. et al. A phase I study of the safety, pharmacokinetics and efficacy of navitoclax plus docetaxel in patients with advanced solid tumors. Futur. Oncol. 17, 2747–2758 (2021).
Cleary, J. M. et al. A phase I clinical trial of navitoclax, a targeted high-affinity Bcl-2 family inhibitor, in combination with gemcitabine in patients with solid tumors. Invest. N. Drugs 32, 937–945 (2014).
Brachet, P. E. et al. A GINECO phase II study of Navitoclax (ABT 263) in women with platinum resistant/refractory recurrent ovarian cancer (ROC). Ann. Oncol. 28, v347 (2017).
Witzens-Harig, M., Memmer, M. L., Dreyling, M. & Hess, G. A phase I/II trial to evaluate the safety, feasibility and activity of salvage therapy consisting of the mTOR inhibitor temsirolimus added to standard therapy of rituximab and DHAP for the treatment of patients with relapsed or refractory diffuse large cell B-cell lymphoma — the STORM trial. BMC Cancer 13, 308 (2013).
Trivedi, N. D. et al. A phase I trial of the mTOR inhibitor temsirolimus in combination with capecitabine in patients with advanced malignancies. Cancer Med. 10, 1944–1954 (2021).
Mayer, E. L. et al. Palbociclib after CDK and endocrine therapy (PACE): a randomized phase II study of fulvestrant, palbociclib, and avelumab for endocrine pre-treated ER+/HER2– metastatic breast cancer. J. Clin. Oncol. 36, TPS1104 (2018).
Lord, C. J., Tutt, A. N. J. & Ashworth, A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 66, 455–470 (2015).
Lopez, J. S. & Banerji, U. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 14, 57–66 (2017).
Wang, L. et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell 173, 1413–1425.e14 (2018).
Acknowledgements
We thank members of the Bernards laboratory and Roderick Beijersbergen for helpful discussion and thoughtful feedback. This work was supported by grant 787925 from the European Research Council (R.B.) and 19-051-ASP from The Mark Foundation (R.B.).
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, contributed substantially to discussion of the content, and wrote the article. L.W. and R.B. reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
R.B. is founder and shareholder of a company that exploits senescence for cancer therapy: Oncosence. R.B. and L.W. are part time employees of Oncosence. L.L. declares no competing interests.
Peer review
Peer review information
Nature Reviews Cancer thanks Jesus Gil, Daniel Munoz-Espin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related Links
ClinicalTrials.gov: https://clinicaltrials.gov/
Glossary
- Senescence-associated β-galactosidase
-
(SA-β-gal). Acidic β-galactosidase activity that occurs in senescent cells; used as a senescence biomarker.
- Myeloid-derived suppressor cells
-
(MDSCs). Myeloid cells that specifically inhibit killing of cells by natural killer cells and CD8+ T cells.
- Proteolysis-targeting chimaera (PROTAC) drug
-
A molecule that is capable of removing specific proteins by targeted proteolysis.
- Immune checkpoint
-
An immune response that prevents the immune system from attacking cells.
- Cancer/testis antigens
-
Antigens expressed primarily in male germ cells and various malignancies.
- Chimeric antigen receptor (CAR) T cell
-
T cells expressing artificial receptors, which can recognize certain proteins on cancer cells.
- Drop-out screens
-
Loss-of-function genetic screens that identify genes whose depletion leads to lethality.
- Senomorphic drugs
-
Drugs that modify the senescence-associated secretory phenotype produced by senescent cells.
Rights and permissions
About this article

Cite this article
Wang, L., Lankhorst, L. & Bernards, R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer 22, 340–355 (2022). https://doi.org/10.1038/s41568-022-00450-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41568-022-00450-9
This article is cited by
-
Therapy-induced senescent tumor cell-derived extracellular vesicles promote colorectal cancer progression through SERPINE1-mediated NF-κB p65 nuclear translocation
Molecular Cancer (2024)
-
The senescence journey in cancer immunoediting
Molecular Cancer (2024)
-
Tobacco smoke condensate-induced senescence in endothelial cells was ameliorated by colchicine treatment via suppression of NF-κB and MAPKs P38 and ERK pathways activation
Cell Communication and Signaling (2024)
-
RAB27B-regulated exosomes mediate LSC maintenance via resistance to senescence and crosstalk with the microenvironment
Leukemia (2024)
-
Cell senescence in liver diseases: pathological mechanism and theranostic opportunity
Nature Reviews Gastroenterology & Hepatology (2024)
