
Abstract
Oncogenic KRAS mutations occur in approximately 30% of lung adenocarcinoma. Despite several decades of effort, oncogenic KRAS-driven lung cancer remains difficult to treat, and our understanding of the regulators of RAS signalling is incomplete. Here to uncover the impact of diverse KRAS-interacting proteins on lung cancer growth, we combined multiplexed somatic CRISPR/Cas9-based genome editing in genetically engineered mouse models with tumour barcoding and high-throughput barcode sequencing. Through a series of CRISPR/Cas9 screens in autochthonous lung cancer models, we show that HRAS and NRAS are suppressors of KRASG12D-driven tumour growth in vivo and confirm these effects in oncogenic KRAS-driven human lung cancer cell lines. Mechanistically, RAS paralogues interact with oncogenic KRAS, suppress KRAS–KRAS interactions, and reduce downstream ERK signalling. Furthermore, HRAS and NRAS mutations identified in oncogenic KRAS-driven human tumours partially abolished this effect. By comparing the tumour-suppressive effects of HRAS and NRAS in oncogenic KRAS- and oncogenic BRAF-driven lung cancer models, we confirm that RAS paralogues are specific suppressors of KRAS-driven lung cancer in vivo. Our study outlines a technological avenue to uncover positive and negative regulators of oncogenic KRAS-driven cancer in a multiplexed manner in vivo and highlights the role RAS paralogue imbalance in oncogenic KRAS-driven lung cancer.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
The human cancer genomic data analysed for the presence of rare HRAS and NRAS variants in this manuscript were derived from the AACR’s Project GENIE (https://www.aacr.org/professionals/research/aacr-project-genie/) Release 9.1-public dataset. All data files that support the findings of this study are available through the Synapse platform (https://www.synapse.org/#!Synapse:syn24179657). Human cancer genomic data analysed for alterations in KRAS-interacting proteins were derived from the TCGA PanCancer Atlas dataset, which is publicly available through cBioPortal at https://www.cbioportal.org/study/summary?id=luad_tcga_pan_can_atlas_2018. The protein templates used to model RAS heterodimers in this study are available through the ProtCID database (KRASG12D: PDB entry 5USJ, HRAS homodimer: PDB entry 3K8Y), and resulting structural data files can be accessed through Zenodo (https://zenodo.org/record/7104280). AP/MS data were derived from ref. 10 (PMID: 32727735) and ref. 11 (PMID: 32929263). Gene expression data were derived from ref. 21 (PMID: 28191885). Pre-existing CRISPR/Cas9 screen data were derived from ref. 15 (PMID: 32238925). All BC sequencing datasets are available through the NCBI’s Sequence Read Archive database under the BioProject accession number PRJNA763983 (https://www.ncbi.nlm.nih.gov/sra/PRJNA763983). Processed data plotted in figures are available in the Extended Data Information. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
The code used for data analysis in this study is available on GitHub (https://github.com/eshuldiner/KrasIP).
References
Karnoub, A. E. & Weinberg, R. A. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 9, 517–531 (2008).
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J. & Der, C. J. Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851 (2014).
Zhou, B., Der, C.J. & Cox, A.D. in Seminars in Cell & Developmental Biology Vol. 58, 60–69 (Elsevier, 2016).
Wennerberg, K., Rossman, K. L. & Der, C. J. The Ras superfamily at a glance. J. Cell Sci. 118, 843–846 (2005).
Hobbs, G. A., Der, C. J. & Rossman, K. L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 129, 1287–1292 (2016).
Stephen, A. G., Esposito, D., Bagni, R. K. & McCormick, F. Dragging Ras back in the ring. Cancer Cell 25, 272–281 (2014).
Brose, M. S. et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 62, 6997–7000 (2002).
Prior, I. A., Lewis, P. D. & Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 (2012).
Papke, B. & Der, C. J. Drugging RAS: know the enemy. Science 355, 1158–1163 (2017).
Kelly, M. R. et al. Combined proteomic and genetic interaction mapping reveals new RAS effector pathways and susceptibilities. Cancer Discov. 10, 1950–1967 (2020).
Broyde, J. et al. Oncoprotein-specific molecular interaction maps (SigMaps) for cancer network analyses. Nat. Biotechnol. 39, 215–224 (2021).
Zhou, Y. & Hancock, J. F. Deciphering lipid codes: K‐Ras as a paradigm. Traffic 19, 157–165 (2018).
Wittinghofer, A. & Pal, E. F. The structure of Ras protein: a model for a universal molecular switch. Trends Biochem. Sci. 16, 382–387 (1991).
Omerovic, J., Hammond, D. E., Clague, M. J. & Prior, I. A. Ras isoform abundance and signalling in human cancer cell lines. Oncogene 27, 2754–2762 (2008).
Han, K. et al. CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 580, 136–141 (2020).
Li, Y.-C. et al. Analysis of RAS protein interactions in living cells reveals a mechanism for pan-RAS depletion by membrane-targeted RAS binders. Proc. Natl Acad. Sci. USA 117, 12121–12130 (2020).
Hingorani, S. R. et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003).
Cai, H. et al. A functional taxonomy of tumor suppression in oncogenic KRAS-driven lung cancer. Cancer Discov. 11, 1754–1773 (2021).
Rogers, Z. N. et al. Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat. Genet. 50, 483–486 (2018).
Rogers, Z. N. et al. A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat. Methods 14, 737–742 (2017).
Chuang, C.-H. et al. Molecular definition of a metastatic lung cancer state reveals a targetable CD109–Janus kinase–Stat axis. Nat. Med. 23, 291–300 (2017).
Ruiz, S., Santos, E. & Bustelo, X. R. RasGRF2, a guanosine nucleotide exchange factor for Ras GTPases, participates in T-cell signaling responses. Mol. Cell. Biol. 27, 8127–8142 (2007).
Brandt, A. C., Koehn, O. J. & Williams, C. L. SmgGDS: an emerging master regulator of prenylation and trafficking by small GTPases in the Ras and Rho families. Front. Mol. Biosci. 8, 542 (2021).
Rowell, C. A., Kowalczyk, J. J., Lewis, M. D. & Garcia, A. M. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J. Biol. Chem. 272, 14093–14097 (1997).
Zhang, F. L. et al. Characterization of Ha-ras, N-ras, Ki-Ras4A, and Ki-Ras4B as in vitro substrates for farnesyl protein transferase and geranylgeranyl protein transferase type I. J. Biol. Chem. 272, 10232–10239 (1997).
Takaya, A. et al. R-Ras regulates exocytosis by Rgl2/Rlf-mediated activation of RalA on endosomes. Mol. Biol. Cell 18, 1850–1860 (2007).
Marais, R., Light, Y., Paterson, H. & Marshall, C. Ras recruits Raf‐1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 14, 3136–3145 (1995).
Campbell, J. D. et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 48, 607–616 (2016).
Sánchez-Rivera, F. J. et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431 (2014).
Kohl, N. E. et al. Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor. Science 260, 1934–1937 (1993).
Rowinsky, E. K., Windle, J. J. & Von Hoff, D. D. Ras protein farnesyltransferase: a strategic target for anticancer therapeutic development. J. Clin. Oncol. 17, 3631–3652 (1999).
Collisson, E. et al. Comprehensive molecular profiling of lung adenocarcinoma: The Cancer Genome Atlas Research Network. Nature 511, 543–550 (2014).
Feldser, D. M. et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 468, 572–575 (2010).
Murray, C. W. et al. An LKB1–SIK axis suppresses lung tumor growth and controls differentiation. Cancer Discov. 9, 1590–1605 (2019).
Tsherniak, A. et al. Defining a cancer dependency map. Cell 170, 564–576.e16 (2017).
Staffas, A., Karlsson, C., Persson, M., Palmqvist, L. & Bergo, M. Wild-type KRAS inhibits oncogenic KRAS-induced T-ALL in mice. Leukemia 29, 1032–1040 (2015).
Ambrogio, C. et al. KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell 172, 857–868.e15 (2018).
Kong, G. et al. Loss of wild-type Kras promotes activation of all Ras isoforms in oncogenic Kras-induced leukemogenesis. Leukemia 30, 1542–1551 (2016).
Burgess, M. R. et al. KRAS allelic imbalance enhances fitness and modulates MAP kinase dependence in cancer. Cell 168, 817–829.e15 (2017).
Young, A., Lou, D. & McCormick, F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 3, 112–123 (2013).
Grabocka, E. et al. Wild-type H-and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell 25, 243–256 (2014).
Zhou, Y. et al. Signal integration by lipid-mediated spatial cross talk between Ras nanoclusters. Mol. Cell. Biol. 34, 862–876 (2014).
Zhou, Y. & Hancock, J. F. Ras nanoclusters: versatile lipid-based signaling platforms. Biochim. Biophys. Acta Mol. Cell Res. 1853, 841–849 (2015).
Henis, Y. I., Hancock, J. F. & Prior, I. A. Ras acylation, compartmentalization and signaling nanoclusters. Mol. Membr. Biol. 26, 80–92 (2009).
Inouye, K., Mizutani, S., Koide, H. & Kaziro, Y. Formation of the Ras dimer is essential for Raf-1 activation. J. Biol. Chem. 275, 3737–3740 (2000).
Muratcioglu, S. et al. GTP-dependent K-Ras dimerization. Structure 23, 1325–1335 (2015).
Lin, W.-C. et al. H-Ras forms dimers on membrane surfaces via a protein–protein interface. Proc. Natl Acad. Sci. USA 111, 2996–3001 (2014).
AACR Project Genie Consortium. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 7, 818–831 (2017).
Terrell, E. M. et al. Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol. Cell 76, 872–884.e5 (2019).
Dankort, D. et al. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 21, 379–384 (2007).
Dietrich, P. et al. Neuroblastoma RAS viral oncogene homolog (NRAS) is a novel prognostic marker and contributes to sorafenib resistance in hepatocellular carcinoma. Neoplasia 21, 257–268 (2019).
Weyandt, J. D. et al. Wild-type Hras suppresses the earliest stages of tumorigenesis in a genetically engineered mouse model of pancreatic cancer. PLoS ONE 10, e0140253 (2015).
To, M. D., Rosario, R., Westcott, P. M., Banta, K. L. & Balmain, A. Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene 32, 4028–4033 (2013).
Weyandt, J. D., Carney, J. M., Pavlisko, E. N., Xu, M. & Counter, C. M. Isoform-specific effects of wild-type Ras genes on carcinogen-induced lung tumorigenesis in mice. PLoS ONE 11, e0167205 (2016).
Jeng, H.-H., Taylor, L. J. & Bar-Sagi, D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 3, 1168 (2012).
Miller, M. S. & Miller, L. D. RAS mutations and oncogenesis: not all RAS mutations are created equally. Front. Genet. 2, 100 (2012).
Ding, S. et al. Comparative proteomics reveals strain-specific β-TrCP degradation via rotavirus NSP1 hijacking a host cullin-3-Rbx1 complex. PLOS Pathog. 12, e1005929 (2016).
Xu, Q. & Dunbrack, R. L. ProtCID: a data resource for structural information on protein interactions. Nat. Commun. 11, 711 (2020).
Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031–3048 (2017).
Shapovalov, M. V. & Dunbrack, R. L. Jr A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19, 844–858 (2011).
Acknowledgements
We thank the Stanford Veterinary Animal Care Staff for expert animal care; Human Pathology/Histology Service Center, Stanford Protein and Nucleic Acid Facility for experimental support; and A. Orantes for administrative support; Members of the Winslow laboratory and I. Prior for helpful comments. R.T. was supported by a Tobacco-Related Disease Research Program (TRDRP) postdoctoral fellowship (27FT-0044), a Stanford Cancer Institute fellowship, a Stanford-Rambam Health Care collaboration grant (259348) and an NCI K99/R00 pathway to independence grant (K99CA256039). C.W.M. was supported by the NSF Graduate Research Fellowship Program and an Anne T. and Robert M. Bass Stanford Graduate Fellowship. J.D.H. was supported by a Stanford University School of Medicine Dean’s Postdoctoral Fellowship and a TRDRP Postdoctoral fellowship (T31FT1619). H.C. was supported by a TRDRP Postdoctoral Fellowship (28FT-0019). N.W.H. was supported by the NSF Graduate Research Fellowship Program. M.I.P. was supported by an NIH F30 fellowship (GM142263). Work in the laboratory of R.L.D. was supported by the Fox Chase Cancer Center Support Grant (NIH P30 CA006927). Work in the laboratory of G.M.W. was supported, in part, by Cancer Center Core Grant CA014195, the Breast Cancer Research Foundation, the Freeberg Foundation and the NIH/National Cancer Institute (Grant R35 CA197687). This work was supported by NIH R01-CA230025 (to M.M.W.), NIH R01-CA231253 (to M.M.W. and D.A.P.), NIH R01-CA234349 (to M.M.W. and D.A.P.), TRDRP 27IP-0052 (to M.M.W.), R01-CA250534 (to P.K.J.), R35-GM122517 (to R.L.D.) and in part by the Stanford Cancer institute support grant (NIH P30-CA124435). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
R.T., E.G.S. and M.M.W. conceived the project and designed the experiments. R.T. led experimental data production with contributions from E.G.S., M.K., C.W.M., J.D.H., L.A., M.K.T., N.W.H., M.I.P., H.C., Y.-C.L. and G.M.W. R.T. and E.G.S. led the data analysis. E.G.S. performed the Tuba-seq analysis. M.K. and P.K.J. performed AP/MS data analysis. M.I.P. and R.L.D. performed RAS–RAS dimer modelling. D.A.P. and M.M.W oversaw the project. R.T., E.G.S. and M.M.W. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
M.M.W. and D.A.P. are co-founders of, and hold equity in, D2G Oncology, Inc. The other authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks Mark Philips, Hongbin Ji and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Prioritization of candidate KRAS-interacting proteins for this study.
a. Flow chart for prioritization of candidate KRAS-interacting proteins for this study. Candidate KRAS-interacting proteins were chosen based on multiple criteria including their interaction with KRAS, their homolog mRNA expression in a mouse model of KrasG12D-driven lung cancer, and the consistency with which they bind different RAS-GTPases. RADIL was added at the last step due to its validated importance in KRAS-mutant human cell lines. b. Candidate proteins interact with KRAS in two protein-protein interaction analyses (Kelly, Kostyrko, Han et al. 2020; Broyde, Simpson, Murray et al. 2020). KRAS-interacting proteins are shown as their log10NSAF and SigMap Scores. c. Homolog mRNA expression (TPM) of candidate KRAS-interacting proteins in a mouse model of KrasG12D-driven lung cancer (Chuang et al. 2017). d. Bubble plot of eight AP/MS experiments with GTP- and GDP-locked mutant GTPases as baits (rows), showing the enrichment of selected candidate KRAS-interacting proteins (columns). Dark borders indicate FDR < 0.05. e. Mutation and copy number alteration frequencies of the 13 candidate genes in lung adenocarcinomas with oncogenic KRAS (N = 152; data from TCGA PanCancer Atlas, Cell 2018).
Extended Data Fig. 2 Tumor barcoding coupled with barcode sequencing (Tuba-seq) can uncover engineered alterations that reduce tumor number and growth.
a-b. Schematic of the Tuba-seq approach to measure the effects of essential gene inactivation on tumor growth. Tumors were initiated with pool of barcoded lentiviral-sgRNA/Cre vectors targeting known essential genes and tumor suppressor Apc (Lenti-sgEssential/Cre) in KT and KT;H11LSL-Cas9 mice (a). Tuba-seq was performed on each tumor-bearing lung 12 weeks after initiation (b). c. Points denote tumor sizes at indicated percentiles for each sgRNA relative to the size of sgInert-containing tumors at the corresponding percentiles. Percentiles that are significantly different from sgInert (two-sided FDR-corrected p < 0.05) are in color. d. The impact of each sgRNA on mean tumor size relative to sgInerts, assuming a log-normal distribution of tumor sizes (LNmean). sgRNAs with two-sided FDR-corrected P < 0.05 are in bold. e. Points denote the impact of each sgRNA on tumor burden relative to sgInerts and normalized to the same statistic in KT mice. Relative burdens significantly different from sgInert (two-sided FDR-corrected p < 0.05) are in color. f. Points denote the impact of each sgRNA on tumor number relative to sgInerts and normalized to the same statistic in KT mice. Relative tumor numbers significantly different from sgInert (two-sided FDR-corrected p < 0.05) are in color. g. Points denote the impact of each sgRNA on tumor number plotted against its impact on LNmean tumor size. The lines at y = 1 and x = 1 indicate no effect relative to sgInert on tumor number and size, respectively. For panels c and e-g: Error bars indicate 95% confidence intervals around point estimates of the test statistic. Confidence intervals and P-values were calculated using a nested bootstrap resampling approach across 9 KT;H11LSL-Cas9 mice and 2 KT mice. sgInerts are in gray and the line at y = 1 indicates no effect.
Extended Data Fig. 3 Inactivation of KRAS-interacting proteins has similar impacts on tumor growth in p53-proficient and p53-deficient contexts.
a. Points denote tumor sizes at indicated percentiles for each sgRNA relative to the size of sgInert-containing tumors at the corresponding percentiles in KT mice. Genes are ordered as in Fig. 1f. Line at y = 1 indicates no effect relative to sgInert. As expected, no percentiles were significantly different from sgInert (two-sided FDR-adjusted p < 0.05). b. The impact of each sgRNA on mean tumor size relative to sgInerts in KT;H11LSL-Cas9 mice, assuming a log-normal distribution of tumor sizes (LNmean). sgRNAs with two-sided P < 0.05 after FDR-adjustment are in bold. c-d. Points denote the impact of each sgRNA on tumor burden (c) and number (d) relative to sgInerts in KT;H11LSL-Cas9 mice, normalized to the corresponding statistic in KT mice to account for representation of each sgRNA in the viral pool. sgInerts are in gray and the line at y = 1 indicates no effect. Relative tumor burdens and numbers significantly different from sgInert (two-sided FDR-adjusted p < 0.05) are in color. e. Points denote tumor sizes at the indicated percentiles for each sgRNA relative to the size of sgInert-containing tumors in KT;p53flox/flox;H11LSL-Cas9 mice. Genes are ordered as in Fig. 1f. The line at y = 1 indicates no effect relative to sgInert. Percentiles that are significantly different from sgInert (two-sided FDR-adjusted p < 0.05) are in color. f-h. Comparison of the impact of each sgRNA on relative LNmean tumor size (f), tumor burden (g) and tumor number (h) in KT;H11LSL-Cas9 and KT;p53flox/flox;H11LSL-Cas9 mice. For all panels: Error bars indicate 95% confidence intervals around point estimates of the test statistics. Confidence intervals and P-values were calculated using a nested bootstrap resampling approach described across 11 KT;H11LSL-Cas9 mice, 6 KT;p53flox/flox;H11LSL-Cas9 mice and 5 KT mice.
Extended Data Fig. 4 Top candidate KRAS-interacting proteins from initial Tuba-seq screen impact multiple metrics of tumor growth in validation cohort.
a. Points denote tumor sizes at indicated percentiles for each sgRNA relative to the size of sgInert-containing tumors at the corresponding percentiles in KT mice. KT mice lack Cas9, thus all sgRNAs are functionally equivalent to sgInerts. Genes are ordered as in Fig. 2d, but note the change in axis scaling. Line at y = 1 indicates no effect relative to sgInerts. As expected, no percentiles were significantly different from sgInert (FDR-adjusted p < 0.05). b. The impact of each sgRNA on mean tumor size relative to sgInerts, assuming a log-normal distribution of tumor sizes (LNmean). sgRNAs with two-sided P < 0.05 after FDR-adjustment are in bold. Note that these data for the sgInerts, sgHras#1-3 and sgNras#1-3 are also plotted in Fig. 2e. c. Points denote the impact of each sgRNA on tumor burden relative to sgInerts in KT;H11LSL-Cas9 mice, normalized to the corresponding statistic in KT mice to account for the representation of each sgRNA in the viral pool. sgInerts are in gray and the line at y = 1 indicates no effect. Relative tumor burdens significantly different from sgInert (two-sided FDR-adjusted p < 0.05) are in color. d. Points denote the impact of each sgRNA on tumor number relative to sgInerts in KT;H11LSL-Cas9 mice, normalized to the corresponding statistic in KT mice to account for representation of each sgRNA in the viral pool. sgInerts are in gray and the line at y = 1 indicates no effect. Relative tumor numbers significantly different from sgInert (two-sided FDR-adjusted p < 0.05) are in color. For all panels: Error bars indicate 95% confidence intervals around point estimates of the test statistic. Confidence intervals and P-values were calculated using a nested bootstrap resampling approach across 20 KT;H11LSL-Cas9 mice and 4 KT mice.
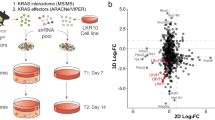
Extended Data Fig. 5 Wild type RAS paralogs constrain the growth of human KRAS-driven cancer cell lines.
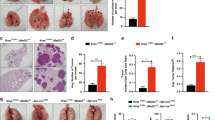
a. RAS family member dependency scores in human lung adenocarcinoma (LUAD) cell lines. b. Effects of RAS gene knockouts in A549 cells. The T-score represents the normalized effect of multiple sgRNAs targeting a gene. A positive T-score indicates a tumor-suppressive effect. The effects of each gene relative to SAFE sgRNAs were tested via Mann–Whitney U test, corrected via Benjamini-Hochberg procedure. (Data source: Kelly, Kostyrko, Han et al. 2020). c. Effects of RAS gene knockouts in KRAS-mutant human LUAD cells in 3D culture. The effects of each gene relative to SAFE sgRNAs were tested via two-sided Benjamini-Hochberg-corrected t-test. (Data source: Han et al. 2020). d. Indel rates in cell lines with the indicated sgRNAs. * denotes sgRNAs used for cell culture and transplantation experiments. e. Re-expression of wild-type HRAS or NRAS suppresses proliferation of HRAS and NRAS double knockout (DKO) HOP62 cells. Cells were seeded in 96-well plates and cultured under limited serum (1%) with or without Doxycycline (Dox). Cell numbers were measured via CCK8 assay. Points are Mean±SD of 16 wells normalized to Day 0. (one-tailed t-test). f. Representative images of subcutaneous tumors 4 weeks after transplantation with H23 cells. Quantification is in Fig. 3h. Scale bar: 2 mm. g. Representative images of Ki67 staining from subcutaneous tumors four weeks after transplantation with H23 cells. Quantification is shown in Fig. 3i. Scale bar: 100 μm. h. Representative images of H&E and human mitochondria staining on lung tumors 4 weeks after intravenous transplantation with H23 cells. Quantification is in Fig. 3j. Scale bar: 500 μm. i. Representative images of Ki67 staining from lung tumors 4 weeks after intravenous transplantation with H23 cells. Quantification is in Fig. 3k. Scale bar: 200 μm.
Extended Data Fig. 6 Wild-type RAS paralogs finetune RAS signaling.
a. Western blot analysis of three RAS paralogs’ expression per 10,000 human and mouse KRAS-driven lung cancer cell lines. Recombinant RAS proteins were used as a standard. b. Western blot analysis of three RAS paralogs’ expression per 20,000 sorted BrafV600E-driven mouse lung cancer cells. Recombinant RAS proteins were used as a standard. c. Quantification of pERKpos cells in KT;H11LSL-Cas9 mice with tumors initiated with Lenti-sgRNA/Cre vectors as indicated in Fig. 4a. Each dot represents a tumor. (one-way ANOVA). d. Quantification of pERKpos cells per field of indicated cells from Fig. 4b. Each dot represents a view field. (one-way ANOVA). SubQ, subcutaneous. e. Western blot analysis of HRAS and NRAS double knockout (DKO) HOP62 cells re-expressing HRAS (TRE-HRAS) or NRAS (TRE-NRAS) under Doxycycline (Dox) treatment. DKO cells were generated as described in Fig. 3a. DKO cells were re-transduced with lentiviral vector expressing TRE-HRAS or TRE-NRAS at high MOI ( > 5) to generate stable re-expressing cells. To re-express HRAS, cells were treated with 10 ng/ml Dox. To re-express NRAS, cells were treated with 50 ng/ml Dox. All cells were cultured under limited serum (1%) for 2 days before protein extraction. HSP90 is blotted as loading control.
Extended Data Fig. 7 HRAS and NRAS directly interact with KRASG12D.
a. Co-immunoprecipitation of HRAS (HA-tagged) and NRAS (Flag-tagged) with KRASG12D (Myc-tagged), imaged by western blotting. 293T cells were co-transfected with Myc-KRASG12D, HA-HRAS, and Flag-NRAS for 24 hours before co-immunoprecipitation. b. Co-immunoprecipitation of HRAS (HA-tagged) with truncated (aa73-aa165) or full length KRASG12D (Myc-tagged), imaged by western blotting. 293T cells were co-transfected with Myc-KRASG12D and HA-HRAS for 24 hrs before co-immunoprecipitation. c. Diagram of the modified ReBiL2.0 system to assess direct KRASG12D-HRAS/NRAS interaction. d. HRAS and NRAS can directly interact with KRASG12D. 293 T cells expressing indicated cLuc- and nLuc- luciferase were cultured in limited serum (1%) for 24 hours and ReBiL2.0 assay was performed. Points are Mean±SD ReBiL2.0 score of 12 wells normalized to cells expressing free luciferase (cLuc-HA/nLuc-HA). (one-way ANOVA). e. Luciferase protein expression in c, imaged by western blotting for the HA-tag. α-Tubulin is loading control. f. Full data from experiment shown in Fig. 5c. 293T cells expressing nLuc-KRASG12D/cluc-KRASG12D or nLuc-C20/cluc-C20 with indicated Myc-tagged RAS-GTPases were cultured in limited serum (1%) for 24 hours and ReBiL2.0 assays were performed. Points are Mean±SD ReBiL2.0 score of 12 wells normalized to cells transduced with empty vector. ns: not significant (one-way ANOVA). g. RAS-GTPases protein expression in e, imaged by western blotting for the Myc-tag. HSP90 is loading control. h. Relative strength of RAS-GTPases in disrupting KRASG12D-KRASG12D interactions in f. Differences in ReBiL2.0 score between empty vector and indicated RAS-GTPases were normalized by their own protein expression via western blotting for the Myc-tag. (one-way ANOVA). i. RAP1A interacts with KRASG12D in two protein-protein interaction analyses (Kelly, Kostyrko, Han et al. 2020; Broyde, Simpson, Murray et al. 2020). j. Co-immunoprecipitation of BRAF with KRASG12D (Myc-tagged), imaged by western blotting. 293T cells were co-transfected with Myc-KRASG12D, BRAF, and with or without HA-HRAS for 24 hours before co-immunoprecipitation. Overexpression (OvE) of HRAS suppressed BRAF co-immunoprecipitation with KRASG12D.
Extended Data Fig. 8 Identification and analysis of rare RAS mutations in oncogenic KRAS-mutant tumors.
a-b. Pan-cancer frequency of HRAS (a) or NRAS (b) mutations in patients from Project GENIE. Mutations that are intergenic, intronic, silent, or in the 3’ or 5’ UTR were excluded. Oncogenic KRAS mutants were defined as tumors having missense mutations in codons 12, 13 or 61. Known oncogenic HRAS (a) or NRAS (b) mutations are highlighted. The dashed line indicates equal mutation frequency in samples with wild-type and mutant KRAS. Non-oncogenic mutations occurring at least once in patients with oncogenic KRAS mutations are annotated. Mutants selected for analysis of ability to disrupt KRASG12D-KRASG12D interactions are in bold. c-d. Characteristics of samples with rare HRAS (c) or NRAS (d) mutations selected for analysis of ability to disrupt KRASG12D-KRASG12D interactions using the ReBiL2.0 system. e. Identification of RAS-RAS interaction-deficient NRAS mutation. 293 T (nLuc-KRASG12D/cluc-KRASG12D) cells expressing wild-type or NRAS mutants were cultured in limited serum (1%) for 24 hours. Points are Mean±SD ReBiL2.0 score of 12 wells normalized to cells transfected with empty vector. ns: not significant. (one-way ANOVA). f. NRAS (wild-type and mutant) protein expression levels in a shown by anti-Myc tag western blot. HSP90 is loading control. g. Western blot of cultured NRAS-null HOP62 cells (HOP62-Cas9-sgNRAS) re-expressing sgRNA-resistant wild-type NRAS or NRASR102Q under Dox treatment. Cells were cultured under limited serum (1%) with or without Dox for 2 days before protein extraction. Re-expression of NRASR102Q had no effect on ERK phosphorylation. GAPDH is loading control. h. Proliferation of cultured NRAS-null HOP62 cells (HOP62-Cas9-sgNRAS) expressing sgRNA-resistant wild-type NRAS or NRASR102Q under Dox treatment. Cells were cultured in limited serum (1%) with or without Dox for 4 days. Cell viability was measured via CCK8 assay and normalized to cells treated with vehicle. Re-expression of NRASR102Q had no effect on cell proliferation. Points are Mean±SD of 10 wells. ns: not significant (one-tailed t-test).
Extended Data Fig. 9 Prediction of RAS-RAS dimer interfaces.
a. Homodimers of RAS present in crystals of HRAS, KRAS, and NRAS in the Protein Data Bank. Dimers were downloaded from the Protein Common Interface Database (ProtCID)58, which clusters interfaces present in different crystals of homologous proteins. The α4-α5 dimer shown is present in 84 entries of HRAS, 13 entries of KRAS, and one entry of NRAS (PDB 5UHV). b. Models of a homodimer of KRASG12D and heterodimers of KRASG12D with HRAS, HRAST50M, and HRASR123C. The α4-α5 HRAS dimer from PDB entry 3K8Y was used as a template. KRASG12D from PDB entry 5USJ was superposed with the program PyMol on one or both monomers of 3K8Y to form the heterodimers and the homodimer respectively. Residues T50 and R123 were mutated with PyMol. R123 is involved in an intrachain salt bridge with residue E143, which also participates in the RAS-RAS interface. Mutation to cysteine results in an uncompensated charge on E143, which may destabilize the RAS-RAS interaction. All four structures were relaxed with the program Rosetta using the FastRelax protocol with the Ref2015 scoring function59. Rosetta uses the backbone-dependent rotamer library of Shapovalov and Dunbrack to repack side chains around the mutated sites60. The resulting energies were: KRASG12D-KRASG12D, -1122.8 kcal/mol; HRAS-KRASG12D, -1144.8 kcal/mol; HRAST50M-KRASG12D, -1135.5 kcal/mol; HRASR123C-KRASG12D, -1130.9 kcal/mol. Residues T50 (magenta) and R123 (orange) are indicated in sticks.
Extended Data Fig. 10 Paired screen in KRAS-driven and BRAF-driven lung cancer models validates HRAS and NRAS as KRAS-specific tumor suppressors.
a-c. Points denote tumor sizes at indicated percentiles for each sgRNA relative to the size of sgInert-containing tumors at the corresponding percentiles in KT;H11LSL-Cas9/+ (a), BrafT;H11LSL-Cas9/+ (b) and KT mice (c). Genes are ordered by 95th percentile tumor size in KT;H11LSL-Cas9/+ mice, with sgInerts on the left. Percentiles that are significantly different from sgInert (two-sided FDR-adjusted p < 0.05) are in color. The negative effects of sgRNAs targeting Fnta and Nme2 in the KT mice (c) are unexpected and indicate a potential bias in the size distributions of tumors with these genotypes. We note that the same bias may be present in the KT;H11LSL-Cas9/+ and BrafT;H11LSL-Cas9/+ data; however, previous experiments showed consistent negative effects on tumor size for these sgRNAs, suggesting that the observed effects in this KT;H11LSL-Cas9/+ cohort are not solely the product of this bias. d. Points denote the impact of each sgRNA on tumor burden relative to sgInerts in KT;H11LSL-Cas9/+ and BrafT;H11LSL-Cas9/+ mice, normalized to the corresponding statistic in KT mice to account for representation of each sgRNA in the viral pool. Relative tumor burdens significantly different from sgInert (two-sided FDR-adjusted p < 0.05) are in color. e. Points denote the impact of each sgRNA on tumor number relative to sgInerts in KT;H11LSL-Cas9/+ and BrafT;H11LSL-Cas9/+ mice, normalized to the corresponding statistic in KT mice to account for representation of each sgRNA in the viral pool. Relative tumor numbers significantly different from sgInert (two-sided FDR-adjusted p < 0.05) are in color. For all panels: Error bars indicate 95% confidence intervals around point estimates of the test statistic. sgInerts are in gray and the line at y = 1 indicates no effect relative to sgInerts. Confidence intervals and P-values were calculated using the nested bootstrap resampling approach described in the Methods across 11 KT;H11LSL-Cas9/+ mice, 14 BrafT;H11LSL-Cas9/+ mice and 10 KT mice.
Supplementary information
Supplementary Tables
Supplementary Table 1. Plasmids used in this study. Supplementary Table 2. sgRNA sequence used in this study. The vectors used for sgRNA expression in each experiment are listed in Supplementary Table 1. Supplementary Table 3. Summary statistics for KrasIP_Essential Tuba-seq experiment. Part 1: relative tumour size; part 2: relative tumour number; part 3: relative tumour burden. Supplementary Table 4. Summary statistics for KrasIP_InititalScreen Tuba-seq experiment. Part 1: relative tumour size; part 2: relative tumour number; part 3: relative tumour burden. Supplementary Table 5. Summary statistics for KrasIP_Validation Tuba-seq experiment. Part 1: relative tumour size; part 2: relative tumour number; part 3: relative tumour burden. Supplementary Table 6. Summary statistics for KrasIP_MultiGEMM Tuba-seq experiment. Part 1: relative tumour size; part 2: relative tumour number; part 3: relative tumour burden.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 5
Unprocessed western blots.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 7
Unprocessed western blots.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 8
Unprocessed western blots.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tang, R., Shuldiner, E.G., Kelly, M. et al. Multiplexed screens identify RAS paralogues HRAS and NRAS as suppressors of KRAS-driven lung cancer growth. Nat Cell Biol 25, 159–169 (2023). https://doi.org/10.1038/s41556-022-01049-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41556-022-01049-w
