
Abstract
Morbilliviruses are among the most contagious viral pathogens of mammals. Although previous metagenomic surveys have identified morbillivirus sequences in bats, full-length morbilliviruses from bats are limited. Here we characterize the myotis bat morbillivirus (MBaMV) from a bat surveillance programme in Brazil, whose full genome was recently published. We demonstrate that the fusion and receptor binding protein of MBaMV utilize bat CD150 and not human CD150, as an entry receptor in a mammalian cell line. Using reverse genetics, we produced a clone of MBaMV that infected Vero cells expressing bat CD150. Electron microscopy of MBaMV-infected cells revealed budding of pleomorphic virions, a characteristic morbillivirus feature. MBaMV replication reached 103–105 plaque-forming units ml−1 in human epithelial cell lines and was dependent on nectin-4. Infection of human macrophages also occurred, albeit 2–10-fold less efficiently than measles virus. Importantly, MBaMV is restricted by cross-neutralizing human sera elicited by measles, mumps and rubella vaccination and is inhibited by orally bioavailable polymerase inhibitors in vitro. MBaMV-encoded P/V genes did not antagonize human interferon induction. Finally, we show that MBaMV does not cause disease in Jamaican fruit bats. We conclude that, while zoonotic spillover into humans may theoretically be plausible, MBaMV replication would probably be controlled by the human immune system.
Similar content being viewed by others


Main
Bats are important reservoir hosts for many viruses with zoonotic potential1. Severe acute respiratory syndrome coronavirus 2, Ebola virus and Nipah virus are examples of such viruses that have caused deadly epidemics and pandemics when spilled over from bats into human and animal populations2,3. Careful surveillance of viruses in bats is critical for identifying potential zoonotic pathogens. However, metagenomic surveys in bats often do not result in full-length viral sequences that can be used to regenerate such viruses for targeted characterization4, at least not without much further effort like 3′ and 5′ rapid amplification of complementary DNA ends. Improvements in sequencing technologies and bioinformatics have enabled more complete genome assemblies. Three metagenomic surveys published in the past year confirm that bats, and to a lesser extent shrews and rodents, are hosts to diverse paramyxoviruses5,6,7 that comprise multiple genera (Jeilongvirus, Morbillivirus and Henipavirus). Metagenomic sequences, however complete, cannot at present yield sufficiently accurate information about viral phenotypes in vitro and in vivo. Detailed virological investigations are still needed to reify taxonomic discoveries.
Morbilliviruses are among the most contagious viral pathogens that infect mammals. While numerous partial sequences of morbilliviruses have been identified in bats and rodents4,5 in metagenomic surveys, no full-length authentic morbillivirus has been isolated or characterized from chiropteran hosts. The morbillivirus genus includes measles virus (MeV), canine distemper virus (CDV), rinderpest virus, phocine distemper virus, cetacean morbillivirus, peste des petis ruminants virus and feline morbillivirus8. A porcine morbillivirus was recently described to be the putative cause of foetal death and encephalitis in pigs9. All morbilliviruses cause severe disease in their natural hosts10,11,12,13,14, and pathogenicity is largely determined by species-specific expression of canonical morbillivirus receptors, encoded by CD150/SLAMF1 (ref. 15) and NECTIN4 (ref. 16).
In this Article, we characterize myotis bat morbillivirus (MBaMV) from a vespertilionid bat species (Myotis riparius) in Brazil, previously identified only from sequence information alone. MBaMV used Myotis spp. CD150 much better than human and dog CD150 in fusion assays. We confirmed this using live MBaMV that was rescued by reverse genetics. Surprisingly, MBaMV replicated in primary human myeloid but not lymphoid cells and did so in a CD150-independent fashion. This is in contrast to MeV, which is known to infect CD150+ human myeloid and lymphoid cells. Furthermore, MBaMV replicated in human epithelial cells and used human nectin-4 almost as well as MeV. Nonetheless, MBaMV P/V do not appear to antagonize human interferon (IFN) induction and signalling pathways, and MBaMV was cross-neutralized, albeit to variable extents, by measles, mumps and rubella (MMR) vaccinee sera. Our results demonstrate the ability of MBaMV to infect and replicate in some human cells that are critical for MeV pathogenesis and transmission. Yet comprehensive evaluation of viral characteristics provides data for proper evaluation of its zoonotic potential.
Results
Isolation of MBaMV sequence
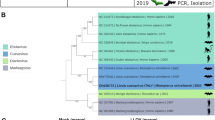
During a metagenomic genomic survey of viruses in bats, we identified a full-length morbillivirus sequence from a riparian myotis bat (M. riparius) in Brazil7. In total, 46 individuals were sampled from the species M. riparius. Of these, 9 were sampled from the Amazon region and 37 were sampled from the Atlantic Forest in southern Brazil. The rectal swab of one individual from the Amazon region was positive for the PDF-3137 virus7. This MBaMV had a genome length of 15,720 nucleotides consistent with the rule of 6 and was composed of 6 transcriptional units encoding the canonical open reading frames (ORFs) of nucleo (N) protein, phospho (P) protein, matrix (M) protein, fusion (F) protein, receptor binding protein (RBP) and large (L) protein (Extended Data Fig. 1a). The sizes of these ORFs are comparable to their counterparts in the other morbilliviruses (Extended Data Table 1). Phylogenetic analysis using the full-length L protein sequence indicated that MBaMV is most closely related to CDV and phocine distemper virus (Extended Data Fig. 1b and Extended Data Table 2).
Paramyxovirus proteins with the most frequent and direct interactions with host proteins, such as P and its accessory gene products (V and C) as well as the RBP, tend to exhibit the greatest diversity17. Morbillivirus P, V and C antagonize host-specific innate immune responses, while its RBP interacts with host-specific receptors. That these proteins are under evolutionary pressure to interact with different host proteins is reflected in the lower conservation of MBaMV P/V/C (31–43%) and RBP (27–32%) with other morbillivirus homologues. This is in contrast to the relatively high conservation (52–76%) of MBaMV N, M, F and L proteins with their respective morbillivirus counterparts (Extended Data Fig. 2).
Species-specific receptor usage
The use of CD150/SLAMF1 to enter myeloid and lymphoid cells is a hallmark of morbilliviruses, and also a major determinant of pathogenicity. CD150 is highly divergent across species, and accounts for the species restricted tropism of most morbilliviruses18. Thus, we first characterized the species-specific receptor tropism of MBaMV. We performed a quantitative image-based fusion assay by co-transfecting expression vectors encoding MBaMV-F and MBaMV-RBP, along with CD150 from the indicated species into receptor-negative Chinese hamster ovary (CHO) cells. MeV-RBP and F formed more syncytia in CHO cells upon human-CD150 (hCD150) co-transfection compared with dog CD150 (dCD150) or bat CD150 (bCD150) (Fig. 1a, top row). In contrast, MBaMV-RBP and F formed bigger and more numerous syncytia upon bCD150 overexpression than hCD150 or dCD150 (Fig. 1a, middle row). CDV-RBP and F formed extensive syncytia with both dCD150 and bCD150, and moderate syncytia with hCD150 and even mock-transfected cells (Fig. 1a, bottom row), suggesting a degree of promiscuity. We quantified these differential syncytia formation results on an image cytometer as described19 (Fig. 1b).

a, Syncytia formation in CHO cells co-transfected with the indicated morbillivirus envelope glycoproteins, species-specific CD150 and Life-act-GFP. Images were taken by the Celigo Imaging Cytometer (Nexcelom) at 48 h post-transfection (hpt) and are computational composites from an identical number of fields in each well. Scale bars, 200 µm. Brightness and contrast settings were identical. b, Quantification of syncytia formation in a. Data are mean ± s.d. from three independent experiments. Indicated adjusted P values are from ordinary one-way ANOVA with Dunnett’s multiple comparisons test. c, VSV-pseudo particle (pp) entry assay showed similar trends. Adjusted P values obtained as in b but only for comparing groups at the highest viral inoculum used (10−1 reciprocal dilution) in three biologically independent experiments (mean ± s.d).
We also evaluated the receptor usage of MBaMV in a vesicular stomatitis virus (VSV)-pseudotype entry assay. VSV-DG[Rluc] bearing MeV-RBP and F entered hCD150-transfected CHO cells better than dCD150-, bCD150- or mock-transfected cells (Fig. 1c) as expected. MBaMV pseudotypes entered only bCD150-transfected CHO cells. CDV-pseudotypes showed good entry into dCD150- and bCD150-transfected but not hCD150-transfected CHO cells. These results are generally consistent with our fusion assay results and support the species specificity of morbilliviruses. CDV has a high propensity to cross species barriers and can cause disease in multiple carnivore families such as large felids (for example, lions and tigers), hyaenids (for example, spotted hyenas), ailurids (for example, red pandas), ursids (for example, black bears), procyonids (for example, raccoons), mustelids (for example, ferrets), viverrids (for example, civets) and even non-carnivore species such as javalinas (peccaries) and rodents (Asian marmots)20. CDV has also been implicated in multiple outbreaks in non-human primates (various Macaca species)21,22,23. The ability of CDV to use bCD150 and dCD150 with equal efficiency suggests potential for efficient transmission from carnivores into some chiropteran species if other post-entry factors do not present additional restrictions.
Generation of MBaMV by reverse genetics
Next, we attempted to generate a genomic cDNA clone of MBaMV that we could rescue by reverse genetics. We synthesized and assembled the putative MBaMV genome in increasingly larger fragments. Two silent mutations were introduced in the N-terminal 1.5 kb of the L gene to disrupt a cryptic ORF in the minus strand (Extended Data Fig. 3) that initially prevented cloning of the entire MBaMV genome. We introduced an additional EGFP transcription unit at the 3′ terminus and rescued this MBaMV-GFP genome using the N, P and L accessory plasmid from MeV (Extended Data Fig. 1a). MBaMV-GFP was initially rescued in BSR-T7 cells but passaged, amplified and titred on Vero-bCD150 cells (Extended Data Fig. 4a). MBaMV formed GFP-positive syncytia containing hundreds of nuclei at 3 days post-infection (d.p.i.) (Fig. 2a) and relatively homogeneous plaques by 7 d.p.i. (Fig. 2b). Transmission electron microscopy (TEM) (Fig. 2c) captured numerous pleiomorphic virions (~100–200 nm) associated with or budding from Vero-bCD150 cells. Some appear to be filamentous particles emerging from a cell. At high magnification, virions were outlined by protrusions suggestive of surface glycoproteins. Ribonucleoprotein-like structures can be found in the interior of the virion shown. These observations are consistent with previous findings from MeV24.

a, Syncytia formation in Vero-bCD150 cells induced by MBaMV 3 d.p.i. Cells formed syncytia involving >100 nuclei upon infection (bright field), which is clearly outlined by virus-expressed GFP (right). Scale bars, 500 µm. This experiment was performed three times, and representative images are depicted. b, MBaMV plaque formation in Vero-bCD150 cells. Cells were infected by ten-fold serially diluted virus stock, incubated with methylcellulose-containing DMEM and stained with crystal violet and neutral red 7 d.p.i. Diameter of well is 22 mm. One well is magnified to show the plaque morphology in detail. c, TEM images of MBaMV virion on the surface of Vero-bCD150 cells at 3 d.p.i. Numerous enveloped virions are budding from or are associated with the plasma membrane (left). Filamentous particles can also be seen (asterisks). Magnified image (right) shows virion and ribonucleoprotein complex (RNP). These images are from one independent experiment.
Evaluation of receptor usage by MBaMV
To understand how well CD150 from various hosts supports MBaMV replication, we tested MBaMV growth in parental Vero-CCL81 cells and isogenic derivatives constitutively expressing CD150 of human, dog or bat. MBaMV formed huge syncytia (Fig. 3a) at 2 d.p.i. in Vero-bCD150 cells and reached peak titres of ~105 PFU ml−1 at 3 d.p.i. (Fig. 3b). MBaMV showed moderate syncytia spread and growth in Vero-dCD150 cells, but peak titres at 5 d.p.i. were ~100-fold lower. No notable virus growth was detected in Vero or Vero-hCD150 cells. These results confirm that MBaMV can use bCD150 but not hCD150 for efficient cell entry and replication. MBaMV appears to use dCD150, albeit to a much lesser extent than bCD150.

a,b, Vero-hCD150, Vero-dCD150, Vero-bCD150 and Vero cells were infected with rMBaMV-EGFP (MOI 0.01). Virus replication and spread were monitored by imaging cytometry (a) and virus titre in the supernatant (b). In a, large syncytia were evident in Vero-bCD150 cells by 2 d.p.i. In b, supernatant was collected every day and the virus titre was determined by a GFP plaque assay (Methods). Data shown are mean ± s.d. from triplicate experiments. c–e, H441 and A549 cells were infected with rMBaMV-EGFP at a low (0.01) or high (0.5) MOI, with virus replication and spread monitored as in a and b: infected H441 and A549 cells at 1, 2 and 5 d.p.i. (D1, D2 and D5) (c); virus growth curves represented by daily titres in the indicated conditions, where data shown are mean titres ± s.d. from triplicate infections (d); the empirical area under the curve (eAUC) was obtained from each growth curve and plotted as a bar graph from three independent experiments (mean ± s.d.) (PRISM v 9.0) (e). The dotted bars represent the growth curves done at an MOI of 0.1 and the filled bars at MOI 0.5. Adjusted P values are indicated (one-way ANOVA Dunnett’s T3 multiple comparison test). f,g, Vero-human nectin-4 cells (Vero-hN4) were infected with MBaMV and MeV (MOI 0.01): MBaMV infected Vero-hN4 at D1, D2 and D4 (f); replicative virus titres for MBaMV and MeV on Vero-hN4 cells over 5 days (mean ± s.d., n = 3) (g). Scale bars in a, c, and f equals 1 mm. All images shown are captured by a Celigo Imaging Cytometer (Nexcelom). Images are computational composites from an identical number of fields in each well. The limit of detection for virus titre determination is 20 PFU ml−1 and is indicated by the dotted line in b, d and g.
MeV uses human nectin-4 as the epithelial cell receptor25,26, which mediates efficient virus shedding from the affected host16,27. CDV also uses human nectin-4 efficiently for entry and growth23. To test if MBaMV can use human nectin-4 in an epithelial cell context, we evaluated the replication kinetics of MBaMV in human lung epithelial cells that express high (H441) or low (A549) levels of nectin-4 (refs. 16,28) (Extended Data Fig. 4b). Surprisingly, MBaMV showed efficient virus spread (Fig. 3c) in H441 cells and reached 104 PFU ml−1 by 6 d.p.i. (Fig. 3d). In contrast, MBaMV showed small GFP foci and ten times lower titre in A549 cells. Comparing the area under the curve (AUC) revealed significant differences in this growth curve metric (Fig. 3e). However, MeV still replicated to higher titres than MBaMV in H441 cells (Fig. 3d-e). This could be due to species specific host factors or differences in IFN antagonism between human and bat morbilliviruses. Thus, we tested MBaMV versus MeV growth in IFN-defective Vero-human nectin-4 cells (Vero-hN4). MBaMV and MeV replicated and spread equally well on Vero-hN4 cells (Fig. 3f,g), validating the ability of MBaMV to use human nectin-4, and suggesting that MBaMV may not be able to counteract human innate immune responses.
Molecular characterization of MBaMV
To better understand the transcriptional profile of MBaMV, we used Nanopore long-read direct RNA sequencing to sequence the messenger RNAs of MBaMV-infected Vero-bCD150 cells at 2 d.p.i. (multiplicity of infection (MOI) 0.01). We found a characteristic 3′–5′ transcriptional gradient where GFP > N > P > M > F > RBP > L (Extended Data Fig. 5a). Morbilliviruses have a conserved intergenic motif (CUU) between the gene end and gene start of adjacent genes ‘AAAA-CUU-AGG’. This intergenic motif was not immediately apparent in the long complex M-F intergenic region of the assembled MBaMV genome. However, the high coverage of this M-F intergenic region (M read-through transcripts) identified the M-F intergenic motif as ‘CGU’ instead of ‘CUU’ (Extended Data Fig. 5b).
The P gene of morbilliviruses is known to generate the V or W genes through the insertion of one or two guanines, respectively, at the conserved editing motif (AAAAGGG)29, which is present in MBaMV. Amplicon sequencing of the P gene editing motif—from the same mRNA pool used above—revealed the frequency of P, V and W mRNA is 42.1%, 51.2% and 2.6%, respectively (Extended Data Fig. 5c), suggesting that the major IFN antagonist (V) is produced. This P-editing ratio is similar to what has been found in previous studies30.
We next evaluated the expression and cleavage of two surface glycoproteins (RBP and F). C-terminal AU-1 tagged F construct showed uncleaved F0 and cleaved F1 (Extended Data Fig. 5d). C-terminal HA-tagged RBP construct showed monomer in addition to oligomers (Extended Data Fig. 5e). MBaMV-RBP showed smear above 110 kDa, which is suggestive of oligomerization. This oligomerization was also seen with MeV-RBP but not with CDV RBP, suggesting differential stability under the subreducing conditions used.
Species tropism of MBaMV
The two suborders of chiropterans (bats), Pteropodiformes (Yinpterochioptera) and Vespertilioniformes (Yangochiroptera), include more than 1,400 species grouped into 6 and 14 families, respectively31. Myotis bats belong to the prototypical Vespertilionidae family that is the namesake of its suborder. Jamaican fruit bats (Artibeus jamaicensis) belong to the same suborder as myotis bats, albeit from a different family (Phyllostomidae). We inoculated six Jamaican fruit bats available in a captive colony via two different routes with MBaMV to assess its pathogenicity in vivo. All bats remained asymptomatic and showed no evidence of developing systemic disease up to 3 weeks post-infection. Nor could we detect any molecular or serological evidence of productive infection (Extended Data Fig. 6). Inspection of Jamaican fruit bat and myotis CD150 sequences revealed key differences in the predicted contact surfaces with RBP (discussed below), which we speculate are responsible for the species-specific restriction seen in our experimental challenge of Jamaican fruit bats with MBaMV.
To identify RBP–CD150 interactions probably involved in determining host species tropism, we compared the amino acid sequences at the putative contact surfaces of morbillivirus RBPs and their cognate CD150 receptors. Using PDBePISA32, we identified three key regions in MeV-RBP (residues 188–198, 498–507 and 524-556, Extended Data Fig. 7a-c) occluding two regions in CD150 (residues 60–92 and 119–131 of human CD150, Extended Data Fig. 8) in the crystal structure of MeV-RBP bound to CD150 (Protein Data Bank (PDB) ID: 3ALW)33. Alignment of key regions in morbillivirus RBPs implicated in CD150 interactions reveals virus-specific differences that suggest adaptation of morbillivirus RBPs to the CD150 receptors of their natural host. Most notably, MBaMV lacks the DxD motif at residues 501–503 (505–507 in MeV) that is present in all morbilliviruses except FeMV (Extended Data Fig. 7). These residues form multiple salt bridges and hydrogen bonds that stabilize MeV-RBP and hCD150 interactions. Their conservation suggest they perform similar roles for other morbilliviruses. On the CD150 side (Extended Data Fig. 8), residues 70–76 and 119–126 are the most variable between host species. Interestingly, Jamaican fruit bat and Myotis CD150 differ considerably in these regions, providing a rationale for the non-productive infection we saw in our Jamaican fruit bat challenge experiments.
Susceptibility of human myeloid and lymphoid cells to MBaMV
Alveolar macrophages and activated T and B cells expressing CD150 are the initial targets for MeV entry and systemic spread. To better assess the zoonotic potential of MBaMV, we compared how well human and bat morbilliviruses can infect human monocyte-derived macrophages (MDMs) and peripheral blood mononuclear cells (PBMCs). Both MeV and MBaMV infected MDMs were clearly GFP+ 24 hours post infection (h.p.i.) (Fig. 4a), but infection was variable between donors and even between different viral stocks on the same donor (Fig. 4b,d). However, MeV infection of MDMs was inhibited by sCD150 whereas MBaMV infection was not (Fig. 4c). MDMs had variable expression of CD150 (10–30% CD150+) and no expression of nectin-4 (Extended Data Fig. 4c), but morbillivirus infection did not appear to be correlated with CD150 expression (Fig. 4d). Conversely, when PBMCs were stimulated with concanavalin-A and IL-2, only MeV robustly infected these cells (Fig. 4e).

a,b, MDMs were infected with MV323-EGFP or MBaMV (1 × 105 IU per sample) and were either fixed by 2% PFA at 24 h.p.i., DAPI stained and imaged (scale bars, 200 µm) (a) or quantified by flow cytometry (b). The percentage of CD68+ GFP+ MDMs from six donors is shown. Open and crossed symbols indicate experiments using lot 1 and lot 2 viruses, respectively. Adjusted P values are from one-way ANOVA with Dunnett’s multiple comparisons test. c, Soluble human CD150 (sCD150) or a CD150 Avi-tag inhibited MeV but not MBaMV infection of macrophages. GFP+ events in untreated controls were set to 100%, and entry under sCD150/ CD150 Avi-tag were normalized to untreated controls. Adjusted P values are from two-way ANOVA with Šídák’s multiple comparisons test. In b and c, data shown are mean ± s.d. from multiple experiments (n = 7 and n = 5, respectively) with individual values also shown. d, Exemplar FACS plots from the summary data shown in b for CD150 staining. e, ConA/IL-2 stimulated PBMCs were infected with MeV or MBaMV (MOI of 0.1) and analysed for GFP expression by flow cytometry at 24 h.p.i.
MBaMV infection may be blocked by human immune defences
MeV-specific antibodies resulting from vaccination can provide cross-protection against CDV infection34. To assess if human sera from MeV-vaccinated individuals could contain cross-neutralizing antibodies to MBaMV, we pooled MMR-reactive human sera and measured their ability to neutralize MeV, MBaMV and CDV in hCD150-, bCD150- and dCD150-expressing Vero cells. Human sera effectively neutralized MeV and MBaMV infection and, to a lesser extent, CDV infection (Fig. 5a, left and centre). Conversely, sera from CDV-infected ferrets neutralized CDV infection much better than MeV or MBaMV (Fig. 5a, right). Sera from the MMR groups 1 and 2 had higher 50% inhibitory concentration (IC50) for MeV and MBaMV than for CDV while CDV-specific sera had a significantly higher IC50 for CDV than for MeV or MBaMV (Fig. 5b). These results indicate that human sera contain cross-neutralizing antibodies for MBaMV.

a, MeV, MBaMV and CDV were incubated with serial dilutions of pooled human sera from MMR-vaccinated individuals (MMR group 1 and group 2) and sera from ferrets infected with CDV. The capacity for sera-treated virus to infect Vero cells expressing the appropriate receptor was measured by imaging infected cells 20 h.p.i., measuring the area of GFP+ cells, and calculating the reduction in infection compared with no sera controls. Neutralization curves were plotted for each virus and corresponding sera group. b, The IC50 from the neutralization curves shown in a were generated for each replicate using a robust fit model and were plotted. Two independent experiments were completed with the pooled human sera neutralization, and one experiment with technical duplicates was completed using the CDV sera (mean ± s.d.). P values were calculated with a one-way ANOVA using a Tukey’s multiple comparisons test. NS, not significant.
The MeV proteins P and V interfere with the innate immune system by disrupting the IFN pathway. Our sequencing results showed that MBaMV infection produced the P and V transcripts (Extended Data Fig. 5c), so we sought to determine if the MBaMV P and V proteins could antagonize the human IFN pathway. We found that cells transfected with MBaMV P or V and treated with IFN did not block interferon stimulatory response element (ISRE) induction, unlike ZIKV NS5, which effectively counteracts the ISRE (Extended Data Fig. 9a). Additionally, cells transfected with MBaMV P or V did not block the induction of IFN when treated with RIG-I, MDA5 or MAVS (Extended Data Fig. 9b–d). These data demonstrate that the MBaMV P and V proteins cannot antagonize the human IFN pathway.
MBaMV is sensitive to morbillivirus RNA-dependent RNA polymerase inhibitors
Potential drug treatments are a critical issue for emerging viruses. Thus, we tested if MBaMV is susceptible to currently available drugs. We have developed two orally bioavailable small compounds targeting the L protein of morbilliviruses, GHP-88309 (ref. 35) and ERDRP-0519 (ref. 36). The differences between MeV and MBaMV across the five functional domains of the L protein are shown schematically in Fig. 6a37. In silico modelling (Fig. 6b) predicts that both drugs should bind similarly to MeV and MBaMV L protein. Closer inspection of the ERDRP-0519 binding pocket (Fig. 6c) shows 1155–1158 YGLE and H1288 residues interacting with ERDRP-0519. These residues directly interact with ERDRP-0519 in MeV L38. Modelling of the GHP-88309 binding pocket (Fig. 6d) reveals involvement of E863, S869, Y942, I1009 and Y1105 residues, which were previously reported as escape mutants of GHP-88309 in MeV35. As predicted, both drugs inhibited MBaMV growth in a dose-dependent manner (Figs. 6e,f). Although the half maximal effective concentration (EC50) of GHP-88309 is lower for MeV than MBaMV, (0.6 µM and 3.0 µM, respectively), GHP-88309 reaches a plasma concentration of >30 µM in animal models, indicating this drug could be an effective inhibitor of MBaMV in vivo.

a, 2D schematic of MBaMV L protein showing the layout of each domain. Conservation between the MeV and MBaMV L protein is shown. Differences between the MeV and MBaMV L proteins are shown as grey lines. b, A 3D homology model of the MBaMV L protein was generated using the structural coordinates of the PIV5 L protein (PDB ID: 6V86). The RNA-dependent RNA polymerase (RdRP), capping, connector, methyltransferase (Mtase) and C-terminal domains (CTD) are coloured blue, green, yellow, orange and pink, respectively. The locations of the top scoring in silico docking poses for ERDRP-0519 and GHP-88309 are boxed, and the compounds are shown as red sticks. c, The top scoring docking pose of ERDRP-0519 in the homology model of MBaMV L protein (red sticks). An overlay of the previously identified docking pose of ERDRP-0519 in a homology model of MeV L protein is shown (yellow sticks)38. Residues identified in previous photoaffinity crosslinking experiments (Y1155, G1156, L1157 and E1158) and H1288 of the HR motif are shown as magenta sticks. d, The top scoring docking pose of GHP-88309 in the homology model of MBaMV L protein (red sticks). An overlay of the previously identified docking pose of GHP-88309 in a homology model of the MeV L protein is shown (yellow sticks)35. Residues identified in MeV resistance profiling studies are shown as magenta sticks. e, The dose–response inhibition growth curves of ERDRP-0519 against MBaMV. Vero-bCD150 cells were infected by MBaMV at MOI of 0.01 for 1 h, then inoculum was replaced by fresh medium containing inhibitor at the indicated concentrations (0–50 µM). 2 d.p.i., viral supernatants were collected and titred on Vero-bCD150 cells as described in Methods. Dots represent the values from three independent experiments. Regression curve (solid line) and 95% confidence interval (CI) (dot line) were generated in PRISM (v.8.0). f, The drug response of GHP-88309 against MBaMV growth. The virus inhibition was conducted identically as for ERDRP0519.
Discussion
Metagenomic viral surveillance studies aided by next-generation sequencing (NGS) have allowed scientists to monitor viruses circulating in animal species and identify potential zoonotic threats5,6,7,39. Surveillance of bat species has been particularly critical. For instance, >60 novel paramyxovirus sequences were identified in a 2012 bat surveillance study, several of which mapped to the Morbillivirus genus4. Recent metagenomic surveys confirm that bats harbour diverse orthoparamyxoviruses5,6,7. While comparing novel virus sequences with known pathogens may help inform the risks associated with future spillover events, this type of in silico modelling based on viral sequences should also be complemented by functional characterization of such viruses. In a previous study, we identified a full-length morbillivirus genomic sequence from M. riparius bats in Brazil7. Here we generated an infectious clone of this virus using reverse genetics. Furthermore, no recombinant virus was detected during the sequencing experiments. With this approach, we circumvented the arduous process of isolating and culturing live virus directly from animals and instead produced MBaMV in the lab.
MBaMV characterized as a morbillivirus
Before this study, there were only seven International Committee on Taxonomy of Viruses-recognized morbilliviruses species, none of which was isolated from bats. While the annotated MBaMV genome aligned with the classic morbillivirus genome organization (N, P/V/C, M, F, RBP and L), it was important to verify that virus generated by reverse genetics successfully recapitulated morbillivirus biology. Fusion assays and entry experiments confirmed that MBaMV preferentially used myotis CD150 over human or dog CD150 to enter transgenic Vero cells (Fig. 3), which fits the paradigm that CD150 is the major determinant of host specificity for morbilliviruses. We also assessed P-editing—a hallmark of paramyxoviruses—and found RNA editing of P-mRNA, creating V-mRNA (single G insertion) or W-mRNA (double G insertion) of MBaMV. Interestingly, the proportion of V-mRNA at 51.2% of total P transcripts is unusually high for orthoparamyxoviruses, resembling the now extinct rinderpest virus more than extant morbilliviruses40.
In their natural hosts, morbillivirus are highly pathogenic and can cause deadly acute infections41. Thus, one reasonable prediction is that MBaMV would cause visible disease in the bat host. However, when we challenged Jamaican fruit bats with MBaMV, we found the virus was not able to cause systemic disease in the bats (Extended Data Fig. 6) and there was no evidence that MBaMV productively infected these bats. This lack of infection could be due to the CD150 differences between the species—CD150 of Jamaican fruit bats and Myotis species is only 70% conserved on the amino acid level (Extended Data Fig. 8). We predict that MBaMV infection is more likely to cause serious disease in the M. riparius species. Alternatively, it is also possible that bat morbilliviruses do not cause severe illness in their hosts since bats possess unique immune systems that allow them to harbour deadly viruses such as Nipah, Ebola and severe acute respiratory syndrome without exhibiting illness42.
Zoonotic potential of MBaMV?
When assessing the zoonotic potential of a novel virus, multiple factors must be considered, including receptor usage, the existence of cross-neutralizing antibodies in human sera, and interactions with the innate immune system. While non-human morbilliviruses are not currently known to jump the species barrier and infect humans, we did find that MBaMV was able to utilize human receptors in vitro to a certain extent. Traditionally, morbilliviruses use CD150 to enter myeloid and lymphoid cells. However, unlike MeV, which infects human macrophages via CD150, MBaMV infects human macrophages in a CD150-independent manner (Fig. 4c)43. This result indicates that a non-CD150/nectin-4 entry receptor for MBaMV exists on human macrophages. In addition, MBaMV replicated well in H441 cells and in Vero cells expressing human nectin-4 (Fig. 3). CDV is also reported to use human nectin-423 and can replicate in H358 cells44. Alarmingly, there have been several outbreaks of CDV in non-human primates, resulting in acute disease or death in the animals34. In one outbreak, mutations were found in the RBP, which rendered CDV-RBP capable of efficiently using primate-CD150 (ref. 23). However, CDV is unlikely to adapt to humans in the presence of cross-reactive MeV immunity. Human sera from MMR-vaccinated individuals was able to cross-neutralize MBaMV infection in vitro (Fig. 5)—this would probably limit MBaMV infection if MMR-vaccinated humans were exposed to the virus. Finally, while MeV P and V proteins antagonize the innate immune response, MBaMV P and V were unable to block the IFN induction or signalling (Extended Data Fig. 9). Taken together, our findings suggest that the zoonotic potential for MBaMV is low due to cross-neutralizing anti-MeV antibodies and innate immune restriction. The former reinforces the need to maintain broad and high coverage of measles vaccination even when the virus has been eliminated in human populations.
Methods
Ethics declaration
Animal study was performed following the Guide for the Care and Use of Laboratory Animals. Animal experiment was approved by the Institutional Animal Care and Use Committee of Colorado State University (protocol number 1090) in advance and conducted in compliance with the Association for the Assessment and Accreditation of Laboratory Animal Care guidelines, National Institutes of Health regulations, Colorado State University policy and local, state and federal laws. Archival CDV hyperimmune ferret sera were obtained from previous animal experiments approved by the Institutional Animal Care and Use Committee of Georgia State University (IACUC number A22035).
Biosafety
All research was performed with Institutional Biosafety Committee approval (protocol number SPROTO202100000065 and preceding approvals) at Biosafety Safety Level 2+ (BSL2+). Morbilliviruses are classified as BSL2 agents. The present work does not come under Dual Use Research of Concern regulations since MBaMV is not listed as one of the 15 agents officially recognized as Dual Use Research of Concern agents. No mutagenic experiments to enhance the tropism of MBaMV were performed. For risk assessment, experiments were performed in a sequential fashion to minimize any risks involved.
First, we examined whether the RBP of MBaMV uses the canonical human lymphoid receptor (CD150), which is the receptor responsible for transmission, using a fusion assay (Fig. 1b) and pseudotyped virus assay (Fig. 1c). Only after confirmation that MBaMV did not use human CD150 in both assays, suggesting that any zoonotic risks were low, did we attempt full-length virus rescue at BSL2+. In addition, our experiments were conducted under our standard operating procedure, which requires that experiments are halted if any unexpected virulent phenotypes are seen and then reported to the IBC. Further risk mitigation factors include the requirement that all personnel have positive MMR titres.
Method to isolate bat morbillivirus sequence
MBaMV was identified as part of a previous metagenomic survey of bats sampled in Brazil and Malaysia. All the metadata associated with this study, including the number and species of bats sampled, can be found in Wells et al.7. The virus was sampled by rectal swab from a bat that was a subadult male (immature, but independent) and apparently healthy. Mitochondrial DNA profiling (MW554523 and MW557650) identified the bat as a riparian myotis (M. riparius). RNA was subjected to NGS analysis, and viral genome (MW557651) was assembled from fastq read files (GSE166170). The bat was captured by mist net, then oral, rectal and urogenital swabs were all collected for RNA extraction. Total nucleic acid was extracted using the Roche MagNA Pure 96 platform following the manufacturer’s protocol, then total nucleic acid was DNase treated (DNase I; Ambion, Life Technologies) and reverse transcribed using SuperScript III (Invitrogen, Life Technologies) with random hexamer primers. The cDNA was treated with RNase H before second-strand synthesis by Klenow fragment (3′ to 5′ exonuclease) (New England Biolabs), then the double-stranded cDNA was sheared into average of 200 bp fragments using a Covaris focused ultrasonicator E210. Sheared cDNA was deep sequenced using the Illumina HiSeq 2500 platform, and reads were bioinformatically de novo assembled using MEGAHIT v1.2.8 after quality control steps and exclusion of host reads using Bowtie2 v2.3.5 (ref. 45). This method was the same as one previously published7.
Generation of phylogenetic tree and conservation matrix table
Amino acid sequences of L proteins were aligned by ClustalW, then the evolutionary history of L proteins was inferred by maximum likelihood method with bootstrap test of 1,000 replicates. All processes were done in MEGA X (ref. 46). For conservation matrix table, amino acid sequences of each gene were aligned by ClustalW, then the conservations were evaluated. The accession numbers used for the alignment are summarized in Extended Data Table 2.
Cells
293T cells (ATCC cat. no. CRL-3216), A549 cells (ATCC cat. no. CCL-185), Vero cells (ATCC cat. no. CCL-81, RRID:CVCL_0059) and BSR T7/5 cells (RRID:CVCL_RW96) were grown in in Dulbecco’s modified Eagle medium (DMEM, Thermo Fisher Scientific) supplemented with 10% foetal bovine serum (FBS, Atlanta Biologicals) at 37 °C. NCI-H441 cells (ATCC cat. no. HTB-174) were grown in RPMI 1640 medium (Thermo Fisher Scientific) with 10% FBS. 293T cells, A549 cells and H441 cells were all certified using the ATCC cell authentication testing service. Vero-hCD150 (Vero-human SLAM) cells are Vero cell derivatives that constitutively express hCD150. Vero-dCD150 cells are Vero cell derivatives that constitutively express HA-dCD150. Vero-hCD150 cells47 and Vero-dCD150 cells48 were provided by Dr Yanagi at Kyushu University and maintained in DMEM with 10% FBS. Vero-bCD150 cells and Vero-hN4 cells were generated as written below and maintained in DMEM with 10% FBS. CHO cells were grown in DMEM/F12 (1:1) medium (Gibco) with 10% FBS.
Plasmids
We cloned the ORF of hCD150, dCD150 and bCD150 (from Myostis brandtii since the CD150 sequence from M. riparius is unknown) into the pCAGGS vector cut by EcoRI (NEB) and NheI-HF (NEB). We introduced HA tag-linker-Igk signal peptides (amino acids corresponding to MVLQTQVFISLLLWISGAYG-YPYDVPDYA-GAQPARSP) at the N-terminus of CD150s as previously reported49. The sequence of hCD150, dCD150 and bCD150 was from NP_003028.1, NP_001003084.1 and XP_014402801.1, respectively. We synthesized codon-optimized gene sequences at GeneArt Gene Synthesis (Invitrogen), generating pCAGGS-Igk-HA-hCD150, pCAGGS-Igk-HA-dCD150 and pCAGGS-Igk-HA-bCD150. We also generated pCAGGS-Igk-HA-bCD150-P2A-Puro, which additionally express puromycin-resistant gene. For pCAGGS-human nectin-4-P2A-puro, synthesized DNA by GeneArt Gene Synthesis (Invitrogen) was cloned into pCAGGS.
The sequence of MBaMV RBP and F ORF were synthesized by GenScript. These were cloned into pCAGGS vector cut by EcoRI and NheI-HF with adding HA tag (RBP gene) or AU1 tag (F gene) in C-terminus, generating pCAGGS-MBaMV-RBP-HA and pCAGGS-MBaMV-F-AU1.
For MeV RBP and F-expressing plasmid, we amplified RBP and F sequence from p(+) MV323-AcGFP with the addition of HA-tag and AU1-tag same as MBaMV-RBP and MBaMV-F, creating pCAGGS-MeV-RBP-HA and pCAGGS-MeV-F-AU1. For CDV RBP and F cloning, we amplified RBP and F sequence from pCDV-5804P plasmid with the addition of HA-tag and AU1-tag, creating pCAGGS-CDV-RBP-HA and pCAGGS-CDV-F-AU1.
Genome coding plasmids for MeV; (p(+) MV323-AcGFP) and CDV; pCDV-5804P were kindly gifted by Dr Makoto Takeda50 and Dr Veronica von Messling, respectively51. We transferred the MeV genome sequence into pEMC vector, adding an optimal T7 promotor, a hammer head ribozyme, and we introduced an eGFP transcriptional unit at the head of the genome (pEMC-IC323-eGFP), which is reported in the previous study19.
For the generation of MBaMV genome coding plasmid, we synthesized pieces of DNA at 2,000–6,000 bp at GenScript with the addition of eGFP transcriptional unit at the head of genome (eGFP-MBaMV). DNA fragments were assembled into pEMC vector one-by-one using in-fusion HD cloning kit (Takara), generating pEMC-eGFP-MBaMV. The N-terminal 1.5 kb of the L gene was initially unclonable. Sequence analysis revealed a putative 86-amino-acid (a.a.) ORF (ORF-X) in the complementary strand. Introduction of two point mutations in this region to disrupt ORF-X without affecting the L amino acid sequence (Extended Data Fig. 4) finally enabled cloning of the full-length genome, suggesting that ORF-X was probably toxic in bacteria.
Recovery of recombinant MBaMV and MeV from cDNA
For the recovery of recombinant MBaMV, 4 × 105 BSR-T7 cells were seeded in six-well plates. The next day, the indicated amounts (written below) of antigenomic construct, helper plasmids (-N, -P and -L from MeV), T7 construct and LipofectamineLTX / PLUS reagent (Invitrogen) were combined in 200 ml Opti-MEM (Invitrogen). After incubation at room temperature for 30 min, the DNA–Lipofectamine mixture was added dropwise onto cells. The cells were incubated at 37 °C for 24 h. The cells containing P0 viruses were trypsinized and passed onto Vero-bCD150 cells (2.0 × 106 cells per flask in one 75 cm2 flask). We collected supernatant 2 days after overlay (P1 virus) and re-amplified MBaMV in fresh Vero-bCD150 cells (>2× T1 75 cm2 flasks). These passage 2 (P2) stocks were titred, frozen down in aliquots and used for all experiments.
The amount of measles plasmids used for rescue is reported in our previous study52: 5 mg antigenomic construct, 1.2 mg T7-MeV-N, 1.2 mg T7-MeV-P, 0.4 mg T7-MeV-L, 3 mg of a plasmid encoding a codon-optimized T7 polymerase, 5.8 ml PLUS reagent and 9.3 ml Lipofectamine LTX.
The rescue of MeV was done exactly same way as MBaMV rescue except that 5 mg of pEMC-IC323eGFP was used for transfection and Vero-hCD150 cells were used for co-culturing.
Titration of viruses and plaque assay
For MBaMV, a monolayer of Vero-bCD150 cells in 12 wells was infected by 500 ml of serially diluted samples for 1 h, followed by medium replacement with methylcellulose containing DMEM. Then, 5 d.p.i., the number of GFP-positive plaque was counted to determine titre. For the plaque assay, infected Vero-bCD150 cells were incubated under methylcellulose-containing DMEM for 7 days. Cells were then stained with 1% crystal violet and 1% neutral red sequentially. For MeV, we used Vero-hCD150 cells and fixed the plates at 4 d.p.i.
Growth analysis
A total of 2.0 × 105 cells per well were seeded in a 12-well plate. Cells were infected by indicated titre of viruses (MOI 0.01 or 0.5) for 1 h, followed by replacement of fresh medium. Viruses were grown for 5 days with medium change every day. Collected supernatants were used for titration.
Generation of Vero-bCD150 cells and Vero-hN4 cells
A total of 4.0 × 105 of VeroCCL81 cells were transfected with 2 mg of pCAGGS-Igk-HA-bCD150-P2A-Puro with Lipofectamine 2000 (Invitrogen); cells were selected under 5 mg ml−1 of puromycin (Gibco) until colonies were visible. Colonies were isolated independently and checked for HA expression using fluorescence-activated cell sorting (FACS). Vero-hN4 cells were generated by transfecting pCAGGS-human nectin-4-P2A-Puro into VeroCCL81 cells, followed by 5 mg ml−1 of puromycin selection, and clone isolation. Surface expression was checked by FACS.
Generation of VSV-pseudotyped virus and entry assay
A total of 6 × 106 cells of 293T were seeded in a 10 cm dish (pre-coated with poly-l-lysine (Sigma)) 1 day before transfection. Twelve milligrams of RBP plus 12 mg of F coding plasmid from MeV, CDV or MBaMV were transfected to cells by PEI MAX (Polysciences). VSV-deltaG-Gluc supplemented by G protein (VSVDG-G*) were infected at an MOI of 10 for 1 h at 8 h post plasmid transfection. Cells were washed with phosphate-buffered saline (PBS) three times, and medium was maintained with Opti-MEM for 48 h. Supernatant was collected and ultracentrifuged at 30,000g for 2 h, and the pellet was re-suspended with 100 µl of PBS53. For the quantification of pseudotyped viral entry, CHO cells in 10 cm dish were transfected with 24 mg of hCD150-, dCD150- or bCD150-expressing plasmid with PEI MAX. CHO cells were passaged onto 96-well plates at 8 h post transfection The pseudotyped-VSV of MeV, CDV or MBaMV were used to infect the CHO cells. Renilla luciferase units (RLUs) were measured by Renilla luciferase assay system (Promega) to quantify the pseudotype virus entry into cells.
Image-based fusion assay
CHO cells were seeded at 50,000 cells in 48-well dish 24 hours before transfection. Cells were transfected with 200 mg of pCAGSS-RBP-HA (of MeV/CDV/MBaMV), 200 mg of pCAGGS-F-AU1(of MeV/CDV/MBaMV), pCAGGS-Igk-HA-CD150 (20 ng human, 5 ng dog, or 20 ng bat), and 50 mg of pEGFP-C1 Lifeact-EGFP (purchased from Addgene) with 2.5 ml of polyethylenimine max (Polysciences). At 36 h post transfection, cells were imaged with a Celigo imaging cytometer (Nexcelom) with the GFP channel, and pictures were exported at the resolution of 5 µm per pixel. The GFP-positive foci (single cell or syncytia) were analysed by ImageJ (developed by NIH), creating the profile of individual GFP-positive foci with size information.
For the evaluation of syncytia size, we first filtered the GFP-positive foci with the size of ≥10 pixel2, which is the median size of GFP area in the well of MeV-F plus LifeactGFP transfection to exclude non-specific background noise. Then we calculated the frequency of syncytia which is defined as the GFP counts of ≥100 pixel2 (10 times of median size of single cells)/total GFP counts of ≥10 pixel2.
Surface expression check of bCD150 in Vero-bCD150 cells and human nectin-4 in Vero-hN4 cells by FACS
A total of 50,000 cells in a 96-well plate were dissociated with 10 µM EDTA in Dulbecco’s PBS, followed by a 2% FBS in Dulbecco’s PBS block. Cells were treated with primary antibody for 1 h at 4 °C, then washed and treated by secondary antibody for 1 h at 4 °C. Vero-bCD150 cells were examined with a Guava easyCyte flow cytometers (Luminex) for the detection of signal. Vero-hN4 cells were subjected to Attune NxT flow cytometer (ThermoFisher Scientific). For primary antibody, mouse monoclonal nectin-4 antibody (clone N4.61, Millipore Sigma) and rabbit polyclonal HA tag antibody (Novus Biologicals) were both used at a 1:1,000 dilution. For secondary antibody, goat anti-rabbit IgG H&L Alexa Fluor 647 (Abcam) and goat anti-mouse IgG H&L Alexa Fluor 647 (Abcam) were used appropriately. FlowJo was used for analysing FACS data and presentation.
Soluble CD150 production and purification
Production and purification of soluble CD150 is as previously reported54. Soluble CD150 is a chimaera comprising the human V (T25 to Y138) and mouse C2 domains (E140 to E239) + His6-tag, which was cloned into pCA7 vector. The expression plasmid was transfected by using polyethyleneimine, together with the plasmid encoding the SV40 large T antigen, into 90% confluent HEK293S cells lacking N-acetylglucosaminyltransferase I (GnTI) activity. The cells were cultured in DMEM (MP Biomedicals), supplemented with 10% fetal calf serum (FCS) (Invitrogen), l-glutamine and non-essential amino acids (Gibco). The concentration of FCS was lowered to 2% after transfection. The His6-tagged protein was purified at 4 days post transfection from the culture medium by using the Ni2+-NTA affinity column and Superdex 200 GL 10/300 gel filtration chromatography (Amersham Biosciences). The pH of all buffers was adjusted to 8.0. Soluble CD150 Fc fusion avitag was purchased from BPS Bioscience, and reconstituted by PBS.
Macrophage experiments
CD14+ monocytes were isolated from leukopaks purchased from the New York Blood Bank using the EasySep Human CD14 positive selection kit (StemCell #17858). For macrophage differentiation, CD14+ monocytes were seeded at 106 cells ml−1 and cultured in R10 medium (RPMI supplemented with FBS, HEPES, l-glutamine and penicillin–streptomycin) with 50 ng ml−1 of granulocyte–macrophage colony-stimulating factor (Sigma Aldrich G5035) in a 37 °C incubator. Media and cytokines were replaced 3 days post seeding. At 6 days post seeding, macrophages were infected with either MeV or MBaMV at 100,000 infectious units (IU) per 500,000 cells and were spinoculated at 300g for 1 h at room temperature. Virus inoculum was removed and cells were incubated in R10 medium with granulocyte–macrophage colony-stimulating factor at 37 °C. For imaging experiments, macrophages were fixed in 4% paraformaldehyde (PFA) at 30 h.p.i. and stained with 4′,6-diamidino-2-phenylindole (DAPI), and fluorescent and bright field images were captured on the Cytation 3 plate reader. For flow cytometry experiments, infected macrophages were stained for viability at 24 h.p.i. (LIVE/DEAD fixable stain kit from Invitrogen L34976), treated with human Fc block (BD Biosciences), stained with antibodies against CD14 (eBioscience clone 61d3; 1:100 dilution) and HLA-DR (eBioscience clone LN3; 1:75 dilution), fixed in 2% PFA, permeabilized with saponin and stained for intracellular CD68 (eBioscience clone Y1/82A; 1:100 dilution) and CD150 (eBioscience; 1:75 dilution). Soluble CD150 (1 mg ml−1) or CD150 Avi-tag (BPS Bioscience) were incubated with MeV or MBaMV for 15 min before infection for inhibition experiments. Stained macrophages were run through an Attune NxT Flow Cytometer, and data were analysed using FlowJo software (v10).
T-cell experiments
PBMCs were isolated from fresh blood donations obtained through the New York Blood Center using density centrifugation and a ficoll gradient. Isolated PBMCs were then resuspended in RPMI medium (10% FBS, 1% l-glutamine and 1% penicillin–streptomycin) and were stimulated for T-cell activation with concanavalin-A (ConA) at 5 µg ml−1 for 72 h. Following, cells were washed once with PBS and stimulated with 10 ng ml−1 of IL2 for 48 h. Cells were subsequently infected at an MOI of 0.2 with MeV or BaMV or were mock infected in 12-well plates at 106 cells ml−1. Cells were collected 24 h.p.i., stained with Invitrogen’s LIVE/DEAD Fixable dead cell far-red dye as per the manufacturer’s protocol, and analysed for eGFP expression by flow cytometry with an Attune NxT Flow Cytometer. Analysis was completed using FCSExpress-7. A total of two donors were utilized for this analysis, with the data from donor 1 shown in Fig. 4.
Western blot for RBP and F protein
A total of 1 × 106 293T cells were seeded onto collagen-coated six-well plate. 293T cells were transfected by 2 mg of pCAGGS, pCAGGS-MBaMV-RBP-HA or pCAGGS-MBaMV-F-AU1 using polyethylenimine max (Polysciences). Cells were washed with PBS, then lysed by RIPA buffer. Collected cytosolic proteins were run on 4–15% poly polyacrylamide gel (Bio-Rad, #4561086) and transferred onto polyvinylidene fluoride membrane (Fisher Scientific, #45-004-113), followed by primary antibody reaction and secondary antibody reaction. Rabbit polyclonal HA tag antibody (Novus Biologicals, #NB600-363; 1:1,000 dilution) and rabbit polyclonal AU1 epitope antibody (Novus Biologicals, #NB600-453; 1:1.000 dilution) was used for primary antibody for HA and AU1 tag detection, respectively. Rabbit monoclonal antibody (Cell Signaling Technology, #2118; 1:1,000 dilution) was chosen as primary antibody to detect GAPDH. Alexa Fluor 647-conjugated anti-rabbit antibody (Invitrogen, #A-21245; 1:2,000 dilution) was used as secondary antibody appropriately. Image capturing was done by Chemidoc MP (Bio-Rad).
Transcriptome analysis of MBaMV
A total of 4.0 × 105 Vero-bCD150 cells were infected by MBaMV at an MOI of 0.01. Cytosolic RNA was collected by 500 ml of TRIzol (Ambion) at 2 d.p.i. Collected cytosolic RNA was sequenced by direct RNA sequence by MinION (Oxford Nanopore Technologies) with some modifications in the protocol. First, we started library preparation from 3 mg of RNA. Second, we used SuperScript IV (Invitrogen) instead of SuperScript III. Sequencing was run for 48 h by using R9.4 flow cells. The fastq file was aligned to MBaMV genome sequence by minimap2, and coverage information was extracted by IGVtools.
Evaluation of P mRNA editing
Infection and RNA extraction was same as above (transcriptome analysis). One microgram of RNA was reverse transcribed by TetroRT (Bioline) with poly-A primer, followed by PCR with primer set of Pedit-f (sequence GGGACCTGTTGCCCGTTTTA) and Pedit-r (sequence TGTCGGACCTCTTACTACTAGACT). Amplicons were processed by using NEBNext Ultra DNA Library Prep kit following the manufacturer’s recommendations (Illumina), and sequenced by Illumina MiSeq on a 2 × 250 paired-end configuration at GENEWIZ. Base calling was conducted by the Illumina Control Software (HCS) on the Illumina instrument. The paired-end fastq files were merged by BBTools. These merged fastq files were aligned to the reference sequence using bowtie2, creating a SAM file, and we counted the number of P-editing inserts.
Neutralization assay
Vero-hCD150, Vero-dCD150 and Vero bCD150 cells were seeded in 96-well plates. Two groups of pooled human sera from people who previously received the MMR vaccine (three individuals per pool), and sera from ferrets vaccinated with CDV (courtesy of Richard Plemper) were heat inactivated for 30 min at 56 °C. Equal amounts of CDV, MeV and MBaMV (20,000 IU ml−1) were incubated with serial dilutions of the heat-inactivated sera for 15 min at room temperature. Virus and sera were then added to the Vero cells with the correct receptor and placed at 37 °C. At 20 h.p.i., the cells were imaged using a Celigo imaging cytometer (Nexelcom) with the GFP channel. Exported images were analysed using ImageJ to measure the extent of viral infection by GFP+ area (MeV and MBaMV), or total GPF+ counts (CDV). The percentage of reduction in infection was calculated by setting the level of infection in the no sera control wells to 100%. The normalized data were plotted using GraphPad Prism, and neutralization curves were generated using non-linear regression with inhibitor concentration versus normalized response. IC50 values were calculated for each replicate using a robust fit model. Five replicates were completed for the MeV and MBaMV neutralization with the pooled human sera, and two replicates were repeated with CDV and the ferret sera.
Research animals
Ferrets were vaccinated with CDV, and serum was collected and pooled from three female ferrets. This serum was used in viral neutralization assays. Although these archival sera were obtained as part of IACUC-approved studies (IACUC protocol A22035), the work has not been published elsewhere. Ferrets were kept in Animal Biosafety Level 1 in an open cage grouped housing environment at 20 °C (40% relative humidity) with weekly cage changes, food (Marshall Farms formulated diet) and water ad libitum, and a photoperiod of 14 h of light and 10 h of dark. These ferrets came immunized from the vendor (Tripple F farms) and were vaccinated (subcutaneously) at 10 weeks of age using a ribosomal DNA canarypox-vectored CDV vaccine, Purevax Ferret Distemper as per the manufacturer’s protocol (prime plus two additional doses at 3 week intervals). Immunized ferrets were bled at 8 months of age. After a single blood draw of 2 ml was collected from the jugular vein, the ferrets were retired with no adverse effects.
Jamaican fruit bats were sourced from the Colorado State University breeding colony that was established from donated zoo bats in 2006. This is a free-flight colony, and reproductive females produce one offspring at about 5–6 month intervals. Bats are fed daily with fresh fruit (cantaloupe, watermelon and banana) supplemented with monkey biscuits and iron-containing bird feed as additional protein and calorie sources. For experimental challenge with virus, bats were housed in a bird cage (76h × 56w × 56d cm) that allowed full extension of wings and movement. Landscape fabric is routinely used as a roosting substrate in cages. The bats were kept under a photoperiod of 12/12 h with ambient temperature maintained at 22 °C and relative humidity ranging from 35% to 50%. Bats were killed by inhalation of 3% isoflurane to effect, followed by thoracotomy.
IFN induction and response assays
For interferon stimulatory gene induction assays, HEK 293T cells were transfected with plasmids coding for ISG54-ISRE-FLuc, TK-RLuc and either empty vector, MBaMV P, MVaMV V or ZIKV MR766 NS5. At 24 h post-transfection, the cells with treated with 100 U of human IFNb (at 100 U ml−1). Cells were lysed 24 h after IFNb treatment, and FLuc and RLuc expression was measured using the Promega Dual luciferase assay. Data were calculated as a ratio of Fluc:RLuc to normalize for transfection efficiency. Two independent experiments with three technical replicates were completed. To measure the antagonism of IFN induction (IFNb promoter activation), HEK 293T cells were transfected with plasmids coding for IFNb-FLuc, TK-RLuc an IFN promoter stimulant (either RIG-I, MDA5 or MAVS), and empty vector, MBaMV P, MBaMV V or HCV NS3/4A (potential IFN antagonists). At 24 h post transfection, cells were lysed and FLuc and RLuc expression was measured by Promega Dual luciferase assay. Data were calculated as a ratio of Fluc:RLuc to normalize for transfection efficiency. Two independent experiments with three technical replicates were completed. For statistical analysis, one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons was performed with Prism.
Bat challenge experiment and evaluation of infection
Six adult male Jamaican fruit bats (Artibeus jamaicensis) were inoculated with 2 × 105 PFU MBaMV-eGFP; three bats were intranasally inoculated and three bats were intraperitoneally inoculated. At 1 week post virus inoculation, bats were subjected to blood and serum collection, visually inspected for GFP expression around the nares, oral cavity and eyes by LED camera in each group (intranasally and intraperitoneally) while being housed in bird cages. At 2 weeks post virus infection, blood, serum and tissues (lung, spleen and liver) were collected from one bat in each group. At 3 weeks post virus infection, blood, serum and tissues (lung, spleen and liver) were collected from one bat in each group. At the point of sample collection, the bats were killed and their tissues processed. Bats were sourced from and housed at the breeding colony at the Colorado State University.
Blood RNA was extracted by TRIzol. RNA was reverse transcribed by Tetro cDNA synthesis kit (Bioline) with the primer of ‘GAGCAAAGACCCCAACGAGA’ targeting MBaMV-GFP genome, then the number of genomes was quantified by SensiFAST SYBR & Fluorescein Kit (Bioline) and CFX96 Touch Real-Time PCR Detection System (Bio-Rad). The primer set for qPCR is ‘GGGGTGCTATCAGAGGCATC’ and ‘TAGGACCCTTGGTACCGGAG’.
Virus neutralization assay was done as follows. Heat-inactivated (56 °C × 30 min) bat serum was serially diluted by three times (starting from five times dilution) and mixed with 2 × 104 PFU ml−1 of MBaMV at a 1:1 ratio for 10 min at room temperature. One-hundred millilitres of mixture was applied to Vero-batCD150 cells in 96 wells. GFP foci were detected and counted by Celigo imaging cytometer (Nexcelom). GFP counts of serum-treated samples were normalized by no-serum-treated well.
Tissues were fixed with 10% buffered formalin and embedded with paraffin, then thinly sliced. GFP immunohistochemistry was performed by using VENTANA DISCOVERY ULTRA. Rabbit monoclonal antibody (Cell Signaling Technology, #2956) was used as a primary antibody, and OMNIMap anti-rabbit-HRP (Roche, #760-4310) was used as a secondary antibody. The GFP signal was visualized by using Discovery ChromoMap DAB Kit (Roche, #760-2513). Tissues were counterstained with haematoxylin to visualize the nuclei. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those previously published. Data collection and analysis were not performed blind to the conditions of the experiments.
In silico docking
In silico docking was performed with MOE 2018.1001 (Chemical Computing Group), as previously described38. A homology model of MBaMV L was created on the basis of the structural coordinates of PIV5-L (PDB ID: 6V86) using the SWISS-MODEL homology modelling server55. Before docking, the model of the MBaMV L protein was protonated and energy minimized. An induced-fit protocol using the Amber10 force field was implemented to dock ERDRP-0519 and GHP-88309 into MBaMV L. For binding of ERDRP-0519, residues Y1155, G1156, L1157, E1158 and H1288, and for binding of GHP-88309, residues E858, D863, D997, I1009 and Y1106, were pre-selected as docking targets, which are predicted to line the docking sites of ERDRP-0519 and GHP-88309, respectively, in MeV L. Top-scoring docking poses were selected and aligned in Pymol to the previously characterized in silico docking poses of the inhibitors to MeV L protein. Sequence alignment of MBaMV and MeV L proteins was performed using Clustal Omega56. Conservation was scored using the AL2CO alignment conservation server57,58.
TEM
Routine TEM processing was done as described. The Vero-bCD150 cells infected by MBaMV for 3 days were washed with PBS and then fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) on ice for 1 h. The cells were scraped off the 100 mm tissue-culture-treated Petri dish and pelleted by low-speed centrifugation (400g for 5 min). The pellet was fixed for 30 min with the same fixative before secondary fixation with 2% osmium tetraoxide on ice for 1 h. The cells were then stained with 2% uranyl aqueous solution en bloc for 1 h at room temperature, dehydrated with a series of increasing ethanol gradients followed by propylene oxide treatment, and embedded in Embed 812 Resin mixture (Electron Microscopy Sciences). Blocks were cured for 48 h at 65 °C and then trimmed into 70 nm ultrathin sections using a diamond knife on a Leica Ultracut 6 and transferred onto 200 mesh copper grids. Sections were counterstained with 2% uranyl acetate in 70% ethanol for 3 min at room temperature and in lead citrate for 3 min at room temperature, and then examined with a JEOL JSM 1400 transmission electron microscope equipped with two CCD cameras for digital image acquisition: Veleta 2K × 2K and Quemesa 11 megapixel (EMSIS) operated at 100 kV.
Statistics and reproducibility
All statistical methods used in analysis are listed in the accompanying figure legends. Analyses were computed using GraphPrad Prism (v10). Data distribution was assumed to be normal, but this was not formally tested. Regarding the bat challenge study, no statistical method was used to pre-determine sample size and no data were excluded from the analysis.
Human subjects research
Normal primary dendritic cells and macrophages used in this project were sourced from ‘human peripheral blood Leukopack, fresh’, which is provided by the commercial provider New York Blood Center. Leukapheresis was performed on normal donors using institutional review board-approved consent forms and protocols by the vendor. The vendor holds the donor consents and the legal authorization that should give permission for all research use. The vendor is not involved in the study design and has no role in this project. Samples were deidentified by the vender and provided to us. To protect the privacy of donors, the vendor does not disclose any donor records. If used for research purposes only, the donor consent applies. Aliquots of pooled immune sera were obtained from a previous anonymous serosurvey study that was qualified as Exemption 4 under NIH Exempt Human Subjects Research guidelines (Icahn School of Medicine at Mount Sinai)59. Serum samples were purchased from Innovative Research as deidentified research reagents. Serum was collected between August and November 2014 in Michigan, from donors aged 18–64 years. For the human serum, MuV-reactive titres were previously determined with enzyme-linked immunosorbent assay against recombinant MuV-F and MuV-HN in addition to neutralization titres using the recombinant vaccine strain of MuV (JL5). The sera samples used in this study were from donors that exhibited neutralization titres above the median titres for this cohort as determined previously59.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw NGS results of bat surveillance, P gene editing and transcriptome by MinION are uploaded at NCBI GEO: GSE166170, GSE166158 and GSE166172, respectively. Unprocessed images from our experiments have been uploaded to Figshare, and links are available in the source data. Assembled MBaMV sequence and pEMC-MBaMVeGFP sequence information are available at NCBI Genbank MW557651 and MW553715, respectively. Cytochrome oxidase I host sequence and cytochrome b host sequence of virus infected bat are available at MW554523 and MW557650. MeV genomic cDNA coding plasmid (pEMC-IC323eGFP) sequence is available at MW401770. Source data are provided with this paper.
References
Wang, L. F. & Anderson, D. E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 34, 79–89 (2019).
Han, H. J. et al. Bats as reservoirs of severe emerging infectious diseases. Virus Res. 205, 1–6 (2015).
Letko, M., Seifert, S. N., Olival, K. J., Plowright, R. K. & Munster, V. J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 18, 461–471 (2020).
Felix Drexler, J. et al. Bats host major mammalian paramyxoviruses. Nat. Commun. https://doi.org/10.1038/ncomms1796 (2012).
Vanmechelen, B. et al. The characterization of multiple novel paramyxoviruses highlights the diverse nature of the subfamily Orthoparamyxovirinae. Virus Evol. 8, veac061 (2022).
Larsen, B. B., Gryseels, S., Otto, H. W. & Worobey, M. Evolution and diversity of bat and rodent paramyxoviruses from North America. J. Virol. 96, e01098–21 (2022).
Wells, H. L. et al. Classification of new morbillivirus and jeilongvirus sequences from bats sampled in Brazil and Malaysia. Arch. Virol. https://doi.org/10.1007/s00705-022-05500-z (2022).
Amarasinghe, G. K. et al. Taxonomy of the order Mononegavirales: update 2017. Arch. Virol. 162, 2493–2504 (2017).
Arruda, B., Shen, H., Zheng, Y. & Li, G. Novel morbillivirus as putative cause of fetal death and encephalitis among swine. Emerg. Infect. Dis. 27, 1858–1866 (2021).
Morens, D. M., Holmes, E. C., Davis, A. S. & Taubenberger, J. K. Global rinderpest eradication: lessons learned and why humans should celebrate too. J. Infect. Dis. 204, 502 (2011).
Donduashvili, M. et al. Identification of peste des petits ruminants virus, Georgia, 2016. Emerg. Infect. Dis. 24, 1576 (2018).
Beineke, A., Puff, C., Seehusen, F. & Baumgärtner, W. Pathogenesis and immunopathology of systemic and nervous canine distemper. Vet. Immunol. Immunopathol. 127, 1–18 (2009).
Couacy-Hymann, E., Bodjo, C., Danho, T., Libeau, G. & Diallo, A. Evaluation of the virulence of some strains of peste-des-petits-ruminants virus (PPRV) in experimentally infected West African dwarf goats. Vet. J. 173, 178–183 (2007).
Bressem, M.-F. V. et al. Cetacean morbillivirus: current knowledge and future directions. Viruses 6, 5145 (2014).
Tatsuo, H., Ono, N., Tanaka, K. & Yanagi, Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406, 893–897 (2000).
Mühlebach, M. et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480, 530–533 (2011).
Thibault, P. A., Watkinson, R. E., Moreira-Soto, A., Drexler, J. F. & Lee, B. Zoonotic potential of emerging paramyxoviruses: knowns and unknowns. Adv. Virus Res. 98, 1–55 (2017).
Ohishi, K., Maruyama, T., Seki, F. & Takeda, M. Marine morbilliviruses: diversity and interaction with signaling lymphocyte activation molecules. Viruses 11, 606 (2019).
Ikegame, S. et al. Fitness selection of hyperfusogenic measles virus F proteins associated with neuropathogenic phenotypes. Proc. Natl Acad. Sci. USA 118, e2026027118 (2021).
Beineke, A., Baumgärtner, W. & Wohlsein, P. Cross-species transmission of canine distemper virus-an update. One Health 1, 49–59 (2015).
Yoshikawa, Y. et al. Natural infection with canine distemper virus in a Japanese monkey (Macaca fuscata). Vet. Microbiol. 20, 193–205 (1989).
Qiu, W. et al. Canine distemper outbreak in rhesus monkeys, China. Emerg. Infect. Dis. J. 17, 1541 (2011).
Sakai, K. et al. Lethal canine distemper virus outbreak in cynomolgus monkeys in Japan in 2008. J. Virol. 87, 1105–1114 (2013).
Nakai, M. & Imagawa, D. T. Electron microscopy of measles virus replication. J. Virol. 3, 187 (1969).
Mateo, M., Navaratnarajah, C. K. & Cattaneo, R. Structural basis of efficient contagion: measles variations on a theme by parainfluenza viruses. Curr. Opin. Virol. https://doi.org/10.1016/j.coviro.2014.01.004 (2014).
Lin, L. T. & Richardson, C. D. The host cell receptors for measles virus and their interaction with the viral hemagglutinin (H) protein. Viruses 8, 1–29 (2016).
Leonard, V. et al. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J. Clin. Invest. 118, 2448–2458 (2008).
Noyce, R. S. et al. Tumor cell marker PVRL4 (Nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 7, e1002240 (2011).
Cattaneo, R., Kaelin, K., Baczko, K. & Billeter, M. A. Measles virus editing provides an additional cysteine-rich protein. Cell 56, 759–764 (1989).
Wignall-Fleming, E. B. et al. Analysis of paramyxovirus transcription and replication by high-throughput sequencing. J. Virol. 93, 1–17 (2019).
Eick, G. N., Jacobs, D. S. & Matthee, C. A. A nuclear DNA phylogenetic perspective on the evolution of echolocation and historical biogeography of extant bats (Chiroptera). Mol. Biol. Evol. 22, 1869–1886 (2005).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Hashiguchi, T. et al. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat. Struct. Mol. Biol. 18, 135–142 (2011).
Kennedy, J. M. et al. Canine and phocine distemper viruses: species barriers. Viruses 11, 944 (2019).
Cox, R. M. et al. Orally efficacious broad-spectrum allosteric inhibitor of paramyxovirus polymerase. Nat. Microbiol. 5, 1232–1246 (2020).
Krumm, S. A. et al. An orally available, small-molecule polymerase inhibitor shows efficacy against a lethal morbillivirus infection in a large animal model. Sci. Transl. Med. 6, 1–11 (2014).
Abdella, R., Aggarwal, M., Okura, T., Lamb, R. A. & He, Y. Structure of a paramyxovirus polymerase complex reveals a unique methyltransferase-CTD conformation. Proc. Natl Acad. Sci. USA 117, 4931–4941 (2020).
Cox, R. M., Sourimant, J., Govindarajan, M., Natchus, M. G. & Plemper, R. K. Therapeutic targeting of measles virus polymerase with ERDRP-0519 suppresses all RNA synthesis activity. PLoS Pathog. 17, e1009371 (2021).
Li, B. et al. Discovery of bat coronaviruses through surveillance and probe capture-based next-generation sequencing. mSphere 5, e00807–e00819 (2020).
Douglas, J., Drummond, A. J. & Kingston, R. L. Evolutionary history of cotranscriptional editing in the paramyxoviral phosphoprotein gene. Virus Evol. 7, veab028 (2021).
De Vries, R. D., Paul Duprex, W. & De Swart, R. L. Morbillivirus infections: an introduction. Viruses 7, 699–706 (2015).
Banerjee, A. et al. Novel insights into immune systems of bats. Front. Immunol. 11, 26 (2020).
Minagawa, H., Tanaka, K., Ono, N., Tatsuo, H. & Yanagi, Y. Induction of the measles virus receptor SLAM (CD150) on monocytes. J. Gen. Virol. 82, 2913–2917 (2001).
Otsuki, N. et al. The V protein of canine distemper virus is required for virus replication in human epithelial cells. PLoS ONE 8, e82343 (2013).
Anthony, S. J. et al. Further evidence for bats as the evolutionary source of middle east respiratory syndrome coronavirus. mBio 8, 1–13 (2017).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Ono, N. et al. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J. Virol. 75, 4399–4401 (2001).
Seki, F., Ono, N., Yamaguchi, R. & Yanagi, Y. Efficient isolation of wild strains of canine distemper virus in Vero cells expressing canine SLAM (CD150) and their adaptability to marmoset B95a cells. J. Virol. 77, 9943–9950 (2003).
Tatsuo, H., Ono, N. & Yanagi, Y. Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J. Virol. 75, 5842–5850 (2001).
Seki, F. et al. The SI strain of measles virus derived from a patient with subacute sclerosing panencephalitis possesses typical genome alterations and unique amino acid changes that modulate receptor specificity and reduce membrane fusion activity. J. Virol. 85, 11871 (2011).
von Messling, V., Zimmer, G., Herrler, G., Haas, L. & Cattaneo, R. The hemagglutinin of canine distemper virus determines tropism and cytopathogenicity. J. Virol. 75, 6418–6427 (2001).
Beaty, S. M. et al. Efficient and robust paramyxoviridae reverse genetics systems. mSphere 2, e00376–16 (2017).
Oguntuyo, K. Y. et al. Quantifying absolute neutralization titers against SARS-CoV-2 by a standardized virus neutralization assay allows for cross-cohort comparisons of COVID-19 sera. mBio 12, 1–23 (2021).
Hashiguchi, T. et al. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl Acad. Sci. USA 104, 19535–19540 (2007).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 (2019).
Pei, J. & Grishin, N. V. AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics 17, 700–712 (2001).
Noda, T. et al. Importance of the 1+7 configuration of ribonucleoprotein complexes for influenza A virus genome packaging. Nat. Commun. 9, 1–10 (2018).
Beaty, S. M. et al. Cross-reactive and cross-neutralizing activity of human mumps antibodies against a novel mumps virus from bats. J. Infect. Dis. 215, 209–213 (2017).
Acknowledgements
S.I. was supported by Fukuoka University’s Clinical Hematology and Oncology Study Group (CHOT-SG). This study was supported in part by NIH grants U19 AI171403 and AI071002 (R.K.P and B.L.), AI149033 (B.L. and J.L.), AI140442 (T.S.), AI134768 (T.S.) and USAID PREDICT (S.J.A, J.H.E., P.D. and E.D.). J.C.C. J.A.A., K.Y.O, A.P. and C.S.S. acknowledge support from T32 AI07647. K.Y.O. was additionally supported by F31 (AI154739). J.A.A was additionally supported by F31 (HL149295). J.C.C. was additionally supported by F32 (HL158173). This work was also supported by Japan Agent for Medical Research and Development (AMED) Grant 20wm0325002h (T.H.), JSPS KAKENHI grant number 20H03497 (T.H.) and Joint Usage/Research Center programme of Institute for Frontier Life and Medical Sciences, Kyoto University (S.I.). T. Yanagi at Kyushu University kindly provided the Vero-hCD150 and Vero-dCD150 cells. M. Takeda (National Institute of Infectious Diseases, Tokyo, Japan) and V. von Messling (German Federal Ministry of Education and Research, Berlin, Germany) kindly provided the genome coding plasmids for MeV and CDV (respectively). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.I., S.J.A. and B.L. conceived this study. S.I. conducted fusion assay, rescuing viruses, growth analysis, RNA sequencing of transcriptome analysis and generation of cell lines written in the study. R.L.F.O. conducted TEM imaging. J.C.C., J.A.A., A.R.P. and J.K.L. performed the macrophage and T-cell experiments and data analysis. J.C.C. performed the sera neutralization assays. K.Y.O. conducted VSV-pseudotype entry assay. R.M.C. and R.K.P. provided ERDRP-0519 and GHP-88309 in addition to in silico modelling of MBaMV-L. H.-P.C. evaluated protein production by western blot. T.H. provided structure-guided insights into conservation of R.B.P. and CD150 binding as well as soluble human CD150 for inhibition assay. K.Y.O. and S.K. evaluated surface expression of morbillivirus receptors. C.S.S. evaluated P-mRNA editing frequency from NGS data. T.S., M.E. and S.Z. performed bat challenge experiment. E.D. conducted bat surveillance in collaboration with J.H.E. and P.D. S.J.A. and H.W. conducted NGS analysis of bat surveillance and retrieved MBaMV sequences. M.J.E. and E.C.V. performed the IFN response and Induction experiments. J.H.E., P.D. and S.J.A. provided insights into viral ecology and zoonotic threats. B.L. supervised this study. S.I., J.C.C., S.J.A. and B.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Roberto Cattaneo, Bert Rimaand and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 The genomic composition of myotis bat morbillivirus (MBaMV) and its phylogeny.
a, Schematic representation of the MBaMV genome and its encoded genes. The recombinant MBaMV-eGFP generated in this study is also shown. eGFP transcriptional unit is depicted as ‘G’ (green box). # indicates the position in L where silent mutations were introduced to prevent expression of putative ORF-X that appeared toxic in bacteria (Extended Data Fig. 3). The nucleotide scale bar is provided for context. b, The entire amino acid sequence of the L protein was used for constructing the phylogenetic tree of representative viruses from the three major subfamilies of Paramyxoviridae. L protein sequences were first aligned by clustalw, then the phylogenetic tree was generated by the maximum likelihood method using MEGA 10 (version 10.1.8). The numbers at each node indicate the fidelity by bootstrap test (1,000 times). MBaMV (red arrowhead) was most closely related to canine distemper virus (CDV) and phocine distemper virus (PDV) until the recent discovery of porcine morbillivirus (PoMV). The scale indicates substitutions per site. The accession numbers for the sequences used in this phylogenetic analysis are shown at Table S2.
Extended Data Fig. 2 Comparison of protein amino acid sequence conservation to known morbilliviruses.
The percent amino acid similarity (% AA) of MBaMV proteins compared to the 7 established morbilliviruses are plotted to visualize the degree of conservation across the genus. N, M, F, and L showed relatively high conservation (50 – 80%) to all morbilliviruses with the exception FeMV. P, V, C and RBP showed 30-50% conservation to all morbilliviruses again with the exception of FeMV. ClustalW was used to align the cognate amino acid sequences from each morbillivirus and the % AA similarity was calculated using default settings. The accession number of the sequences used are indicated in Table S1.
Extended Data Fig. 3 Deletion of putative cryptic toxic ORF in MBaMV L gene.
The first 1,500 nt of the L gene was refractory to cloning. Inspection of the L gene sequence revealed a putative ORF of 86 aa in the complementary (-) strand of the first 1,500 nt of the L gene (a, original sequence and b, ORF-X). To determine if this putative ORF might be toxic to the bacteria used for cloning the MBaMV genome, we made two mutations that were silent in the L ORF but which abolished the initiator methionine(s) in this putative ORF-X. Red shaded nucleotides correspond to third codon of methionine in ORF-X (a, Deleted ORF-X). This “ORF-X KO” MBaMV genomic clone was the eventual construct that was rescued. The amino acid sequence of ORF-X is shown in (b).
Extended Data Fig. 4 Vero cells stably expressing permissive receptors for MBaMV.
Vero cells stably expressing Myotis brandtii CD150 (bCD150) and human nectin-4 were made as described in methods. a, FACS staining for surface expression of HA-tagged bCD150 on Vero-bCD150 cells. The HA-tag was placed immediately after the signal peptide sequence. b, shows the human nectin-4 surface expression by FACS. Parental Vero cells and H441 (human lower airway epithelial) cells were used as negative and positive controls for human nectin-4, respectively. Greater than 50% of Vero-Nectin-4 cells express higher levels of human nectin-4 than H441 cells. c, Monocyte derived macrophages were harvested and stained for surface human nectin-4, confirming that macrophages do not express nectin-4. Vero-Nectin-4 cells were used as a positive control.
Extended Data Fig. 5 Molecular characterization of MBaMV transcripts.
Vero-bCD150 cells were infected with MBaMV (MOI = 0.01) and cytosolic RNA collected at 2 dpi were subjected to direct RNA sequencing targeting mRNAs using the MinION platform (Oxford Nanopore Technology). 93,917 reads / total 797,133 reads are aligned to MBaMV. a, Read coverage showed that MbaMV infection and replication exhibited the 3’ to 5’ transcriptional gradient typical of paramyxoviruses. The boundary of each transcriptional unit can also be appreciated by the steep drop in reads that coincide with the tri-nucleotide intergenic sequence. Median (IQR) sequencing coverage ranges are 6509 (N, 5368-7465), 3640 (P, 3193-4047), 3018 (M, 2533-3656), 929 (F, 722-1096), 535 (RBP, 491-633) and 10 (L, 5-33). b, The read coverage graph around the M-F intergenic region is magnified to show the uncommon intergenic motif of ‘CGU’. The intergenic motif between all other genes is ‘CUU’. c, Amplicon sequencing around the P gene editing site shows the frequency of P gene-mRNA editing. 51.2% of P gene mRNAs acquired a single ‘G’ insertion at the editing site, creating V mRNA. The numbers shown are the average of three independent experiment with error bars representing S.D. (d-e) show the expression of surface glycoproteins (RBP and F) of MBaMV, MeV, and CDV by Western blot. 5 μg expression plasmids (pCAGGS, pCAGGS-RBP-HA, pCAGGS-F-AU1) were transfected into 293 T in 6-well plate. Cytosolic proteins were collected and run on acrylamide gel. AU1 staining (d) showed full-length F protein (F0; blue arrowhead) and cleaved F1 (red asterisk) product. HA staining (e) showed expression of RBP monomer (blue arrowhead) around 60 – 70 kDa in addition to oligomer. COXIV was used as loading control for blot. The experiments in d) and e) were completed twice, with similar results.
Extended Data Fig. 6 Summary of bat challenge experiment.
a, 6 Jamaican fruit bats (Artibeus jamaicensis) were inoculated with MBaMV (2×105 PFU/body) intranasally (I.N.) or intraperitoneally (I.P.). Blood, serum, and tissues (lung, liver, and spleen) were collected as indicated. GFP expression in the tissues were checked by GFP-IHC. b, MeV infected cell pellet (positive control) showed strong GFP IHC signal. c, bat-derived tissues showed no GFP signal. The bat challenge experiment was completed one time. All scale bars set to 50 µM.
Extended Data Fig. 7 Amino acid sequence alignment of morbillivirus receptor binding proteins (RBP).
The RBP of 8 morbilliviruses and other representative paramyxoviruses (Sendai virus (SeV), Nipah virus (NiV), Mumps virus (MuV)) were aligned by clustalw. Alignments around the CD150 contact residues are shown separately in (a)180-200 aa, (b) 490-510 aa, and (c) 520-560 aa. Highly and moderately conserved residues in these regions are shaded in black and gray, respectively. Occluded residues at the RBP-CD150 interface from the MeV-marmoset CD150 crystal structure were identified by PDBePISA and indicated by the peach-colored shading above the alignment. Hydrogen bonds (deep blue), salt bridges (red), hydrogen bonds + salt bridge (purple) in these occluded regions are indicated.
Extended Data Fig. 8 Amino acid sequence alignment of CD150 from cognate morbillivirus hosts.
CD150 from the 8 mammals that are the natural hosts of the indicated morbilliviruses are aligned by clustalw. Alignments around the RBP contact residues (aa60- 179) are shown. Highly and moderately conserved residues in these regions are shaded in black and gray, respectively. Occluded residues at the RBP-CD150 interface (PDB: 3ALZ) were identified by PDBePISA and indicated by the peach-colored shading above the alignment. Hydrogen bonds (deep blue), salt bridges (red), hydrogen bonds + salt bridge (purple) in these occluded regions are indicated. The color rendering is identical to Extended Data Figure 7.
Extended Data Fig. 9 MBaMV accessory genes do not antagonize human IFN induction.
a) HEK 293 T cells were co-transfected with an ISG54-ISRE-FLuc plasmid and either MBaMV P, MBaMV V, MeV V, MeV P, or ZIKV NS5. Cells were treated with human IFNb and ISRE induction was measured by luciferase assay. HEK 293 T cells were co-transfected with an IFNb-FLuc plasmid, an IFN promoter stimulant b) RIG-I, c) MDA5, and d) MAVS, and MeV P, MeV V, MBaMV P, MBaMV V, or HCV NS3/4 A. IFN induction was measured by luciferase assay in 2 independent experiments with 3 technical replicates (mean + /- S.D.). The specific p values (* p < 0.5; **** p =< 0.0001) were calculated using a one-way ANOVA with a Dunnett’s multiple comparisons test.
Supplementary information
Supplementary Information
Flow cytometry gating figure.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ikegame, S., Carmichael, J.C., Wells, H. et al. Metagenomics-enabled reverse-genetics assembly and characterization of myotis bat morbillivirus. Nat Microbiol 8, 1108–1122 (2023). https://doi.org/10.1038/s41564-023-01380-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-023-01380-4
