
Abstract
Microbial rhodopsins are photoreceptor proteins that convert light into biological signals or energy. Proteins of the xanthorhodopsin family are common in eukaryotic photosynthetic plankton including diatoms. However, their biological role in these organisms remains elusive. Here we report on a xanthorhodopsin variant (FcR1) isolated from the polar diatom Fragilariopsis cylindrus. Applying a combination of biophysical, biochemical and reverse genetics approaches, we demonstrate that FcR1 is a plastid-localized proton pump which binds the chromophore retinal and is activated by green light. Enhanced growth of a Thalassiora pseudonana gain-of-function mutant expressing FcR1 under iron limitation shows that the xanthorhodopsin proton pump supports growth when chlorophyll-based photosynthesis is iron-limited. The abundance of xanthorhodopsin transcripts in natural diatom communities of the surface oceans is anticorrelated with the availability of dissolved iron. Thus, we propose that these proton pumps convey a fitness advantage in regions where phytoplankton growth is limited by the availability of dissolved iron.
Similar content being viewed by others

Main
Microorganisms can convert sunlight into biological signals or metabolic energy using rhodopsins1,2,3,4,5. Microbial (type 1) rhodopsins are integral membrane photoreceptor proteins with seven or eight transmembrane helices6,7,8 that bind a β-carotene-derived retinal chromophore1,2,3,9. They are an abundant family of light-harvesting proteins10 and comprise a large diversity that is found in all domains of life5,11. Most microbial rhodopsins appear to bind all-trans-retinal to absorb light in the wavelengths of ~400–600 nm and catalyse a wide range of different biological functions3,6,12,13,14,15.
Since the discovery of the light-driven proton pumps bacteriorhodopsin in the archaeon Halobacterium salinarum16 and proteorhodopsin from an uncultured marine Gammaproteobacterium17, genomic and metagenomic analyses have revealed a wealth of rhodopsins from microbes ranging from Asgard archaea18 to eukaryotic microorganisms, including all major algal groups and even giant viruses19,20,21,22,23. Consequently, microbial rhodopsins are proposed to be major marine light capturers and estimates suggest they may absorb as much light as chlorophyll-based photosynthesis in the sea24,25.
The first eukaryotic microbial rhodopsins with proven proton-pumping functions were discovered in the fungal pathogen Leptosphaeria maculans26 and in the marine green alga Acetabularia acetabulum27 and ascribed to fungal and algal proton pump families with bacteriorhodopsin-like DTD motifs, respectively2. Subsequent work identified a substantial number of distantly related proton pumps from the xanthorhodopsin family in other marine algae such as dinoflagellates20,28,29,30, haptophytes19 and diatoms19,31,32,33. Moreover, recent structural characterization of fungal rhodopsins suggests that archaeal and eukaryotic proton pumps share a common ancestor34. However, their biological role in eukaryotes, including phytoplankton, has remained elusive.
Over large parts of the ocean, growth of eukaryotic phytoplankton is regulated by the micronutrient iron35, with the majority of the cellular iron demand being associated with photosynthesis36. To cope with iron limitation, phytoplankton have evolved a range of iron-sparing mechanisms, including replacement of iron in various catalysts37,38, acquisition of multiple forms of iron and modification of their photosynthetic architecture39,40,41. Resolving these mechanisms and their prevalence across different oceanic regions is critical to understanding how changes in iron availability impact phytoplankton growth and therefore ecosystem productivity.
Iron limitation has been proposed to induce expression of xanthorhodopsins, on the basis of preliminary results from gene expression studies using cultured phytoplankton and metatranscriptomes from natural communities42,19,32,43. Chlorophyll-based photosynthesis is limited by iron availability, thus it was suggested that xanthorhodopsins provide a selective advantage under these conditions44. Accordingly, rhodopsin proton pumps identified in diatoms have been proposed to play a light-harvesting role leading to the production of adenosine triphosphate (ATP) when iron availability limits chlorophyll-based photosynthesis32,43. Here we critically examine this hypothesis by applying an integrative approach to study the function of FcR1, a xanthorhodopsin gene copy variant from the cold-adapted diatom Fragilariopsis cylindrus. Expression in Xenopus oocytes allowed us to investigate electrophysiological characteristics of FcR1, while recombinant expression of FcR1 in the heterologous diatom host Thalassiosira pseudonana, which does not encode rhodopsin, coupled with bioassay experiments provided evidence that xanthorhodopsins support diatom growth under iron limitation. Furthermore, relative expression of xanthorhodopsin-encoding genes from surface ocean eukaryotic phytoplankton communities was found to correlate with a nutrient limitation index indicating iron limitation. Taken together, our results suggest that xanthorhodopsins support primary productivity contributions of algae in ~35% of the surface ocean where phytoplankton growth is limited by dissolved iron availability.
Results
Xanthorhodopsin gene copy variants in F. cylindrus
The proportion of nucleotides that differ between both gene copy variants (FcR1/2) of xanthorhodopsin in F. cylindrus was estimated to be 5.8%. Although their amino acid sequence is 100% identical, FcR1 has an N-terminal extension of 30 amino acids, including a potential signal peptide that is cleaved in the mature protein. Full-length alignments of FcR1/2 with characterized rhodopsins showed conserved residues responsible for a proton pump. The characteristic lysine residue (K-261) provides the retinal Schiff base linkage and acidic residues are found at positions of the proton acceptor (D-121) and donor (E-132) sites (Fig. 1a) homologous to K-216, D-85 and D-96 in the prototypic archaeal rhodopsin proton pump, bacteriorhodopsin (BR). AlphaFold2-based three-dimensional (3D) structure prediction45 (Fig. 1b) confirmed that FcR1/2 consists of seven transmembrane helices with an antiparallel beta sheet and extended extracellular loop between helix II and helix III (Fig. 1a,b) as shown for fungal rhodopsins34,46, and high structural similarity to a rhodopsin from the extremophile permafrost bacterium Exiguobacterium sibiricum42 based on a structure homology search47.

a, Secondary structure prediction of FcR1 with key residues highlighted: proton acceptor D-121 and donor E-132 (black), K-261 (pink) forming a Schiff base link with retinal, and retinal binding pocket (Y-119, W-122, V-126, L-129, A-160, I-161, G-165, W-181, A-184, M-185, F-188, W-226, Y-229, P-230, Y-233, Y-253 and S-260) (light grey). The N-terminal extension of 30 amino acids of FcR1 compared to FcR2 is indicated as a dotted line. Potential N-terminal chloroplast signal peptide with conserved ‘ASAFAP’ (red circles) and lysine-rich motif (orange diamonds) in the N terminus of FcR1 are highlighted. Grey boxes correspond to transmembrane helices as predicted by AlphaFold2 structure prediction in b. b, Overall 3D view of the FcR1 protomer in the membrane as predicted by AlphaFold2. The experimental structure of PDB entry 4HYJ42 was used for structural alignment with FcR1 to position the retinal ligand, and the final model was generated by removing the structure of PDB entry 4HYJ. Estimated hydrophobic–hydrophilic membrane boundaries are shown as light blue lines.
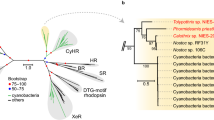
The essential retinal chromophore for microbial opsins can be provided via a putative biosynthetic pathway including a β-carotene cleavage enzyme (β-carotene 15,15’-dioxygenase, EC 1.13.11.63), of which we annotated two putative isoforms in the F. cylindrus genome sequence (BCMO1_1 JGI ID: 228160, Uniprot: A0A1E7EZR0, GenBank: OEU11319; BCMO1_2 JGI ID: 246831) and also in the sequenced diatoms Phaeodactylum tricornutum (Uniprot: B7G4J2) and T. pseudonana (Uniprot: B8C6K5). Additional light-harvesting antennas as shown for fenestrated xantho- and proteorhodopsins48,49 may further be provided by competing carotenoid biosynthetic and xanthophyll cycle enzymes. Using RNA-seq transcriptome analysis, we showed that the biosynthetic genes were significantly expressed over an array of nine experimental conditions (Extended Data Fig. 1). A phylogenetic analysis focusing on eukaryotic microbial rhodopsins confirmed that FcR1 belongs to the xanthorhodopsin family (Fig. 2). The phylogenetic tree further showed that eukaryotic rhodopsins generally followed the current established evolution of the group, but also gave evidence of horizontal gene transfer. Diatom, haptophyte and uncultured marine stramenopiles rhodopsins formed coherent monophyletic clades, whereas dinoflagellate rhodopsins appeared paraphyletic with two main clusters (Fig. 2).

Broad taxonomic classification of rhodopsin sequences is indicated and the FcR1 sequence is highlighted with an F. cylindrus icon. Nodes with multiple rhodopsin sequences of the same taxon were collapsed and accessions of the longest sequence are shown together with numbers of collapsed sequences. The tree is rooted in the proteorhodopsin clade for display purposes. Asterisks denote potentially falsely-annotated rhodopsin sequences in the database due to culture contamination. (Credit for F. cylindrus icon to Charlotte Eckmann.).
The absolute quantification of both FcR gene copies (FcR1/2) showed a higher gene expression (by more than two orders of magnitude) in iron-limited F. cylindrus when their expression levels were combined (Fig. 3a). However, FcR1 was only (and strongly) induced during iron-limited growth, whereas FcR2 was detected during all tested conditions (Fig. 3b). In agreement with quantitative PCR with reverse transcription (RT–qPCR) results, full-length FcR1 complementary DNA (cDNA) transcripts could only be amplified from F. cylindrus grown under iron limitation (Extended Data Fig. 2), whereas FcR2 could additionally also be amplified from other experimental conditions. The strong induction of the FcR1 transcript under iron limitation was also reflected at the protein level (Fig. 3c). Note that probably due to the similar protein sizes between FcR1 and FcR2 after cleavage of the putative N-terminal signal peptide of FcR1 (Fig. 1a), we were unable to resolve separate bands for both proteins. Protein expression confirmed the RT–qPCR results (Fig. 3c).

a, Absolute RT–qPCR gene transcript quantification of FcR1/2 under nine different growth conditions. Statistics are displayed as mean ± s.d. (n = 6) and individual datapoints are colour-coded by condition. b, Variant-specific RT–qPCR gene expression analysis of FcR1 and FcR2 under the same nine growth conditions as in a. Relative variant expression of total FcR expression is shown. Statistics are displayed as mean ± s.d. (n = 6) and individual datapoints are colour-coded by variant. c, Representative western blot analysis of FcR1/2 protein levels in iron-limited (−Fe) and iron-replete (+Fe) F. cylindrus using a custom FcR antibody from three independent experiments. The star indicates the FcR1/2 protein band with a predicted molecular weight between 29–32 kDa. Reversible Ponceau S staining before antibody probing was used as a loading control (right side). Molecular weight (MW) marker lanes are shown in different illumination modes. Note that probably due to FcR1 and FcR2 proteins having similar size after cleavage of the putative FcR1 N-terminal signal peptide (Fig. 1a), separate bands were not resolved for both proteins.
Biophysical characterization of FcR1/2
FcR1 and 2 were characterized and compared to the prototypic archaeal rhodopsin proton pump BR by expression in Xenopus oocytes. On the basis of the conserved residues and structural models, we expected both FcRs to act as proton pumps. Indeed, we observed photocurrents of up to 5 nA in some oocytes expressing FcR1 wild type during initial two-electrode voltage clamp (TEVC) measurements (Extended Data Fig. 3a) but no photocurrents for FcR2, thus we focused on optimization of FcR1 expression in oocytes for reproducible biophysical characterization (Fig. 4a,b and Extended Data Fig. 3b,c).

a, Schematics of DNA constructs used for TEVC analysis in X. laevis oocytes. Orange, amino acids 1–105 of rat gastric H+/K+-ATPase β-subunit fragment (β-linker); sky blue, amino acids 1–10 of C. subellipsoidea rhodopsin (CsR); bluish green, N-terminally truncated FcR1 (FcR1Δ27); blue, full-length FcR2; vermillion, the plasma membrane trafficking signal from Kir2.1 K+ channel (T); yellow, enhanced yellow fluorescent protein (eYFP); reddish purple, the endoplasmic reticulum export signal from Kir2.1 K+ channel (E); grey, FLAG-tag (FLAG). b, Voltage dependence of the green light-induced pump signal of FcR1 and FcR2. Currents were recorded at incremental membrane potential steps of 20 mV from −100 mV (grey) to +20 mV (vermillion). A 530 nm laser was used for illumination. c, Comparison of FcR1 and FcR2 photocurrents. Statistics are displayed as mean ± s.d. (n = 10) and the P value displayed was determined by a two-sided unpaired t-test. d, Action spectrum of FcR1. FcR1 photocurrent was measured in NMG buffer with pH 7.5 at a membrane potential of −20 mV. The photocurrent was normalized to the FcR1 photocurrent with 516 nm light illumination and is shown as arbitrary units. Statistics are displayed as mean ± s.d. (n = 3) and the blue line shows smoothed conditional means (loess smooth) of photocurrents, including their 0.95 confidence intervals (grey). e, FcR1 photocurrents in Ori BaCl2 pH 7.5, NMG pH 7.5 and NMG Asp pH 7.55 buffers and at membrane potentials from −120 mV to +30 mV. The photocurrent was normalized to the FcR1 photocurrent in NMG Asp pH 7.55 buffer at a membrane potential of +30 mV and is shown as arbitrary units. A 532 nm laser was used for illumination. Statistics are displayed as mean ± s.d. (n = 4). f, FcR1 photocurrent measured in NMG buffer with pH 5.6, 7.5 and 8.8 at membrane potentials from −120 mV to +30 mV. The photocurrent was normalized to the FcR1 photocurrent in NMG pH 8.8 buffer at a membrane potential of +30 mV and is shown as arbitrary units. A 532 nm laser was used for illumination. Statistics are displayed as mean ± s.d. (n = 3). g, FcR1 photocurrents at different temperatures (n = 5). Oocytes were incubated in ND96 buffer containing 1 µM all-trans-retinal, and 532 nm green light was used for illumination. The blue line shows smoothed conditional means (loess smooth) of photocurrents, including their 0.95 confidence intervals (grey). h, Comparison of FcR1 and BR photocurrents at different temperatures and light intensities. Photocurrents were tested at 10°C (ripe plum, circles), 20°C (bluish green, squares) and 30°C (light yellow, triangles) and different light intensities (n = 3). Oocytes were incubated in ND96 buffer containing 1 µM all-trans-retinal and 532 nm green light was used for illumination. Photocurrents were normalized and are shown as arbitrary units. i, Green-light-induced photocurrents of FcR1 R118H mutant in three different extracellular buffers: NMG Asp pH 7.7, NMG pH 7.6 and Ori BaCl2 pH 7.6. Currents were recorded at incremental membrane potential steps of 30 mV from −120 mV to −30 mV. Statistics are displayed as mean ± s.d. (n = 5). j, Comparison of stationary photocurrents of FcR1 wild type (WT, n = 5) and FcR1 R118H (n = 4) in NMG buffers of different pHs at a membrane potential of 0 mV. Statistics are displayed as mean ± s.d. k, Green-light-induced photocurrents of FcR1 R118H mutant in NMG buffers of different pHs. Currents were recorded at incremental membrane potential steps of 20 mV from −100 mV to +20 mV. Statistics are displayed as mean ± s.d. (n = 5).
Upon illumination with green or blue light, FcR1/2 responded with a typical ion pump signal, showing no significant difference between the two proteins when applying the same optimized expression construct (Fig. 4b,c). Given the spectral tuning of microbial rhodopsins in the marine environment, we analysed the spectral range promoting maximal pump activity of FcR1 by recording its action spectrum. FcR1 showed a bell-shaped action spectrum (Fig. 4d), with maximal pumping activity in the green spectrum (excitation at ~516 nm). Interestingly, the photocurrents of FcR1 were significantly higher than those of BR50 but showed a similar dependence on all-trans-retinal (Extended Data Fig. 3d).
To test whether FcR1 solely pumps protons, we analysed photocurrents in Xenopus oocytes in buffers of the same pH (7.5) but containing different ions. The FcR1 photocurrents were measured at different potentials in all three buffers, showing no difference (Fig. 4e). We also tested the pH dependency of the pump activity. FcR1 showed the expected behaviour of an outward-directed proton pump, with lowest pumping activity at negative voltages in a pH 5.5 buffer and highest pumping activities at positive voltages in a pH 8.8 buffer (Fig. 4f). Together, our data illustrate that the FcR1 photocurrent is solely caused by protons.
We further tested the activity of FcR1 at low temperatures because of the psychrophilic nature of F. cylindrus. Photocurrents of FcR1 at 2°C were reduced to about half of its photocurrent at 19°C (Fig. 4g). Reducing temperatures from 30°C to 10°C, the photocurrent of FcR1 at 40 mW mm−2 was reduced to only 68.02% ± 0.01, whereas the photocurrent of BR was reduced to 41.25% ± 0.04 (Fig. 4h). In addition, simultaneous pairwise comparisons of the half-maximal photocurrents (K0.5) indicated that K0.5 did not vary for FcR1 but decreased in response to decreasing temperatures for BR, indicating that FcR1 photocurrents were less sensitive to a reduction in temperature, as expected for a cold-adapted enzyme.
During molecular cloning, we unintentionally recovered a point mutation of arginine (R) to histidine (H) in helix III at position 118 (R118H, Fig. 1a). Since this residue is conserved in different microbial rhodopsins and is known to be important for proton transfer, we kept the FcR1 (R118H) mutant for analysis of its photocurrents. Interestingly, the R118H mutant behaves like a proton channel allowing bidirectional transport of protons (Fig. 4i–k and Extended Data Fig. 3e). It displayed comparable current amplitudes in buffers with different anions and cations at similar pH levels (Fig. 4i and Extended Data Fig. 3e). Under conditions of approximately pH 7.6, we observed an inward proton flux at more negative potentials for FcR1 R118H (Fig. 4i–k), whereas FcR1 WT only demonstrated outward proton transport upon illumination (Fig. 4e,f,j). The reverse potential of FcR1 R118H was determined to be approximately −15 mV at pH 7.6 (Fig. 4i). In addition, the current amplitude and reverse potentials of FcR1 R118H displayed pH dependence (Fig. 4k). Together, our findings show that FcR1 R118H functions as a light-gated H+ channel. The discovery of a light-gated, H+-specific channel holds potential for optogenetic applications aiming at pH manipulation. Building on these findings, we successfully engineered light-gated proton channels derived from fungal rhodopsins by homologous point mutations46.
Characterization of FcR1 using reverse genetics and phenotyping
To identify the subcellular localization of FcR1, we expressed C-terminal eGFP fusion constructs of the full-length FcR1 and FcR1 N terminus (FcR1::eGFP, FcR1N-term::eGFP) in P. tricornutum and T. pseudonana using the biolistics method51,52. Using differential interference contrast (DIC) and epifluorescence microscopy, the fluorescence signals of FcR1::eGFP and FcR1N-term::eGFP transformants were observed to be overlapping with the chlorophyll fluorescence from the plastids of both species, suggesting FcR1 subcellular localization in the plastid (Fig. 5a,b). These results, together with our gene expression data and previous studies, support the hypothesis that rhodopsins play a role in supporting algal growth when chlorophyll-based photosynthesis is iron-limited32. Furthermore, maximum activity of FcR1 under green light indicates that growth benefits may be conveyed in this light spectrum. Thus, phenotyping experiments with F. cylindrus were conducted using a two-factor design: iron-limited and iron-replete growth at green and red light, each to test the interaction and effect of different light spectra and iron nutrition on growth. Iron-limited F. cylindrus showed significant growth differences under green versus red light, whereas no significant differences were observed for either green or red light under iron-replete growth (Fig. 5c–e). In comparison, using P. tricornutum as control, because it lacks rhodopsin, no difference in iron-limited growth under green or red light was observed (Extended Data Fig. 4). These results corroborate the hypothesis that the green light-driven FcR1 proton pump facilitates growth when photosynthesis is iron-limited.

a,b, Localization of FcR1::eGFP fusion protein after expression in the pennate diatom P. tricornutum (a) and in the centric diatom T. pseudonana (b), including untransformed wild-type control strains (PtWT and TpWT, respectively). Shown are Normarski DIC, green fluorescence protein (GFP) and red chlorophyll autofluorescence (Alexa568). Representative micrographs of five independent experiments are shown. Scale bars, 5 µm. c, Growth of iron-limited F. cylindrus under green and red LEDs (n = 3). d, Growth of nutrient-replete F. cylindrus under green and red LEDs (n = 3). e, Spectral wavelength of green and red LEDs (n = 1). f, Growth of T. pseudonana rhodopsin knock-in gain-of-function (KI) mutant (TpFcR1, sky blue) and wild-type (TpWT, reddish purple) under iron limitation (150 nM) and constant white light (38 µmol photons m−2 s−1). Exponential growth phase to determine growth rates is highlighted as a grey-shaded box. g, Growth of T. pseudonana rhodopsin knock-in gain-of-function (KI) mutant (TpFcR1, sky blue) and wild type (TpWT, reddish purple) in nutrient-replete medium and constant white light (38 µmol photons m−2 s−1). Exponential growth phase to determine growth rates is highlighted as a grey-shaded box. h, Growth rates of T. pseudonana rhodopsin knock-in mutant (TpFcR1, sky blue) and T. pseudonana wild type (TpWT, reddish purple) under constant white light (38 µmol photons m−2 s−1) during iron-limited and nutrient-replete growth. i, Maximum cell abundance of T. pseudonana rhodopsin knock-in mutant (TpFcR1, sky blue) and T. pseudonana wild type (TpWT, reddish purple) under constant white light (38 µmol photons m−2 s−1) during nutrient-replete and iron-limited growth. Statistics are displayed as mean ± s.d. (n = 5) and the P values displayed were determined using two-sided Wilcoxon rank-sum test (h,i). (Credit for diatom illustrations of F. cylindrus (c,d) and T. pseudonana (f,g) to Charlotte Eckmann).
To provide additional evidence for FcR1 being responsible for enhanced growth of diatoms under iron limitation, we generated a rhodopsin gain-of-function cell line using the model T. pseudonana because it lacks rhodopsin and, similar to F. cylindrus, is part of a broadly distributed genus. T. pseudonana was selected over P. tricornutum for testing the effect of recombinant expression of FcR1 on growth of diatoms under iron limitation because it is less tolerant to iron limitation, making growth phenotyping at low iron concentrations more controllable. We cloned an untagged version of FcR1 into T. pseudonana (TpFcR1) because tagging abolished its proton-pumping activity. We then used diagnostic PCR to verify its integration and expression. The TpFcR1KI mutant grew faster under iron limitation and white light compared with the T. pseudonana wild type (P = 0.00794) and reached a higher biomass yield in stationary phase in both iron-limited and iron-replete media (P < 0.01) (Fig. 5f,g,i).
Xanthorhodopsin in eukaryotic metatranscriptomes from pole-to-pole
To evaluate the potential importance of proton-pumping xanthorhodopsins in supporting algal growth in iron-limited surface oceans, we performed a multiple regression analysis of the normalized abundance of xanthorhodopsin transcripts from 82 upper-ocean eukaryotic metatranscriptomes obtained from pole to pole53,54 with the environmental variables temperature, salinity, nitrate (N), phosphate (P), silicate (Si) and iron (Fe) (Fig. 6a). Missing values for observed N, P and Si concentrations were estimated from the World Ocean Atlas55,56 on the basis of sampling date, and concentrations of dissolved Fe were estimated from recent biogeochemical modelling efforts (Fig. 6b). Overall, the highest rhodopsin transcript abundances were observed at low latitudes (40° N–40° S) (Fig. 6b), often characterized by stratified upper-ocean waters with relatively low nutrient concentrations including dissolved iron.

a, Metatranscriptome sampling stations are scaled on the basis of estimated concentration of dissolved iron. b, Normalized environmental transcript abundances of eukaryotic rhodopsins as a function of latitude. The dark blue line shows smoothed conditional means (loess smooth) of rhodopsin transcript abundances. c, Correlation of normalized environmental rhodopsin transcript abundances and estimated dissolved iron concentration from a global ocean model. d, Correlation of estimated iron stress index and environmental diatom rhodopsin transcript abundances. The P values displayed were determined using a two-tailed t-test and Benjamini–Hochberg adjusted for multiple comparisons. e, Molecular physiological model of diatom rhodopsin.
Application of a generalized linear model (GLM) showed that sampling location in the Southern Hemisphere explained most of the variance in eukaryotic microbial rhodopsin transcript abundances (P < 0.001), with Si (P = 0.001) and Fe (P = 0.029) being the nutrients significantly affecting eukaryotic microbial rhodopsin transcript abundances. A backwards stepwise algorithm to select variables from the initial GLM based on Akaike Information Criterion (AIC) values showed that sampling location in the Southern Hemisphere remained a significant factor for rhodopsin transcript abundances (P < 0.001), with both Si (P = 0.001) and Fe (P = 0.034) remaining significant. In addition, in the reduced minimal GLM, temperature appeared as a significant predictor of rhodopsin transcript abundances (P = 0.011) (Extended Data Fig. 5). Spearman’s rank correlations between eukaryotic rhodopsin transcript abundances and the selected environmental variables showed a positive correlation with temperature (r(82) = 0.37, P = 0.001) and negative correlation with Si (r(82) = −0.36, P = 0.001), but the relationship with Fe lacked statistical support (r(82) = −0.04, P = 0.736).
Given the significance of silicate as a variable, and since diatoms differ from most other phytoplankton taxa in requiring silicic acid (Si(OH)4) to biomineralize their frustules, we reasoned that in addition to uncertainties in estimated environmental variables, our analysis may be confounded by including microbial rhodopsins from all eukaryotes, not just diatoms. We therefore used phylogenetic placement of metatranscriptome sequences onto a custom rhodopsin reference tree (Fig. 2) to identify putative diatom rhodopsins. A total of 58,316 translated rhodopsin metatranscripts were placed on the tree, of which 24,377 (41.8%) mapped to dinoflagellate rhodopsins and 3,439 (5.9%) to diatoms. We then calculated a limitation index from N:Fe ratios (log10(N/Fe)) which has emerged as an effective empirical means to determine N limitation, Fe limitation or N–Fe co-limitation57. Values close to zero indicate N–Fe co-limitation, whereas log10(N/Fe) > 0 indicates Fe limitation and log10(N/Fe) < 0 indicates N limitation of the phytoplankton community. In contrast to results for all eukaryotic microbial rhodopsins, Spearman’s rank correlation analysis between diatom rhodopsin transcript abundances and environmental factors showed a negative correlation with Fe (r(82) = −0.27, P = 0.025), and positive correlations with Si (r(82) = 0.23, P = 0.047) and the log10(N/Fe) limitation index (r(82) = 0.43, P = 2 × 10−4) (Fig. 6d; no significant correlation was found with temperature (P = 0.08)). Taken together, our results suggest that diatom rhodopsins play a particularly important physiological role in shaping their adjustments to iron stress in the Southern Ocean, which is chronically depleted in dissolved Fe35,58.
Discussion
This study provides evidence that light-driven xanthorhodopsins enhance the growth and biomass of diatom species in iron-limited surface oceans. As the latter represent ~35% of the global ocean surface, our results contribute to the understanding of drivers of marine ecosystem productivity. Thus, alongside previously identified strategies of modifications to the relative abundance of photosystem I and II39,40,41 and replacement of iron-containing ferredoxin and cytochrome c6 with metal-free flavodoxin and copper-containing plastocyanin, respectively37,38, xanthorhodopsins appear to provide another evolutionary strategy for accommodating growth under iron-limiting conditions. In contrast to a previous 'omic study on a variety of eukaryotic phytoplankton59, we additionally find a statistically supported correlation of metatranscriptome-derived diatom xanthorhodopsin abundances with dissolved Fe concentrations. This discordance may be due to sampling of different geographic regions, or methods for identifying diatom rhodopsins60. Moreover, while previous studies on microbial rhodopsins from diatoms and dinoflagellates such as Pseudo-nitzschia granii61 and Polarella glacialis62, respectively, identified proton-pumping activity, we provide experimental evidence for their biological role and importance for adaptation to environmental conditions.
Our studies are particularly relevant for the Southern Ocean, which is both the largest iron-limited aquatic ecosystem and among the most productive, supporting the largest populations of consumers such as krill, fish, penguins and whales. The genome of F. cylindrus, as a keystone primary producer in the Southern Ocean, has revealed that genomic complexity, including the divergence of alleles and paralogues, probably contributes to success in this extreme environment33. Our results exemplify how the FcR1 xanthorhodopsin generally provides a fitness advantage for primary producers in iron-limited waters. They also illustrate how sequence divergence of two gene copy variants relates to coping with specific environmental conditions, although the biological role of FcR2 remains elusive. The two gene copy variants rendered different expression patterns in our experiments, and their presumed sub- or neo-functionalization appears to be related to a single non-synonymous nucleotide polymorphism that introduces a stop codon at the FcR2 N terminus, resulting in an N-terminal truncation of 30 amino acids. Otherwise, the amino acid sequences of both variants are identical, including the transmembrane regions and conserved residues, and both can function as proton pumps.
Although native subcellular targeting signal sequences have been predicted for algal rhodopsins63, our study provides experimental evidence for plastid localization as evidenced through subcellular targeting analysis. Nevertheless, additional work will be needed to determine the precise localization inside the complex plastids of diatoms. Because this is essential information for determining the mechanism by which it enhances growth, we developed a conceptual model to discuss the potential mechanisms that lead to the observed growth enhancement under iron limitation (Fig. 6e). Two scenarios appear possible based on its biophysical characterization in Xenopus oocytes: (1) localization in the thylakoid membrane and (2) localization in one of the outer plastid membranes. The essential all-trans-retinal would be available for either localization because it can be synthesized from β-carotene by a putative plastid-targeted β-carotene dioxygenase (Extended Data Fig. 1). For scenario (1), FcR1 would presumably be oriented so that the thylakoid lumen would be akin to the extracellular side in a prokaryotic cell. Under green light, FcR1 would then transport protons from the plastid stroma into the thylakoid lumen independently of cellular iron availability to generate a trans-thylakoidal proton gradient driving chloroplast ATP synthases. Hence, FcR1 and homologous proton-pumping rhodopsins would compensate for the iron limitation impact of a reduced proton gradient in the photosynthetic electron transport chain, which is strongly dependent on iron availability (for example, 24 Fe atoms per individual photosynthetic electron transport chain) (Fig. 6e). Under scenario (2), if FcR1 is localized in one of the four outer plastid membranes oriented towards the cytosol, it would probably contribute to generating a proton motive force facilitating the uptake of dissolved iron and therefore supporting photosynthetic electron transport under iron scarcity. Both scenarios contribute to increased production of ATP under iron-limited growth if iron uptake is suboptimal, which depends on the range of iron transporters, their association with prevalent forms of available iron and the cellular iron demand.
In conclusion, we have characterized two gene copy variants (FcR1/2) of xanthorhodopsin from the cold-adapted diatom F. cylindrus. We find that the structural evolution of FcR1 has enhanced its efficiency under lower temperatures on the basis of temperature-dependent changes in the photocurrents, a finding that aligns well with biophysical measurements conducted with a proteorhodopsin HSG119 from an Antarctic marine bacterium64. The two variants exhibit differences, with FcR1 being expressed only under iron-limited conditions and experimentally demonstrated green-light-activated proton transport. Collectively, these insights suggest a benefit of rhodopsin-enhanced phototrophy for diatoms, especially in iron-limited regions supporting recent findings43. Future challenges include understanding the functional role of the variant FcR2, which is more ubiquitously expressed, and the intricacies of rhodopsin phototrophy relative to chlorophyll-based photosynthesis during iron limitation. Recognizing the inherent intertwining with ocean warming and our findings on cold optimization of the xanthorhodopsins characterized herein, we postulate that the importance of diatom rhodopsins in iron-limited oceans may increase as oligotrophic areas expand and iron availability is modified in response to ongoing global change65,66, external inputs67 and ocean acidification68.
Methods
Protein sequence and prediction analysis
The amino acid sequence of FcR was deduced from the F. cylindrus genome sequence33. Protein alignments with characterized microbial rhodopsins were performed using the MUSCLE algorithm implemented in Geneious (v.5.6)69. Predictions of subcellular location were performed with SignalP v.4.0 and TargetP (v.1.1)70. Putative transmembrane domains and secondary (2D) structure predictions were performed using CCTOP71 and visualized with Protter (v.1.0)72 with manual modifications. A predicted (3D) structure of FcR1 was generated using ColabFold73. The Protein Data Bank (PDB, http://rcsb.org)74 was scanned for homologous structures using a Dali search47. The experimental structure of PDB entry 4HYJ42, a top five search hit, was manually chosen on the basis of highest quality according to PDB indicators and used for structural alignment with FcR1 using ChimeraX75 to position the retinal in the predicted FcR1 structure. The final FcR1 model was generated by removing the structure of PDB ID 4HYJ and visualized using ChimeraX.
Phylogenetic analysis and placement of rhodopsin metatranscripts
Rhodopsin sequences focusing on eukaryotic xanthorhodopsin protein sequences were collected from the literature2,11,76 and supplemented with manually curated sequences from BlastP search results of the FcR1 protein sequence against NCBI databases77, a custom database (A. Rozenberg and O. Beja, unpublished), as well as similar sequences at an amino acid sequence identity threshold of ≥50% based on their membership in the UniRef50 cluster built using FcR1 as seed sequence (UniRef50_A0A7S2RIV1)78. The selected set of proteins was further supplemented with a manually curated set of outgroup sequences including characterized members of the proteorhodopsin subfamily.
To assess the physiological significance of proton-pumping xanthorhodopsins, we searched predicted proteins from 82 upper-ocean eukaryotic metatranscriptome sampling sites53,54 that had been functionally annotated using the Pfam database79 by the US Department of Energy (DOE) Joint Genome Institute (JGI) for sequences annotated as bacteriorhodopsin-like proteins (Pfam: PF01036).
To only select the rhodopsin sequences attributed to diatoms in the proteins predicted from metatranscriptomes, we used a phylogenetic placement approach as a filter to retain sequences for downstream analyses: the massively parallel Evolutionary Placement Algorithm (EPA-ng). In the rhodopsin reference dataset, one sequence labelled as a diatom (from the MMETSP0851 transcriptome) appeared to be mislabelled as a probable contamination from a dinoflagellate. Sequences were aligned using MAFFT with default parameters80 and positions with 25% or more gaps were masked. The maximum-likelihood (ML) reference reconstruction was built using RAxML (v.8.2)81 under the LG + G4 model (selected using IQ-TREE82 with the corrected AIC). A total of 58,316 metatranscriptomic sequences assigned as proteorhodopsin in amino acid space were then aligned against the unmasked reference tree sequences using the –addfragment option in MAFFT83. Then, these aligned metatranscriptomic sequences were phylogenetically placed onto the reference ML reconstruction using EPA-ng (v.0.3.6)84, which employs a RAxML evolutionary placement algorithm under the LG model. Only the sequences that were placed onto the diatom branches of the reference tree were kept for downstream analyses.
Phytoplankton strains, media and growth conditions
The phytoplankton strains of F. cylindrus (strain CCMP 1102) and T. pseudonana (strain CCMP 1335) used were obtained from the Provasoli-Guillard National Centre for Marine Algae and Microbiota (NCMA (formerly CCMP), https://ncma.bigelow.org/). P. tricornutum (strain UTEX 646) was obtained from the University of Texas at Austin (UTEX) Culture Collection of Algae85.
F. cylindrus cell culture experiments were performed as described previously31,33,86. In addition to the previously described experimental treatments including (1) optimal growth (4°C, nutrient replete, 24 h light at 35 μmol photons m−2 s−1), (2) freezing temperatures (−3°C, nutrient replete, 24 h light at 35 μmol photons m−2 s−1), (3) elevated temperatures (11°C, nutrient replete, 24 h light at 35 μmol photons m−2 s−1), (4) elevated carbon dioxide (4°C, 1,000 ppm CO2, 24 h light at 35 μmol photons m−2 s−1), (5) iron starvation (4°C, −Fe, 24 h light at 35 μmol photons m−2 s−1) and (6) prolonged darkness (4°C, nutrient replete, 7 d darkness), we subjected F. cylindrus to (7) growth at half-saturation with silicate (4°C, 0.3 μM silicate, 24 h light at 35 μmol photons m−2 s−1) as well as (8) red (4°C, nutrient replete, 24 h light at 35 μmol photons m−2 s−1, 550–700 nm filter) and (9) blue light illumination (4°C, nutrient replete, 24 h light at 35 μmol photons m−2 s−1, 480–540 nm filter). Experimental F. cylindrus cultures were sampled for RNA preparations during mid-exponential phase (~500,000 cells per ml). Different light environments were created by wrapping culture vessels in commercial colour filters (Lagoon Blue 172 and Sunset Red 025, LEE Filters) or illuminating cultures with red and green LED lamps (Crompton). Light spectra were determined using a spectroradiometer (SR9910, Macam Photometrics). For batch culturing of F. cylindrus under half saturation with silicate, the nutrient was regularly resupplied to a final concentration of 0.3 μmol l−1 during the experiment. Before the batch culturing, the half-saturation constant Km of F. cylindrus for silicate was determined by growing cells over a concentration range of 0.01–100 μmol l−1 silicate (Supplementary Fig. 1).
T. pseudonana cell culture experiments were performed analogous to that of F. cylindrus using iron-free Aquil* medium87, adding trace metals stock solution without iron that was prepared separately (1 mM FeCl3 × 6H2O, 100 μM EDTA) and added at final iron concentrations. The medium was supplemented with all-trans-retinal (Sigma-Aldrich) at a final concentration of 3 μmol l−1 from a 10 mM retinal stock dissolved in 100% dimethyl sulfoxide. T. pseudonana cultures were grown in a 20°C incubator under 24 h light at 38 µmol photons m−2 s−1 white light. Cells were maintained and grown in the iron-replete Aquil* media (1,000 nmol l−1 iron). Before iron-limitation experiments, the T. pseudonana cultures were transferred into iron-free Aquil* media and incubated for 5 d. The iron-starved cultures were then transferred into either iron replete (1,000 nmol l−1 iron, 3 μmol l−1 retinal) or iron-limited Aquil* media (150 nmol l−1 iron, 3 μmol l−1 retinal) at an initial concentration of ~8 × 105 cells per ml. Five replicates each were used for control and treatment groups.
Cell abundance over time was estimated by a Multisizer Coulter counter (Beckman Coulter). Specific growth rates per day (μ) were calculated from the linear regression of the natural log of cell abundances (cells per ml) over time during the exponential growth phase or (when using only two sampling points) according to the following equation:
where Nt1 denotes the cell abundance at timepoint t1, Nt2 the cell abundance at timepoint t2 and Δt the time difference between the two timepoints88.
The maximum quantum yield of photosystem II (Fv/Fm) was measured using pulse-amplitude-modulated (PAM) fluorometry, using a Phyto-PAM fluorometer equipped with a Phyto-ED measuring head (Walz). The in vivo quantum yields were determined in each culture and calculated using PhytoWin software (v.2.00a, Walz) from fluorescence readings of dark-acclimated samples.
RNA extraction from F. cylindrus
Total RNA was extracted using guanidinium thiocyanate-phenol-chloroform extraction89 and TRI reagent (Sigma-Aldrich), followed by DNase I (Qiagen) treatment (1 h, 37°C) and purification using RNeasy MiniElute cleanup kits (Qiagen) according to manufacturer instructions. The purity of RNA was checked on a NanoDrop spectrophotometer (Thermo Fisher) and integrity using 2% denaturing formaldehyde gels or an Agilent 2100 Bioanalyzer (Agilent). RNA concentrations were determined in duplicate readings using a NanoDrop spectrophotometer.
RT–qPCR
RT–qPCR to determine F. cylindrus rhodopsin gene expression levels was performed using a two-tube RT–qPCR protocol90.
First-strand cDNA synthesis was performed using Superscript II reverse transcriptase (Invitrogen) utilizing Anchored Oligo(dT)20 primer (Invitrogen) or Oligo(dT)20 primer (Invitrogen). Reverse transcription of 500 ng of total RNA was carried out in 50 μl reactions at 42°C for 50 min, followed by inactivation at 70°C for 15 min. As a control for DNA contamination, RNA was pooled from each biological replicate and first-strand synthesis reaction mix was added, omitting reverse transcriptase.
Oligonucleotides (Supplementary Table 1) were designed towards the 3’ end of the gene of interest using the web-based RealTimeDesign software (Biosearch Technologies, http://www.biosearchtech.com/realtimedesign) aiming for an amplicon length of 80–150 bp (optimum 115 bp), a GC content of amplicon and primer of 30–80%, a primer length of 18–30 bp and a primer melting temperature TM of 63–68°C. BLAST searches of the primer sequences against the F. cylindrus genome sequence (https://mycocosm.jgi.doe.gov/Fracy1/Fracy1.home.html) were performed and, if necessary, primer sequences were modified manually to ensure maximum specificity. Oligonucleotides were assessed for TM, hairpins and primer dimers using the web-based tool OligoAnalyzer 3.1 (Integrated DNA Technologies, http://eu.idtdna.com/analyzer/Applications/OligoAnalyzer), parameterized with oligo concentrations of 0.4 μM, Na+ of 50 mM, Mg++ of 5.5 mM and deoxynucleotide triphosphates of 0.5 mM. Primers were synthesized by Eurofins Genomics.
For qPCR reactions and second-strand amplification, 5 μl of a 10-fold diluted reverse transcriptase reaction mix was supplemented with 20 μl 2× SensiMix SYBR Green NoROX master mix (Bioline). Forward and reverse primers were added at a concentration of 200 nM. Amplifications were performed in white 96-well plates on a CFX96 real-time system (Bio-Rad) using the following conditions: initial denaturation at 95°C for 10 min, followed by 40 amplification and quantification cycles of 15 s at 95°C, 15 s at 59°C, 10 s at 72°C. Finally, a melting curve analysis (65°C to 95°C, increments of 0.5°C, dwelling time 5 s) was carried out to check for primer dimers and non-specific amplification. For each primer pair, the reliability of qPCR was demonstrated by five-to-six-point standard curves made by amplification from 1:10 serial dilutions of reverse transcription reactions. Standards for absolute RT–qPCR gene expression analysis were generated as follows: target sequences were amplified using conventional PCR from cDNA or plasmid templates, separated by agarose gel electrophoresis and purified (illustra GFX PCR DNA and Gel Band Purification kit, GE Healthcare). The concentration of agarose gel-purified target sequences was determined in duplicate readings using a NanoDrop spectrophotometer (Thermo Fisher) and diluted to 1:10,000. Subsequently, six-point standard curves were determined for specific target sequences by qPCR amplification from 1:10 serial dilutions of the prepared 10,000× dilution. The absolute amount of cDNA in the samples was calculated on the basis of the equation obtained for logarithmic regression lines for standard curves.
Variant-specific qPCR
Variant-specific qPCR to discriminate between expressions of nearly identical gene copies in F. cylindrus was performed91. Briefly, the specificity of the PCR amplification was conferred by placing the 3’-end of a forward or the reverse variant-specific primer directly over a single nucleotide polymorphism but matching one or the other variant. Then variant-specific qPCR was performed in two separate reactions using a common primer and either an FcR1-specific primer or an FcR2-specific primer. Although in theory, only completely matching primers should be extended and only the matching variant sequence should get amplified, there would be amplification of the mismatched variant but with lesser efficiency91. The more frequent variant would reach the cycle threshold (Ct) at an earlier qPCR amplification cycle (that is, having a smaller Ct) and the difference in Ct values between the two separate qPCR reactions, the ΔCt, provides a measure of the variant frequency. The allele frequency was calculated91 using the following equation:
where \(\Delta {C}_{\rm{t}}=({C}_{\rm{t}}\,{\rm{of}\,{{variant}}}_{1}\,{\rm{specific}\,{qPCR}})-({C}_{\rm{t}}\,{\rm{of}\,{{variant}}_{2}\,{specific}\,{qPCR}})\) describes the difference in Ct values between the two qPCR reactions.
Generally, variant-specific primers were designed as described above. However, in addition to their specific design to match only one of the variant sequences at its 3’-terminal nucleotide, additional nucleotide mismatches located three bases from the 3’-end of the variant-specific primer were incorporated to improve amplification specificity as performed previously92,93. The variant-specific primers used in this study are described in Supplementary Table 2.
To validate our variant-specific qPCR, a prepared standard mix containing known ratios of plasmid DNAs encoding cloned full-length sequences of either FcR variant was used. Therefore, pPha-T1 plasmids (pPha-T1 FcR1::GFP and pPha-T1 FcR2::GFP) were linearized (KpnI restriction digest, 37 °C, 3 h) to avoid strong biases by circular (supercoiled) plasmid standards94. DNA concentrations of purified linearized plasmid (illustra GFX PCR DNA and Gel Band Purification kit, GE Healthcare) were determined in three technical replicates using a NanoDrop spectrophotometer (Thermo Fisher). Subsequently, 10 μl of purified linearized plasmids were concentrated until dry using a centrifugal evaporator (miVac DNA concentrator, Genevac), and 10 nM standard solutions of each plasmid were set up with molecular-grade water. Subsequently, 0.001 nM (1 pM) plasmid standards were made using 1:10 serial dilutions of 10 nM standards. Standard mixtures of 100 μl were made up from both 1 pM plasmid standards to contain FcR variants with known copy frequencies of 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and 0.95, which were used as templates for qPCR as described above but with an annealing temperature of 60°C.
Production of F. cylindrus rhodopsin antiserum
Antiserum against FcR1/2 was produced using synthetic antigenic peptides (Standard immunization programme, Eurogentec). Polyclonal antibodies to FcR1/2 were raised against the peptides GELAKTGSPFNDAYR (extracellular loop between helix II and helix III, residues 104–118) and KSLALKNGNEETPLL (C terminus, residues 275–289) and used to immunize two different rabbits each. A cysteine residue was coupled to the C and N termini of the antigenic peptides for coupling to m-maleimidobenzoic acid N-hydroxysuccinimide ester conjugated to keyhole limpet haemocyanin carrier protein. IgGs from the rabbit sera were affinity purified after coupling to Toyopearl AF-amino resin, washing with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) and elution with 100 mM glycine pH 2.5. Immunoreactivity against the antigen was assessed by subsequent western blotting, obtaining best results with the unpurified antiserum.
Protein extraction and western blot analysis
Membrane protein extractions were prepared from 100 ml of mid-exponential F. cylindrus cell cultures (~5 × 105–1 × 106 cells per ml) collected by centrifugation (3,000 g, 10 min at 4°C) using the Mem-PER Plus membrane protein extraction kit (Thermo Fisher) according to manufacturer instructions. The total protein concentrations were determined using a bicinchoninic acid (BCA) assay (Pierce BCA protein assay kit, Thermo Fisher). A protein sample of 30–50 μg was mixed with 6× Laemmli SDS–PAGE sample loading buffer (375 mM Tris-HCl pH 6.8, 10% (w/v) SDS, 60% glycerol, 0.06% bromophenol blue) supplemented with reducing agent β-mercaptoethanol in a total volume of 40–50 μl and directly loaded on 12% SDS–PAGE gels without boiling to avoid membrane protein oligomerization. In addition, 10 μl of a molecular weight marker (Precision Plus Protein Dual Color Standards, Bio-Rad) were loaded and proteins were transferred to nitrocellulose or PVDF membranes using a Criterion Blotter (Bio-Rad). Equal sample loading was checked by total protein staining, as recommended95, with Ponceau S stain (0.5% (w/v) Ponceau S, 1% acetic acid) for 20 min at room temperature and rinsing with milli-Q water. The membrane was blocked with 5% milk in PBS containing 0.1% (w/v) Tween 20 (PBST) for at least 1 h at room temperature or overnight. The FcR1/2 protein was detected by blotting the membrane with a 1:1,000 dilution of rabbit polyclonal anti-FcR1/2 antiserum with 5% milk in PBST for 1 h at room temperature, then washed three times with PBST for 10 min, followed by blotting with a 1:10,000 dilution of secondary horseradish peroxidase-conjugated anti-rabbit IgG. Three additional washes with PBST of 10 min were done before visualizing immune complexes with the Amersham enhanced chemiluminescence kit (Thermo Fisher) and a LAS-3000 imager (Fujifilm).
PCR amplification of F. cylindrus rhodopsin variants
A full-length product of the FcR1 variant was amplified from cDNA, which was synthesized from RNA of iron-limited F. cylindrus, using touchdown PCR amplification with the forward primer 5’-CCTTTTACCGTACAATGCGAGAG-3’ and reverse primer 5’-CAAAATCTGACACTAGGCCCTACC-3’ and successive annealing temperatures of 72°C (5 cycles), 70°C (5 cycles) and 62°C (30 cycles). Touchdown PCR reactions were performed with Phusion DNA polymerase (Finnzymes) according to manufacturer recommendations but using a 1:1 mixture of HF and GC buffer as well as addition of 3% dimethyl sulfoxid and 1 μg μl−1 BSA.
The full-length FcR2 variant was amplified from ~100 ng cDNA template with proofreading DNA polymerase (Pfu, Fermentas), using the forward primer 5’-ATGATCAGC GGAACTCAATTCAC-3’ and reverse primer 5’-AAGGAGAGGAGTTTCTTCGTTTC-3’. A 50 μl reaction contained 0.4 pM of each primer, 0.2 pM of each deoxynucleotide triphosphate, 1× Pfu buffer and 10 mM MgSO4. The amplification profile was as follows: 4 min initial denaturation at 95°C, followed by 35 cycles of 95°C for 45 s, 55°C for 45 s, 72°C for 90 s extension and final extension at 72°C for 5 min.
After purification of agarose gel fragments, gene-specific primers containing restriction sites were used for directed cloning and subcloning of FcR gene constructs into vectors for heterologous expression in diatoms and X. laevis oocytes (Supplementary Figs. 2–5). Expression vectors were transformed and maintained in E. coli. Plasmids from E. coli were isolated by the alkaline extraction method96 using commercial plasmid prep kits. The orientation and accuracy of expression constructs was verified by small-scale capillary sequencing.
Plasmids for in vitro RNA synthesis
The DNA fragments of FcR1 and FcR2 were amplified by PCR with primers carrying the corresponding restriction sites and digested before being inserted into the oocyte expression vectors pGEMHE (Supplementary Fig. 2) carrying different tags. The sequences were confirmed by sequencing. NheI-linearized plasmid DNA was used for the in vitro generation of complementary RNA (cRNA) following the protocol of the AmpliCap-MaxT7 High Yield Message Maker kit (Epicentre Biotechnologies) for oocyte injection.
TEVC experiments
Conserved residues of ion-transporting rhodopsins can also be present in sensory rhodopsins21,97,98. Hence, it is necessary to provide evidence for their proton-pumping activity by experimental characterization using heterologous expression systems such as Xenopus oocytes99,100,101. To test whether both FcR gene copies function as proton pumps, we expressed them individually in Xenopus oocytes for electrophysiological characterization using TEVC measurements. Indeed, photocurrents were observed in some oocytes expressing FcR1 wild type upon illumination with a 532 nm diode-pumped solid state (DPSS) laser. Despite the observed photocurrents of FcR1, targeting the protein to the plasma membrane and its expression in Xenopus oocytes had to be optimized by applying protein engineering strategies including membrane trafficking signal sequences. We found that N-terminal fusion of a rat gastric H+/K+-ATPase β-subunit fragment (amino acids 1–105, ref. 102) to an N-terminally truncated FcR1 enhanced targeting to the oocyte plasma membrane. We also found that C-terminal eYFP fusions and epitope tagging either abolished or strongly reduced FcR1 photocurrents in oocytes, which we solved by inserting the Golgi apparatus trafficking signal from the Kir2.1 K+ channel between the truncated FcR1 and eYFP sequence. Altogether, we achieved the best results by a combination of both, fusing the H+/K+-ATPase β-subunit fragment together with amino acids 1–10 from Coccomyxa subellipsoidea rhodopsin (CsR) to the N terminus of the truncated FcR1 and fusing the Golgi apparatus trafficking (T) signal from the Kir2.1 K+ channel, an eYFP (Y) tag and the Kir2.1 endoplasmic reticulum export (E) signal to its C terminus.
For expression of all constructs, 30 ng of cRNA were injected into Xenopus oocytes. cRNA-injected oocytes were incubated in ND96 solution (in mM, 96 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 5 HEPES, pH 7.4) containing 1 µM all-trans-retinal at 16°C. Photocurrents were measured 2 or 3 d after injection with the light source from a 530 nm LED (Thorlabs) or a 532 nm DPSS laser (Changchun New Industries Optoelectronics). The light intensities were measured with a PLUS 2 power and energy metre (LaserPoint Srl). Electrophysiological measurements were performed at room temperature (20–23°C) (otherwise indicated in the legend) with a TEVC amplifier (TURBO TEC-05, npi electronic). The bath solutions are indicated in figure legends. Electrode capillaries (Ф = 1.599 mm, wall thickness 0.178 mm, Hilgenberg) were filled with 3 M KCl, with tip openings of 0.2–1 MΩ. A USB-6221 DAQ device (National Instruments) and WinWCP (v.5.5.3, Strathclyde University, United Kingdom) were used for data acquisition.
Action spectrum
Light of different wavelengths was obtained from a PhotoFluor II light source (89 North) together with narrow bandwidth interference filters (Edmund Optic) of different wavelengths. The light intensities at different wavelengths were measured with a Laser Check photometer (Coherent Technologies).
Oocyte imaging
The expression levels of rhodopsin proteins in oocytes were estimated from the C-terminally fused YFP 2 d after cRNA injection. The images of Xenopus oocytes were obtained using a confocal laser scanning microscope (LSM 5 Pascal, Zeiss) equipped with a Plan-Neofluar ×10/0.5 objective (Zeiss). The fluorescence images were processed using the LSM 5 image browser and the pictures were exported for insertion into figures.
Subcellular localization of FcR1 in diatoms
The Fragilariopsis rhodopsin gene was cloned into the P. tricornutum expression vector pPha-T1 (ref. 103) for analysis of subcellular targeting using green fluorescent protein (GFP) labelling104. PCR-amplified products FcR1 were purified (illustra GFX PCR DNA and Gel Band Purification kit, GE Healthcare) from 1.2% TAE agarose gels (40 mM Tris acetate, 1 mM EDTA, 0.5 μg ml−1 ethidium bromide) and ligated into the P. tricornutum transformation vector StuI-GFP pPha-T1 (refs.103,105) (Supplementary Figs. 3 and 4) using blunt-ended non-directional ligation with StuI (Eco147I) restriction enzyme. Restriction digest with StuI and ligation was performed in a single tube. Therefore, a 20 μl reaction mix (1 μl vector, 5 μl insert, 1 μl StuI restriction enzyme, 2 μl PEG 4000, 2 μl ATP/DTT (10 mM/100 mM), 2 μl restriction enzyme buffer, 1 μl T4 ligase; adjusted to 20 μl with molecular-grade water) was incubated overnight at room temperature until the reaction was inactivated by heating to 65°C for 20 min. After cooling down to room temperature, empty vector molecules were digested by adding 1 μl StuI restriction enzyme to the inactivated reaction mix and incubating for 1.5 h at 37°C. The digest of empty StuI-GFP pPha-T1 vector molecules was inactivated by heating to 65°C for 20 min. After cooling down to room temperature, 7 μl of the ligation reaction mix was transformed into CaCl2-competent E. coli DH5α cells and selected on ampicillin (100 μg ml−1). Colony PCR using a combination of insert and vector primers was used to screen for plasmids with correct orientation of insert. In addition, FcR1 was amplified and cloned into the vector pTpFCP-GFP/fcpNat (Supplementary Fig. 5) for constitutive expression in T. pseudonana51,106 with primers carrying the corresponding restriction sites. Plasmid and primer sequences were managed with Geneious v.2023.0 (Biomatters, https://www.geneious.com).
Nuclear transformation of P. tricornutum and T. pseudonana was performed using a Biolistic PDS-1000/He particle delivery system (Bio-Rad) fitted with 1,350 psi rupture discs as described previously51,104,107. For the selection and cultivation of P. tricornutum transformants, 75 μg ml−1 Zeocin (InvivoGen) was added to the solid 1.2% agar medium. To detect green fluorescence signals from GFP-transformed diatom cell lines, flow cytometry (FACScalibur, BD) with the standard optical filter configuration was used. Therefore, green fluorescence was measured in the FL1 channel using a 515–545 nm emission filter. FL1 histograms were used to identify transformed (peak ~103) and non-transformed (peak ~101) cells. Milli-Q water was used as a sheath fluid and all analyses were performed using a low flow rate (~20 μl min−1). Triggered on green fluorescence, 10,000 events were collected. An event rate between 100 and 400 cells per second was used to avoid coincidence and when needed, samples were diluted in 0.2-μm-filtered artificial seawater before analysis. To confirm the presence of GFP and analyse the morphology of the cells, upright wide-field fluorescence microscopy (Axioplan 2 IE imaging microscope equipped with a CCD Axiocam camera, Zeiss) was performed. Chloroplasts were identified by red autofluorescence of chlorophyll a/c during excitation at 562 ± 20 nm (Alexa568 filter set). The excitation and emission of filters used during microscopic analyses are listed in Supplementary Table 3.
Generation of T. pseudonana FcR1 knock-in cell line
T. pseudonana wild-type cells (5 × 107 cells per ml) were collected from the exponential growth phase using 0.2 μm Isopore polycarbonate membrane filters (MilliporeSigma). Biolistic transformation of the FcR1 construct followed a previously published protocol107. Colonies were selected on nourseothricin agar plates (100 μg ml−1 nourseothricin/clonNAT) (Werner BioAgents) prepared with half-salinity Aquil* medium.
Analogous to ref. 108, RT–qPCR and DNA sequencing of the PCR product was performed to detect and validate successful transformation and insertion of the FcR1 gene in T. pseudonana. A cell culture volume of 80 ml was collected by centrifugation at the end of the exponential growth phase. Collected cells were frozen in liquid nitrogen and lysed by adding 1 ml of Trizol pre-heated to 60°C and using a bead beater mill (BioSpec) for 2 min at room temperature, followed by incubation on ice for 1 min. Total RNA was extracted and purified using the Direct-zol RNA kit and RNA Clean and Concentrator kit (Zymo) according to manufacturer instructions. Before RNA cleanup, genomic DNA was removed by DNase treatment with RNase-free DNase (Zymo) at 37°C for 60 min. A total of 0.2 µg of RNA was used for reverse transcription using the SuperScript IV first-strand synthesis system (Thermo Fisher) according to manufacturer instructions. PCR was then performed with primer pair FR271123 F1 (5’-CGTTTGGCCATTCCTTCATATC-3’) and rhodopsin screen R (5’-GAACCGGAGATTCCCATCATAG-3’). A 50 μl RT–qPCR was prepared according to REDTaq ReadyMix PCR Reaction Mix manufacturer instructions (Sigma-Aldrich) and performed in a PCR cycler (Bio-Rad) with initial denaturation for 1 min at 95°C, followed by 35 cycles of 30 s at 95°C, annealing for 30 s at 52°C and elongation for 1 min at 72°C, with a final elongation for 5 min at 72°C. The PCR product was confirmed on a 1% agarose gel and analysed by sequencing (Eurofins Genomics). The FcR1 knock-in mutant cell line was maintained in half-salinity Aquil* medium containing 100 μg ml−1 nourseothricin.
Statistics and reproducibility
Statistical analysis was performed in R (v.4.2.1)109. For comparison between specific sets of datapoints, we used parametric and non-parametric testing procedures as indicated in the text. OriginPro v.2020 (OriginLab) was used for analysis of electrophysiological data. A multiple regression analysis using a GLM was performed analogous to that in ref. 110. Briefly, we first calculated the abundance of environmental transcripts annotated with bacteriorhodopsin-like protein domains (Pfam: PF01036) and normalized these as a percentage of total transcripts mapped in each metatranscriptome sample. A GLM was then fitted using the glm function of the R stats package (v.4.2.1) using percentages of rhodopsin domains as response variable and temperature, salinity, hemisphere and the transformed nutrients (log(phosphate+0.01), log(nitrate+0.01), log(silicate+0.01) and log(iron+0.01)) as covariates. We then performed a backward elimination of factors by AIC using a stepwise algorithm with the step function of the R stats package. Regression summaries were exported using the jtools R package (v.2.2.0)111. Data analysis and visualization were performed in R tidyverse112 and using ggplot2 (ref.113) and ggtree114.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-5429. Utilized RNA-seq and metatranscriptome datasets released with previous studies are available in the ArrayExpress database under accession number E-MTAB-5024 and at https://genome.jgi.doe.gov/ (https://doi.org/10.25585/1488054). Additional data and metadata including the rhodopsin reference alignment used to taxonomically assign translated rhodopsin metatranscripts, protein structure prediction and vector maps are available at https://doi.org/10.5281/zenodo.8322266. All other data and biological materials are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
de Grip, W. J. & Ganapathy, S. Rhodopsins: an excitingly versatile protein species for research, development and creative engineering. Front. Chem. 10, 879609 (2022).
Rozenberg, A., Inoue, K., Kandori, H. & Béjà, O. Microbial rhodopsins: the last two decades. Annu. Rev. Microbiol. 75, 427–447 (2021).
Ernst, O. P. et al. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 114, 126–163 (2014).
Grote, M., Engelhard, M. & Hegemann, P. Of ion pumps, sensors and channels—perspectives on microbial rhodopsins between science and history. Biochim. Biophys. Acta 1837, 533–545 (2014).
Govorunova, E. G., Sineshchekov, O. A., Li, H. & Spudich, J. L. Microbial rhodopsins: diversity, mechanisms, and optogenetic applications. Annu. Rev. Biochem. 86, 845–872 (2017).
Gao, S. et al. Optogenetic manipulation of cGMP in cells and animals by the tightly light-regulated guanylyl-cyclase opsin CyclOp. Nat. Commun. 6, 8046 (2015).
Ikuta, T. et al. Structural insights into the mechanism of rhodopsin phosphodiesterase. Nat. Commun. 11, 5605 (2020).
Tian, Y., Gao, S., von der Heyde, E. L., Hallmann, A. & Nagel, G. Two-component cyclase opsins of green algae are ATP-dependent and light-inhibited guanylyl cyclases. BMC Biol. 16, 144 (2018).
Spudich, J. L., Yang, C.-S., Jung, K.-H. & Spudich, E. N. Retinylidene proteins: structures and functions from archaea to humans. Annu. Rev. Cell Dev. Biol. 16, 365–392 (2000).
Finkel, O. M., Béjà, O. & Belkin, S. Global abundance of microbial rhodopsins. ISME J. 7, 448–451 (2013).
Nagata, T. & Inoue, K. Rhodopsins at a glance. J. Cell Sci. 134, jcs258989 (2021).
Pinhassi, J., Delong, E. F., Béjà, O., González, J. M. & Pedrós-Alió, C. Marine bacterial and archaeal ion-pumping rhodopsins: genetic diversity, physiology, and ecology. Microbiol. Mol. Biol. Rev. 80, 929–954 (2016).
Govorunova, E. G., Sineshchekov, O. A. & Spudich, J. L. Emerging diversity of channelrhodopsins and their structure–function relationships. Front. Cell. Neurosci. 15, 800313 (2022).
Inoue, K., Tsukamoto, T. & Sudo, Y. Molecular and evolutionary aspects of microbial sensory rhodopsins. Biochim. Biophys. Acta 1837, 562–577 (2014).
Mukherjee, S., Hegemann, P. & Broser, M. Enzymerhodopsins: novel photoregulated catalysts for optogenetics. Curr. Opin. Struct. Biol. 57, 118–126 (2019).
Oesterhelt, D. & Stoeckenius, W. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat. New Biol. 233, 149–152 (1971).
Beja, O. et al. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289, 1902–1906 (2000).
Inoue, K. et al. Schizorhodopsins: a family of rhodopsins from Asgard archaea that function as light-driven inward H+ pumps. Sci. Adv. 6, eaaz2441 (2020).
Marchetti, A. et al. Comparative metatranscriptomics identifies molecular bases for the physiological responses of phytoplankton to varying iron availability. Proc. Natl Acad. Sci. USA 109, E317-E325 (2012).
Lin, S., Zhang, H., Zhuang, Y., Tran, B. & Gill, J. Spliced leader-based metatranscriptomic analyses lead to recognition of hidden genomic features in dinoflagellates. Proc. Natl Acad. Sci. USA 107, 20033–20038 (2010).
Sineshchekov, O. A. et al. Rhodopsin-mediated photoreception in cryptophyte flagellates. Biophys. J. 89, 4310–4319 (2005).
Needham, D. M. et al. A distinct lineage of giant viruses brings a rhodopsin photosystem to unicellular marine predators. Proc. Natl Acad. Sci. USA 116, 20574–20583 (2019).
Philosof, A. & Béjà, O. Bacterial, archaeal and viral-like rhodopsins from the Red Sea. Environ. Microbiol. Rep. 5, 475–482 (2013).
Gómez-Consarnau, L. et al. Microbial rhodopsins are major contributors to the solar energy captured in the sea. Sci. Adv. 5, eaaw8855 (2019).
Hassanzadeh, B. et al. Microbial rhodopsins are increasingly favoured over chlorophyll in high nutrient low chlorophyll waters. Environ. Microbiol. Rep. 13, 401–406 (2021).
Waschuk, S. A., Bezerra, A. G. Jr, Shi, L. & Brown, L. S. Leptosphaeria rhodopsin: bacteriorhodopsin-like proton pump from a eukaryote. Proc. Natl Acad. Sci. USA 102, 6879–6883 (2005).
Tsunoda, S. P. et al. H+-pumping rhodopsin from the marine alga Acetabularia. Biophys. J. 91, 1471–1479 (2006).
Slamovits, C. H., Okamoto, N., Burri, L., James, E. R. & Keeling, P. J. A bacterial proteorhodopsin proton pump in marine eukaryotes. Nat. Commun. 2, 183 (2011).
Okamoto, O. K. & Hastings, J. W. Novel dinoflagellate clock-related genes identified through microarray analysis. J. Phycol. 39, 519–526 (2003).
McCarren, J. & DeLong, E. F. Proteorhodopsin photosystem gene clusters exhibit co-evolutionary trends and shared ancestry among diverse marine microbial phyla. Environ. Microbiol. 9, 846–858 (2007).
Strauss, J. A Genomic Analysis Using RNA-seq to Investigate the Adaptation of the Psychrophilic Diatom Fragilariopsis Cylindrus to the Polar Environment. PhD thesis, Univ. of East Anglia (2012).
Marchetti, A., Catlett, D., Hopkinson, B. M., Ellis, K. & Cassar, N. Marine diatom proteorhodopsins and their potential role in coping with low iron availability. ISME J. 9, 2745–2748 (2015).
Mock, T. et al. Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature 541, 536–540 (2017).
Zabelskii, D. et al. Structure-based insights into evolution of rhodopsins. Commun. Biol. 4, 821 (2021).
Tagliabue, A. et al. The integral role of iron in ocean biogeochemistry. Nature 543, 51–59 (2017).
Raven, J. A., Evans, M. C. W. & Korb, R. E. The role of trace metals in photosynthetic electron transport in O2-evolving organisms. Photosynth. Res. 60, 111–150 (1999).
Roche, J. L., Murray, H., Orellana, M. & Newton, J. Flavodoxin expression as an indicator of iron limitation in marine diatoms. J. Phycol. 31, 520–530 (1995).
Peers, G. & Price, N. M. Copper-containing plastocyanin used for electron transport by an oceanic diatom. Nature 441, 341–344 (2006).
Strzepek, R. F., Maldonado, M. T., Hunter, K. A., Frew, R. D. & Boyd, P. W. Adaptive strategies by Southern Ocean phytoplankton to lessen iron limitation: uptake of organically complexed iron and reduced cellular iron requirements. Limnol. Oceanogr. 56, 1983–2002 (2011).
Strzepek, R. F. & Harrison, P. J. Photosynthetic architecture differs in coastal and oceanic diatoms. Nature 431, 689–692 (2004).
Coale, T. H. et al. Reduction-dependent siderophore assimilation in a model pennate diatom. Proc. Natl Acad. Sci. USA 116, 23609–23617 (2019).
Gushchin, I. et al. Structural insights into the proton pumping by unusual proteorhodopsin from nonmarine bacteria. Proc. Natl Acad. Sci. USA 110, 12631–12636 (2013).
Andrew, S. M. et al. Widespread use of proton-pumping rhodopsin in Antarctic phytoplankton. Proc. Natl Acad. Sci. USA 120, e2307638120 (2023).
Raven, J. A. Functional evolution of photochemical energy transformations in oxygen-producing organisms. Funct. Plant Biol. 36, 505–515 (2009).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Panzer, S. et al. Modified rhodopsins from Aureobasidium pullulans excel with very high proton-transport rates. Front. Mol. Biosci. 8, 750528 (2021).
Holm, L. Dali server: structural unification of protein families. Nucleic Acids Res. 50, W210–W215 (2022).
Kopejtka, K. et al. A bacterium from a mountain lake harvests light using both proton-pumping xanthorhodopsins and bacteriochlorophyll-based photosystems. Proc. Natl Acad. Sci. USA 119, e2211018119 (2022).
Chazan, A. et al. Phototrophy by antenna-containing rhodopsin pumps in aquatic environments. Nature 615, 535–540 (2023).
Nagel, G., Möckel, B., Büldt, G. & Bamberg, E. Functional expression of bacteriorhodopsin in oocytes allows direct measurement of voltage dependence of light induced H+ pumping. FEBS Lett. 377, 263–266 (1995).
Poulsen, N., Chesley, P. M. & Kröger, N. Molecular genetic manipulation of the diatom Thalassiosira pseudonana (Bacillariophyceae). J. Phycol. 42, 1059–1065 (2006).
Apt, K., Grossman, A. & Kroth-Pancic, P. Stable nuclear transformation of the diatom Phaeodactylum tricornutum. Mol. Gen. Genet. 252, 572–579 (1996).
Martin, K. et al. The biogeographic differentiation of algal microbiomes in the upper ocean from pole to pole. Nat. Commun. 12, 5483 (2021).
Mock, T. Sea of Change: Eukaryotic Phytoplankton Communities in the Arctic Ocean. (United States Department of Energy Joint Genome Institute (JGI), 2014); https://doi.org/10.25585/1488054
Boyer, T. P. et al. World Ocean Atlas 2018. Nutrients (NOAA National Centers for Environmental Information (United States), 2018); https://repository.library.noaa.gov/view/noaa/49137
García, H. E. et al. World Ocean Atlas 2018 Vol. 4 (NOAA Atlas NESDIS 84, 2019).
Browning, T. J. et al. Nutrient co-limitation at the boundary of an oceanic gyre. Nature 551, 242–246 (2017).
Ryan-Keogh, T. J., Thomalla, S. J., Monteiro, P. M. S. & Tagliabue, A. Multidecadal trend of increasing iron stress in Southern Ocean phytoplankton. Science 379, 834–840 (2023).
Caputi, L. et al. Community-level responses to iron availability in open ocean plankton ecosystems. Glob. Biogeochem. Cycles 33, 391–419 (2019).
Coesel, S. N. et al. Diel transcriptional oscillations of light-sensitive regulatory elements in open-ocean eukaryotic plankton communities. Proc. Natl Acad. Sci. USA 118, e2011038118 (2021).
Yoshizawa, S. et al. Light-driven proton pumps as a potential regulator for carbon fixation in marine diatoms. Microbes Environ. 38, ME23015 (2023).
Volkov, D. New Chloride and Proton Light-driven Pumps: Functional Characterization and Crystallization (PhD thesis, RWTH Aachen Univ., 2017).
Sushmita, K., Sharma, S., Kaushik, M. S. & Kateriya, S. Algal rhodopsins encoding diverse signal sequence holds potential for expansion of organelle optogenetics. Biophys. Physicobiol. 20, e201008 (2023).
Kim, S.-H., Jung, B., Hong, S. G. & Jung, K.-H. Temperature dependency of proton pumping activity for marine microbial rhodopsin from Antartic Ocean. Sci. Rep. 10, 1356 (2020).
Raven, J. A. & Beardall, J. Influence of global environmental change on plankton. J. Plankton Res. 43, 779–800 (2021).
Hutchins, D. A. & Boyd, P. W. Marine phytoplankton and the changing ocean iron cycle. Nat. Clim. Change 6, 1072–1079 (2016).
Tang, W. et al. Widespread phytoplankton blooms triggered by 2019–2020 Australian wildfires. Nature 597, 370–375 (2021).
Shi, D., Xu, Y., Hopkinson, B. M. & Morel, F. M. M. Effect of ocean acidification on iron availability to marine phytoplankton. Science 327, 676–679 (2010).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Emanuelsson, O., Brunak, S., von Heijne, G. & Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2, 953–971 (2007).
Dobson, L., Reményi, I. & Tusnády, G. E. CCTOP: a consensus constrained topology prediction web server. Nucleic Acids Res. 43, W408–W412 (2015).
Omasits, U., Ahrens, C. H., Müller, S. & Wollscheid, B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 (2014).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Burley, S. K. et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 49, D437–D451 (2021).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Vader, A., Laughinghouse, H. D. IV, Griffiths, C., Jakobsen, K. S. & Gabrielsen, T. M. Proton-pumping rhodopsins are abundantly expressed by microbial eukaryotes in a high-Arctic fjord. Environ. Microbiol. 20, 890–902 (2018).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 50, D20–D26 (2022).
UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Katoh, K. & Frith, M. C. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics 28, 3144–3146 (2012).
Barbera, P. et al. EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst. Biol. 68, 365–369 (2019).
Starr, R. C. & Zeikus, J. A. UTEX—the culture collection of algae at the University of Texas at Austin: 1993 list of cultures. J. Phycol. 29, 1–106 (1993).
Paajanen, P. et al. Building a locally diploid genome and transcriptome of the diatom Fragilariopsis cylindrus. Sci. Data 4, 170149 (2017).
Sunda, W. G., Price, N. M. & Morel, F. M. M. in Algal Culturing Techniques (ed Anderson, R.) 35–63 (Elsevier, 2005).
Guillard, R. R. L. in Handbook of Phycological Methods: Culture Methods and Growth Measurements (ed Stein, J. R.) 289–311 (Cambridge Univ. Press, 1973).
Chomczynski, P. & Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat. Protoc. 1, 581–585 (2006).
Nolan, T., Hands, R. E. & Bustin, S. A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559–1582 (2006).
Germer, S., Holland, M. J. & Higuchi, R. High-throughput SNP allele-frequency determination in pooled DNA samples by kinetic PCR. Genome Res. 10, 258–266 (2000).
Gupta, M., Yates, C. R. & Meibohm, B. SYBR Green-based real-time PCR allelic discrimination assay for beta2-adrenergic receptor polymorphisms. Anal. Biochem. 344, 292–294 (2005).
Wilkening, S. et al. Determination of allele frequency in pooled DNA: comparison of three PCR-based methods. Biotechniques 39, 853–858 (2005).
Hou, Y., Zhang, H., Miranda, L. & Lin, S. Serious overestimation in quantitative PCR by circular (supercoiled) plasmid standard: microalgal pcna as the model gene. PLoS ONE 5, e9545 (2010).
Moritz, C. P. Tubulin or not tubulin: heading toward total protein staining as loading control in western blots. Proteomics 17, 10.1002/pmic.201600189 (2017).
Birnboim, H. C. & Doly, J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7, 1513–1523 (1979).
Bieszke, J. A., Spudich, E. N., Scott, K. L., Borkovich, K. A. & Spudich, J. L. A eukaryotic protein, NOP-1, binds retinal to form an archaeal rhodopsin-like photochemically reactive pigment. Biochemistry 38, 14138–14145 (1999).
Jung, K.-H., Trivedi, V. D. & Spudich, J. L. Demonstration of a sensory rhodopsin in eubacteria. Mol. Microbiol. 47, 1513–1522 (2003).
Miller, A. J. & Zhou, J. J. Xenopus oocytes as an expression system for plant transporters. Biochim. Biophys. Acta 1465, 343–358 (2000).
Grewer, C., Gameiro, A., Mager, T. & Fendler, K. Electrophysiological characterization of membrane transport proteins. Annu. Rev. Biophys. 42, 95–120 (2013).
Pike, S., Matthes, M. S., McSteen, P. & Gassmann, W. Using Xenopus laevis oocytes to functionally characterize plant transporters. Curr. Protoc. Plant Biol. 4, e20087 (2019).
Kleinlogel, S. et al. A gene-fusion strategy for stoichiometric and co-localized expression of light-gated membrane proteins. Nat. Methods 8, 1083–1088 (2011).
Zaslavskaia, L. A., Lippmeier, J. C., Kroth, P. G., Grossman, A. R. & Apt, K. E. Transformation of the diatom Phaeodactylum tricornutum (Bacillariophyceae) with a variety of selectable marker and reporter genes. J. Phycol. 36, 379–386 (2001).
Kroth, P. G. in Methods in Molecular Biology (ed van der Giezen, M.) 257–267 (Humana Press, 2007).
Gruber, A. et al. Protein targeting into complex diatom plastids: functional characterisation of a specific targeting motif. Plant Mol. Biol. 64, 519–530 (2007).
Scheffel, A., Poulsen, N., Shian, S. & Kröger, N. Nanopatterned protein microrings from a diatom that direct silica morphogenesis. Proc. Natl Acad. Sci. USA 108, 3175–3180 (2011).
Hopes, A. et al. Genome editing in diatoms using CRISPR-Cas to induce precise bi-allelic deletions. Bio Protoc. 7, e2625 (2017).
Faktorová, D. et al. Genetic tool development in marine protists: emerging model organisms for experimental cell biology. Nat. Methods 17, 481–494 (2020).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2022).
Ye, N. et al. The role of zinc in the adaptive evolution of polar phytoplankton. Nat. Ecol. Evol. 6, 965–978 (2022).
Long, J. A. jtools: Analysis and Presentation of Social Scientific Data (CRAN, 2022).
Wickham, H. et al. Welcome to the tidyverse. J. Open Source Softw. 4, 1686 (2019).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (CRAN, 2016).
Yu, G. Data Integration, Manipulation and Visualization of Phylogenetic Trees (CRC Press, 2022).
Acknowledgements
We thank S. Förster (University of Würzburg) and K. Martin (University of East Anglia), as well as J. Morrissey, A. Tanaka, F. R. J. Vieira, J. J. Pierella Karlusich and C. Bowler (Ecole Normale Superieure) for support at the initial phase of the project; A. Rozenberg and O. Beja from the Technion-Israel Institute of Technology (Haifa, Israel) for provision of rhodopsin sequences from a custom database; F. Hartig from the University of Regensburg (Regensburg, Germany) and M. Lenz from GEOMAR Helmholtz Centre for Ocean Research Kiel (Kiel, Germany) for advice on advanced statistical analysis. The PhD studentship of J.S. was funded by the School of Environmental Sciences at UEA. Funding was further provided by grants from the Natural Environment Research Council (NE/I001751/1 to T.M. and NE/K013734/1 to T.M. and J.S.). J.S. was additionally supported by a fellowship from the EMBL Interdisciplinary Postdoc (EIPOD) programme under the Marie Sklodowska-Curie Actions COFUND programme (grant number 664726), GEOMAR Helmholtz Centre for Ocean Research Kiel and the Gordon and Betty Moore Foundation (Grant 3788) to A.Z.W. G.N. acknowledges support from the Louis-Jeantet Prize for medicine.
Author information
Authors and Affiliations
Contributions
J.S. and T.M. conceptualized and designed the project. T.M., G.N., A.Z.W. and J.S. acquired funding. J.S., L.D., S.G., C.Z., A.K., A.H., R.U. and A. Tagliabue conducted the investigations. J.S., S.G., A. Toseland, C.B. and C.Z. curated and analysed the data. J.S., S.G., A.K. and A.H. developed the methodology. T.M., G.N., C.L. and A.Z.W. administered and supervised the project. J.S., C.L., S.G., C.Z. and C.B. created the visualizations. E.F.J. contributed additional experimental analyses to address reviewers’ comments. J.S. prepared the original draft. T.M., A.Z.W., A. Tagliabue, A.K. and J.S. reviewed and edited the paper with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Oded Beja and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Natural retinal biosynthetic pathway from isoprenoid precursors in Fragilariopsis cylindrus.
Bar charts show RNA-seq gene expression of genes involved in putative biosynthetic pathway of all-trans-retinal in Fragilariopsis cylindrus including the cytosolic MEV and plastid DXP/MEP (grey box) during nine replicated experimental conditions (n = 3). Color scale represents absolute gene expression values (FPKM) on a relative scale per gene/gene copy. Gene expression barcharts for identified enzymes and isoenzymes are shown together with Joint Genome Institute (JGI) protein identifiers. Data are presented as mean values +/- s.d. To distinguish gene models belonging to divergent allelic pairs encoding for a single enzyme, they are labelled with suffixes (‘_1’ and ‘_2’). Chemical compounds are represented as circles (red) and enzymes are represented as rounded rectangles (light blue) together with their essential cofactors. Abbreviations: BCMO, β-carotene 15,15’-monooxygenase; CHYB, β-carotene 3-hydroxylase; DMAPP, dimethylallyl diphosphate; DMATT, dimethylallyltranstransferase; FAD, flavin adenine dinucleotide; Fe2+ , ferrous iron; FMN, flavin mononucleotide; FPKM, fragments per kilobase of exon per million mapped fragments; FPP, farnesyl diphosphate; FPPS, farnesyl diphosphate synthase; GGPP, geranylgeranyl diphosphate; GGPPS, geranylgeranyl diphosphate synthase; GPP, geranyl diphosphate; IDI, IPP:DMAPP isomerase; IPP, isopentenyl diphosphate; LCYB, lycopene β-cyclase; MEP, Methylerythritol phosphate; MEV, mevalonic acid; Mg2+ , divalent magnesium ion; Mn2+ , divalent manganese ion; PDS, phytoene desaturase; PSY, phytoene synthase; ZDS, ζ-carotene desaturase; Z-ISO, ζ-carotene isomerase.
Extended Data Fig. 2 Amplification of FcR1 from cDNA template.
(a) Molecular cloning of FcR1 from F. cylindrus cDNA generated from cell cultures grown under low iron. +Fe, F. cylindrus cDNA from nutrient replete cell cultures, -Fe, F. cylindrus cDNA from iron-limited cell cultures. A representative gel of three independent experiments is shown.
Extended Data Fig. 3 Biophysical characterization of FcR1 and comparison to bacteriorhodopsin.
(a) Photocurrent of FcR1 wild-type without N and C terminal modifications measured using two-electrode voltage clamp in two Xenopus laevis oocytes. (b) Schematics of DNA constructs used for two-electrode voltage clamp analysis in X. laevis oocytes. Orange, amino acids 1–105 of rat gastric H+ /K+ -ATPase β-subunit fragment (β-linker); sky blue, amino acids 1–10 of Coccomyxa subellipsoidea rhodopsin (CsR); bluish green, N-terminally truncated FcR1 (FcR1Δ27); yellow, enhanced yellow fluorescent protein (eYFP); gray, FLAG-tag (FLAG). (c) Photocurrents of different FcR1 expression constructs. Statistics are displayed as mean ± s.d. (n = 4) together with individual data points. (d) FcR1 and bacteriorhodopsin (BR) photocurrents with and without additional all-trans retinal (Ret). +Ret (orange), incubation of oocytes in ND96 buffer containing 1 µM Ret. –Ret (green), no additional Ret in ND96 buffer. +1dpi Ret (light blue), 1 µM Ret was added to ND96 buffer one day post oocyte injection (1 dpi). Photocurrent was measured at a membrane potential of -30 mV in Ori NMG pH 7.5 buffer. A 532 nm green light laser was used for illumination. Statistics are displayed as mean ± s.d. (n = 4). (e) Representative green light-induced photocurrent traces of FcR1 R118H mutant in three different extracellular buffers: NMG Asp pH 7.7, NMG pH 7.6 and Ori BaCl2 pH 7.6. Currents were recorded at incremental membrane potential steps of 30 mV from -120 mV to +30 mV.
Extended Data Fig. 4
Iron-limited growth of diatom Phaeodactylum tricornutum wild-type strain UTEX646 under green and red light.
Extended Data Fig. 5 Generalized Linear Model results.
We fitted a generalized linear model (estimated using ML) to predict rhodopsin abundance and derived a reduced minimal model based on Akaike Information Criterion (AIC) values using a backwards stepwise algorithm. Both models’ explanatory power is substantial (R2 = 0.40). Standardized parameters were obtained by fitting the models on a standardized version of the dataset. 95% Confidence Intervals (CIs) and p-values were computed using a Wald t-distribution approximation.
Supplementary information
Supplementary Information
Supplementary Tables 1–3 and Figs. 1–5.
Source data
Source Data Fig. 1
AlphaFold2 structure prediction of FcR1 with retinal in crystallographic information file format.
Source Data Fig. 2
Newick tree file for maximum-likelihood phylogenetic tree.
Source Data Fig. 3
Source data.
Source Data Fig. 4
Statistical source data for Fig. 4a–k.
Source Data Fig. 5
Statistical source data for Fig. 5c–i.
Source Data Fig. 6
Statistical source data for Fig. 6a–d.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Source data.
Source Data Extended Data Fig. 3
Statistical source data for Extended Data Fig. 3a–e.
Source Data Extended Data Fig. 4
Source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Strauss, J., Deng, L., Gao, S. et al. Plastid-localized xanthorhodopsin increases diatom biomass and ecosystem productivity in iron-limited surface oceans. Nat Microbiol 8, 2050–2066 (2023). https://doi.org/10.1038/s41564-023-01498-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-023-01498-5
This article is cited by
-
Iron-limitation light switch
Nature Microbiology (2023)
